

Abstract
Gorham-Stout disease (GSD) is a rare condition in which spontaneous, progressive resorption of bone occurs. There are no previous reports of patients with fatal progression of GSD with skull base osteomyelitis (SBO) and lateral medullary syndrome (LMS). We present the case of a 27-year-old man diagnosed with GSD with involvement of the maxillofacial bones and skull base. The patient developed SBO; LMS resulted from progressive osteolysis, and the patient died of associated brainstem stroke. Careful follow-up with special emphasis on the early detection of intracranial complications is critical in patients presenting with progressive GSD with involvement of the skull base.
Keywords: denosumab, sirolimus, sphenoiditis, Wallenberg syndrome, lateral medullary syndrome
Introduction
Gorham-Stout disease (GSD) is a form of lymphangiomatosis characterized by diffuse lymphatic vessel proliferation accompanied by progressive osteolysis. The symptoms of GSD are variable; lesions can arise in the skull, maxillofacial skeleton, arms, legs, pelvis, spine, and ribs (1). Mortality depends on the area affected. Patients with GSD of the maxillofacial bones are at a particularly high risk of death because of the possibility of disease progression to the brain and spine (2). Kim et al. reviewed 64 reports of GSD in the maxillofacial region among articles published from 1928 through 2015. In that review, there were 7 cases of disease-specific death, resulting in a mortality rate of 10.94% (3). Four patients died of lung-related problems, two of spinal involvement, and one of intracranial extension. The patient with intracranial extension suffered from osteolytic lesions of the parietal bone, zygoma, ramus, and vertebra (4). However, there are no previous reports of patients with GSD who died of cerebral infarction associated with skull base and maxillofacial lesions.
While standard treatments for GSD are lacking, previous reports have described treatment with surgery, radiation therapy, and medications (bisphosphonates, thalidomide, and interferon) (1). Recently, the efficacy of sirolimus for various vascular anomalies including GSD has been reported, and clinical trials of sirolimus are ongoing (5).
We herein report the case of a patient with GSD involving the skull base and maxillofacial bones who presented with brainstem infarction secondary to rapid osteolysis.
Case Report
A 27-year-old man presented with painless mobility of the right maxillary incisors. He reported no history of trauma. A physical examination revealed no signs of local inflammation or systemic disease. All laboratory values were normal. Computed tomography (CT) demonstrated osteolysis of the right maxilla. A histological study of a maxillary biopsy specimen revealed the presence of abundant fibrous tissue. Six years later, the maxilla was rebiopsied because of progressive disease. Histopathology showed resorption of typical bone structures, which had been replaced with thin-walled endothelium-lined capillaries of vascular or lymphatic origin. D2-40 immunostaining delineated the endothelium of the lymphatic channels, so a diagnosis of GSD was made.
The patient was treated with localized radiotherapy (40 Gy) and bisphosphonates. However, CT revealed osteolysis spreading from the right maxilla to the frontal bone, the temporal bone, and the zygomatic bone (Fig. 1). Extraosseous soft-tissue masses compressed the right facial nerve and trigeminal nerve, leading to facial paresis and pain. One year later, the patient was transferred to Gifu University Hospital for alternative treatment. We decided to treat him with an mTOR inhibitor, sirolimus. This treatment was approved by the review board at Gifu University Hospital. Written informed consent was obtained from the patient. Sirolimus treatment (2 mg/day) was started. Six months later, the patient was hospitalized for the control of sharp pain resulting from nerve damage.
Figure 1.
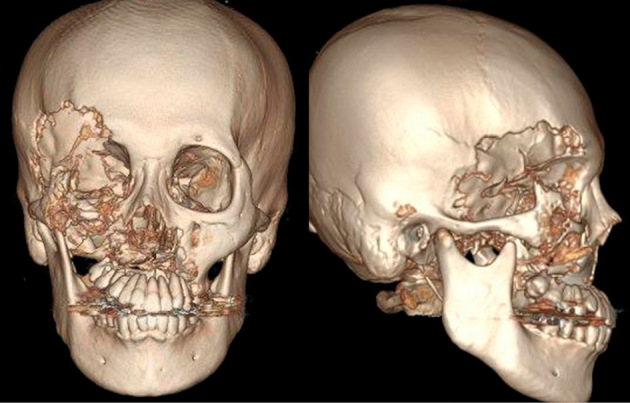
Three-dimensional volume rendering images of computed tomography shows massive osteolysis of the right maxillofacial region in the maxilla, the frontal bone, the temporal bone, and the zygomatic bone.
The patient received a steroid and morphine for pain control. We initiated treatment with denosumab (120 mg, subcutaneous injection) in addition to sirolimus to halt the progression of the osteolysis. However, the osteolysis progressed and extended to the skull base. Axial CT one month after hospitalization showed progression of massive osteolysis in the right central skull base (Fig. 2). Magnetic resonance imaging (MRI) of the skull base after one month of hospitalization revealed extraosseous soft-tissue masses filling the sphenoid sinus (Fig. 3A). Nasal endoscopy revealed a connection between the sphenoid sinus and the oral cavity. Biopsy from the soft-tissue mass was negative for neoplasia.
Figure 2.

Axial CT image showing progression of bone destruction in the skull base. (A) At the time of hospitalization, there is indistinct osteolysis in the skull base. (B) One month after hospitalization, axial CT shows progression of massive osteolysis in the right central skull base.
Figure 3.

(A) Axial T1-weighted image after 1 month of hospitalization reveals extraosseous soft-tissue masses filling the sphenoid sinus. (B) Axial fat-suppressed contrast-enhanced T1-weighted image shows moderately enhanced bone marrow of the skull base and extraosseous soft-tissue masses.
The patient complained of sudden vertigo, dysphagia, and left-sided paresthesia after six weeks of hospitalization. Neurological examinations revealed both tactile and thermal/pain hyperesthesia on the right side of his face, and thermal/pain hyperesthesia in his left limbs. Horner’s syndrome on the right side and limb ataxia in the left upper and lower limbs were evident. Subsequently performed diffusion-weighted images demonstrated a hyperintense area in the right lateral medulla, consistent with an acute infarct (Fig. 4A). Magnetic resonance angiography failed to identify any arterial dissection. Coagulation workup and echocardiograms were all normal. The patient was diagnosed with a brainstem stroke with lateral medullary syndrome (LMS) and was started on aspirin.
Figure 4.

(A) Diffusion-weighted image after the onset of lateral medullary syndrome (LMS) shows a hyperintense area in the right lateral medulla. (B) A few days after the onset of LMS, fluid attenuation inversion recovery image shows hyperintense areas in the right temporal lobe, the pons, and the bilateral middle cerebellar peduncles. The hyperintense white matter of the right temporal lobe is caused by the pressure of the extraosseous soft-tissue masses.
Contrast-enhanced T1-weighted MRI demonstrated moderately enhanced bone marrow of the skull base and extraosseous soft-tissue masses (Fig. 3B). Laboratory data indicated high levels of inflammation, with an elevated white blood cell count (14,000/μL) and C-reactive protein level (8.72 mg/dL). Microbiological testing of a nasopharyngeal swab revealed Pseudomonas aeruginosa. Therefore, skull base osteomyelitis (SBO) was diagnosed. Antibiotic therapy was initiated empirically with meropenem. A few days after the onset of LMS, disturbance of consciousness progressed. Fluid-attenuated inversion recovery imaging demonstrated hyperintense areas in the right temporal lobe, the pons, and the bilateral middle cerebellar peduncles (Fig. 4B). The treatments were not effective; the patient’s condition progressively worsened, and he died after several weeks.
Discussion
We herein report a case of GSD with involvement of the maxillofacial bones and skull base. Although GSD often progresses slowly and insidiously, this patient’s osteolysis progressed more rapidly than we expected. The osteolytic lesion eventually contributed to his death. The tissue of the osteolytic skull base lesion was observed to change into densely structured, fibrous connective tissue. The replacement of bone by connective tissue is the characteristic finding in GSD, which makes the diagnosis difficult based on histological examinations (1,3). Furthermore, the soft-tissue masses were fragile, and the sphenoid sinus was connected to the oral cavity in our patient, which increased susceptibility to bacterial infection via the oral cavity.
In our patient, extensive osteolysis of the skull base caused sphenoiditis with both SBO and LMS. SBO was diagnosed based on the following findings: laboratory results showing high levels of inflammation, the presence of Pseudomonas aeruginosa in the nasopharynx, multiple cranial nerve palsies that appeared later, and abnormal bone marrow signal intensities on MRI (6). Ng et al. described a case in which a 10-year-old immunocompetent patient developed LMS secondary to SBO arising from sphenoiditis (7). SBO is rarely caused by paranasal infections (8). However, it is difficult to diagnosis the disease properly because the only clinical symptom may be headache, with cranial neuropathies occurring later. In our patient, MRI clearly revealed the abnormal bone marrow of the skull base with hyperintensity on fat-suppressed contrast-enhanced T1-weighted images, which was highly suggestive of SBO. If the diagnosis is delayed, this condition has high morbidity and mortality. Therefore, patients with GSD who have osteolytic lesions of the skull base should be monitored closely for any signs of SBO.
LMS, also known as Wallenberg syndrome, is a vascular syndrome occurring in the posterior circulation of the brainstem. Although the precise pathogenesis of LMS secondary to SBO is unknown, it likely results from direct extension of SBO, with thrombophlebitis causing vascular occlusion and lateral medullary infarct, rather than a thromboembolic event, as is usually seen in LMS (7). Our patient had neurological symptoms before the diagnosis of SBO. He required prolonged antibiotic therapy for SBO but eventually experienced fatal LMS. We assumed that vascular occlusion from thrombophlebitis had already occurred when the skull base became inflamed. Therefore, the early diagnosis and treatment are important to decrease the mortality and morbidity of these conditions.
In the present case, denosumab and sirolimus were prescribed. However, these drugs were ineffective. Some studies suggest that osteoclast progenitor cells in GSD patients may be more sensitive to osteoclast-inducing factors and receptor activator of nuclear factor κ-B ligand (RANKL) than control cells (9). The inhibition of RANKL with denosumab might be useful in these patients; however, there are no reports of denosumab treatment for GSD. Recently, several studies have reported the efficacy of sirolimus, a mammalian target of rapamycin inhibitor, against complex vascular anomalies (5,10). Adams et al. reported the efficacy of sirolimus for complex vascular anomalies in a phase 2 clinical trial. In that trial, all three GSD patients had a partial response at the end of a six-month course. However, the case number is small at present, so further studies are needed to elucidate the efficacy of sirolimus for GSD. In the present case, these drugs were unable to prevent the progression of osteolysis. Sirolimus acts as an anti-angiogenic drug; however, the drug does not inhibit osteolysis or cause new bone formation. Therefore, it is difficult to control the progression of osteolysis once fatal lesions arise. Aggressive GSD, as in the present case, should be treated in the early stages of the disease. It is currently difficult to detect aggressive GSD at an early stage and therefore difficult to administer treatment early in the course of the disease. Several studies have examined whether lymphangiogenic and osteoclastogenic factors can serve as biomarkers of activity of GSD. These studies have shown that VEGF-A and IL-6 concentrations can be high in patients with active GSD and that their concentrations can fall following treatment with various therapies (1). Thus, these biomarkers may enable the early diagnosis of aggressive GSD.
To our knowledge, this is the first case report of GSD in a patient with SBO and LMS. Osteolysis of the skull base and maxillofacial region are alarming GSD lesions. Careful follow-up with special emphasis on the early detection of intracranial complications is critical.
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional guidelines on human experimentation (Akifumi Nozawa, Michio Ozeki, Tomohiro Hori, Hiroki Kato, Naoyuki Ohe, Toshiyuki Fukao) and with the Helsinki Declaration of 1975, as revised in 2008.
The patient has consented to the submission of the case report for submission to the journal.
The authors state that they have no Conflict of Interest (COI).
Financial Support
The present study was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (25461587); a Health and Labour Science Research Grant for Research on Intractable Diseases from the Ministry of Health, Labour and Welfare of Japan received by M.O.; and Practical Research Project for Rare/Intractable Diseases from Japan’s Agency for Medical Research and Development, AMED (17lk0201055h0002).
Acknowledgments
The authors thank Drs. Norio Kawamoto, Takeshi Kimura, Kaori Kanda, Shiho Yasue, and Yoriko Matsuzawa from Gifu University, and Dr. Masataka Takahashi from the University of Tokyo Hospital.
References
- 1. Dellinger MT, Garg N, Olsen BR. Viewpoints on vessels and vanishing bones in Gorham-Stout disease. Bone 63: 47-52, 2014. [DOI] [PubMed] [Google Scholar]
- 2. Perschbacher S, Perschbacher K, Pharoah M, Bradley G, Lee L, Yu E. Gorham's disease of the maxilla: a case report. Dentomaxillofac Radiol 39: 119-123, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim MK, Hong JR, Kim SG, Lee SK. Fatal progression of Gorham disease: a case report and review of the literature. J Oral Maxillofac Surg 73: 2352-2360, 2015. [DOI] [PubMed] [Google Scholar]
- 4. Lee S, Finn L, Sze RW, Perkins JA, Sie KC. Gorham Stout syndrome (disappearing bone disease): two additional case reports and a review of the literature. Arch Otolaryngol Head Neck Surg 129: 1340-1343, 2003. [DOI] [PubMed] [Google Scholar]
- 5. Adams DM, Trenor CC 3rd, Hammill AM, et al. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics 137: e20153257, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chandler JR, Grobman L, Quencer R, Serafini A. Osteomyelitis of the base of the skull. Laryngoscope 96: 245-251, 1986. [DOI] [PubMed] [Google Scholar]
- 7. Ng J, Connolly DJ, Rittey CD, Mordekar SR. Skull base osteomyelitis leading to lateral medullary syndrome in a child. Eur J Paediatr Neurol 11: 111-114, 2007. [DOI] [PubMed] [Google Scholar]
- 8. Grobman LR, Ganz W, Casiano R, Goldberg S. Atypical osteomyelitis of the skull base. Laryngoscope 99: 671-676, 1986. [DOI] [PubMed] [Google Scholar]
- 9. Hirayama T, Sabokbar A, Itonaga I, Watt-Smith S, Athanasou NA. Cellular and humoral mechanisms of osteoclast formation and bone resorption in Gorham-Stout disease. J Pathol 195: 624-630, 2001. [DOI] [PubMed] [Google Scholar]
- 10. Nozawa A, Ozeki M, Kuze B, Asano T, Matsuoka K, Fukao T. Gorham-Stout disease of the skull base with hearing loss: dramatic recovery and antiangiogenic therapy. Pediatr Blood Cancer 63: 931-934, 2016. [DOI] [PubMed] [Google Scholar]