

Abstract
Chemotherapeutic agents generally suffer from off-target cytotoxicity in noncancerous cell types, leading to undesired side effects. As a result, significant effort has been put into identifying compounds that are selective for cancerous over noncancerous cell types. Our laboratory has recently developed a series of near-infrared (NIR) fluorophores containing a phosphinate functionality at the bridging position of a xanthene scaffold, termed Nebraska Red (NR) fluorophores. Herein, we report the selective cytotoxicity of one NR derivative, NR744, against HeLa (cervical cancer) cells versus NIH−3T3 (noncancerous fibroblast) cells. Mechanistic studies based on the NIR fluorescence signal of NR744 showed distinct subcellular localization in HeLa (mitochondrial) versus NIH−3T3 (lysosomal) that resulted from the elevated mitochondrial potential in HeLa cells. This study provides a new, NIR scaffold for the further development of reagents for targeted cancer therapy.
Keywords: apoptosis, cancer, cytotoxicity, fluorescence probes, phosphinate
Graphical Abstract
On target:
A near-infrared fluorophore can selectively induce cell death in cancer cells. The induction of apoptotic markers as well as the inhibition of migration is demonstrated. This scaffold could be used for the further development of reagents for targeted cancer therapy.
Introduction
Cancer is the second leading cause of death in the USA, with nearly one in every four deaths being caused by this disease.[1] Indeed, the US Food and Drug Administration (FDA) has approved more than 200 anticancer drugs over the past few decades. However, one factor that continues to complicate treatment is the off-target toxicity observed for commonly used chemotherapeutic drugs.[2] Recently, drug-discovery campaigns have turned towards the development of targeted molecules with tissue-specific activities.[3] Such compounds could potentially be used at higher dosages with less off-target effects. At the same time, significant attention has been focused on the development of NIR fluorophores for applications in in vivo imaging and image-guided surgery.[4] These NIR probes circumvent issues associated with the light scattering and endogenous background fluorescence of biological species. For example, two FDA-approved NIR contrast agents, indocyanine green (ICG) and methylene blue (MB), have been extensively employed to visualize tumors.[5] However, neither of these dyes shows significant toxicity to the labeled cells. Furthermore, although some chemotherapeutic drugs exhibit weak or moderate fluorescence under illumination in the visible region of the spectrum,[6] none displays NIR absorption and emission. Thus, we sought to combine the benefits of selective toxicity and NIR imaging into one molecular scaffold.
One factor that can be used to discriminate between cancerous tissue and surrounding normal tissue is altered metabolic function, known as the Warburg effect.[7] Corresponding increases in mitochondrial membrane potential (MMP, Δm) and the production of reactive oxygen species in cancer cells have been well documented.[8] For example, the mitochondria of cancer cells display an increased MMP of ≈ 60 mV compared to normal epithelial cells.[9] These factors led to the discovery that delocalized lipophilic cations (DLCs) are selectively taken up into the mitochondria of cancer cells and, in certain cases, elicit a cytotoxic response.[10] Interestingly, the fluorophore known as rhodamine-123 (Rh-123, Scheme 1) is the first reported DLC with selective toxicity to carcinoma cells.[10a,b] Since this observation, several elegant strategies, based on altered MMP, for the delivery of compounds to cancer cells have been described.[11] However, there remains a need to identify smallmolecule fluorophores with NIR fluorescence excitation and emission that display selective toxicity to cancer cells. Such molecules would provide a starting-point for the generation of simplified reagents for both the visualization and potential treatment of tumors.
Scheme 1.
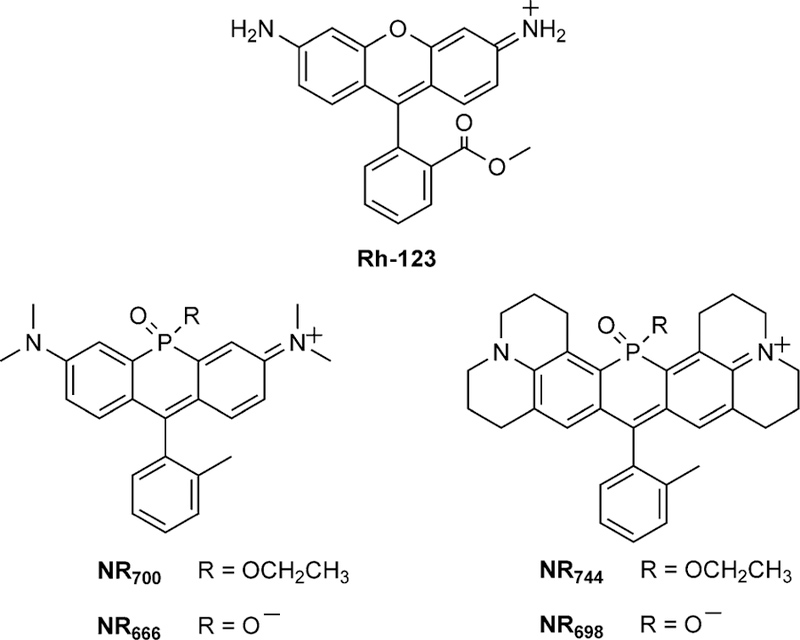
Structures of Rh-123 and NR derivatives.
Our laboratory has recently reported a series of NIR fluorophores based on the incorporation of a phosphinate functionality at the bridging position of rhodamine-based fluorophores (Scheme 1).[12] These fluorophores display bright NIR fluorescence and provide a scaffold for the development of chemical biology tools such as self-reporting delivery reagents[12] or turn-on fluorescent probes.[13] Herein, we report our observation of the selective mitochondrial localization and toxicity of NR744 in cancer cells. As with previously described DLCs,[14] NR744 localization is dependent upon MMP. However, NR744 provides a robust NIR signal that can be used to visualize dye uptake (λex = 744 nm, λem = 764 nm, ε= 8× 104, and Φ= 0.16). In addition, the chemical reactivity of phosphorous allows for the potential, long-term development of improved reagents for selectively imaging and subsequently treating cancer cells.
Results and Discussion
Before constructing imaging agents from NR fluorophores, we sought to fully define the cellular localization and toxicity of our initial NR derivatives by using HeLa cells as a model cell line. During these experiments, we observed that three of the NR derivatives were nontoxic up to 50 µm, whereas NR744displayed clear toxicity at low-micromolar concentrations (Figure S1 in the Supporting Information). The phosphinate-containing compounds (NR666 and NR698) did not cross the cell membrane,[12] whereas the phosphinate ethyl ester-containing derivatives NR700 and NR744 localized to lysosomes (Figure S2) or the mitochondria (Figure 1 A, Pearson’s coefficient R = 0.92), respectively. Comparing NR698 and NR744, we hypothesized that the phosphinate ethyl ester of NR744 facilitates cell uptake. In addition, we have previously shown that the ester group of NR744 is stable to hydrolysis in buffer at pH 7.4,[12] maintaining a net positive charge in the molecule that might contribute to mitochondrial localization. Moreover, more than 80 % of NR744 survived 48 h of incubation in culture medium containing 50 % fetal bovine serum (FBS) at 37 °C (Figure S3). The localization of NR700 to lysosomes indicates that the lipophilicity of NR744 might also play a role in mitochondrial localization.[15] However, it should be noted that the phosphinate ethyl ester of NR700 undergoes spontaneous hydrolysis in aqueous solutions to yield NR666 (with a half-life of 27 min at pH 7.4),[12] potentially altering the net charge of the molecule in cells. To further investigate the mechanism of NR744 localization, HeLa cells were treated with carbonyl cyanide m-chlorophenyl hydrazone (CCCP, a known proton uncoupler) or oligomycin A (an ATP synthase inhibitor). Importantly, CCCP is known to decrease MMP whereas oligomycin A increases it.[16] Thus, if NR744 localization is dependent upon MMP, decreased mitochondrial localization in CCCP-treated cells and increased mitochondrial accumulation in oligomycin A-treated cells would be anticipated. Gratifyingly, a clear decrease in mitochondrial localization was observed in the presence of CCCP, and oligomycin A treatment led to a sharp increase in mitochondrial accumulation of NR744 (Figure 1 B). These data indicate that localization of NR744 in cells with a normal MMP might differ from that observed in HeLa cells. Accordingly, we incubated NR744 with NIH-3T3 cells and observed localization to lysosomes (Figure 1 C, Pearson’s coefficient R = 0.93). Together, these data indicate that NR744 toxicity in HeLa is due to localization to the mitochondria and that the localization pattern of NR744 changes based on the MMP.
Figure 1.
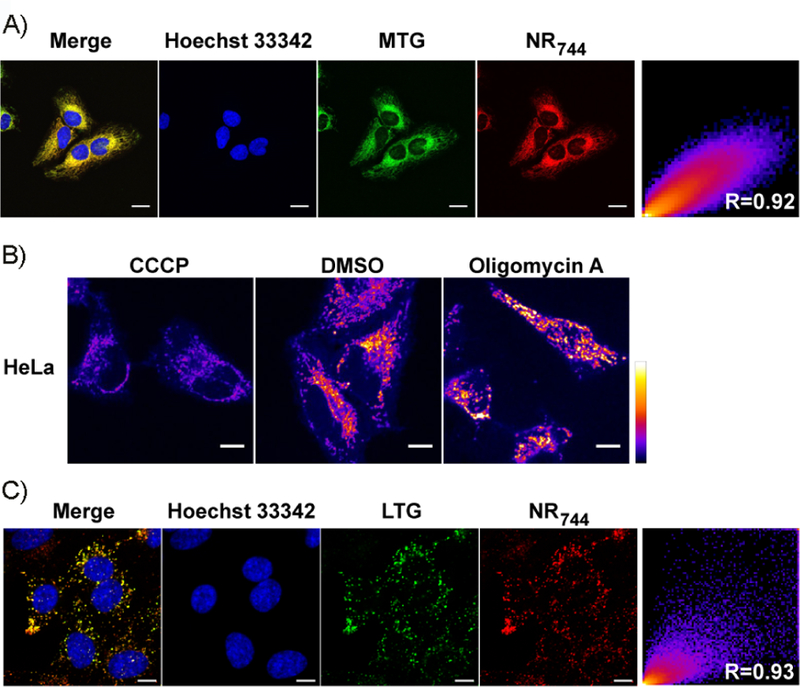
NR744localization. A) Co-localization of NR744and MitoTracker Green (MTG) in HeLa cells. B) NR744 localization in HeLa cells is dependent upon the mitochondrial membrane potential. NR744 (5 µm) and CCCP(1 µm), DMSO, or oligomycin A (2 µgmL−1) were incubated with cells for 30 min. Cells were washed and immediately imaged under identical conditions. C) Co-localization of NR744 with LysoTracker Green (LTG) in NIH-3T3 cells. All scale bars: 10 µm.
We next asked whether the toxicity of NR744 would be altered in cells with different MMPs. At relatively high concentrations (> 25 µm, Figure S4), toxicity in NIH-3T3 cells was observed. However, using lower concentrations of NR744 (5 µm) enabled discrimination between NIH-3T3 (90.3 ± 3.2 % viability) and HeLa cells (57.9 ± 2.2 % viability) after incubation with NR744 for 24 h (Figures S1 and S4). We observed an enhancement in selectivity upon increasing the incubation time of NR744 to 48 h, yielding IC50 values of 453 nm and 12.6 µm, respectively for HeLa and NIH-3T3 cells (Figure 2). Under these conditions, selective cell death in HeLa cells could be induced while NIH-3T3 cells remained viable. Additionally, NR744 was found to be toxic across a panel of cancerous cell lines including MCF-7 (breast), HepG2 (liver), and HCT-116 (colon; Figure S5).
Figure 2.
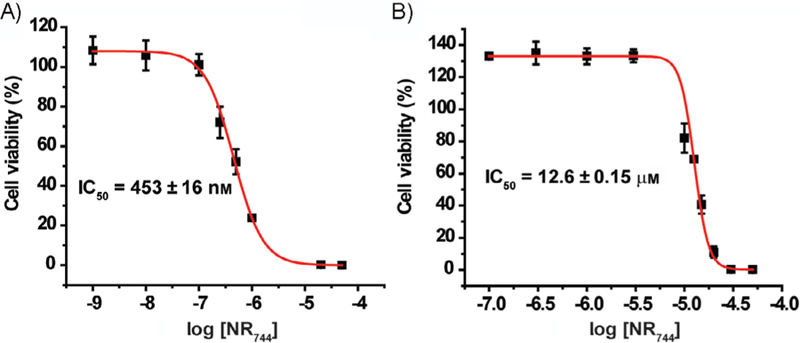
Selective toxicity of NR744. Cells were incubated with NR744 at various concentrations for 48 h. Increased toxicity in A) HeLa versus B) NIH-3T3 cells is observed. Error bars represent the standard deviation of triplicate experiments.
Encouraged by these results, we evaluated NR744 toxicity in a co-culture system containing both HeLa and NIH-3T3 cells. In order to distinguish each cell type, we first labeled pure populations of cells with fluorescent dyes that are retained for ≈ 3– 6 generations. This differential labeling with violet (HeLa) or red (NIH-3T3) markers allowed cell viability to be analyzed by using flow cytometry to probe the well-established apoptotic marker, phosphatidylserine[17] in each cell type within the co-cultures. Cells were labeled with cell-tracking dyes and subsequently co-cultured in the presence or absence of NR744. Each cell type was gated by using its respective cell-tracker dye fluorescence channel, and viability was probed by using an annexin V–Alexa 488 conjugate, to determine the extent of phosphatidylserine present on the cell surface.[18] Importantly, no appreciable difference in phosphatidylserine labeling was observed in cells treated with DMSO (Figures 3 and S6); however, distinct 77 and 270 % increases in phosphatidylserine exposure were observed for HeLa cells when incubated with NR744 (Figures 3 and S6). This difference in the induction of apoptosis could also be replicated in cells that were independently cultured and treated with NR744 (Figure S7). Thus, NR744 is capable of selectively inducing apoptosis in cancerous cells in the presence of noncancerous cells.
Figure 3.
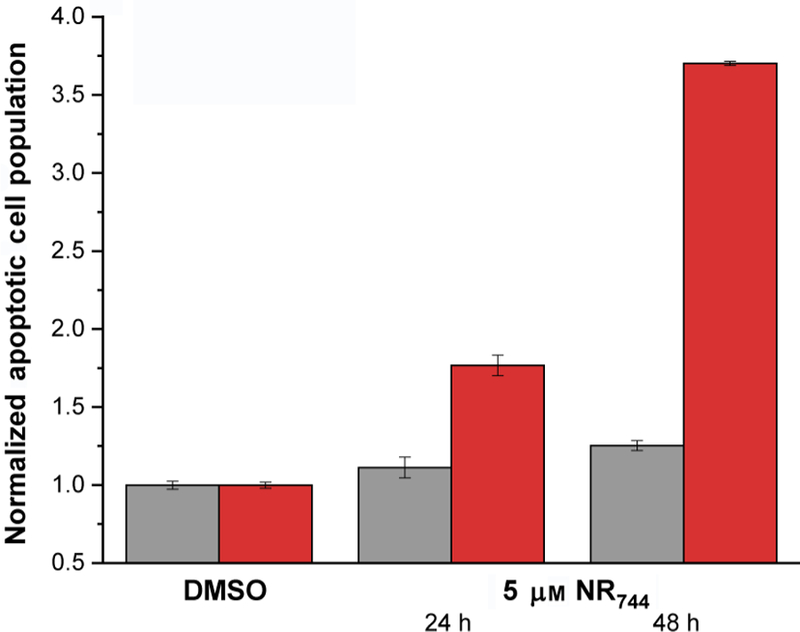
Annexin V labeling in co-cultures of HeLa ( ) and NIH-3T3 (
) and NIH-3T3 ( ) cells visualized by flow cytometry. HeLa and NIH-3T3 cells were co-cultured in the presence or absence of 5 µm NR744 for the indicated times, and annexin V labeling was evaluated by gating the appropriate cell tracker color for each cell population. Error bars represent the standard deviation of triplicate experiments.
) cells visualized by flow cytometry. HeLa and NIH-3T3 cells were co-cultured in the presence or absence of 5 µm NR744 for the indicated times, and annexin V labeling was evaluated by gating the appropriate cell tracker color for each cell population. Error bars represent the standard deviation of triplicate experiments.
Lastly, we sought to determine the effect of NR744 on cell migration in a wound-healing assay.[19] Cell migration is a critical processes involved in the normal physiology of immune responses and tissue homeostasis as well as disease-associated processes such as metastasis.[20] Confluent monolayers of cells were wounded and subsequently cultured in the presence of 5 µm NR744 for 24 h. Over this time period, NIH-3T3 cells displayed complete healing of the wound, with essentially no difference between treated and control cultures (Figure 4 A). However, a significant inhibition of wound closure was observed for HeLa cells cultured in the presence of NR744 (17 % closure) versus solvent (36 % closure, Figure 4 B). Taken together, these data demonstrate that NR744 can selectivity inhibit both the growth and motility of cancerous cells.
Figure 4.
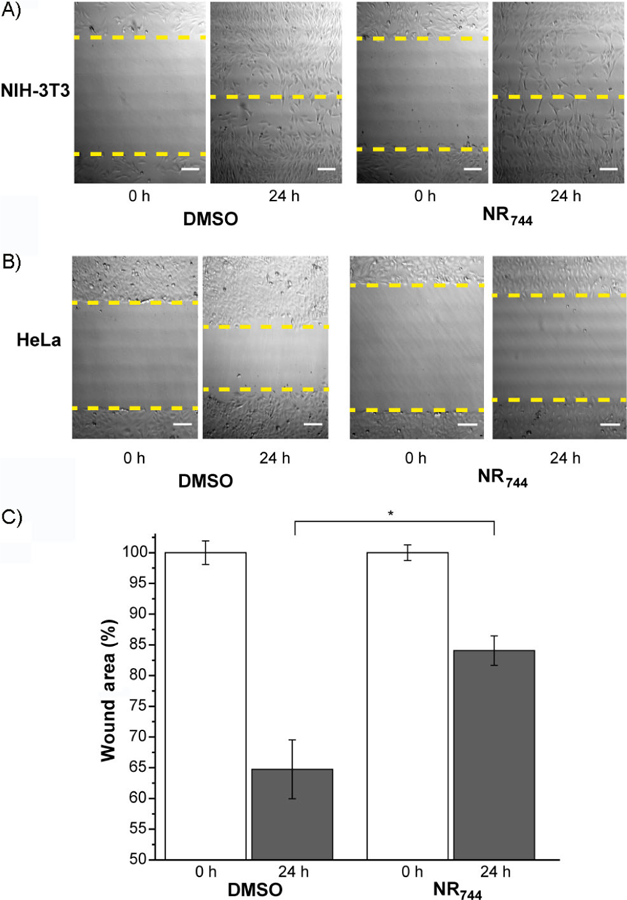
Wound healing in HeLa and NIH-3T3 cells treated with NR744. Representative figures of wound closure for A) NIH-3T3 cells and B) HeLa cells after incubation with blank (DMSO) or NR744 (5 µm) for the indicated times. C) Quantification of the percentage of open wound area in HeLa cells. Error bars represent the standard deviation of triplicate experiments; * p < 0.05.
Conclusion
In summary, we have reported the selective toxicity of a phosphinate-containing NIR fluorophore in cells with increased mitochondrial membrane potential. Selectivity was confirmed by multiple readouts including ATP production, quantification of apoptotic markers, and wound healing. In addition, the molecular mechanism of NR744 toxicity was investigated; this indicated that NR744 toxicity is due to increased MMP in HeLa cells, similar to previously reported delocalized lipophilic cations. In the long term, NR744 can be used as a starting-point to develop improved tools for imaging and potential therapeutic applications. For example, further modification by adding heavy atoms could potentially afford a NIR photosensitizer.[21] In addition, the unique reactivity of phosphorous,[12] coupled with the extensive literature surrounding xanthene-based turn-on fluorescent probes,[22] could provide a means to further increase the selectivity of NR744 for certain cell types.
Experimental Section
Fluorophore synthesis and characterization:
The synthesis and characterization of fluorophores used in this study have been reported previously.[12]
NR744 hydrolytic stability:
A solution of NR744 (5 µm) in 0.5 % DMSO was prepared in phosphate-buffered saline (PBS), Dulbecco’s modified Eagle’s medium (DMEM) with 10 % FBS, or DMEM with 50 % FBS. Solutions were aliquoted (2 mL) into the center three wells of a 24-well clear-bottom plate. In the wells surrounding the samples, sterilized water (2 mL) was added to prevent evaporation. The plate was then placed in a mammalian cell-culture incubator at 37°C under a 5 % CO2, humidified atmosphere. Samples (100 µL) were removed at 0, 14, 24, 38, and 48 h and mixed with acetonitrile (100 µL) containing 0.1 % TFA. Samples were centrifuged at 21130 g for 2 min. HPLC separation was conducted with a Waters 1525 Binary HPLC pump and a 2489 UV/Vis detector by using a gradient of 30–95 % acetonitrile containing 0.1 % trifluoroacetic acid (TFA) over 45 min in water containing 0.1 % TFA. Samples (80 µL) were injected into the HPLC and monitored at 700 nm. The area of the peak corresponding to NR744 was determined for each sample and normalized to the 0 h time point.
Cell culture:
NIH-3T3 (ATCC, CRL-1658), HeLa (ATCC, CCL-2), MCF-7 (ATCC, HTB-22), and HepG2 (ATCC, HB-8065) cells were grown in DMEM (Life Technologies, 10566016) supplemented with 10 % FBS (Life Technologies, 10082147) and 1 x Anti-Anti (Life Technologies, 15240062). HCT-116 cells (ATTC, CCL-247) were cultured in McCoy’s 5A (Gibco, 16600082) containing 10 % FBS and 1 x Anti-Anti. Cells were maintained at 378C under a 5 % CO2, humidified atmosphere.
Confocal microscopy and data analysis:
Cells were grown to ≈ 80 % confluency on glass-bottomed, 12-well plates (MatTek, P12G-1.5–14-F). After the culture medium had been aspirated, the cells were washed three times with prewarmed Dulbecco’s phosphate-buffered saline (DPBS; Life Tech, 14040117) and incubated with the indicated concentration of NR744, along with the indicated subcellular organelle tracker, for 30 min in culture medium. Cells were then washed with DPBS three times and imaged in DPBS. Confocal microscopy was performed with a Nikon A1R-TiE live-cell imaging confocal system. Laser lines used include blue (405 nm), green (488 nm), and far-red (640 nm). ImageJ software was used for data analysis.
Cell toxicity assays:
Cells were seeded in a 96-well plate (2 × 104 cells in each well) and were incubated with the indicated concentration of NR744 in culture medium containing 0.5 % DMSO. After the indicated time period, cell viability was assessed by using the CellTiter-Glo 2.0 Assay (Promega) according to the manufacturer’s protocol.
Wound-healing assays:
The assay was performed according to a previously reported protocol.[19] Briefly, cells were cultured on a 60 mm petri dish to 80 % confluency. A P200 pipet tip was used to create the wound, and the cells were washed and incubated with culture medium containing the indicated concentration of NR744. Images of wounded cells were immediately taken. After 24 h of incubation, the same spot was imaged. ImageJ software was used for data analysis.
Flow cytometry assays:
Cells (6 × 106) were washed with DPBS before resuspension in DPBS (1 mL) containing cell tracker violet (20 µm; for HeLa cells) or cell tracker red (20 µm; for NIH-3T3 cells). After 30 min of incubation, excess dye was removed by centrifugation (100 g, 5 min), and cells were seeded into individual wells or co-cultured in 12-well plates at ≈ 30–40 % confluency. After 24 h, the culture medium was removed, and cells were washed three times with DPBS. Cells were then incubated in culture medium containing NR744 for the indicated time periods. Apoptotic cell percentages under different conditions were analyzed by annexin V– Alexa Fluor 488 (Life Technologies, V13241) labeling according to the manufacturer’s protocol. When analyzing annexin V staining, different cell populations were gated by using cell tracker fluorescence (violet and red).
Supplementary Material
Acknowledgements
We thank the Morrison Microscopy Core Facility for assistance with the confocal fluorescence microscopy. We also thank the Flow Cytometry Service Center for assistance with the flow cytometry-related experiments. This work was funded by the NIH (R35GM119751) and the University of Nebraska–Lincoln. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting information and the ORCID identification numbers for the authors of this article can be found under https://doi.org/10.1002/cbic.201800811.
References
- [1].Health, United States, 2016: With Chartbook on Long-Term Trends in Health, National Center for Health Statistics, Hyattsville, MD, 2017. [PubMed] [Google Scholar]
- [2].Zheng YQ, Ji XY, Yu BC, Ji KL, Gallo D, Csizmadia E, Zhu MY, Choudhury MR, De La Cruz LKC, Chittavong V, Pan ZX, Yuan ZN, Otterbein LE, Wang BH, Nat. Chem 2018, 10, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, Overington JP, Nat. Rev. Drug Discovery 2017, 16, 19–34; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mamoshina P, Volosnikova M, Ozerov IV, Putin E, Skibina E, Cortese F, Zhavoronkov A, Front. Genet 2018, 9, 242; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Schneider G, Schmidt-Supprian M, Rad R, Saur D, Nat. Rev. Cancer 2017, 17, 239–253; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sun J, Wei Q, Zhou Y, Wang J, Liu Q, Xu H, BMC Syst. Biol 2017, 11, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Ding F, Zhan Y, Lu X, Sun Y, Chem. Sci 2018, 9, 4370–4380; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Podgorski K, Terpetschnig E, Klochko OP, Obukhova OM, Haas K, PLoS One 2012, 7, e51980; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cosco ED, Caram JR, Bruns OT, Franke D, Day RA, Farr EP, Bawendi MG, Sletten EM, Angew. Chem. Int. Ed 2017, 56, 13126–13129; Angew. Chem. 2017, 129, 13306 – 13309; [DOI] [PubMed] [Google Scholar]; d) Gorka AP, Nani RR, Schnermann MJ, Acc. Chem. Res 2018, 51, 3226–3235. [DOI] [PubMed] [Google Scholar]
- [5].a) Alander JT, Kaartinen I, Laakso A, Patila T, Spillmann T, Tuchin VV, Venermo M, Valisuo P, Int. J. Biomed. Imaging 2012, 2012, 940585; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim W, Applegate BE, Opt. Lett 2015, 40, 1426–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Aaron JJ, Trajkovska S, Curr. Drug Targets 2006, 7, 1067–1081. [DOI] [PubMed] [Google Scholar]
- [7].Vander Heiden MG, Cantley LC, Thompson CB, Science 2009, 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Smith RA, Hartley RC, Cocheme HM, Murphy MP, Trends Pharmacol. Sci 2012, 33, 341–352; [DOI] [PubMed] [Google Scholar]; Murphy MP, Biochem. J 2009, 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Murphy MP, Biochim. Biophys. Acta Bioenerg 2008, 1777, 1028–1031. [DOI] [PubMed] [Google Scholar]
- [10].a) Bernal SD, Lampidis TJ, Summerhayes IC, Chen LB, Science 1982, 218, 1117–1119; [DOI] [PubMed] [Google Scholar]; b) Bernal SD, Lampidis TJ, McIsaac RM, Chen LB, Science 1983, 222, 169–172; [DOI] [PubMed] [Google Scholar]; c) Smith RA, Hartley RC, Murphy MP, Antioxid. Redox Signaling 2011, 15, 3021–3038. [DOI] [PubMed] [Google Scholar]
- [11].a) Milane L, Trivedi M, Singh A, Talekar M, Amiji M, J. Controlled Release 2015, 207, 40–58; [DOI] [PubMed] [Google Scholar]; b) Chen ZP, Li M, Zhang LJ, He JY, Wu L, Xiao YY, Duan JA, Cai T, Li WD, J. Drug Targeting 2016, 24, 492–502; [DOI] [PubMed] [Google Scholar]; c) Zielonka J, Joseph J, Sikora A, Hardy M, Ouari O, VasquezVivar J, Cheng G, Lopez M, Kalyanaraman B, Chem. Rev 2017, 117, 10043–10120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhou X, Lai R, Beck JR, Li H, Stains CI, Chem. Commun 2016, 52, 12290–12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chai XY, Xiao J, Li M, Wang CM, An HY, Li C, Li YT, Zhang DZ, Cui XY, Wang T, Chem. Eur. J 2018, 24, 14506–14512. [DOI] [PubMed] [Google Scholar]
- [14].a) Sun XC, Wong JR, Song KL, Hu JL, Garlid KD, Chen LB, Cancer Res 1994, 54, 1465–1471; [PubMed] [Google Scholar]; b) Fantin VR, Berardi MJ, Scorrano L, Korsmeyer SJ, Leder P, Cancer Cell 2002, 2, 29–42; [DOI] [PubMed] [Google Scholar]; c) Koya K, Li Y, Wang H, Ukai T, Tatsuta N, Kawakami M, Shishido, Chen LB, Cancer Res 1996, 56, 538–543. [PubMed] [Google Scholar]
- [15].Shen F, Chu S, Bence AK, Bailey B, Xue X, Erickson PA, Montrose MH, Beck WT, Erickson LC, J. Pharmacol. Exp. Ther 2008, 324, 95–102. [DOI] [PubMed] [Google Scholar]
- [16].a) Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD, Essays Biochem 2010, 47, 53–67; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Felle H, Bentrup FW, Biochim. Biophys. Acta Biomembr 1977, 464, 179–187. [DOI] [PubMed] [Google Scholar]
- [17].Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM, J. Immunol 1992, 148, 2207–2216. [PubMed] [Google Scholar]
- [18].Tait JF, Gibson D, Arch. Biochem. Biophys 1992, 298, 187–191. [DOI] [PubMed] [Google Scholar]
- [19].Liang CC, Park AY, Guan JL, Nat. Protoc 2007, 2, 329–333. [DOI] [PubMed] [Google Scholar]
- [20].Vicente-Manzanares M, Webb DJ, Horwitz AR, J. Cell Sci 2005, 118, 4917–4919. [DOI] [PubMed] [Google Scholar]
- [21].Gorman A, Killoran J, O’Shea C, Kenna T, Gallagher WM, O’Shea DF, J. Am. Chem. Soc 2004, 126, 10619–10631. [DOI] [PubMed] [Google Scholar]
- [22].Chen X, Pradhan T, Wang F, Kim J, Yoon J, Chem. Rev 2012, 112, 1910–1956. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.