

Abstract
Carbohydrates are structurally diverse, synthetically challenging molecules with vital biological roles. Our inability to efficiently synthesize carbohydrates hinders our ability to probe how their structural differences affect biological function. Thus, the site-selective functionalization of carbohydrates is an active area of research. We previously reported that a cation-lone pair interaction can direct site-selective acylation in O-glycoside trans-1,2-diols using a chiral catalyst. We speculated that we could selectively functionalize S-glycosides via a similar interaction. Surprisingly, the sterically encumbered adamantyl group directed site-selective acylation at the C2 position of S-glycosides through dispersion interactions between the adamantyl C-H bonds and the π-system of the cationic acylated catalyst, which may have broad implications in many other chemical reactions. Because of their stability, chemical orthogonality, and ease of activation for glycosylation, the site-selective acylation of S-glycosides streamlines oligosaccharide synthesis and will have wide applications in complex carbohydrate synthesis.
Keywords: acylation, carbohydrates, glycosylation, S-glycosides, site-selective acylation
Graphical Abstract
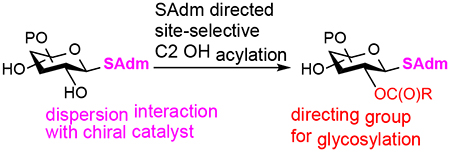
Carbohydrates have indispensable roles in almost all biological processes, including growth, development, inflammation and infection.[1,2] As such, researchers constantly seek to better understand how structural differences influence their biological functions. Because of their structural complexity, carbohydrates are synthetically challenging targets. Our inability to efficiently synthesize carbohydrates then hinders our ability to probe how structural differences affect their biological functions.
A preeminent goal of carbohydrate chemistry is to develop site-selective functionalization methods for the synthesis of carbohydrate building blocks and complex glycans. In many cases, 1,2- and 1,3-diols need to be differentiated within a given monosaccharide. Historically, site-selective methods have targeted cis-1,2-diols[3] because of the inherent reactivity differences between equatorial and axial hydroxyl groups and the ability to amplify this reactivity difference by chelation with metals, such as boron,[4] tin[5] and iron.[6] Conversely, the intrinsic selectivity can be overridden using achiral[7] and chiral[8] catalysts. Peptide catalysts can also be used to differentiate hydroxyl groups, but this method often lacks generality.[9] While significant progress has been made in respect to cis-1,2-diols, the site-selective functionalization of prevalent trans-1,2-diols is much less developed and more challenging.[3][10]
We[11] previously reported the site-selective acylation of many trans-1,2-diols in O-glycosides directed by a cation-lone pair interaction using paired chiral benzotetramisole (BTM) catalysts (Scheme 1A).[12] While considering how to extend the scope of this method, our attention was drawn to the fact that O-glycosides and S-glycosides are isoelectronic. We hypothesized that we could acylate trans-1,2-diols in S-glycosides in a similar manner (Scheme 1B). Should our hypothesis be correct, we would be able to site-selectively functionalize an additional subset of carbohydrates and significantly streamline the synthesis of oligosaccharides (Scheme 1C).
Scheme 1:

Site-selective acylation of O- and S-glycosides.
S-glycosides have found widespread use in oligosaccharide synthesis. They are easy to make, stable to various protecting group manipulations, orthogonal to many other glycosyl donors, and easily converted into other glycosyl donors, such as glycosyl halides. Herein we report our development of a site-selective acylation method for S-glycosides, which will further add to their synthetic utility. Surprisingly, we found that the sterically demanding S-adamantyl group was the best directing group for C2 OH acylation. Density functional theory (DFT) calculations indicated that dispersion interactions between the adamantyl C-H bonds and the electron-deficient π-system of the cationic acylated catalyst directed the site-selectivity in S-glycosides.
To determine a suitable directing group for site-selective acylation, we synthesized S-glycosides with different thioalkyl or thioaryl substituents. We then used these S-glycosides in our previously reported site-selective acylation protocol in the presence of chiral BTM catalysts (Table 1).[11] Intrinsic site-selectivity was obtained by using 4-dimethylaminopyridine (DMAP) as an achiral catalyst. Initial thioglycoside donors with ethyl and phenyl thioalkyl substituents only led to minimal selectivity enhancements, as determined by 1H NMR spectroscopy of the crude reaction mixture (entries 1 and 2). To increase the electron-donating ability of the S-glycoside, we synthesized a donor with a para-methoxyphenyl substituent (entry 3). While this donor could override the intrinsic selectivity, the selectivity gains were modest. To our surprise, the bulky tert-butyl group showed a significant increase in selectivity, increasing the C2:C3 acylated product ratio to 11:1 (entry 4). With an inclination that sterically more encumbered substituents may be the best directing groups, we synthesized adamantyl 4,6-O-benzylidene-1-thio-β-D-galactopyranoside 6e. We were pleased to observe >20:1 selectivity for the C2-acylated product 7e and to identify S-adamantyl (SAdm) as the superior directing group for site-selective acylation and future oligosaccharide synthesis.
Table 1.
Screening of S-glycosides substituents to determine the best directing group for site-selective acylation.
 | ||||
|---|---|---|---|---|
| Entry | Substrate | R | (R)-BTM C2:C3:C2+C3 | DMAP C2:C3:C2+C3 |
| 1 | 6a | ethyl | 5 : 1 : 0.6 | 2 : 2 : 1 |
| 2 | 6b | phenyl | 4 : 1 : 0.6 | 2 : 1 : 1 |
| 3 | 6c | p-methoxyphenyl | 5 : 1 : 0.5 | 1 : 2.5 : 1 |
| 4 | 6d | terf-butyl | 11 : 1 | 1 : 1 |
| 5 | 6e | adamantyl | >20 : 1 : 1 | 3 : 1 : 1 |
In our previous studies with O-glycosides, the stabilizing interactions between the oxygen lone pair and the positively charged π-system of the acylated catalyst were identified as the dominant factor that controlled the site-selectivity. If this lone-pair/π interaction indeed directed the acylation of S-glycosides, then we would expect the ethyl and phenyl thioalkyl substituents (6a and 6b) to be superior for C2-acylation due to the steric accessibility of the lone pairs. The fact that the sterically encumbered adamantyl substituent (6e) led to the best result is inconsistent with this site-selectivity model.
We performed DFT calculations to understand what factors contributed to the high C2-selectivity with adamantyl-substituted S-glycoside 6e. Because the site-selectivity of the reaction is determined in the acyl transfer from the acylated (R)-BTM catalyst to the S-glycoside, we calculated the possible isomers of the acylation transition states with 6e at the ωB97XD/6–311++G(d,p)//M06–2X/6–31G(d)/SMD level of theory. The most stable C2- and C3-acylation transition states (C2-TS and C3-TS) are shown in Figure 1. The C2-acylation transition state (C2-TS) is 3.5 kcal/mol lower in energy than the C3-acylation transition state (C3-TS), which is consistent with the high experimentally observed C2-selectivity. The lone pair/π interactions are relatively weak in both the C2- and C3-acylation transition states. In C2-TS, the vertical distance between the sulfur atom and the BTM π system (d4 = 3.86 Å) is too long for an effective stabilizing interaction. In C3-TS, the C4-oxygen atom is not placed above the BTM catalyst.
Figure 1.

Most stable transition states of the C2- and C3-acylation of S-glycoside 6e.
Instead, C-H/π interactions between the adamantyl group and the BTM π system and π/π interactions between the benzylidene protecting group and the BTM are observed in C2-TS and C3-TS, respectively. To investigate the relative strength of the C-H/π and π/π interactions, we computed the dispersion energy (ΔEdispersion) between the S-glycoside substrate and the BTM catalyst in the transition states (see SI for details). The calculations indicated a substantial amount of dispersion interactions in C2-TS due to contributions from multiple C-H/π interactions (d1-d3 ≈ 2.4~2.6 Å). On the other hand, the substrate-catalyst dispersion interaction in C3-TS is 3.7 kcal/mol weaker. These results suggest the greater stability of the C2-acylation transition state (C2-TS) is largely due to the more favorable C-H/π interactions between the S-adamantyl group and the BTM π-system.
We were then interested in determining the relative strength of the cation-lone pair interaction (as previously reported) versus the dispersion interactions (this work). We used 8 as a substrate capable of both a cation-lone pair interaction between the benzylidene C4 oxygen atom and the (R)-BTM catalyst and dispersion interactions between the adamantyl group and the (R)-BTM catalyst. We determined the cation-lone pair interaction outcompetes the dispersion interactions, though not by much, leading the C3-acylated product 8b to be slightly favored in comparison to the C2-acylated product 8a (Scheme 2). However, by inserting a para-nitrobenzylidene protecting group in 9, the C2-acylation product 9a is favored over 9b because the nitro group electronically deactivates the benzylidene C4 oxygen atom lone pair. We thus concluded that the relative strength of the cation-lone pair interactions is similar to dispersion interactions.
Scheme 2:

Comparison of the relative strength of a cation-lone pair and dispersion interactions.
a) Conditions: (R)-BTM (10 mol %), isobutyric anhydride (2.5 eq), iPr2NEt (3.0 eq), CHCl3, rt, Ar
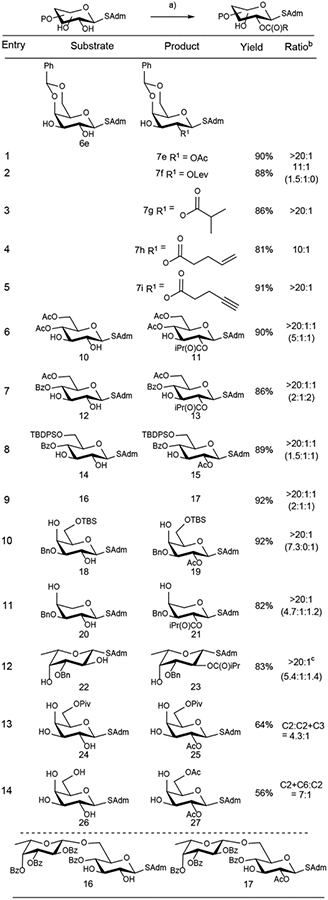
After having a better understanding of the origin of the selectivity, we proceeded to examine the scope of this method (Table 2). Using the mixed anhydride method, various acyl groups can be introduced to final products 7e-7i, depending on the choice of the carboxylic acid (entries 1–5). The magnitude of site-selectivity was >20:1 for acylation at the C2 hydroxyl of S-galactoside 6e, in most cases, by employing the (R)-BTM catalyst, and isolated yields ranged from 81–91%. This method showed a tolerance for carboxylic acids of varying complexities, from acetic acid to those bearing alkene, alkyne, and ketone functionalities, which will be useful for further transformations.
Table 2:
Scope of saccharides amenable to site-selective acylation.
|
Conditions: (R)-BTM (10 mol%), carboxylic acid (2.5 eq), Piv2O (2.5 eq), iPr2NEt (3.0 eq), CHCl3, rt, Ar; b) C2:C3 or C2:C3:C2+C3 for C2/C3-diols; C2:C4 or C2:C4:C2+C4 for C2/C4-diols; () are the ratios when DMAP (10 mol%) was used as a catalyst; c) (S)-BTM was used as a catalyst for L-sugar.
β-S-Glucoside with a 4,6-O-benzylidene protecting group (e.g. 8) did not show any selectivity for the acylation of the C2 OH group, due to the competing cation-lone pair interaction,[11] which facilitates acylation of the C3 OH group as previously discussed. We envisioned that applying an electron-withdrawing protecting group on the C4 OH would disable the cation-lone pair interaction between the C4 oxygen lone pair and the cationic acylated catalyst. Indeed, selective C2 OH acylation was observed for β-S-glucosides 10, 12, and 14, with acetyl, benzoyl, and silyl protecting groups on the C4 and C6 OH groups (entries 6–8). The C2-selective acylation can be further extended to disaccharide 16 (entry 9).
In all of the above β-S-glucosides, low site-selectivity was observed when (S)-BTM catalyst was employed as the catalyst. This is what one would predict with our previously proposed model[11] because the lone pair on the C2-oxygen or C3-oxygen can interact with the cationic acylated catalyst when the C3 or C2 OH undergoes acylation, respectively.
We next examined the site-selectivity for the acylation of C2,C4–1,3-diols in galactoside 18, arabinopyranoside 20 and fucopyranoside 22 (entries 10–12). In all cases, the C2 OH group was selectively acylated using chiral BTM catalyst. Selective acylation of the C2 OH group was also observed in the presence of achiral DMAP catalyst, which reflects the intrinsically more reactive nature of the equatorial OH group over the axial OH group in these cases. However, the much higher selectivity observed using the (R)-BTM catalyst indicates that the dispersion interactions between the acylated catalyst and the SAdm group is a significant contributor to the site-selectivity. We were pleased to find that good site-selectivity was observed for triol 24 and tetrol 26 (entries 13 and 14). No site-selectivity was observed using DMAP as the catalyst (see SI for details), but when using (R)-BTM as the catalyst, the C2 acylated product 25 was favored over the C2+C3 bisacylated compound in a 4.3:1 ratio. When tetrol 26 with a free primary hydroxyl group was subjected to the procedure, a ratio of 7:1 was observed for the formation of C2+C6 bisacylated product and C2 OH monoacylated product, suggesting that C2 OH was acylated first in the presence of the chiral catalyst.
After successful development of the site-selective acylation method for S-glycosides, we envisioned that the acylated products could be readily activated and used as glycosyl donors for glycosylation. S-glycosides have been widely used for oligosaccharide synthesis because they are easily synthesized, benchtop stable, orthogonal to many other glycosyl donors, and easily activated for glycosylation.[13] Unfortunately, many of the thiols used to make S-glycosides result in pervasive and unpleasant odors. 1-Adamantanethiol, however, a solid thiol with less offensive odor compared to liquid thiols, making it attractive for the synthesis of S-glycosides.[14] Furthermore, adamantyl-substituted S-glycosides avoid unwanted aglycon transfer reactions observed between trichloroimidate glycosyl donors and thioglycoside acceptors,[15] and they are more reactive than common thioglycoside donors, including S-phenyl.[16] Thus, adamantyl-substituted S-glycosides can not only direct site-selective acylation, but they are also preferred glycosyl donors to make complex glycans.
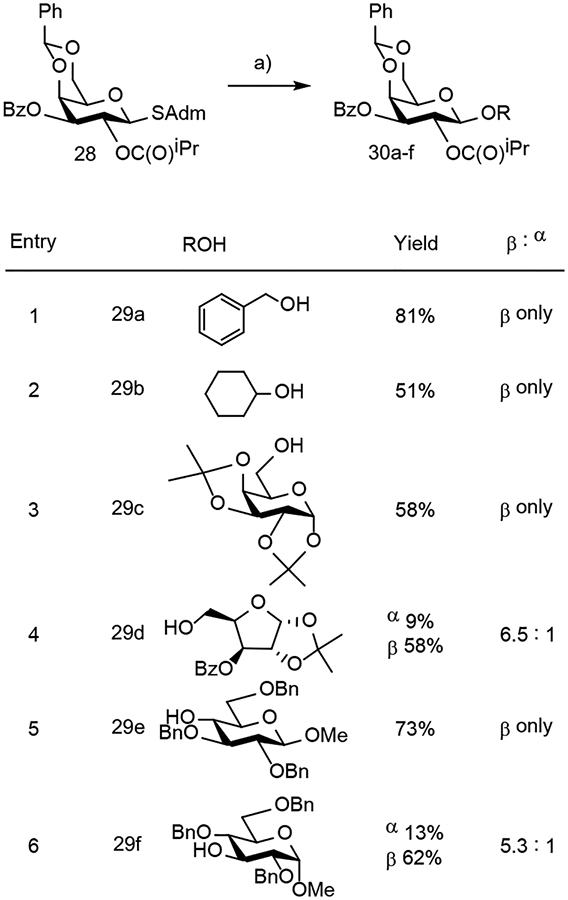
Using adamantyl-substituted S-glycosides with a C2-acyl group provided an inherent driving force for the formation of a β-glycosidic bond via neighboring group participation. C2 acyl product 7g was protected at C3 with a benzoyl group to provide 28 as our model glycosyl donor (Table 3). In four of the six glycosylations, only the β-anomer was obtained, and the other two glycosylations still produced the β-anomer as the major product. These results demonstrate the ability of S-glycosides to serve as glycosyl donors for glycosyl acceptors of varying complexity and preferentially form β-glycosidic bonds.
Table 3:
Demonstration of S-glycosides’ ability to promote β-glycosidic bond formation through the presence of a C2 acyl group.
|
Conditions: ROH 29a-f (1.3–2 eq), NIS (2 eq), TMSOTf (1.1 eq), CH2Cl2/CH3CN (2:1), −78°C to 0°C
We next evaluated the site-selective acylation method’s ability to streamline the assembly of oligosaccharides. The presence of particular glycolipids can stimulate natural killer T cells to release cytokines, which then trigger proinflammatory and immunomodulatory responses.[17] One glycolipid of particular importance is isoglobotrihexosylceramide, which has been shown to be an antigen for natural killer T cells.[18] Our site-selective acylation protocol provides a streamlined method to obtain the trisaccharide core 31 in isoglobotrihexosylceramide (Scheme 3). Both our cation-lone pair directed and S-adamantyl directed acylation can selectively acylate the C2 hydroxyl in one step to go from 33 to 34 and 6e to 7f, respectively. Unfortunately, the cation-lone pair directed methodology required late stage anomeric position manipulations. S-adamantyl directed acylation using S-glycosides, however, eliminates late-stage hydrolysis and installation of an anomeric leaving group, meaning we can directly glycosylate 36 to yield 31. We anticipate that this sequence of site-selective acylation and direct glycosylation can be used to construct oligosaccharides with various linkages.
Scheme 3.

Comparison of method efficiency to prepare oligosaccharides using either cation-n directed C2 OH acylation with O-glycosides or site-selective acylation using readily activated S-glycosides.
Reaction conditions: a) (R)-BTM, LevOH, Piv2O, iPr2Et, CHCl3, rt, 85%; b) 32, TMSOTf, CH2Cl2, 0°C-rt, 83%; (c) CAN, CH3CN, H2O, 70%; d) CCl3CN, DBU, 64%; (e) 29e, TMSOTf, CH2Cl2, 0°C-rt, 81%; f) (R)-BTM, LevOH, Piv2O, iPr2Et, CHCl3, rt, 88%; g) 32, TMSOTf, CH2Cl2, 0°C-rt, 62%; (h) 29e, NIS, TMSOTf, CH2Cl2:CH3CN (2:1), −78°C–0°C, 69%
Using chiral catalysts and the adamantyl directing group, we have developed a site-selective acylation protocol for 1,2-trans-diols, 1,3-diols, triol, and even tetrol in various S-glycosides. DFT calculations indicated that dispersion interactions between the C-H bonds of the adamantyl group and the electron-deficient π-system of the cationic acylated catalyst are the major contributor to the high degree of site-selectivity. This dispersion interaction-directed selectivity may have broad implications in many other chemical reactions. The S-adamantyl group directed C2 OH selective acylation method eliminates tedious protection and deprotection steps often needed for site-selective functionalization and results in a stable, chemically orthogonal, readily activated glycosyl donor. The utility of the method was demonstrated in the streamlined synthesis of oligosaccharides. We anticipate this method will find applications for the synthesis of many more complex carbohydrates.
Supplementary Material
Acknowledgements
W. T. and P. L thank NIH (U01GM125290) and S. A. B. thanks the NIH Chemistry-Biology Interface Training Grant (T32 GM008505) for financial support. We would also like to thank the Analytical Instrumentation Center and the Medicinal Chemistry Center within the UW-Madison School of Pharmacy for their assistance.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under the following link.
References
- [1].Bertozzi CR, Kiessling LL, Science 2001, 291, 2537. [DOI] [PubMed] [Google Scholar]
- [2].Gijsen HJM, Qiao L, Fitz W, Wong C-H, Chem. Rev 1996, 96, 443. [DOI] [PubMed] [Google Scholar]
- [3].Recent relevant reviews include:; a) Wang H–Y, Blaszczyk SA, Xiao G, Tang W, Chem. Soc. Rev 2018, 47, 681. [DOI] [PubMed] [Google Scholar]; b) Lawandi J, Rocheleau S, Moitessier N, Tetrahedron 2016, 72, 6283. [Google Scholar]; c) Ueda Y, Kawabata T. In Topics in Current Chemistry; Kawabata T, Ed.; Springer International Publishing AG: Gewerbestrasse, 2016; Vol. 372, p 203. [DOI] [PubMed] [Google Scholar]; d) Giuliano MW; Miller SJIn Topics in Current Chemistry; Kawabata T, Ed.; Springer International Publishing AG: Gewerbestrasse, 2016; Vol. 372, p 157. [DOI] [PubMed] [Google Scholar]
- [4].a) Mancini RS, Lee JB, Taylor MS, Org. Biomol. Chem 2017, 15, 132; [DOI] [PubMed] [Google Scholar]; b) Mancini RS, Lee JB, Taylor MS, J. Org. Chem 2017, 82, 8777; [DOI] [PubMed] [Google Scholar]; c) Fukuhara K, Shimada N, Nishino T, Kaji E, Makino K, E. J. Org. Chem 2016, 902; [Google Scholar]; d) Gouliaras C, Lee D, Chan L, Taylor MS, J. Am. Chem. Soc 2011, 133, 13926; [DOI] [PubMed] [Google Scholar]; e) Lee D, Taylor MS, J. Am. Chem. Soc 2011, 133, 3724; [DOI] [PubMed] [Google Scholar]; f) Oshima K, Kitazono E, Aoyama Y, Tetrahedron Lett 1997, 38, 5001. [Google Scholar]
- [5].a) Grindley TB, Adv. Carbohydr. Chem. Biochem 1998, 53, 17; [DOI] [PubMed] [Google Scholar]; b) David S, Hanessian S, Tetrahedron 1985, 41, 643. [Google Scholar]
- [6] a).Ren B, Ramström O, Zhang Q, Ge J, Dong H, Chem. Eur. J 2016, 22, 2481; [DOI] [PubMed] [Google Scholar]; b) Ren B, Lv J, Zhang Y, Tian J, Dong H, ChemCatChem 2017, 9, 950. [Google Scholar]
- [7].Peng P, Linseis M, Winter RF, Schmidt RR, J. Am. Chem. Soc 2016, 138, 6002. [DOI] [PubMed] [Google Scholar]
- [8].Sun X, Lee H, Lee S, Tan KL, Nat. Chem 2013, 5, 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Tong ML, Huber F, Taghuo Kaptouom ES, Cellnik T, Kirsch SF, Chem. Commun 2017, 53, 3086; [DOI] [PubMed] [Google Scholar]; b) Allen CL, Miller SJ, Org. Lett 2013, 15, 6178; [DOI] [PubMed] [Google Scholar]; c) Sanchez-Rosello M, Puchlopek ALA, Morgan AJ, Miller SJ, J. Org. Chem 2008, 73, 1774; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Griswold KS, Miller SJ, Tetrahedron 2003, 59, 8869. [Google Scholar]
- [10].b) Yanagi M, Imayoshi A, Ueda Y, Furuta T, Kawabata T, Org. Lett 2017, 19, 3099; [DOI] [PubMed] [Google Scholar]; c) Rocheleau S, Pottel J, Huskić I, Moitessier N, N. Eur. J. Org. Chem 2017, 646–656. [Google Scholar]
- [11].Xiao G, Cintron-Rosado GA, Glazier DA, Xi B-M, Liu C, Liu P, Tang W, J. Am. Chem. Soc 2017, 139, 4346. [DOI] [PubMed] [Google Scholar]
- [12].Birman VB, Li X, Org. Lett 2006, 8, 1351. [DOI] [PubMed] [Google Scholar]
- [13].a) Lian G, Zhang X, Yu B, Carb. Res 2015, 403, 13; [DOI] [PubMed] [Google Scholar]; b) Stick RV, Williams SJ, Carbohydrates: The Essential Molecules of Life, Elsevier, Oxford, 2009, p. 157. [Google Scholar]
- [14].Dohi H, Nishida T, Trends Glycosci. Glyc 2014, 26, 119. [Google Scholar]
- [15].Li ZT, Gildersleeve JC, J. Am. Chem. Soc 2006, 128, 11612. [DOI] [PubMed] [Google Scholar]
- [16].a) Lahmann M, Oscarson S, Can. J. Chem 2002, 80, 889; [Google Scholar]; b) Crich D, Li W, J. Org. Chem 2007, 72, 7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Bendelac A, Savage PB, Teyton L, Annual Rev. Immunol 2007, 25, 297; [DOI] [PubMed] [Google Scholar]; b) Matsuda JL, Mallevaey T, Scott-Browne J, Gapin L, Curr. Op. Immunol 2008, 20, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhou D et al. Science, 2004, 306, 1786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.