

Abstract
Background
Colorectal cancer (CRC) represents a major public health problem as the second leading cause of cancer-related mortality in the United States. Of an estimated 140,000 newly diagnosed CRC cases in 2018, roughly one-third of these patients will have a primary tumor located in the distal large bowel or rectum. The current standard of care approach includes curative-intent surgery, often following preoperative (neoadjuvant) radiotherapy (RT) to increase rates of tumor downstaging, clinical and pathologic response, as well as improving surgical resection quality. However, despite advancements in surgical techniques, as well as sharpened precision of dosimetry offered by contemporary RT delivery platforms, the oncology community continues to face challenges related to disease relapse.
Methods
Ongoing investigations are aimed at testing novel radiosensitizing agents and treatments that might exploit the systemic antitumor effects of RT utilizing immunotherapies. If successful, these treatments may usher in a new curative paradigm for rectal cancers such that surgical interventions may be avoided. Importantly, this disease offers an opportunity to correlate matched paired biopsies, radiographic response and molecular mechanisms of treatment sensitivity and resistance with clinical outcomes.
Results
Herein, the authors highlight the available evidence from preclinical models and early-phase studies, with an emphasis on promising developmental therapeutics undergoing prospective validation in larger-scale clinical trials.
Conclusions
This review by the NCI’s Radiation Research Program Colorectal Cancer Working Group provides an updated comprehensive examination of the continuously evolving State of the Science regarding radiosensitizer drug development in the curative treatment of CRC.
Keywords: precision radiation medicine, radiation therapy, radiosensitization, chemoradiotherapy, radiation biology, rectal cancer, immunotherapy, targeted therapeutics, abscopal effect, colorectal cancer
Table of Contents precis:
This review by the NCI’s Radiation Research Program Colorectal Cancer Working Group provides an updated comprehensive examination of the continuously evolving State of the Science regarding radiosensitizer drug development in the curative treatment of CRC. Herein, the authors highlight the available evidence from preclinical models and early-phase studies, with an emphasis on promising developmental therapeutics undergoing prospective validation in larger-scale clinical trials.
Background
Colorectal cancer (CRC) represents the second leading cause of cancer-associated deaths in the United States, with an estimated 135,430 new cases and 50,260 cancer-related deaths in 2018.[1] Of these cases, nearly one-third represent tumors arising in the distal portion of the large bowel, the rectum, where surgical removal may require a permanent colostomy. In many patients, pre-operative treatment with chemoradiotherapy (chemoRT) is a mainstay of therapy that supports increased tumor downstaging, fewer colostomies and reduced local recurrence. Previous attempts to intensify therapy through radiosensitization with resultant improvement in tumor sterilization have failed to improve outcomes in comparison to concurrent fluoropyrimidine use.
Strategic development of novel radiosensitizers represents a clinical unmet need and has been a focus of the National Cancer Institute’s (NCI) Radiation Research Program.[2] The NCI’s Radiation Research Program has organized disease-specific Working Groups comprised of experts from across academics, industry, government, cancer disciplines, clinical care and basic cancer biology. The Colorectal Cancer Working Group has systematically catalogued and prioritized agents and interventions that may help improve outcomes for patients with rectal cancer. These efforts provide guidance to investigators involved in pre-clinical testing and have minimized duplication of effort in clinical trial design and development.
This manuscript provides a summary update of the State of the Science related to radiosensitizer development in clinical trials for CRC. Importantly, this field has expanded to include both the traditional sensitizer of radiation for improved local response, as well as agents that can be systemically catalyzed by radiation. This latter group includes immunotherapies, vaccines and immune checkpoint inhibitors that have the potential to revolutionize the management of many diseases. Given the rapidly changing landscape of discovery and development, this manuscript provides a contemporary vantage point of the field and relevant clinical studies that form the basis for ongoing and future clinical trials.
Principles of Radiosensitization
Refinements in surgical technique with the adoption of the total mesorectal excision (TME), incorporation of modern chemotherapy, and advances in timing and dosimetry of radiotherapy (RT), have demonstrated a meaningful impact on local tumor control, however, distant relapse remains the leading cause of mortality in this patient population, with approximately 35% developing metastatic relapse within 5 years of trimodality treatment.[3]
Following neoadjuvant chemoRT, pathological complete response (pCR), defined as no histopathologic evidence of residual cancer cells, has been extensively studied as a standard measurement tool of tumor regression. In the current era of consistently low local tumor recurrence rates, the goal of increasing the pCR rate is driven from where we aspire to see the field move towards. First, the ability to achieve a pCR serves as an easily defined and pragmatic metric of anticancer activity. Inherent to achieving a pCR, we infer that the tumor and/or the treatment provided limited opportunities for chemo- and radioresistance mechanisms to develop. As such, a higher pCR rate is a useful short-term signal of anti-cancer activity involving novel treatment combinations and sequencing approaches in the neoadjuvant setting. However, it remains a poor surrogate for long term outcomes including survival, which is why more continuous variables of tumor downstaging such as the neoadjuvant rectal (NAR) score, more accurately predict long-term outcomes.[4]
Second, a growing area of investigation and clinical practice involves non-operative management (NOM). In these situations, patients who achieve a clinical complete response (cCR) after neoadjuvant therapy, as determined by physical exam, radiological and endoscopic evaluation, might be given the opportunity for a delayed or deferred operation. Only the subset of cCR patients who actually have a pCR can be legitimately spared an operation. Thus, the use of pCR as an endpoint in the testing of neoadjuvant strategies may help establish which patient population or treatment approach could more legitimately support a non-operative approach as part of future trials and clinical care.
The goal of combining systemic agents with radiation is multifold. In addition to sensitizing the effects of radiation, the ideal drug combination would result in maximizing tumor cell kill while simultaneously minimizing the effects on surrounding normal tissues. This concept of achieving a high therapeutic index is critical in the design of novel drug-radiation combinations.
Improving therapeutic ratio can be accomplished using three general principles: (1) spatial cooperation (2) synergistic tumor cell kill (3) normal tissue protection.[5–7] Spatial cooperation involves using radiation to control local disease and chemotherapy to control distant micrometastatic disease. Synergistic tumor cell kill involves enhancing the tumoricidal effects of radiation (radiosensitizers) through additive or synergistic inclusion with another agent (e.g., concurrent continuous infusion 5-fluorouracil [5-FU] or capecitabine). Normal tissue protection can be accomplished by modulating the cellular response to RT and mitigating radiation injury to normal tissues, thereby allowing a higher cumulative RT dose to be delivered. The FDA approved radioprotector, amifostine, is an organic thiophosphate compound that acts as a free radical scavenger.[8] Although phase II trials in rectal cancer suggest that each of these approaches are feasible, only synergistic tumor cell killing with concurrent chemoRT is routinely practiced.[9–11]
Major classes of radiosensitizers currently used in clinical practice include cell cycle modulators, signal transduction inhibitors and DNA damaging agents. 5-FU was one of the first drugs to be combined with RT and remains the backbone of chemoRT therapy in rectal cancer. Large randomized trials have established that it is superior to either chemotherapy or RT alone.[12, 13] The synergistic effect of fluoropyrimidines with radiation is due to its ability to redistribute cells into S-phase, as well as deplete nucleotide pools, which decrease the capacity for DNA repair. The radiosensitizing effects of fluoropyrimidines depend on continuous exposure of tumor cells to the drug. Continuous intravenous infusion of 5-FU is non-inferior to capecitabine, an orally bioavailable fluoropyridine, with either being used with RT in the neoadjuvant treatment of rectal cancer.[14] Capecitabine is extensively absorbed from the intestine to undergo a three-step enzymatic conversion to 5-FU and avoids the need for an indwelling venous catheter.
There is a diversity of new agents in development that are significantly different than historical radiosensitizers (Figure 1). Some include those that focus on hypoxia modification or change the intrinsic radiosensitivity of the tumor. Another new paradigm involves immunotherapy whereby RT is being used to potentiate the effects of the drug.[15] Because ionizing radiation can cause immunogenic cell death, modulate antigen presentation by cancer cells; and alter the microenvironment within the radiated field, it has been demonstrated that local radiation can enhance responses to immunomodulating agents at sites distant from the area being treated (i.e., the “abscopal effect”).[16–18] These are exciting areas of drug development, a number of which are detailed below.
Figure 1.

Targeted cellular pathways and new radiosensitizers in development.
Rationale for Radiation-Targeted Therapy Combinations
MEK inhibition
The Cancer Genome Atlas and other studies have demonstrated the importance of the canonical RAS-RAF-MAPK pathway in CRC with frequencies of KRAS, NRAS, and BRAF gain of function mutations being ~40%, 10%, and 5%, respectively.[19, 20] RAS isoforms (e.g. K-, N-, and H-RAS) encode small (~21 kD), membrane-bound GTP-binding proteins that are involved in promoting proliferation, migration, invasion, de-differentiation, and inhibition of apoptosis.[21, 22] Preclinical studies demonstrate that hyper-activation or overexpression of RAS can lead to development of intrinsic radioresistance, defined through clonogenic cell survival assays. Conversely, KRAS downregulation by siRNA or chemical inhibitors of farnesylation, radiosensitize tumor cells.[23–25] While RT activates RAS-MAPK signaling in KRAS mutant cells, inhibition of MAPK signaling by MEK inhibition can attenuate survival after RT.[26–28] Emerging clinical evidence also supports the finding of relative RT resistance with tumors harboring KRAS mutations.[20, 29–31] [32] Trametinib, a potent and selective MEK1/2 inhibitor that is FDA-approved for metastatic or unresectable BRAFV600E/K mutant melanoma, has been shown to have anti-proliferative activity in human KRAS or BRAF mutant CRC cell lines with reductions in ERK activation.[33] Multiple groups have shown that MEK1/2 inhibitors in RAS mutant carcinoma preclinical models result in effective radiosensitization (with or without additive effects of 5FU), resulting in tumor cell death and tumor growth delay through increased apoptosis, DNA damage induced cell death, reduced DNA repair, and reduction in hypoxic response.[26, 34–37] [33, 38] However, MEK inhibition benefits do not appear to be dictated exclusively by genomic subtype (e.g. specifically KRAS or BRAF mutant).[39]
As such, phase I clinical studies have been developed. In the first, a clinical trial testing AZD6244 in rectal cancer with chemoRT was terminated early due to unspecified dose-limiting toxicities (NCT01160926).[40] However, a second study using trametinib was designed in conjunction with standard chemoRT with 5-FU for patients with locally advanced rectal cancer (NCT01740648). There was a 5-day lead-in of single-agent trametinib followed by the combination of trametinib and infusional 5-FU (225 mg/m2/day, Monday to Friday) with RT (50.4Gy) using 3 dose cohorts of trametinib: 0.5 mg, 1 mg, and 2 mg (oral, daily Monday to Friday). A total of 18 evaluable patients demonstrated no grade 4 toxicities, with only 1 patient having a dose-limiting toxicity of diarrhea, which was attributed to chemoRT. Twelve patients received the maximum tolerated dose (MTD) of 2 mg. Analysis of the whole patient cohort showed tumor downstaging of 72%, with pathological complete response (pCR) seen in 17% patients. In the cohort of patients treated at the MTD (2 mg), 75% had tumor down-staging and 25% achieved pCR suggesting at least additive benefit and good tolerance.[41] Correlative analysis was performed on serial tumor biopsies obtained at pre-treatment, after the 5-day trametinib lead-in, and the surgical specimen. Preliminary data shows a trend for dose-dependent decrease in the level of phosphorylated-ERK protein level in tumor cells, as detected by immunohistochemistry, confirming pharmacodynamic inhibition of the RAS-ERK pathway. Future studies in larger patient cohorts are proposed to determine if this activity is clinically meaningful.
Protein Kinase C
Midostaurin is a multi-target kinase inhibitor and a staurosporine analog. It was initially developed as a protein kinase C inhibitor in solid tumors and more recently obtained FDA- approval in fms-related tyrosine kinase 3 (FLT3) mutated acute myeloid leukemia.[42] In a study by Liu and colleagues,[43] 32 cancer cell lines were screened for radiosensitization effects using 18 targeted therapeutic agents. Short-term radiosensitization factors (SRF2Gy) and calculated clonogenic survival assay-based dose enhancement factors (DEFSF0.1) were derived. Midostaurin was found to increase radiosensitization in KRAS mutant cell lines. Interestingly, greater sensitization was seen in codon 12/13 mutations, rather than codon 61. The radiosensitization effects were also more pronounced in cells with high expression of the stem cell marker CD133, typically deemed a more radioresistant phenotype. The efficacy appeared to be mediated through PKCα, as similar in vitro results were seen with a pure PKC inhibitor.
Based on this data, a phase I study was initiated whereby patients with MRI or ultrasound defined T3 or T4 or node+ adenocarcinoma of the rectum without evidence of metastatic disease were treated with neoadjuvant chemoRT (50.4 Gy in 28 fractions combined with infusional 5-FU [225 mg/m2/day]) continuously with midostaurin twice daily (BID) on days of RT (NCT01282502). The midostaurin dose was escalated in a 3+3 design: dose level 1 was 50 mg BID, dose level 2 was 75 mg BID followed by a dose expansion cohort. Surgery was performed 6–9 weeks following the completion of chemoRT. Genotyping was performed using whole exome sequencing, with either blood or adjacent normal tissue normal control.
This phase Ib study enrolled 19 patients with clinical stage II and III rectal cancer. All patients completed therapy and underwent surgery achieving an R0 resection with a pCR rate of 16% (3/19). Maximum tolerated dose (MTD) was 75 mg BID. At least one grade 3–4 toxicity occurred in 9/19 (47%) of patients, with the most common grade ≥3 events observed being lymphopenia (26%, 5/19) and rash (16%, 3/19). With a median follow up of 3 years, two patients had recurrence- both had synchronous local and distant. DFS at 3 years was 89.5% (CI 75.7–100%), and 3 year OS was 87.7% (CI 71.6–100%). Sixteen tumors underwent mutational sequencing with the genotype of the pCR cases being RAS (2/3) mutant or unknown. Overall, the combination of midostaurin with chemoRT was well tolerated, and final genotyping results and correlative analysis of RAS status and pCR are eagerly awaited.[44]
Heat Shock Protein 90
Heat Shock Protein 90 (HSP90) is a chaperone involved in folding, stabilization and intracellular trafficking of client proteins.[45] HSP90 client proteins include key proteins involved in cellular growth, apoptosis, immune function, epithelial to mesenchymal transition (EMT), and angiogenesis. HSP90 is overexpressed in several malignancies; hence the interest in HSP90 in cancer biology.[45] It has been demonstrated that HSP90 protein is upregulated in CRC tissue as compared to matched normal colon tissue.[46] HIF-1α, STAT-3, and NF-κB are transcriptional factors that influence radioresistance and are also chaperone proteins of HSP90. Agents in this class (i.e., ganetespib) are small molecule inhibitors of HSP90.[47] The addition of ganetespib to 5-FU and RT significantly inhibited growth of CRC cell lines, decreased the colony formation and inhibited in vivo tumor growth as compared to untreated controls as well as those treated with 5-FU plus radiation.[48] Ganetespib inhibited EMT, invasion and angiogenesis through blocked activation of transcriptional factors HIF-1α, STAT-3, and NF-κB.[46, 49, 50]
With this pre-clinical data, a phase I study of ganetespib in combination with chemoRT was conducted in patients with stage II-III rectal cancer.[51] Ganetespib run-in was started at day −14, given twice weekly for 2 weeks and accompanied by a pre- and post run-in biopsies. Patients then completed standard chemoRT (50.4 Gy in 28 fractions over 5.5 to 6 weeks administered with concurrent oral capecitabine at 825 mg/m2 PO BID) with ganetespib followed by surgery. Four dose levels of ganetespib were studied from 60 mg/m2 to 120 mg/m2. The recommended phase II dose was established at 100 mg/m2 once weekly given on days 1, 8, 15, 29, and 36 concurrently with chemoRT. Fifteen patients were enrolled on the trial with baseline endoscopic staging of T3N0 (5), T3N1 (5), and T3N2 (5). Fourteen patients underwent successful surgical resection (3 APR and 11 LAR). The only Grade 3 toxicity seen was diarrhea, and Grade 2 toxicities were fatigue, nausea/vomiting, radiation dermatitis, elevated AST/ALT, and hand/foot syndrome. Pharmacokinetics did not reveal any drug- drug interaction between ganetespib and capecitabine. The pCR was 21% (3/14 patients), and 2 patients had <1cm tumor at surgery, with an overall Neoadjuvant Rectal (NAR) Score of 7.9. By comparison, the NAR score for capecitabine and radiation is approximately 15, with lower scores indicating more treatment activity.[52] Correlative studies analyzing post-treatment tumor tissue showed down-regulation of mRNA of HIF-1 α STAT-3, and VEGF.[46] Despite these encouraging early results, ganetespib is no longer being developed; however, there are several other HSP90 inhibitors in early stages of clinical development. Identification of another HSP90 inhibitor for incorporation into the setting of rectal cancer is a rational future pursuit.
Epidermal and Vascular Endothelial Growth Factor Pathways
Simultaneous targeting of the vascular endothelial growth factor (VEGF) and epidermal growth factor receptor (EGFR) pathways could potentially enhance the effects of RT. In a study on human head and neck cancer xenografts, the combination of the VEGF inhibitor, AZD2171, the EGFR tyrosine kinase inhibitor (TKI), gefitinib, with RT led to much greater inhibition of tumor growth compared to RT alone, or compared to the two-drug combination.[53] This study provided a sound rationale for dual inhibition of the VEGF and EGFR pathways in combination with RT, particularly in CRC where both pathways are effective targets in advanced disease.
Investigators at the University of Texas MD Anderson Cancer Center conducted a phase I trial in patients with clinical stage II-III rectal adenocarcinoma, evaluating preoperative chemoRT (50.4 Gy in 28 fractions with concurrent capecitabine), the anti-VEGF-A antibody, bevacizumab, and the EGFR TKI, erlotinib.[54] Patients were administered bevacizumab (5 mg/kg every two weeks) with escalating dose levels of capecitabine (650–825 mg/m2 BID) and erlotinib (50–100 mg daily). The combination was well-tolerated with only 1 Grade 3 acute toxicity (hypertension), and 3 Grade 3 post-operative complications. Among 18 evaluable patients, the pCR rate was 44% with an additional 44% of patients with ≤ 10% viable tumor in the surgical specimen.
In a similar phase I/II trial at the Massachusetts General Hospital, 32 patients with stage II-III (T3–4 and/or node-positive) rectal cancer were treated with chemoRT (50.4 Gy in 28 fractions combined with infusional 5-FU [225 mg/m2/day]), bevacizumab (5 mg/kg every two weeks), and escalating doses of erlotinib (50–100 mg daily).[55] The rate of Grade 3–4 toxicity was higher in this trial (47%), however, the pCR rate was similarly robust at 33%.
Taken together, the consistent results of high pCR rates from these two independent clinical trials makes the data on EGFR and VEGF dual inhibition particularly interesting. However, proposals to develop these agents further were met with little enthusiasm given that a major limitation to these early clinical trials related to the lack of biomarker or correlative studies. While promising, future studies are needed to evaluate the clinical and biologic predictors of response to dual VEGF/EGFR targeting in combination with chemoRT.
Poly(ADP-ribose) polymerase (PARP)
DNA damage is continuously repaired by an integrated network of repair mechanisms. Double-stranded DNA breaks, which typically occur with radiation, recruit homologous recombination and non-homologous end-joining pathways for repair.[56] Single-strand breaks utilize another pathway including nucleotide-excision repair, base-excision repair and mismatch repair. Poly (ADP-ribose) polymerase (PARP) is a nuclear enzyme that rapidly recognizes and binds to single strand breaks and facilitates single strand DNA repair. Since there are at least two sets of mechanisms leading to DNA repair, the disruption of one pathway may not lead to cell death; however, when both pathways are disrupted the accumulated genomic instability leads to loss of viability. This concept, known as synthetic lethality, is supported by clinical studies in BRCA deficient cancers and triple negative breast cancer.[57, 58] PARP-1 inhibitors have been shown to enhance the efficacy of RT by interfering with compensatory DNA repair.[59, 60] The PARP inhibitor, veliparib (ABT-888) in combination with RT in HCT-116 CRC xenograft model, showed a near doubling in median survival compared to RT alone while ABT-888 alone was no better than vehicle.[61] A similar synergistic effect was demonstrated when veliparib was combined with 5-FU and RT compared to 5FU and RT alone in preclinical CRC xenografts.[62]
A phase I study testing the safety and tolerability of veliparib with capecitabine and RT in locally advanced rectal cancer (NCT01589419) enrolled patients with stage II-III rectal cancer. They received standard neoadjuvant chemoRT with capecitabine (825 mg/m2 BID) on days of RT (50.4 Gy in 28 fractions) with veliparib added at varying dose cohorts. Surgery was performed 5–10 weeks after completion of therapy. Of the 32 patients who received treatment per study protocol, 30 were included in the final analysis. Seventy-three percent of patients had histologic downstaging at the time of surgery, including a pCR rate of 28%. The NAR score for patients treated in the full dose cohort was 12.8. There were two documented DLTs (diarrhea and skin toxicity), neither of which compromised therapy. Twenty-eight percent of patients experienced any Grade 3 of 4 AE with diarrhea (9%) being the most common. There were no surgical complications attributed to the treatment. There was no clinical benefit to dose escalation beyond the recommended for phase II dose level (400mg PO BID) and pharmacokinetic data confirmed no interference with fluoropyrimidine therapy.[63] Importantly, there were no data provided on differential responses or toxicities based upon patient homologous recombination deficiency (HRD). Based on the promising phase I study results, veliparib as a radiosensitizer is the first experimental arm being explored in the NRG-GI002 randomized phase II trial platform in neoadjuvant treatment of locally advanced rectal cancer (NCT02921256) (Figure 2 and section below). Of note, more potent PARP inhibitors have recently become clinically available and may offer additional opportunities to refine this hypothesis in rectal cancer.
Figure 2.
Study schema for randomized phase II arms of NRG-GI002 (NCT02921256). Additional arms added through protocol amendments with comparison to the ongoing control arm.
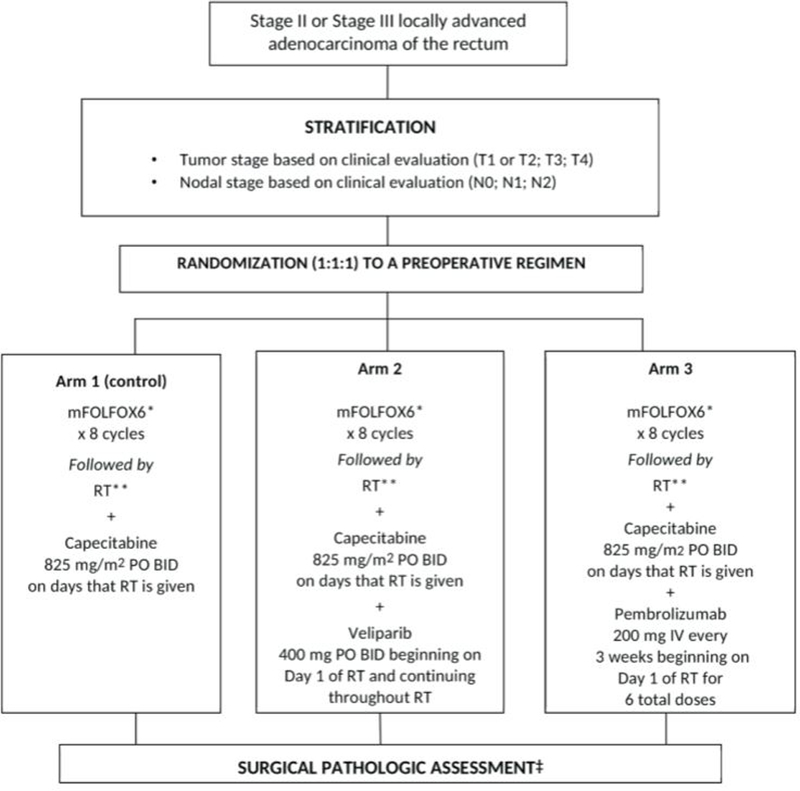
* modified FOLFOX6 regimen: oxaliplatin 85mg/m2 IV Day 1 + leucovorin 400mg/m2 IV Day 1 + 5-FU 400mg/m2 IV bolus followed by 5-FU 2400 mg/m2 continuous infusion over 46 hours every 2 weeks for 8 cycles
** RT starts 3–4 weeks following last dose of mFOLFOX6
4500 cGy in 25 fractions over 5 weeks + 540 cGy boost in 3 fractions: IMRT is allowed
‡ Surgery performed 8–12 weeks following last dose of radiotherapy
Ropidoxuridine (IPdR)
5-iodo-2-pyrimidinone-2′deoxyribose (Ropidoxuridine; IPdR) is a halogenated pyrimidine nucleoside that is a prodrug of the halogenated pyrimidine analog, iododeoxyuridine (IUdR). IPdR is administered orally with high bioavailability, and is metabolized to the active drug, IUdR, by an aldehyde oxidase principally in the liver. This metabolic profile is crucial to the improved therapeutic index of oral IPdR compared to continuous infusions of IUdR. The biochemical and cellular interactions of RT and fluoropyrimidines are felt to result from inhibition of the enzyme thymidylate synthetase (TS) by the fluoropyrimidine monophosphate metabolite, FdUMP, leading to decreased (or unbalanced) nucleotide pools needed for DNA synthesis and decreased DNA repair following RT damage. The intracellular IPdR monophosphate metabolite, IdUMP, can inhibit (by binding) TS, leading to increased fluoropyrimidine-mediated radiosensitization as well as enhancing IPdR-mediated radiosensitization secondary to IUdR-DNA incorporation. Thus, oral IPdR added to chemoRT is postulated to increase the pCR rate and overcome radioresistance.
After a first-in-human Phase 0 trial of a single dose of IPdR in patients with advanced malignancies confirmed the presence of adequate plasma levels of the active drug, IUdR, from the oral prodrug, IPdR, were achieved, the Phase I treatment trial was developed.[64] This dose finding study includes patients with GI cancers (mostly CRC) being treated with palliative RT. Patients receive escalating doses of once daily IPdR for 28 days with RT to 37.5 Gy (2.5 Gy daily fractions) starting on Day 8 of IPdR administration. Thus far, 16 patients have been treated without dose-limiting systemic toxicity.
Interestingly, in prior clinical trials of continuous infusion IUdR, steady-state IUdR plasma levels of >1μM were associated with myelosuppression and GI toxicities. In the current Phase I study (NCT02381561), patients who received 28 days of IPdR (1200 mg daily) had plasma levels of IUdR in the range of 2–5μM. While pharmacodynamic assessments are ongoing, the lack of toxicity seen with IPdR doses that achieve IUdR plasma levels of >1μM supports the observed improved therapeutic index seen in pre-clinical studies. Final results from this study are anxiously awaited to determine the clinical activity of this new biochemical intervention.
Rationale for Radiation-Immunotherapy Combination
Increasing evidence has been mounting to suggest that the immune system plays a major role in controlling tumor progression in CRC.[65] Microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic CRC is highly sensitive to checkpoint inhibition alone.[66, 67] Thus, current CRC combinatorial studies using RT with immunotherapy has focused on microsatellite stable (MSS) patients. The Immunoscore®, first described by Galon et al, was calculated based on the combined analysis of CD8+ plus CD45RO+cells in the center of the tumors and at the invasive margins, and was found to correlate with prognosis.[68–70] Interestingly, patients with greater numbers of tumor infiltrating CD8 and granzyme B-expressing T-cells (activated T-cells) had better survival compared to those with tumors that had lower numbers of infiltrating CD8 cells. Chemotherapy and RT can increase the immunogenic properties of tumor cells by enhancing MHC class I expression, thereby increasing their vulnerability to cytotoxic lymphocytes. Another frequent effect of treatment-associated DNA damage is the increased expression of death receptors, enabling lysis of the tumor cells by Fas/CD95 ligand and TRAIL-positive immune effectors.[71] Immunogenic cell death induced by RT involves the cell surface exposure of calreticulin, the release of the TLR4 agonist HMGB1, and ATP that trigger dendritic cell engulfment of dying cells, antigen presentation, and production of interleukin (IL)-1β, ultimately leading to activation of CD8+ T cells.[72–74] RT can also program the differentiation of iNOS+ M1 macrophages in the tumor microenvironment, supporting T cell recruitment into tumor tissue and successful tumor immune rejection through an NO-dependent mechanism.[75] [74, 76] Thus, the synergy between RT and immunomodulatory therapies is based on RT’s own immunomodulatory effects including inducing immunogenic cell death, releasing antigens for T cell priming, increasing MHC expression and antigen processing, upregulation of immunogenic cell surface markers, improving T cell homing to tumor sites, shifting the polarization of tumor associated macrophages, and destruction of immunosuppressive stromal cells in the tumor microenvironment.[72]
Anti-PD-1/PD-L1
Strong PD-L1 expression was observed in 30% of CRC and was found to correlate with infiltration by CD8 (+) lymphocytes which did not express PD-1.[77] Both RT and 5-FU induce the expression of PD-L1 on tumor cells through IFNγ production by CD8(+) T cells leading to an immune-suppressive environment and promoting PD-L1-mediated T-cell apoptosis.[78, 79] In a CRC xenograft model, concomitant but not sequential administration of fractionated RT in combination with anti-PD-1 or PD-L1 antibodies generated efficacious CD8(+) T-cell responses leading to better tumor response and survival compared to either modality alone.[78] These results could explain the adaptive RT resistance by tumors through expression of PD-L1. In fact, pre and post-chemoRT pathology from 93 matched-pair rectal cancer patients demonstrated CD8+ stromal tumor infiltrating lymphocyte (STL) density doubled after chemoRT, whereas FOXP3+ STL counts remained stable. High post-chemoRT CD8 + STL density was associated with better prognosis and a high pre-treatment CD8/FOXP3 intraepithelial tumor infiltrating lymphocyte ratio was a predictor of favorable tumor regression.[80] Additional pre-clinical studies have highlighted the immunomodulatory enhancing effects of RT with PD-1/PD-L1 checkpoint blockade demonstrating improved local, disease free and abscopal effects.[81–85]
Segal NH, et al. conducted a non-randomized phase II study of pembrolizumab (anti-PD-1 antibody) plus RT in MSS CRC (NCT02437071).[86] Patients with refractory metastatic disease underwent palliative RT followed by pembrolizumab (200 mg IV every 3 weeks). The primary objective was overall response rate (ORR) in a non-radiated lesion. Twenty-two patients were enrolled and one partial response was observed (ORR, 4.5%). The combination was very well tolerated.
There are currently two active multi-center clinical trials investigating PD-1/PD-L1 blockade as an immunomodulator leveraging radiation as the immune sensitizer in MSS rectal cancer. The NSABP FR-2 study is a phase II, open label study testing the safety and efficacy of durvalumab (anti-PD-L1) monotherapy following standard chemoRT in patients with stages II-IV rectal cancer (NCT03102047). This study uses a “window of opportunity” design whereby the immunotherapy is incorporated into the space where no treatment traditionally takes place after chemoRT and before surgical resection. This window represents the sweet spot for maximal tumor cytoreduction, optimal neoantigenic expression, and immunologic capacity when concurrent suppressive therapy is absent. Pembrolizumab is being tested as a concurrent adjunct to chemoRT in an experimental arm of the NRG-GI002 clinical trial (NCT02921256) (Figure 2 and section below). Pembrolizumab is administered concurrently with and following chemoRT. The primary endpoint for both studies is tumor downstaging (NAR Score) with correlative analyses to assess immunogenic response and tumor infiltrating lymphocyte (TIL) density.
The complimentary designs of these studies should allow determination of the optimal sequencing of immunotherapy with RT as well as the assessment of many potential biomarkers in the tumor microenvironment and the periphery. If successful, these studies may serve as a foundation to build upon given the rapid development of immune checkpoint inhibitors and alternate co-stimulatory and inhibitory molecules. Importantly, these studies include a number of exploratory analyses of immune biomarkers in an effort to inform the design of future studies and identify patients who may derive the most benefit from immunotherapy and RT.
Dual Inhibition of PD-(L)1 and CTLA-4
Some data suggests that the combination of RT with a single checkpoint inhibitor may not be adequate to activate or maintain activation of the immune system in MSS CRC. Anti-CTLA4 predominantly inhibits Treg cells, thereby increasing the CD8+ T cell to Treg ratio (CD8+/Treg). RT enhances the diversity of the T-cell receptor (TCR) repertoire of intratumoral T cells. This preclinical observation suggested that dual immune checkpoint blockade is required to best induce synergistic antitumor immunity with RT. Twyman-Saint Victor and colleagues reported that resistance to RT with anti-CTLA4 treatment was due to upregulation of PD-L1 on tumor cells, resulting in T-cell exhaustion in a murine melanoma model.[87] RT in combination with dual checkpoint blockade with anti-CTLA4 and anti-PD-L1/PD-1 showed significant synergistic antitumor activity in murine melanoma and pancreatic cancer models.[87] Importantly, sequencing of these biologic interventions may matter and need to be fully explored to optimize clinical outcomes.
There are two studies currently testing the role of dual immunotherapy checkpoint inhibitors with RT in advanced pMMR/MSS CRC. NSABP FC-9 is an ongoing phase II study of durvalumab plus tremelimumab following palliative hypofractionated RT in patients with refractory metastatic CRC (NCT03007407). The primary objective is to determine the anti-tumor efficacy of the dual immune checkpoint blockade via assessment of tumor response at unirradiated target lesions (i.e., a quantifiable abscopal effect). Following three doses of hypofractionated palliative RT (9 Gy x 3 on days −2, −1, and day 0 prior to cycle 1), patients receive both tremelimumab (75 mg IV) and durvalumab (1500 mg IV) on Day 1 for 4 cycles. Beginning with cycles 5 through 12, patients receive durvalumab alone (1500 mg IV) on Day 1 of each 28-day cycle. The sample size requires 21 evaluable patients to demonstrate a statistically significant increase in the objective response rate. This trial is nearly done enrolling patients.
Similarly, the NCI-sponsored ETCTN 10021 study is a phase II trial of durvalumab and tremelimumab with high or low-dose RT in patients with metastatic CRC or non-small cell lung cancer (NCT02888743). The CRC cohort of this trial will enroll similar patients as NSABP FC-9, but randomizes between provision of low-dose RT (0.5 Gy twice a day for 2 days) and 8 Gy daily for 3 days. Both modalities are followed by durvalumab and tremelimumab for cycles 1–4 and durvalumab monotherapy from cycle 5 onward. Together, these two trials are particularly well poised to explore the relative contributions of dual checkpoint inhibitor and perhaps an optimal way to deliver the antigenic release from RT.
Oncolytic Virus (T-VEC)
Talimogene laherparepvec (T-VEC; Imlygic™) is a herpes simplex-1 (HSV-1) derived oncolytic virus designed to selectively replicate in tumor cells causing cell lysis and to provoke anti-tumor immunity.[88, 89] T-VEC was modified from a clinical isolate of HSV-1 by disrupting the genes for neurovirulence (ICP34.5) and for immune-evasion (ICP47) allowing for tumor specific replication and increasing the local immune reaction, respectively. The latter is further increased by addition of a transgene for GM-CSF. T-VEC is administered as an intralesional injection by an initial priming dose for seroconversion followed by serial injections every 2 weeks at higher doses.[89] After injection and subsequent replication of T-VEC, cell death and lysis of cancer cells occurs with infection of surrounding cells in subsequent waves. The addition of GM-CSF leads to recruitment of immune cells and induction of T-cell mediated antitumor immunity while decreasing the number of regulatory T cells. Accumulating data also suggest the possibility of abscopal effects in distant metastatic lesions resulting from sustained systemic antitumor immunity from cytotoxic T cells that recognize tumor-specific antigens at the site of injection.[88] In an initial phase I study demonstrating safety, T-VEC obtained approval by multiple regulatory agencies for stages III and IVM1a melanoma showing improvement in overall survival.[90] Early clinical trials have combined T-VEC with RT in various tumor types, including a phase I/II study in stages III-IVB squamous cell cancer of the head and neck with promising outcomes.[91] Preclinical data showed increased activity of T-VEC in CRC cell lines compared to those from other primary sites (Kaufman et al, unpublished data). Furthermore, data suggests that RAS-driven cancer cells are more permissible to viral entry, thus offering a potential therapeutic advantage for this approach in a cohort of CRC with such mutations.[88, 92, 93]
In an ongoing NCI-sponsored (ETCTN 10058) dose-finding phase I trial (NCT03300544), patients with low-lying rectal adenocarcinomas (≤ 6 cm of anal verge) requiring neoadjuvant treatment are administered serial intratumoral T-VEC injections (x 4) via endoscopy in two dose escalation cohorts in combination with neoadjuvant chemotherapy (FOLFOX) followed by chemoRT (50.4 Gy in 28 fractions with capecitabine 825 mg/m2 bid on days of RT) (Figure 3). An expansion cohort is planned at the MTD with a total accrual of up to 18 patients. The primary objective is to determine the MTD of the combination and key secondary objectives include determining NARl score and MRI-response of the combination. Paired biopsies and blood samples will be obtained for correlative studies. Future studies may evaluate the role of T-VEC in combination with checkpoint inhibitors resectable rectal adenocarcinoma given strong data suggesting up regulation of PD-L1 after T-VEC injection.[94] Together, these novel approaches will further expand our collective understanding of how CRC immunogenicity might be successfully induced by RT, checkpoint inhibitors and oncolytic viruses.
Figure 3.
Sequence of interventions associated with neoadjuvant rectal cancer clinical trial (NCT03300544) incorporating TVEC.
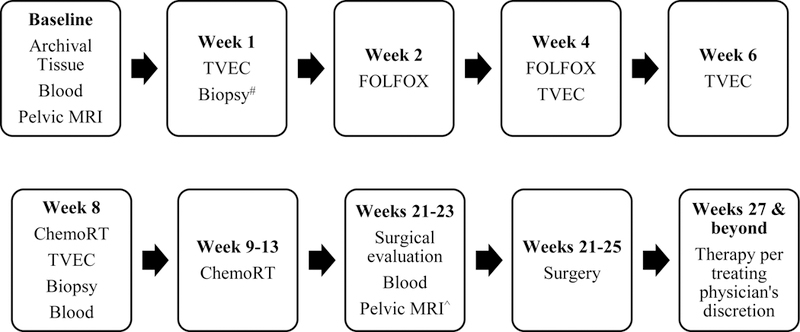
#Patients with inadequate archival tissue for integrated biomarker analysis
^As part of pre-surgical evaluation or at a corresponding time point in patients not undergoing surgery
+Dose escalation will be done using a 3+3 design for a total of 15 patients that will be treated at MTD
TVEC = talimogene laherparepvec
Additional Agents of Clinical Interest
Table 1 includes a number of candidate agents under clinical investigation to further improve outcomes in rectal cancer. Each of these agents is being tested in conjunction with the current chemoRT standard of care in rectal cancer (i.e., concurrent fluoropyrimidine with standard doses and fractionation of RT) and directly compared to that standard approach or to historical outcomes using that standard.
Table 1.
Contemporary clinical trials incorporating novel agents in combination with radiation in the treatment of colorectal cancer
| Target / Mechanism | Agent(s) | ClinicalTrial.gov identifier |
|---|---|---|
| VEGF TKI | Cediranib | NCT01160926 |
| VEGF MoAb + EGFR TKI | Bevacizumab + Erlotinib | NCT00307736 |
| Pan-target (VEGF) TKI | Sorafenib | NCT01376453 |
| Pan-target (VEGF) TKI | Lenvatinib | NCT02935309 |
| Protein kinase C | Midostaurin | NCT01282502 |
| Heat shock protein 90 | Ganetespib | NCT01554969 |
| PARP | Veliparib |
NCT01589419; NCT02921256 |
| MEK | AZD 6244; Trametinib |
NCT01160926; NCT01740648 |
| Pyrimidine analog | IUdR | NCT02381561 |
| Topoisomerase | Nano-particle Irinotecan (CRLX101) | NCT02010567 |
|
Programmed death receptor 1 (PD-1) |
Pembrolizumab | NCT02921256 |
|
Programmed death receptor ligand 1 (PD-L1) |
Durvalumab | NCT03102047 |
| Programmed death receptor ligand 1 (PD-L1) and CTLA4 | Durvalumab + Tremilimumab |
NCT02888743 NCT03007407 |
| Oncolytic virus | T-VEC | NCT03300544 |
Abbreviations: VEGF = vascular endothelial growth factor; TKI = tyrosine kinase inhibitor; MoAb = monoclonal antibody; EGFR = epidermal growth factor receptor; PARP = poly (ADP-ribose) polymerase; MEK = mitogen-activated protein kinase enzyme; IUdR = iododeoxyuridine; PD-1 = programmed death receptor 1; PD-L1 = programmed death receptor ligand 1; CTLA4 = cytotoxic T-lymphocyte-associated protein 4; T-VEC = talimogene laherparepvec
Future Directions in the Clinical Development of Novel Sensitizers
Despite the last few decades of clinical trials, pCR rates following neoadjuvant chemoRT in rectal cancer continue to hover in the low 20% range. To improve outcomes for patients and truly offer organ preservation or non-operative management, higher complete tumor sterilization rates are needed. Better understanding of the molecular signals, oncogenic drivers, and the surrounding tumor microenvironment involved in treatment sensitivity and persistent disease resistance may hold the may key to future progress. Areas of fruitful exploration include investigating combinations of DNA damage response inhibitors for tumors with DNA damage repair defects, as well as combinations of immune enabling drugs concurrently or after radiation in a pre-op window of opportunity approach. Incorporation of radiogenomics and more novel functional imaging techniques to assist in maximizing cancer control while minimizing acute and chronic toxicity are poorly resourced priorities. As such, pre-clinical hypothesis driven discovery is essential to support a subsequent scientifically rational clinical trial, but early collaboration between industry and academic partners can leverage the NCI Formulary and established CTEP CRADAs as part of a longitudinal effort towards clinical testing and ultimate FDA registration.
In order to facilitate development of novel radiosensitizers in rectal cancer, several barriers must be overcome. Table 2 identifies some of these challenges and offers potential solutions. While not exhaustive, if adequately resourced this inventory could significantly accelerate progress. For example, development of robust translational research requires access to match-paired tumor samples, serial collection of blood specimens and high quality radiographic imaging. Overcoming data sharing restrictions and biospecimen unavailability represents a major research infrastructure need that can be facilitated through coordination and collaboration of the above referenced clinical trials and with NCI support and resources. Future trials testing innovative neoadjuvant rectal cancer hypotheses should incorporate standardized data collection, patient reported outcomes and biospecimen procurement at consistent time points with centralized public access to the data and molecular determinants. Such guidance could be organized and resourced by the NCI through the NCTN, ETCTN and extramural research administrative supplement mechanisms. This centrally coordinated approach across trials will help to accelerate discoveries and validate mechanisms of treatment resistance that can be further exploited. Lastly, drug development with RT must have a clear pathway towards FDA registration to support industry investment in this treatment priority. The incorporation of high quality RT delivery as a component of a registrational pathway for new therapies is essential and can only be done in conjunction with high quality control, provision of RT delivery review, and traditional data monitoring of investigational agent toxicity in the context of radiation effects. Such quality control represents critical safeguards so that outcomes in a clinical trial are attributable to the novel investigation rather than differences in treatment delivery.
Table 2.
Select barriers and potential solutions to accelerate rectal cancer treatment outcomes as a component of neoadjuvant clinical trials.
| Barrier | Potential Solution(s) | Comments |
|---|---|---|
| Lack of translational and clinically annotated biospecimens for discovery and biomarker development | Collection of matched paired biospecimens including primary tumor | Mandate and resource tissue collection in all neoadjuvant clinical trials at key time points with central catalogue for investigator use |
| Limited ability to monitor treatment response or detect minimal residual disease | Circulating tumor DNA analyses | Mandate and resource blood collection for all neoadjuvant clinical trials at key time points |
| Limited ability to monitor treatment response or confirm tumor resolution | Radiographic imaging, including radiomic assessments | I. Establish and mandate standard imaging requirements and protocols for imaging performance, acquisition and centralized collection |
| II. NCI sponsored RAVE data collection and TRIAD imaging storage supports these functions, when utilized | ||
| Data silos | Data sharing requirements and use of NCI-sponsored metadatabases | I. This is being actively addressed through data sharing requirements for NCI-sponsored trials including utilization of RAVE |
| II. Other silos include individual academic center and industry-sponsored studies | ||
| Quality control in radiotherapy delivery | I. Consistently required technology and dosimetry standards built into trial protocols | The NCI supported Imaging and Radiation Oncology Core (IROC) supports these functions, when utilized |
| II. Central review of treatment plans | ||
| III. Clinical Trial site credentialing | ||
| Perceived lack of clear pathway for FDA registration | Template guidelines for registration with FDA and industry stakeholders | Precedent exists with temozolomide and cetuximab |
To help facilitate coordinated and rapid clinical testing, NRG-GI002 was developed by the NCI (NCT02921256). This NCTN multi-arm randomized phase II trial is designed as a master protocol with parallel experimental arms and correlative biomarkers. The study allows testing of a variety of novel hypotheses with neoadjuvant chemo and chemoRT in a consistent and homogenous high-risk patient population. Patients with locally advanced rectal cancer undergo total neoadjuvant therapy (TNT) with systemic chemotherapy (FOLFOX x 8 cycles) followed by chemoRT and then surgical resection (Figure 2). Each hypothesis is added as a protocol amendment as an experimental arm directly compared to a continuously enrolling control arm. The primary endpoint is a reduction in the Neoadjuvant Rectal (NAR) Cancer Score, an externally validated pathologic surrogate endpoint for OS and DFS in rectal cancer.[52, 95]
Conclusions
Rectal cancer represents nearly a third of CRC patients and has the added challenge of optimizing treatment to achieve organ preservation. The incorporation of novel radiosensitizers to increase the rate of tumor sterilization and/or induce an immunogenic response to improve overall survival represents contemporary efforts of investigators. The NCI’s Radiation Research Program Colorectal Cancer Working Group represents a collaborative unit of content experts and stakeholders that facilitate the recognition, prioritization and development of new therapies in this regard. Patients, and the field, will benefit from a coordinated approach to identify and test hypotheses with the most promising pre-clinical rational coupled with well controlled and randomized clinical validation.
Funding:
None
Footnotes
Author financial disclosures and conflicts of interest:
Thomas J. George- Research Support: Bristol-Myers Squibb, Merck, AstraZeneca/MedImmune, Lilly, Bayer, Incyte, Tesaro, Pharmacyclics, Seattle Genetics.
Aaron J. Franke- None
A. Bapsi Chakravarthy- None
Prajnan Das- Consultant: Eisai and Adlai Nortye.
Arvind Dasari- None
Bassel F. El-Rayes- Research Support- Merck, BMS, Synta Pharmaceuticals, Exilixis.
Theodore S. Hong- None
Timothy J. Kinsella- None
Jerome C. Landry- None
James J. Lee- None
Arta M. Monjazeb- Research Support: Genentech, Astra-Zeneca, Merck, Incyte, Dynavax, BMS
Samuel A. Jacobs – None
David Raben- None
Osama E. Rahma- None
Terence M. Williams- None
Christina Wu- Research Support: Vaccinex, Bristol Myers Squibb, Boston Biomedical Inc, Lycera, Seattle Genetics
C. Norman Coleman, Bhadrasain Vikram & Mansoor M. Ahmed- None
Contributor Information
A. Bapsi Chakravarthy, Department of Radiation Oncology, Vanderbilt-Ingram Cancer Center, Vanderbilt University Medical Center, Nashville, TN..
Prajnan Das, Department of Radiation Oncology, University of Texas MD Anderson Cancer Center, Houston, TX..
Arvind Dasari, Department of GI Medical Oncology, University of Texas MD Anderson Cancer Center, Houston, TX.
Bassel F. El-Rayes, Department of Hematology and Oncology, Winship Cancer Institute, Emory University, Atlanta, GA..
Theodore S. Hong, Department of Radiation Oncology, Massachusetts General Hospital, Harvard, Boston, MA.
Timothy J. Kinsella, Department of Radiation Oncology, Rhode Island Hospital-Brown University Alpert Medical School, Providence, RI.
Jerome C. Landry, Department of Radiation Oncology, Winship Cancer Institute, Emory University, Atlanta, GA.
James J. Lee, Division of Hematology-Oncology, Department of Medicine, UPMC Hillman Cancer Center, University of Pittsburgh School of Medicine, UPMC Cancer Pavilion, Pittsburgh, PA
Arta M. Monjazeb, Division of Radiation Oncology, UC Davis Comprehensive Cancer Center, Sacramento, CA
Samuel A. Jacobs, NSABP Foundation/NRG Oncology, Pittsburg, PA
David Raben, Department of Radiation Oncology, University of Colorado Denver School of Medicine, Aurora, CO..
Osama E. Rahma, Center for Immuno-oncology, Department of Medical Oncology, Dana Farber Cancer Institute and Harvard Medical School, Boston, MA.
Terence M. Williams, The Ohio State University, Department of Radiation Oncology, Columbus, OH.
Christina Wu, Department of Hematology and Oncology, Winship Cancer Institute, Emory University, Atlanta, GA..
References
- 1.Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA: a cancer journal for clinicians 2017. [DOI] [PubMed]
- 2.Lin SH, George TJ, Ben-Josef E, et al. Opportunities and challenges in the era of molecularly targeted agents and radiation therapy. Journal of the National Cancer Institute 2013;105(10):686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franke AJ, Parekh H, Starr JS, et al. Total neoadjuvant therapy: a shifting paradigm in locally advanced rectal cancer management. Clinical colorectal cancer 2018;17(1):1–12. [DOI] [PubMed] [Google Scholar]
- 4.George TJ, Allegra CJ, Yothers G. Neoadjuvant rectal (NAR) score: a new surrogate endpoint in rectal cancer clinical trials. Current colorectal cancer reports 2015;11(5):275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steel GG, Peckham MJ. Exploitable mechanisms in combined radiotherapy-chemotherapy: the concept of additivity. Int J Radiat Oncol Biol Phys 1979;5(1):85–91. [DOI] [PubMed] [Google Scholar]
- 6.Seiwert TY, Salama JK, Vokes EE. The concurrent chemoradiation paradigm--general principles. Nat Clin Pract Oncol 2007;4(2):86–100. [DOI] [PubMed] [Google Scholar]
- 7.Moding EJ, Kastan MB, Kirsch DG. Strategies for optimizing the response of cancer and normal tissues to radiation. Nat Rev Drug Discov 2013;12(7):526–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moding EJ, Kastan MB, Kirsch DG. Strategies for optimizing the response of cancer and normal tissues to radiation. Nature reviews Drug discovery 2013;12(7):526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katsanos KH, Briasoulis E, Tsekeris P, et al. Randomized phase II exploratory study of prophylactic amifostine in cancer patients who receive radical radiotherapy to the pelvis. Journal of Experimental & Clinical Cancer Research 2010;29(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunst J, Semlin S, Pigorsch S, et al. Intermittent use of amifostine during postoperative radiochemotherapy and acute toxicity in rectal cancer patients. Strahlentherapie und Onkologie 2000;176(9):416–421. [DOI] [PubMed] [Google Scholar]
- 11.Koukourakis MI, Giatromanolaki A, Tsoutsou P, et al. Bevacizumab, capecitabine, amifostine, and preoperative hypofractionated accelerated radiotherapy (HypoArc) for rectal cancer: a Phase II study. International Journal of Radiation Oncology• Biology• Physics 2011;80(2):492–498. [DOI] [PubMed] [Google Scholar]
- 12.Prolongation of the disease-free interval in surgically treated rectal carcinoma. Gastrointestinal Tumor Study Group. N Engl J Med 1985;312(23):1465–72. [DOI] [PubMed] [Google Scholar]
- 13.Wolmark N, Wieand HS, Hyams DM, et al. Randomized trial of postoperative adjuvant chemotherapy with or without radiotherapy for carcinoma of the rectum: National Surgical Adjuvant Breast and Bowel Project Protocol R-02. J Natl Cancer Inst 2000;92(5):388–96. [DOI] [PubMed] [Google Scholar]
- 14.Allegra CJ, Yothers G, O’Connell MJ, et al. Neoadjuvant 5-FU or capecitabine plus radiation with or without oxaliplatin in rectal cancer patients: a phase III randomized clinical trial. Journal of the National Cancer Institute 2015;107(11):djv248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaipl US, Multhoff G, Scheithauer H, et al. Kill and spread the word: stimulation of antitumor immune responses in the context of radiotherapy. Immunotherapy 2014;6(5):597–610. [DOI] [PubMed] [Google Scholar]
- 16.Postow MA, Callahan MK, Barker CA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. New England Journal of Medicine 2012;366(10):925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ngwa W, Irabor OC, Schoenfeld JD, et al. Using immunotherapy to boost the abscopal effect. Nature Reviews Cancer 2018. [DOI] [PMC free article] [PubMed]
- 18.Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. JNCI: Journal of the National Cancer Institute 2013;105(4):256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487(7407):330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duldulao MP, Lee W, Nelson RA, et al. Mutations in specific codons of the KRAS oncogene are associated with variable resistance to neoadjuvant chemoradiation therapy in patients with rectal adenocarcinoma. Ann Surg Oncol 2013;20(7):2166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer 2004;4(12):937–47. [DOI] [PubMed] [Google Scholar]
- 22.Russo M, Di Nicolantonio F, Bardelli A. Climbing RAS, the everest of oncogenes. Cancer discovery 2014;4(1):19–21. [DOI] [PubMed] [Google Scholar]
- 23.Sklar MD. The ras oncogenes increase the intrinsic resistance of NIH 3T3 cells to ionizing radiation. Science 1988;239(4840):645–7. [DOI] [PubMed] [Google Scholar]
- 24.Brunner TB, Cengel KA, Hahn SM, et al. Pancreatic cancer cell radiation survival and prenyltransferase inhibition: the role of K-Ras. Cancer Res 2005;65(18):8433–41. [DOI] [PubMed] [Google Scholar]
- 25.Bernhard EJ, McKenna WG, Hamilton AD, et al. Inhibiting Ras prenylation increases the radiosensitivity of human tumor cell lines with activating mutations of ras oncogenes. Cancer Res 1998;58(8):1754–61. [PubMed] [Google Scholar]
- 26.Williams TM, Flecha AR, Keller P, et al. Cotargeting MAPK and PI3K signaling with concurrent radiotherapy as a strategy for the treatment of pancreatic cancer. Molecular cancer therapeutics 2012;11(5):1193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dent P, Reardon DB, Park JS, et al. Radiation-induced release of transforming growth factor alpha activates the epidermal growth factor receptor and mitogen-activated protein kinase pathway in carcinoma cells, leading to increased proliferation and protection from radiation-induced cell death. Molecular biology of the cell 1999;10(8):2493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiao L, Yacoub A, McKinstry R, et al. Pharmocologic inhibitors of the mitogen activated protein kinase cascade have the potential to interact with ionizing radiation exposure to induce cell death in carcinoma cells by multiple mechanisms. Cancer Biol Ther 2002;1(2):168–76. [DOI] [PubMed] [Google Scholar]
- 29.Mak RH, Hermann G, Lewis JH, et al. Outcomes by tumor histology and KRAS mutation status after lung stereotactic body radiation therapy for early-stage non-small-cell lung cancer. Clin Lung Cancer 2015;16(1):24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grimminger PP, Danenberg P, Dellas K, et al. Biomarkers for cetuximab-based neoadjuvant radiochemotherapy in locally advanced rectal cancer. Clin Cancer Res 2011;17(10):3469–77. [DOI] [PubMed] [Google Scholar]
- 31.Russo AL, Ryan DP, Borger DR, et al. Mutational and clinical predictors of pathologic complete response in the treatment of locally advanced rectal cancer. J Gastrointest Cancer 2014;45(1):34–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong TS, Wo JY, Borger DR, et al. Phase II study of proton-based stereotactic body radiation therapy for liver metastases: Importance of tumor genotype. JNCI: Journal of the National Cancer Institute 2017;109(9). [DOI] [PubMed] [Google Scholar]
- 33.Yamaguchi T, Kakefuda R, Tajima N, et al. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol 2011;39(1):23–31. [DOI] [PubMed] [Google Scholar]
- 34.Lin SH, Zhang J, Giri U, et al. A high content clonogenic survival drug screen identifies mek inhibitors as potent radiation sensitizers for KRAS mutant non-small-cell lung cancer. J Thorac Oncol 2014;9(7):965–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Estrada-Bernal A, Chatterjee M, Haque SJ, et al. MEK inhibitor GSK1120212-mediated radiosensitization of pancreatic cancer cells involves inhibition of DNA double-strand break repair pathways. Cell Cycle 2015;14(23):3713–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shannon AM, Telfer BA, Smith PD, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) enhances the radiation responsiveness of lung and colorectal tumor xenografts. Clin Cancer Res 2009;15(21):6619–29. [DOI] [PubMed] [Google Scholar]
- 37.Chung EJ, Brown AP, Asano H, et al. In vitro and in vivo radiosensitization with AZD6244 (ARRY-142886), an inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 kinase. Clin Cancer Res 2009;15(9):3050–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urick ME, Chung EJ, Shield WP 3rd, et al. Enhancement of 5-fluorouracil-induced in vitro and in vivo radiosensitization with MEK inhibition. Clinical cancer research : an official journal of the American Association for Cancer Research 2011;17(15):5038–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dry JR, Pavey S, Pratilas CA, et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Cancer Res 2010;70(6):2264–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.https://clinicaltrials.gov/ct2/show/results/NCT01160926. Last Accessed: 07–20-2018.
- 41.Wu CS-Y, Williams TM, Wei L, et al. Phase I study of trametinib with neoadjuvant chemoradiation (CRT) in patients with locally advanced rectal cancers (LARC). In: American Society of Clinical Oncology; 2017. [Google Scholar]
- 42.Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. New England Journal of Medicine 2017;377(5):454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Q, Wang M, Kern AM, et al. Adapting a drug screening platform to discover associations of molecular targeted radiosensitizers with genomic biomarkers. Molecular Cancer Research 2015;13(4):713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong TS, Wo JY-L, Ryan DP, et al. Phase Ib study of neoadjuvant chemoradiation (CRT) with midostaurin, 5-fluorouracil (5-FU) and radiation (XRT) for locally advanced rectal cancer: Sensitization of RAS mutant tumors. Journal of Clinical Oncology 2018;36(15_suppl):e15674–e15674. [Google Scholar]
- 45.Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol 2017;18(6):345–360. [DOI] [PubMed] [Google Scholar]
- 46.Nagaraju GP, Park W, Wen J, et al. Antiangiogenic effects of ganetespib in colorectal cancer mediated through inhibition of HIF-1alpha and STAT-3. Angiogenesis 2013;16(4):903–17. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Trepel JB, Neckers LM, et al. STA-9090, a small-molecule Hsp90 inhibitor for the potential treatment of cancer. Curr Opin Investig Drugs 2010;11(12):1466–76. [PubMed] [Google Scholar]
- 48.He S, Smith DL, Sequeira M, et al. The HSP90 inhibitor ganetespib has chemosensitizer and radiosensitizer activity in colorectal cancer. Invest New Drugs 2014;32(4):577–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagaraju GP, Long TE, Park W, et al. Heat shock protein 90 promotes epithelial to mesenchymal transition, invasion, and migration in colorectal cancer. Mol Carcinog 2015;54(10):1147–58. [DOI] [PubMed] [Google Scholar]
- 50.Nagaraju GP, Alese OB, Landry J, et al. HSP90 inhibition downregulates thymidylate synthase and sensitizes colorectal cancer cell lines to the effect of 5FU-based chemotherapy. Oncotarget 2014;5(20):9980–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.El-Rayes BF SCA, Diaz R, Sullivan PS, Shaib WL, Landry JC Phase I study of ganetespib (G), capecitabine (C), and radiation (RT) in rectal cancer. In: ASCO Proceedings. Chicago, IL, 2015: Abstract 33, p. 3596 J Clin Oncol [Google Scholar]
- 52.George TJ Jr, Allegra CJ, Yothers G. Neoadjuvant rectal (NAR) Score: a new surrogate endpoint in rectal cancer clinical trials. Current colorectal cancer reports 2015;11(5):275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bozec A, Formento P, Lassalle S, et al. Dual inhibition of EGFR and VEGFR pathways in combination with irradiation: antitumour supra-additive effects on human head and neck cancer xenografts. Br J Cancer 2007;97(1):65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Das P, Eng C, Rodriguez-Bigas MA, et al. Preoperative radiation therapy with concurrent capecitabine, bevacizumab, and erlotinib for rectal cancer: a phase 1 trial. Int J Radiat Oncol Biol Phys 2014;88(2):301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blaszkowsky LS, Ryan DP, Szymonifka J, et al. Phase I/II study of neoadjuvant bevacizumab, erlotinib and 5-fluorouracil with concurrent external beam radiation therapy in locally advanced rectal cancer. Ann Oncol 2014;25(1):121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takahashi A, Ohnishi T. Does γH2AX foci formation depend on the presence of DNA double strand breaks? Cancer letters 2005;229(2):171–179. [DOI] [PubMed] [Google Scholar]
- 57.O’shaughnessy J, Osborne C, Pippen JE, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. New England Journal of Medicine 2011;364(3):205–214. [DOI] [PubMed] [Google Scholar]
- 58.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers. New England Journal of Medicine 2009;361(2):123–134. [DOI] [PubMed] [Google Scholar]
- 59.Brock WA, Milas L, Bergh S, et al. Radiosensitization of human and rodent cell lines by INO-1001, a novel inhibitor of poly (ADP-ribose) polymerase. Cancer letters 2004;205(2):155–160. [DOI] [PubMed] [Google Scholar]
- 60.Chalmers A, Johnston P, Woodcock M, et al. PARP-1, PARP-2, and the cellular response to low doses of ionizing radiation. International Journal of Radiation Oncology• Biology• Physics 2004;58(2):410–419. [DOI] [PubMed] [Google Scholar]
- 61.Donawho CK, Luo Y, Luo Y, et al. ABT-888, an orally active poly (ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clinical cancer research 2007;13(9):2728–2737. [DOI] [PubMed] [Google Scholar]
- 62.Shelton JW, Waxweiler TV, Landry J, et al. In vitro and in vivo enhancement of chemoradiation using the oral PARP inhibitor ABT-888 in colorectal cancer cells. International Journal of Radiation Oncology• Biology• Physics 2013;86(3):469–476. [DOI] [PubMed] [Google Scholar]
- 63.Michael M, Mulcahy MF, Deming DA, et al. Safety and tolerability of veliparib combined with capecitabine plus radiotherapy in patients with locally advanced rectal cancer (LARC): Final results of a phase Ib study. In: American Society of Clinical Oncology; 2015. [Google Scholar]
- 64.Kummar S, Anderson L, Hill K, et al. First-in-human phase 0 trial of oral 5-iodo-2-pyrimidinone-2′-deoxyribose in patients with advanced malignancies. Clinical Cancer Research 2013;19(7):1852–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maas M, Beets-Tan RG, Lambregts DM, et al. Wait-and-see policy for clinical complete responders after chemoradiation for rectal cancer. Journal of clinical oncology 2011;29(35):4633–4640. [DOI] [PubMed] [Google Scholar]
- 66.https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm560167.html. Last Accessed June 13, 2018.
- 67.https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/125554Orig1s034ltr.pdf. Last Accessed June 13, 2018.
- 68.Pages F, Kirilovsky A, Mlecnik B, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. Journal of Clinical Oncology 2009;27(35):5944–5951. [DOI] [PubMed] [Google Scholar]
- 69.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006;313(5795):1960–1964. [DOI] [PubMed] [Google Scholar]
- 70.Bigot F, Castanon E, Baldini C, et al. Prospective validation of a prognostic score for patients in immunotherapy phase I trials: the Gustave Roussy Immune Score (GRIm-Score). European Journal of Cancer 2017;84:212–218. [DOI] [PubMed] [Google Scholar]
- 71.Devaud C, John LB, Westwood JA, et al. Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy. Oncoimmunology 2013;2(8):e25961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Esposito A, Criscitiello C, Curigliano G. Immune checkpoint inhibitors with radiotherapy and locoregional treatment: synergism and potential clinical implications. Curr Opin Oncol 2015;27(6):445–51. [DOI] [PubMed] [Google Scholar]
- 73.Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell 2010;140(6):798–804. [DOI] [PubMed] [Google Scholar]
- 74.Golden EB, Pellicciotta I, Demaria S, et al. The convergence of radiation and immunogenic cell death signaling pathways. Front Oncol 2012;2:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klug F, Prakash H, Huber PE, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013;24(5):589–602. [DOI] [PubMed] [Google Scholar]
- 76.Postow MA, Callahan MK, Barker CA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med 2012;366(10):925–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Droeser RA, Hirt C, Viehl CT, et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. European journal of cancer 2013;49(9):2233–2242. [DOI] [PubMed] [Google Scholar]
- 78.Dovedi SJ, Adlard AL, Lipowska-Bhalla G, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer research 2014;74(19):5458–5468. [DOI] [PubMed] [Google Scholar]
- 79.Zhang P, Su D-M, Liang M, et al. Chemopreventive agents induce programmed death-1-ligand 1 (PD-L1) surface expression in breast cancer cells and promote PD-L1-mediated T cell apoptosis. Molecular immunology 2008;45(5):1470–1476. [DOI] [PubMed] [Google Scholar]
- 80.Shinto E, Hase K, Hashiguchi Y, et al. CD8+ and FOXP3+ tumor-infiltrating T cells before and after chemoradiotherapy for rectal cancer. Annals of surgical oncology 2014;21(3):414–421. [DOI] [PubMed] [Google Scholar]
- 81.Deng L, Liang H, Burnette B, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 2014;124(2):687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dovedi SJ, Adlard AL, Lipowska-Bhalla G, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res 2014;74(19):5458–68. [DOI] [PubMed] [Google Scholar]
- 83.Park SS, Dong H, Liu X, et al. PD-1 Restrains Radiotherapy-Induced Abscopal Effect. Cancer Immunol Res 2015;3(6):610–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sharabi AB, Nirschl CJ, Kochel CM, et al. Stereotactic Radiation Therapy Augments Antigen-Specific PD-1-Mediated Antitumor Immune Responses via Cross-Presentation of Tumor Antigen. Cancer Immunol Res 2015;3(4):345–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lim YJ, Koh J, Kim S, et al. Chemoradiation-Induced Alteration of Programmed Death-Ligand 1 and CD8+ Tumor-Infiltrating Lymphocytes Identified Patients With Poor Prognosis in Rectal Cancer: A Matched Comparison Analysis. International Journal of Radiation Oncology* Biology* Physics 2017;99(5):1216–1224. [DOI] [PubMed] [Google Scholar]
- 86.Segal NH, Kemeny NE, Cercek A, et al. Non-randomized phase II study to assess the efficacy of pembrolizumab (Pem) plus radiotherapy (RT) or ablation in mismatch repair proficient (pMMR) metastatic colorectal cancer (mCRC) patients. Journal of Clinical Oncology 2016;34(15_suppl):3539–3539. [Google Scholar]
- 87.Twyman-Saint Victor C, Rech AJ, Maity A, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015;520(7547):373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 2015;14(9):642–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Harrington KJ, Puzanov I, Hecht JR, et al. Clinical development of talimogene laherparepvec (T-VEC): a modified herpes simplex virus type-1-derived oncolytic immunotherapy. Expert Rev Anticancer Ther 2015;15(12):1389–403. [DOI] [PubMed] [Google Scholar]
- 90.Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 2015;33(25):2780–8. [DOI] [PubMed] [Google Scholar]
- 91.Harrington KJ, Hingorani M, Tanay MA, et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res 2010;16(15):4005–15. [DOI] [PubMed] [Google Scholar]
- 92.Kohlhapp FJ, Kaufman HL. Molecular Pathways: Mechanism of Action for Talimogene Laherparepvec, a New Oncolytic Virus Immunotherapy. Clin Cancer Res 2016;22(5):1048–54. [DOI] [PubMed] [Google Scholar]
- 93.Yang H, Peng T, Li J, et al. Treatment of colon cancer with oncolytic herpes simplex virus in preclinical models. Gene Ther 2016;23(5):450–9. [DOI] [PubMed] [Google Scholar]
- 94.Piasecki JRJ, Le T. Talimogene laherparepvec activates systemic T-cell-mediated anti-tumor immunity In: AACR, 2015: Abstract 75. Cancer Research
- 95.Fokas E, Fietkau R, Hartmann A, et al. Neoadjuvant rectal score as individual-level surrogate for disease-free survival in rectal cancer in the CAO/ARO/AIO-04 randomized phase III trial. Annals of Oncology 2018;29(7):1521–1527. [DOI] [PubMed] [Google Scholar]