

Abstract
The discovery and elucidation of genetic codes have profoundly changed not only biology but also many fields of science and engineering. The fundamental building blocks of life comprises of four simple deoxyribonucleotides and yet their combinations serve as the carrier of genetic information that encodes for proteins that can carry out many biological functions due to their unique functionalities. Inspired by nature, the functionalities of DNA molecules have been used as a capping ligand for controlling morphology of nanomaterials and such a control is sequence dependent, which translates into distinct physical and chemical properties of resulting nanoparticles. Herein, we provide an overview on the use of DNA as engineered codes for controlling the morphology of metal nanoparticles, such as gold, silver and Pd-Au bimetallic nanoparticles. Fundamental insights into rules governing DNA controlled growth mechanisms are also summarized, based on understanding of the affinity of the DNA nucleobases to various metals, the effect of combination of nucleobases, functional modification of DNA, the secondary structures of DNA and the properties of the seed employed. The resulting physical and chemical properties of these DNA encoded nanomaterials are also reviewed, while perspectives into the future directions of DNA mediated nanoparticle synthesis are provided.
Keywords: DNA, metal nanoparticles, shape-control, sequence-specificity
1. Introduction:
The synthesis and characterization of colloidal metal nanoparticles is an area that has garnered much interest over the past years, because the properties that these nanoparticles exhibit at the nanoscale have demonstrated enormous potential in applications that span many diverse areas such as medicine,[1–5] photonics,[6–8] catalysis,[9–15] sensing[16–19] and electronics.[20, 21] The arrangement of atoms in such a nanoscale-confined environment has played a major role in determining the physical and chemical properties of these metal nanoparticles.[21–25] Hence, the ability to precisely control their morphologies, such as shape and surface structures is a major avenue to realize their full potential in a myriad of applications.
Among the methods to control the nanoparticle morphologies, solution-based synthesis methods involve regulation of kinetic and thermodynamic parameters.[26, 27] These parameters are often interdependent and can be meticulously tuned by the selection of different precursors,[28, 29] temperatures,[30, 31] solvents,[32, 33] pH,[34–37] additives,[21, 23, 25, 38, 39] and capping ligands.[40, 41]Among all those parameters, the capping agent has been the center of interest. The capping ligand is often involved in a range of functions, including binding to a specific facet of the nanocrystals, influencing precursor reduction and diffusion, and providing overall colloidal stability.[41–45] For example, commonly used capping ligands, such as cetyltrimethylammonium bromide/chloride (CTAB/CTAC), citrate, polyvinylpyrrolidone (PVP) and polyols, have shown the ability to bind specific facets of noble metals such as {100} or {111} independently or when complemented with other ligands such as halides ions (for example, Br− is known to stabilize the {100} facet).[46–48] While these capping ligands have been successful in controlling morphologies of metal nanoparticles, the lack of systematic variations and fine tuning of their structures, charges or functional groups largely limits their ability to accurately control nanoparticle morphology.
To overcome the above limitation, materials scientists and engineers have resort to biomolecules[49] that have been perfected through evolution, such as amino acids,[42] peptides,[50–52] protein,[44, 53] virus capsids[54] and nucleic acids[55] to be used in the synthesis of nanomaterials. Among them, the unique structure of DNA showcases several tunable properties such as charge, length, affinity and functional groups,[56] and these properties have been exploited extensively in the field of self-assembly,[57, 58] sensing and imaging.[17, 59, 60] Recently, they have also been utilized as capping ligands to precisely control the morphologies of nanomaterials during the synthesis.[61, 62]
This review aims to summarize the recent progress in sequence specific DNA-mediated control of metal nanoparticle morphologies. We will briefly discuss the DNA-mediated synthesis of metal nanoparticles, and focus on highlighting the importance of sequence dependent “DNA-encoded” morphological control of metals at the nanoscale. The process by which the DNA of different sequences employed determines the final shape of metal nanoparticles is quite complex and yet very elegant. The challenges thus faced in understanding such a system, along with solutions and promising approaches will be discussed. Finally, the functional properties of these DNA encoded metal nanoparticles will be presented to show promises of this class of materials for many applications.
1.1. Why use DNA in the synthesis of colloidal metal nanoparticles?:
DNA has traditionally been considered as a storehouse of genetic information, which consists of different combinations of four deoxyribonucleotides (G, A, T and C). The elucidation of genetic codes is a major milestone in biology that allows the DNA to encode biological functions. From a material chemist’s perspective, the structure of DNA consisting of the sugar–phosphate backbone and the nitrogenous bases can be considered as a well-designed blend of hydrophobic and hydrophilic functional groups. The property of single stranded DNA (ssDNA) to be able to bind metal ions was used extensively in the synthesis of Ag nanoclusters.[63–65] Shortly after, studies proved that the fluorescence emission from these clusters can be tuned by altering the DNA sequence used for the nanocluster synthesis,[66, 67] highlighting the importance of the DNA sequences in material synthesis. The methodology was then quickly extended to other noble metal particles and adopted for different applications, such as in sensing.[68]
Functional modifications, such as amine, biotin and thiol, can be incorporated into the DNA sequence without effecting the base pairing ability of that particular sequence. Such modifications are especially useful in integrating DNA with other materials such as gold nanoparticles[69, 70] and silica particles.[71, 72] The backbone can also be modified to phosphorothioate (PS) to maintain the negative charge or to peptide nucleic acids (PNA) that exhibits a neutral backbone. The four canonical nucleobases can also be replaced with modified bases, with either one of functional groups in the canonical nucleobases deleted, substituted, or replaced with a new functional group. For example, HdA, IdA, HdC, mdC, AdT and AdG are among many nucleobases analogues that can be synthesized (Figure 1). While certain organic polymers used in metal nanoparticle synthesis can be designed to have charges and functional groups distributed on their backbone, the length or molecular weight of such polymers cannot be controlled as precisely as with DNA. The superior control over the structure of DNA makes it a powerful candidate in directing the shape of the nanomaterials, and can possibly lead to generating novel morphologies and properties with very fine control.
Figure 1.
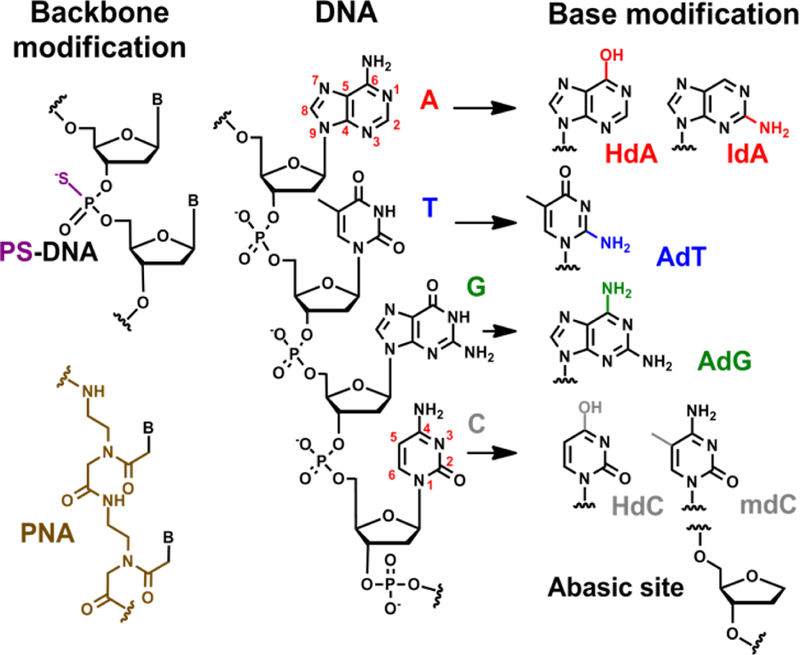
Schematic demonstrating a few amongst many functional modifications that can be made to the different components of DNA, i.e. the phosphate backbone and the nucleobases.
2. Discovery of DNA codes for controlled synthesis of metal NPs:
The process by which the DNA genetic code holds accurate information about the translation of DNA into RNA and then into amino acids for proteins is highly sophisticated. It is known that the sequence plays a pivotal role in determining the end product; even a single mutation can result in a missing, misfolded or malfunctioning protein. By the virtue of intricate functionality of DNA and its transcribed products, molecules interact specifically, making possible the synthesis of highly complex molecules. Inspired by this process in nature, we have explored how a specific sequence of DNA can influence the final morphologies and surface structure of a particular nanomaterial when present in the synthesis. In other words, we aimed to discover and elucidate DNA “codes” for morphologies of abiological nanomaterials. The ultimate goal is to be able to predict the final morphology of the nanomaterials simply based on the specific DNA sequences and nanomaterial precursors used, exactly like how we are now able to predict protein structures with genetic codes. While we are far from being able to predict the final nanostructures at this point, considerable progress has been made to understand such synthetic systems. A DNA- mediated synthesis involves the DNA directed deposition of metal on a pre-synthesized seed. To provide information regarding the morphology transition, growth mechanisms and properties, electron microscopy-based imaging techniques such as SEM and TEM (Scanning Electron Microscopy and Transmission Electron Microscopy) have been used to help characterize the final shape of the metal nanoparticles. Furthermore, time-based electron microscopy studies have shed light on how the nucleation and growth of the seed takes place in the presence of DNA. In addition, absorption spectra of the system has allowed not only the monitoring of the SPR properties of the metal nanoparticles but also analysis of the structure-function relationship between the morphology of the nanoparticle and the SPR during the course of the growth. How each of these techniques provide information on the DNA encoded nanoparticles will be described in details in the later sections that discuss the growth mechanisms. In this section, we will describe these systems, classified by the seed species employed in the synthesis.
2.1. DNA encoded synthesis based on spherical gold nanoparticles (AuNPs) seed:
Spherical AuNPs are easily among the most well studied Au-based nanomaterials in terms of synthesis and characterization.[73–76] The surface of these AuNPs have been modified extensively with ligands, including DNA,[77] for self-assembly[69, 70] and other applications.[78] The affinities of DNA to the surface of spherical AuNPs have also been studied to reveal that dT has a lower binding affinity as compared to dA, dC or dG.[79] The translation of the DNA affinity into the final morphology of particles when oligomers of DNA with different sequences were used, showed remarkable differences.[80] While 30-mer of dT resulted in spherical AuNPs slightly larger in size, 30-mer of dA and dC resulted in nano-flower (AuNF) (Figure 2A). The uneven growth characteristic of the AuNF that dA and dC imposed can be attributed to the higher binding affinity of the nitrogenous nucleobases to the surface of the spherical gold seed. In contrast, a sequence like oligo-T with low binding affinity only manages to colloidally stabilize the final nanoparticle allowing just a simple overgrowth to a bigger spherical nanoparticles, without influencing the shape. These uneven features had a direct influence on the plasmonic properties of the nanoparticles that were red shifted, where the AuNFs formed by oligo-A had a more red shifted peak compared to oligo-C capped particles. As a result, the AuNF colloidal solutions were blue in color. While the affinity of the DNA did make a difference in the final shape, the adsorbed and implanted DNA on the particle surface contribute to the colloidal stability of the AuNPs.
Figure 2.
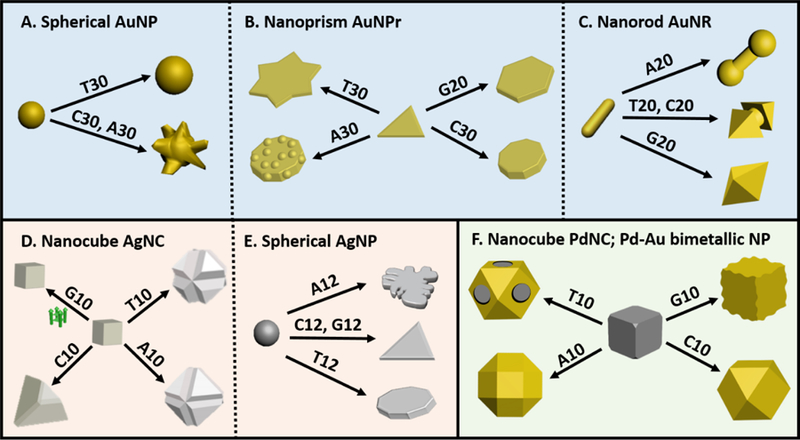
Schematic representing the different nanoparticle systems studied via DNA mediated shape control approach. Systems using gold nanoparticles, specifically spherical AuNP (1A), Au Nanoprism (1B) and Au Nanorod (1C); silver nanoparticles, specifically, Ag Nanocube (1D) and Spherical AgNP (1E); and Bimetallic nanoparticles (Pd-Au) (1F).
2.2. DNA encoded synthesis based on gold nano prisms (AuNPr) seed:
The previous study with spherical AuNPs established that DNA-mediated shape control was indeed possible. As an extension to demonstrate the precise level of nanoparticle shape tunability, AuNPr was used as the seed, because the seed presented different facets than spherical AuNPs. The AuNPr was synthesized using a previous protocol[81] and their overgrowth was monitored in the presence of 30-mer of dA, dC, dT and a 20-mer of dG. The anisotropic nature of the seed, makes the monitoring of shape evolution easier from the surface and the edges. The presence of each oligonucleotide resulted in a unique shape. Specifically, rough-round plates, six-pointed stars, round plates and hexagonal plates were formed in the presence of A30, T30, C30 and G20, respectively (Figure 2B and 3B).[82] The {111} surface was preserved in each case except in the formation of the roughing of the surface in the presence of A30. In addition, the growth in the presence of different lengths of the oligonucleotides (5, 10 and 20-mer of G and 30-mer of other oligonucleotides) yielded similar morphologies, suggesting that the sequence of the oligonucleotides, rather than the length, plays an important role in morphology control. However, the synthesis in the presence of monomeric deoxyribonucleotides, i.e. dAMP, dTMP, dGMP and dCMP led to formation of unstable structures that aggregated due to lack of colloidal stability. Therefore, the length of the oligonucleotides should be long enough (generally above 5 nucleotides) to maintain colloidal stability of the nanostructure of desired morphology.
Figure 3.

A). Circular Dichroism (CD) measurements demonstrating the disruption and formation of the i-motif and the G-quadruplex during the overgrowth of Ag nanocubes in the presence of C10 and G10 respectively. (Parts of the figure reproduced with permission.[85] Copyright 2014 American Chemical Society). B). Scheme showing different AuNP morphologies grown from Au prism seeds in the presence of different DNA sequences (the sizes ~200 nm). (Reproduced with permission.[82] Copyright 2012 Wiley-VCH). C). Overgrowth of AuNR overgrowth using different DNA sequence block combinations, monitored by UV/Vis absorption and SEM. Scale bars=20 nm. (Reproduced with permission.[84] Copyright 2015 Wiley-VCH)
2.3. DNA encoded synthesis based on gold nanorods (AuNR) seed:
AuNRs are anisotropic nanoparticles that exhibit unique absorption and scattering properties in the near infrared (NIR) region. Owing to the anisotropic nature, AuNRs exhibit both longitudinal and transverse localized surface plasmon resonance (referred to as L-LSPR and T-LSPR, respectively) characterized by the AuNR aspect ratio[46] and these anisotropic optical properties have been utilized for optical[23] and biomedical applications.[83] The overgrowth of the AuNRs in the presence of homo-oligomeric 20-mer of DNA not only lead to different geometries, but also result in plasmonic spectral tuning.[84] In the case of A20, the AuNR overgrowth yielded a dumbbell shaped particle (Figure 2C). The tips of the nanorod had more deposition resulting in the red shift of the L-LSPR. The increase in the thickness of the rod slightly shifts the T-LSPR wavelength band. C20 and T20 both resulted in the formation a cracked octahedron structure with difference in the gap between the two pointed features. While the L-LSPR bands for structures formed by both oligonucleotides were blue-shifted, this particular difference in gap allows the L-LSPR peaks to be different. Specifically, the shorter the gap, as observed in the presence of C20 (17±3 nm gap as compared to 24±3 nm in the case of T20), the more is the blue shift. When the gap is close to zero, the L-LSPR is blue-shifted to the extent that the L-LSPR merges with the T-LSPR to give a single unified LSPR peak, which is observed in the presence of G20. The system highlights how subtle changes in the deposition profile can directly influence not only the geometric but also physical properties.
2.4. DNA encoded synthesis based on silver nanocubes (AgNCs) seed:
The interaction of DNA with metal NPs is expected to change depending on the identity of the metal. While most studies have been focused on gold-based systems, it is important to be able to expand the metal NPs to make the above approach more generalizable. DNA-mediated Ag overgrowth on AgNCs as seeds, was the first system to demonstrate the DNA sequence dependent nature of Ag nanoparticle growth.[85] Not surprisingly, the trend of affinity of the different oligonucleotides to Ag was different from that of Au. A previous study has reported the trend of the affinity to AgNPs to be in the order, C > G > A > T.[86] The presence of A10 and T10 resulted in formation of stellated structures, specifically truncated octahedra with varying degrees of truncations depending on the sequence. The presence of T10 induced smaller particles with larger truncations, while A10 induces larger particles with smaller edge-truncations. The overgrowth of AgNCs in the presence of C10 formed truncated tetrahedra and the presence of G10 interestingly, did not allow any overgrowth of the seed (Figure 2D). The stabilization of the {111} facet by the DNA containing T, A and C resulted in the gradual disappearance of the {100} facet that was initially the dominant facet on the AgNC seed. Further investigations suggested the involvement of formation or destruction of secondary structures formed by the DNA sequences during the course of the reaction, indicating the structurally dynamic nature of the DNA strands in the growth solution. The importance of having to consider the substantial role of secondary structure of DNA molecules in control of NP morphologies was demonstrated in this study.
2.5. DNA encoded synthesis based on spherical silver nanoparticles (AgNPs) seed:
Most recently, Yang and coworkers have reported DNA-mediated shape evolution of relatively small spherical AgNPs, in the presence of citrate ligand, to anisotropic morphologies such as nanoprisms. Oligo-G and oligo-C mediated the formation of nanoprisms, while oligo-T and oligo-A resulted in formation of nanodiscs and 2-D nano flower bouquets, respectively (Figure 2E). The spherical AgNP seed is generally known to have multiple {111} and {100} facets.[87] The presence of citrate allows the spherical seed to form multiple twinned particles. Similar to the case of AgNCs which are enclosed mainly by {100} facets and {111}, {110} sites, the DNA had a tendency to bind to the {111} facet of the spherical seed and further stabilize it. Hence, the {111} facet dominates the final 2-D structures. Although the oligo-C would be expected to have a dominant effect on the overgrowth of the Ag spherical nanoparticles, the most dominant effect was produced by oligo-A sequence, due to the stronger binding affinity of oligo-A with higher energy facets (twinned surface) and the lower mobility of the sequence on {111} crystal face, leading to the growth of 2D triangular silver bouquets.
2.6. DNA encoded synthesis of Pd-Au bimetallic nanoparticles:
The generalizability of the DNA-encoded nanomaterials synthetic approach was further demonstrated, when A10, T10, G10 and C10 was used to control the morphology of bimetallic nanoparticles, specifically Pd-Au nanoparticles.[88] Pd nanocubes were used as the seed, and in the presence of DNA a co-reduction of Pd and Au precursors was performed. The nanocube seed is characterized mainly by six {100} faces and additionally {110} and {111} sites, i.e. the truncation of edges and the corners, respectively. The higher surface energy of the {110} and {111} sites directs both binding of oligonucleotides sequences and deposition of incoming metal. Those sequences with lower affinity to the seed such as T10 cannot effectively passivated the high energy sites which allows deposition on the high energy sites to form a Pd-Au core-frame structure, i.e. the metal deposits along the corners and the edges. Those with higher binding affinity, such as A10 bind to the higher energy sites and passivate the surface to lower their energy, resulting in a complete coverage of the seed. Therefore rhombi cuboctahedron, cuboctahedron and undulated Pd-Au shell over the seed were formed in the presence of A10, C10 and G10, respectively (Figure 2F). Metal atom diffusion on the seed also plays an important role in directing the morphology and is also simultaneously influenced by the DNA binding affinities. Interestingly, A10, the sequence with highest binding affinity to gold, can stabilize Au surface with higher energy, to allow the formation of smaller nanocrystallites that eventually coalesce into bigger particles that deposit onto the seed. This phenomenon is not observed in the case of the other DNA bases.
The ability of DNA to control the morphology of Pd-Au nanoparticles was further tested when the seed was changed from a low-index faceted nanocube to a high-indexed concave nanocube. The concave cube was different in that it had extended edges and corners in the <111> and the <110> direction while the {100} facets were retained at the center.[89] DNA was ultimately successful in controlling the final morphology of the Pd@Au core-shell nanocrystals but the effect of the high-energy faceted seed cannot be ignored.[90] The presence of T10 caused the formation of a gold shell with least deposition of gold along the {100} facet and more deposition in the <111> and <110> directions. The G10-encoded structures had rounded vertices, while the edges of C10-encoded structure formed octapods with octahedron-like structures consisting of prominent {111} surfaces. Finally, A10 resulted in the formation of a gold shell with an overall rough surface. The initial metal deposition is dictated almost entirely by the seed’s high surface energy. After an initial deposition, DNA controls the morphology in a sequence dependent manner. Similar to the cubic system, A10 enable the formation of smaller nanocrytsallites before the deposition onto the seed. The fine control displayed in tuning the morphology of the shell demonstrates the immense potential that DNA has in shaping hybrid materials that possess disparate properties.
3. Insights into DNA-encoded metal nanoparticle morphologies:
It is evident, from the above examples, that different sequences of the DNA, reflected by their different chemical composition, and hence the structural properties, play an important role in defining the final morphology of the metal nanoparticles. The interaction between the DNA molecules and the metal atoms can be elucidated to a large extent using chemical and physical methods. Understanding the complexity of the DNA structure and their interaction with metal nanoparticles on a molecular level allows for deeper insights into DNA-mediated nanoparticle growth mechanisms.
3.1. The structure of DNA:
As mentioned earlier, the sugar-phosphate backbone and the nucleobases constitute to the framework of DNA. The nucleotide formed by sugar-phosphate group and one of the four nucleobases is the recurring unit in a given sequence (Figure 1). Each nucleotide unit changes with respect to the nucleobase. The majority of the studies that involve a DNA-encoded synthesis use DNA molecules that are homo-oligomeric in nature, i.e. the nucleobase of the recurring nucleotide unit is kept the same for each individual study. Choosing these homo-oligomeric DNA aids deciphering the structural elements of each nucleobase involved in stabilizing a particular facet.
Although naturally derived, the ssDNA structurally has many aspects that easily make it qualify as a capping agent. First, the phosphodiester backbone contributes to the negative charge and thus the hydrophilicity. The negative charge is expected to induce charge repulsion between different nanoparticles thus providing colloidal stability. Therefore, the extended oligomeric form of DNA is indeed necessary for the stability of metal nanoparticle. Nanoparticles synthesized with just mononucleotides tended to aggregate.[82] Additionally, the minimum length of DNA required for imparting both colloidal stability and shape control was determine to be 5 bases, beyond which the length of DNA does not produce length-specific effects. Second, in comparison to the backbone, the aromatic rings of purines and pyrimidines in the nucleobases are more hydrophobic. Depending on the pKa values of the nucleobases, the pH and composition of the working solution, the nucleobases are either protonated or deprotonated.[91] The potential for metal binding stems from the structure of the DNA molecules. The nucleobase ring N and the keto O atoms that flank the purines and pyrimidines are involved in metal complexation.[56] The exocyclic amine groups also help in metal interaction, ostensibly through coordination.
In the most general structural form of double-stranded DNA (dsDNA), the nucleobases on both strands intertwine in an anti-parallel manner, through Watson-Crick hydrogen bonding to form a helical structure. The base-pairing interaction is specific in that, adenine and guanine (the purines) form hydrogen-bonds with thymine and cytosine (the pyrimidines), respectively. In this way, the functional group-rich nucleobases in dsDNA are mostly masked from interactions with the surrounding environment. As a result, the dsDNA’s sequence has little role to play in a DNA-encoded control of NP morphologies and almost all DNA-encoded studies reported so far use ssDNA instead. dsDNA mediated nanoparticle synthesis has been studied separately, wherein the sequence of DNA plays no role.[92]
3.1.1. Affinity of the DNA bases to various metals:
The interaction of DNA with metal ions has been extensively studied.[93–97] While this interaction is important, what carries more weight in a DNA-encoded control NP morphology is the interaction of DNA with metal surfaces. A variety of techniques have been used in the past to probe the nucleobase affinity for gold based surfaces.[98] Among the first examples, Mirkin and coworkers reported a temperature-programmed desorption (TPD) and reflection−absorption infrared (RAIR) spectroscopic study to calculate the heats of desorption (ΔHdes) of the individual nucleobases and nucleosides on a gold surface.[99, 100] The general trend for desorption revealed that pyrimidines tend to desorb more easily as compared to purines. More specifically, the trend observed with respect to the ΔHdes was guanine (G) > adenine (A) > cytosine (C) > thymine (T), which was in agreement with theoretical studies performed later.[101] An isothermal titration calorimetry (ITC) investigation of PNA-based monomer analogue binding to AuNPs showed the affinity order C > G > A > T.[102] Relevant to DNA-encoded synthesis of nanoparticles where single stranded homo-oligomeric DNA is used, Whitman and coworkers performed more detailed studies on the interaction of ssDNA on gold using Fourier transform infrared (FTIR) spectroscopy and X-ray photoelectron spectroscopy (XPS).[103] Competitive adsorption experiments on Au surfaces showed relative adsorption affinity of DNA nucleobases to be A > C ≥ G > T. This trend of affinity has been observed in investigating the underlying mechanisms of growth of AuNPs in presence of ssDNA sequences and shall be discussed in a later section.[104] A more recent study by Liu et al. involving cyclic voltammetry as a method to probe the adsorption of unmodified ssDNA at low pH showed that A and C have more effective adsorption to the gold surface compared to T and G due to the protonation of functional residues and higher affinity to gold surface.[105]
Reports of interaction of DNA nucleobases with other noble metal surfaces or nanoparticles are rare. Besides gold, only a few reports of interaction of DNA with AgNPs have been reported. The earlier study probed the nucleobase affinities based on surface plasmon resonance spectroscopy (SPRS) and the signal intensity of surface-enhanced Raman scattering (SERS) and determined the trend to be C > G > A > T.[86] The second study used solely colorimetric method to investigate the aggregation kinetics of AgNPs in the presences of nucleobases to obtain a different trend of A > T > C ≥ G.[106] The trends in binding strengths varied greatly between the two studies, probably due to the differences in experimental conditions and properties of AgNPs, such as their geometries, pre-existing capping ligands, and sizes). Thus, the study of DNA interaction with metal surfaces as opposed to nanoparticles is capable of providing more consistent trend in binding affinities, as demonstrated in the case of DNA interaction with gold surfaces. Despite the differences, what these studies highlight is that the conditions of the growth can potentially affect the trend of nucleobase affinities for metal nanoparticles and thus the outcome of a DNA-mediated growth. Additionally, to make DNA-mediated synthesis of noble-metal nanoparticles a more generalizable or predictable protocol, it is imperative to understand and study the affinities of the nucleotides to various surfaces of different metals. A few studies have reported DNA adsorption studies onto metal oxide particles,[107, 108] a class of materials that are slightly different from metal particles in that the metals exist in a non-zero oxidation state.
3.1.2. The effect of combination of nucleobases:
Clearly, the interaction of DNA depends on the nucleobases that are present in the sequence, which is a result of the interacting functional groups on the nucleobases present. While the use of homo-oligomers of DNA does provide for a very fine control of nanoparticle morphology, systematical programming the binding affinities of DNA can be achieved by combining nucleotides, which can further expand the tunable range of nanoparticle morphology. For a given length of a DNA, there is sufficient diversity available with respect to just nucleotide arrangement. A classic case is the used of combination sequences in the overgrowth of Au nanoprism. The use of homo-oligomers result in shapes that are very disparate. The use of nucleotide combinations help access morphologies whose features lie in between the morphologies resulting from any two homo-oligomers.[82] As shown in Figure 3B, surface effects by nucleotides includes roughening by C and A, edge thickening by G and flattening by T. Shape effects are more obviously observed when the homo-oligomers were used, i.e. round plates (A, C), hexagonal (G) and six pointed star (T).
A different, yet important aspect of tuning optical properties via nanoparticle shape control is evidenced when sequence combinations are used in the over-growth of Au nanorods. The study reveals A and G produce dominant effects with respect to shape-control in combination with C and T. The oligomeric G and A gave a blue-shifted L-LSPR at 551 nm and red-shifted L-LSPR at 856 nm, respectively. Increasing the A base in the combination sequence (starting from G20, A10G20, A15G15, A20G10 to A20) involving the two dominating bases produces particles that have L-LSPR peak red-shifted from 551 nm to 856 nm (Figure 3C). This implies that the L-LSPR can be precisely tuned between a given wavelength range by using the right sequence combination.
3.1.3. Functional modification of DNA:
Specific modification of a functional group in a DNA can bring about more predictable changes. For example, the thiol-containing capping ligands including alkyl-thiolated DNA are widely used to functionalize gold nanomaterials[109, 110] to impart special functionalities given the soft nature of the thiol ligands that have a higher affinity for the soft gold metal. Similarly, incorporation of a phosphorothioate (PS) modification to the phosphodiester backbone can increase its affinity for AuNPs.[111] In the case of AuNR overgrowth in the presence of homo-oligomeric DNA, the use of A20 induces a red-shift to the L-LSPR of the final nano-structures as mentioned above. Further increase of the DNA affinity by incorporating PS modifications (1PSA20, 2PSA20, 4PSA20) resulted in particles with L-LSPR red-shifted dramatically from 856 nm (only A20) to 1011 nm (4PSA20) with increasing number of PS modifications. Structurally, the gap of the final dumb-bell shaped particles increased from 8±2 nm (only A20) to 28±6 nm (4PSA20) which accordingly red-shifted the L-LSPR to the NIR-II region (Figure 4).
Figure 4.
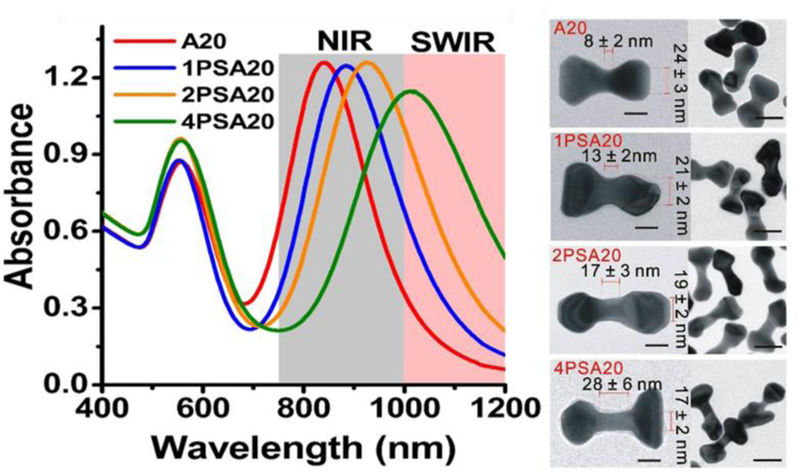
Left: Plasmonic properties of the AuNRs overgrown with A20 DNA and A20 with 1, 2 and 4 PS-modifications. Right: TEM images of the as synthesized nanoparticles. Scale bars are 20 nm (high magnification TEM image) and 50 nm (low magnification TEM image). (Reproduced with permission.[84] Copyright 2015 Wiley-VCH)
The PS modification is one of the wide variety of functionalities that can be incorporated into a DNA sequence. The incorporation of functional groups can be through the backbone, just like the PS modification or via the nucleobase itself. Functional modifications may also be employed to elucidate mechanisms in a DNA mediated synthesis. For example, the amine and carbonyl groups present on the nucleobases play a significant role in binding to metal surfaces. Removal of these groups would help account for the exact role of the base in the growth. Similarly, addition of new functional groups such as amine or hydroxyl group can help tune the nucleobase affinity and thus the morphology of the particle. With the PS modification being able to influence morphologies of nanoparticles, it is clear that the backbone can also play a significant role in engaging with the metal surface. The highly charged phosphodiester backbone can be replaced by a neutrally charged backbone, such as using PNA, that simply connects the nucleobases but keeps the sequence soluble in aqueous medium. The incorporation of modified DNA will not only help elucidate the roles of each functional group but will further enrich the DNA code library for generating nanoparticles with new morphologies.
3.1.4. Secondary Structures of DNA:
Single stranded DNA of a particular sequence has the ability to fold into structures that involves the intra-molecular or inter-molecular interactions such as hydrogen bonding, between the nucleobases present in the sequence. The secondary structures arising from homo-oligomers of DNA, i.e., formed by the interaction between identical nucleobases are more relevant to the present discussion. DNA that are rich in G or C have the ability to form unusual secondary structures under certain conditions. For example, the G-rich DNA forms G-quadraplexes which involves G-quartets that are planar in nature and generally assemble in presence of a stabilizing cation such as K+.[112, 113] The C-rich DNA forms the i-motif of C.C+ base pairs involving one or multiple nucleic acid strands.[112] The protonation of the N3 of C plays a crucial role in the structure formation, thus making it pH dependent. Specifically, the i-motif is more stable at lower pH values, such as below physiological pH.[114] The dependence of secondary structures formation on the species present in the surrounding environment has led to the design of many switchable DNA-based sensors.[115–117]
A typical DNA-mediated nanoparticle growth solution would provide for an acidic environment which is simultaneously populated with metal cations, owing to the metal precursor in solution.[118] Hence, the formation of secondary structures in solution is a possibility and may be of potential importance. The nature of a folded ssDNA or a structure that comes together by inter-strand interactions is bulkier when compared to a single unfolded ssDNA. Although the nucleobases are masked due to their supramolecular preoccupation, the as-formed secondary structure may still show potential to interact with the nanoparticle seed’s surface. Thus far, the formation of G-quadraplex and i-motif has been observed in growth solutions where the overgrowth of silver nanocubes was studied in the presence of short oligomers of DNA (A10, T10, G10 and C10) (Figure 3A).[85] The formation of G-quadruplex occurs only after having added the silver precursor and reductant, indicating that the Ag+ ions played a vital role in structure assembly. The Ag+ ion sequestration by the secondary structure consequently rendered the precursor unavailable for further reduction on the cubic seed surface, thus the morphology of the seed remains unchanged. The bulky secondary structure additionally may be involved in the surface passivation of the seed preventing any Ag0 deposition. On the contrary, the i-motif structure was formed in solution before the addition of the precursor or reductant. Owing to the initial slightly acidic environment, the formation of C-H+-C base pairing is favored. On addition of precursor and reductant, the high binding affinity of Cytosine to Ag+ disrupts the C-H+-C base pairing to form a C-Ag+-C pairing. The Ag+ is still susceptible to reduction and thus destabilization of the secondary structure takes place. The formation and destabilization of these structures in growth solution can be readily monitored using Circular Dichroism (CD) Spectroscopy.
3.2. Properties of the Seed Employed:
All the DNA-encoded synthetic protocols for morphology control involve a seed-mediated synthesis. Using a seed is known not only to increase monodispersity and uniformity of the final nanostructures,[119] but also influence the metal deposition both in monometallic[120, 121] and bimetallic[122] nanomaterials. Although DNA with different sequences by themselves are able to direct morphologies very differently from each other irrespective of the nanoparticle seed, the function of the seed cannot be overlooked. The most important step in a DNA-mediated synthesis is the choice of seed, which determines what surfaces the DNA will potentially interact with. By rationally choosing the seed, one would be able to obtain desired information regarding the exact influence of the seed and the DNA on the final morphology. In this respect, there are two main factors that need to be considered while studying the interaction of the seed; 1) the facets that the seed encloses and its composition and 2) the capping ligands that pre-exist on the surface of the seed.
3.2.1. Facets of the seed:
It is indeed challenging to predict the interaction between DNA and a certain metal. However, it is even more challenging to predict the affinities of DNA to different facets of a nanoparticle of the same metal. The noble metal nanoparticle systems that have been studied for DNA-mediated growth crystallize in the face-centered cubic (fcc) lattice. The most common facets that the seeds employed enclose are the low-indexed facets, i.e. {111}, {100} and the {110} facets, the surface energies of which increase in the same order. Most of the systems that have been studied contain only these three surface facets. For example, in the over growth of AuNR, the growth is determined by the fact that strong binding affinity DNA will bind to {110} and {100} facets on the AuNR surface (Figure 5A).[84] For nucleobases with strong affinity such as adenine oligomers, the DNA binds to the sides of the rods, which has {110} and {100} facets, resulting in preferential growth at the ends and thus formation of dumbbell shape. For nucleobases with relatively weaker binding affinity (T20, C20 and G20), the growth initiates from the ends and will grow to favor the formation of {111} facet, resulting in the formation of sharp tips at the ends of the nanorods. Thus, the binding affinity of DNA to {100} and {110} will determine how much the sides will continue to grow. The weaker binding affinity of the DNA results in more growth on the sides, transiting through a cracked octahedron shape and eventually form an octahedron as observed for G20. The C20 and T20 have slightly stronger binding affinity, probably owing to the secondary structure formation in case of G20, resulting in termination of the growth at cracked octahedron shapes.
Figure 5.

A). Schematic representing the overgrowth of Au nanorod seeds in the presence of DNA. The facets of the seed play an important role in the DNA binding events. (Reproduced with permission.[84] Copyright 2015 Wiley-VCH). B). The growth progression of the Au nanoprism in the presence of T30, starting from (a) nanoprism to (b) nonagon to (c) hexagon to a final (d) six-pointed star with corresponding model of the NP and diffraction pattern (insets). Blue and red lines on the models indicate the distance measured from the center to the corners or sides of the particles, respectively, plotted in the bottom right corner. HR-TEM image of the hexagonal. Scale bar is 2 nm. Red box shows the location where the higher-resolution image was observed. (Reproduced with permission.[104] Copyright 2015 American Chemical Society)
The AuNPr consists of two planar {111} surfaces and a twinned plane in-between.[81] The selected area electron diffraction (SAED) patterns and high resolution TEM studies taken in the [111] zone axis of the particle contained the forbidden 1/3(422) reflections, which confirmed the presence of twin defect (Figure 5B). The defects increase the surface energy of the edge of the prism, allowing the preferential binding of high affinity DNA molecules such as A30. The growth in the presence of T10 proceeds with the deposition on this twinned edge due to the sequence’s low binding affinity. Thus, during the growth progression, the {111} surface is retained at every phase. It is therefore important to characterize the seed, as the relative surface energies of the facets plays an important role in determining the extent of DNA binding. A more interesting area is to study how DNA interacts with high-indexed facets.[123]
3.2.2. Surface functionality of the seed:
Before the DNA can have access to the facets that the nanoparticle seed displays, it has to be able to surpass the shell of pre-existing capping ligands on the seed’s surface. Common ligands that stabilize seeds include CTAB, CTAC, PVP, and citrate. Each of these ligands is capable of individually interacting with DNA. For example, CTAB as a positively charged molecule tends to have electrostatic interaction with negatively charged DNA molecules to form polyplexes. This property has been extensively used in DNA isolation protocols in biology.[124–126] In the case of citrate, the negative charge of the ligand has the potential to repel DNA from the nanoparticle surface.[127] AgNCs have pre-existing PVP on the surface. Although the ligand is neutrally charged, its extended polymeric nature makes the Ag surface inaccessible to DNA. It is hence important to remove the excess ligand that exists in solution and on the surface of the seed to ensure proper interaction between DNA and the nanoparticle surface. However, it should be noted that it is important to maintain a balance where there is enough capping ligand to stabilize the seed in solution, but not in excess to disrupt the DNA’s interaction with the seed. Addition of DNA proceeding the washing of excess ligand ensures the particle stability upon DNA binding, similar to a ligand replacement mechanism. Nanoparticles containing ligands that are covalently or very tightly bound as in the case of thiol-based ligands to Au based particles, would be expected to make DNA binding to the nanoparticle surface quite unfavorable.
3.3. Proposed Mechanisms:
3.3.1. DNA mediated growth of gold nano prisms (AuNPr):
From the discussions thus far, it is clear that DNA-mediated nanoparticle growth involves many different factors that affect the final outcome. The factors range from a simple DNA sequence to the more complicated surface properties of the seed. In order to elucidate the mechanism, Tan et al.[104] chose to study the morphological evolution of AuNPr to different shapes[82] in the presence of DNA molecules of different sequences. Arresting the growth of the nanoparticles at different time points to understand the morphological evolution can give us plenty of information about the intermediates of the reaction and potentially help us elucidate the role of DNA. The growth of the AuNPr in the presence of DNA was arrested at different time points by 3-mercaptopropanoic acid (MPA), which is known to quench the reduction of gold precursor.[128] The intermediates of morphological evolution of the prism in the presence of T30 were observed to be the final morphologies formed in the presence of the remaining 3 homo-oligonucleotides (A30, G20 and C30). Initially, the prism growth took place from the sides and evolved from a nonagon shape to a hexagonal shape to a final six-pointed star. This observation implied that the DNA was capable of binding to the sides of the AuNPr and kinetically trapping the intermediates, the extent of which depended on the DNA binding affinity (Figure 6). A30 and C30 arrested the growth at the nonagon shape and G20 arrested the growth at the hexagonal intermediate. The intermediates that were trapped at an early stage had a smaller diameter as compared to the later intermediates. Hence, the particle diameter followed a trend where particles with T30 had the largest diameter, while particles with A30 and C30 had the smallest diameter. Since the samples were subjected to the same amount of gold precursor, the amount of precursor that was not consumed in the diameter increment is now available for growth in nanoparticle thickness. Hence, the second phase of growth progression is the growth in thickness, which depends upon the amount of precursor available for reduction. The particle thickness obviously had an opposite trend to diameter. Particles synthesized in the presence of A30 had the highest thickness and particles with T30 had the lowest particle thickness.
Figure 6.

Schematic representing the proposed mechanism of growth of Au nanoprisms influenced by DNA in two stages: Shape control and thickness growth. (Reproduced with permission.[104] Copyright 2015 American Chemical Society)
The above experiments have identified three factors that DNA operates through: 1) binding of DNA to the precursor, 2) DNA binding affinity to the NP surface and 3) density of DNA on the seed’s surface. Cyclic voltammetry studies revealed that DNA binding to the precursors only influences the diffusion of the precursor towards Au {111} surface, which is indicated by the change in cathodic current with change in DNA sequence in the order, T (which allows fastest precursor diffusion) > G > C > A. This complies with the second factor, i.e., the DNA binding affinity trend to Au where A > C > G > T. Although dependent on the binding affinity, the density of DNA on the seed’s surface by itself plays a significant role. An experiment where the concentration of T30 was increased to consequently increase the strand density on the seed limited the lateral growth of the prism, i.e. decreased the diameter and increased the thickness. Increasing the T30 DNA concentration to up to 10 times resulted in hexagonal particles, suggesting the kinetic trapping of hexagonal intermediate owing to the increased DNA density that compensates for the low affinity of the sequence. The surface property of the nanoparticles with A30 is rough in nature as compared to a smooth round particle formed in the presence of C30. The roughness may be attributed to the mobility of the base on the Au surface, specifically Au {111}. The calculated mobility of the bases follows the order A < G = T < C.[129] The low mobility of A on the Au surface allows Au deposition only in areas where the A30 strand is absent, thereby producing a rough surface. On hybridizing T30 functionalized 5 nm AuNP with A30 particles resulted in the localization of the 5 nm AuNPs in the cervices of the rough particle, consistent with the base mobility-based hypothesis.
3.3.2. DNA-mediated growth of Pd-Au bimetallic nanoparticles:
A more complicated system involving DNA-encoded growth of two different metal nanoparticles tells a mechanistic story that not only complements well with the monometallic system described above, but also provides new insights into how DNA operates at a more intricate level. The Pd cubic seed which is used as a seed in this study contains the {111} and {110} sites, whose residing atoms lack a fulfilled coordination number, which makes them relatively higher in energy. The DNA binding affinity and DNA density on the seed influence two major factors on the incoming metal atoms, 1) the deposition and 2) the diffusion. The relative rate of metal atom deposition and diffusion determines the morphology of the shell formed [130, 131] and the DNA of different sequences tunes both rates (Figure 7A). The low binding affinity of the T10 does not substantially passivate the seed’s surface, promoting the deposition of the incoming metal on the high energy sites, as observed in the SEM. Hence, the rate of deposition is much higher than the rate of metal diffusion on the seeds surface, ultimately constricting the shell formation to the corners and edges, forming a Pd-Au core-frame structure. C10 on the other hand, does passivate the high energy sites due to higher binding affinity and allows the rates of deposition to be lower than the rate of diffusion, to form a complete cuboctahedron shell.
Figure 7.

A). Schematic representation of the proposed mechanism of growth of Pd−Au bimetallic nanostructures influenced by different sequences of DNA. The atomic models represent the cross-section of the 3-D models along the black box in each individual case. B). Top: Kinetic UV−vis absorption spectra of nanoparticle growth in the presence of T10. Bottom: The plot of absorbance vs time at the λmax values for each of the particles. (Reproduced with permission.[88] Copyright 2016 American Chemical Society)
G10 tends to form a secondary structure with the metal precursors and thus the seed’s surface is passivated with a rigid structure that inherently has low mobility. The rate of atom deposition is higher than the rate of atom diffusion, which initially promotes the formation of islands on the cubic core. The deposition therefore occurs in areas where the rigid secondary structure is absent. Eventually, after metal atom diffusion events, the formation of an undulated Pd-Au shell takes place. Further increase in G10 concentration completely passivates the surface and does not allow any growth on the palladium cube. A10 has high binding affinity with both the seed and the precursor. Thus, the high energy sites on the seed are passivated and the metal deposition and diffusion takes place uniformly to form a rhombi cuboctahedron. The A10 sequence also binds to the precursor to form smaller unstable nanocrystallites that undergo inter-particle fusion and subsequently deposit onto the seed, a major step in an aggregative growth mechanism or Ostwald ripening.[132] The high density of A10 on the seed and its binding to the precursor both equally contribute to this mechanism. The UV-vis kinetic study, where the growth in presence of different DNA sequences was monitored in-situ by the absorption profiles revealed that A10 displays a sigmoidal growth curve in comparison to the more common exponential growth curve exhibited by the other bases (Figure 7B). The sigmoidal growth curve is characteristic of the aggregative growth mechanism.[133, 134]
Further investigation of a similar system consisting of a concave palladium seed instead of a cubic seed, revealed the key influence of the core in a DNA-mediated synthesis.[135] The existence of high-energy sites on the seed influences the initial deposition of gold onto the seed surface in the presence of DNA with low or medium binding affinity, such as T10 and G10, C10, respectively, resulting in an overall increased growth rate. DNA with higher binding affinity, such as A10, still retains the ability to bind to the precursor and form smaller nanocrystallites, validated by the sigmoidal curve observed in the kinetic absorption studies. In the absorbance vs. time kinetic profiles, the increase in absorbance takes place within ~5 min for the concave cube vs. ~10 min in the presence of the simple cube, which agrees with the increased growth rate. In all of the cases, the rate of deposition is greater than the rate of diffusion owing to high surface energy of the protruding edges and corners of the concave seed. The deposition along the <111> direction was almost always greater than Au deposition along the <100> direction, implying that γ increased in the following order: {100} < {110} < {111}. A major influence of the seed was observed in the time-based SEM studies on the morphological evolution. With a simple cubic core, the influence of DNA was evident from the start of growth, whereas, the initial growth in the concave cube core was heavily dictated by the surface energy of the seed. Therefore, when the growth was arrested at an initial time phase or when less gold precursor was used, the morphology that resulted in the presence of T10, G10 and C10 were almost indistinguishable, consistent with their similar SPR properties. However, after the initial surface energy passivation by gold deposition, the influence of the DNA in the further deposition of gold comes in.
The fundamental study of the mechanisms involved in DNA-mediated synthesis of metal nanoparticles helps learn the deciding factors in what exemplifies fine-control of nanomaterial morphology. Although the salient principles under which DNA-mediated shape-control occurs are often interdependent, certain parameters such as seed facets, pre-existing capping ligands, seed composition, can be decoupled and further studied.
4. Surface properties of DNA-coded nanomaterials:
Some of the most distinct advantages of nanotechnology are its interesting surface, optical and chemical properties that sparked widespread interest for its applications in the fields of biology,[136, 137] electronics,[138] medicine,[139, 140] batteries,[141] solar cells,[142] and sensors.[60] When bulk materials are shrunk to the nanoscale, it takes on different optical and chemical properties. Over the past decade, there have been tremendous advancements to design and synthesize new nanoscale materials to engineering solutions to different problems. Hence, it is important to understand the properties of the novel DNA-mediated nanoparticles with different morphologies and explore how these materials can be employed for the above applications.
4.1. Molecular recognition:
Surface engineering of nanomaterials is the bridge between nanotechnology and biology. For nanomaterials to be used effectively in biology, there is a need for effective and efficient modification of biocompatible ligands, allowing the nanoparticles to be soluble and stable in the complex biological medium. However, surface modification of nanomaterials has generally been met with some level of difficulty[70, 143] and complexity.[144] While it is commonly argued that successful surface modification of nanomaterials allowed effective use of these nanomaterials in biological applications, these synthetic routes have often been either complicated or inefficient in conjugation techniques. The majority of nanoparticle synthesis uses the liquid-solid-solution (LSS) phase transfer synthetic route to yield semiconducting, upconversion, conducting polymer, organic optoelectronic semiconducting and magnetic nanoparticles.[145] Nanomaterials synthesized via these routes often require post modification since the capping ligand is generally hydrophobic in nature and not biologically feasible for applications. Gold and silver nanoparticles on the other hand require post modifications after synthesis to load DNA via a tedious and time consuming salt ageing process[146] or acid treatment.[127, 147] Newer, faster freeze directed protocols have been reported but are successful only for Au nanoparticles coated with citrate ligands.[110, 148]
The discovery of DNA-mediated morphological growth of mono or bimetallic nanomaterials offers a completely novel approach for an effective DNA modification to render metallic nanostructures not only water soluble but more importantly with a bio-recognition capability via a one-step synthesis reaction. The DNA mediated morphological growth of nanoparticles resulted in the retention of bio-recognition properties of DNA, while DNA itself being used as a ligand to stabilize the nanoparticle.[80, 85, 104] It was observed that by incubating A30-encoded nanoparticles (i.e., rough-round Au particles from Au nanoprism) with 5 nm AuNPs functionalized complementary DNA (T30), satellite nanostructures were formed. Controls were performed with 5 nm gold nanoparticles functionalized non-complementary DNA (A30) resulted in no satellites nanoassemblies (Figure 8A (I)). Interestingly, as reported by Wu et al,[85] even though only 10 bases of deoxynucleotides were used in the DNA mediated growth of silver nanostructures, satellite nano-assemblies were observed to form in the presence of complementary DNA functionalized 5 nm gold nanoparticles (Figure 8A (II)). These results provide a strong evidence, that only a few bases within the oligonucleotides were used as functional ligand to stabilize the morphological growth of mono-metallic nanoparticles and majority of the DNA is left exposed to the surrounding, allowing the nanostructure be functional for further DNA hybridization. Thiolated DNA functionalized on a nanoparticle surface can be replaced with shorter alkyl thiols such as mercaptoethanol, on the other hand it has been demonstrated that for AuNFs synthesized in the presence of DNA, it is difficult to replace the surface DNA using shorter thiols, validating the robust nature of the functionalization.[80] Hence, these intriguing findings allows the synthesis of nanoparticles with bio-recognition abilities without post synthesis modification.
Figure 8.

The post-synthetic properties of DNA-encoded particles. A). (I).Top: TEM images of AuNF synthesized with A30 assembled with 5 nm Au nanoparticles functionalized with thiolated T30 (Left) and with a thiolated non-complementary sequence (Right). Bottom: AuNPs synthesized with T30 assembled with 5nm Au nanoparticles functionalized with thiolated A30 (Left) and with a thiolated non-complementary sequence (Right). (Parts of the figure reproduced with permission.[80] Copyright 2010 American Chemical Society). (II). Au nanoparticle grown from nanoprism seed in the presence of A30 assembled with AuNP functionalized with thiolated non-complementary sequence (Left) and thiolated T30 (Right). (Figure reproduced with permission.[104] Copyright 2015 American Chemical Society). B). Dark field image of the CHO cells treated with AuNF particles (Left) and without nanoparticle treatment (Right). (Figure reproduced with permission.[80] Copyright 2010 American Chemical Society). C). The SERS spectra of 4-MBT modified on the surfaces of different Pd@Au core-shell nanoparticles grown from a concave Pd seed with various DNA sequences. (Figure reproduced with permission.[135] Copyright 2018 Springer Nature).
4.2. Surface plasmon resonance:
Metal nanoparticles exhibit a unique property known as surface plasmon resonance (SPR) wherein the conduction electrons on the surface of the nanoparticle interact with incident light. The SPR depends heavily on the shape and the size of the nanoparticle and this specific property can be used for several applications such as photocatalysis, surface-enhanced Raman-based detection, and biomedical imaging.[149, 150] DNA mediated growth of mono-metallic nanoparticles has also allowed the effective tuning of surface plasmonic properties of nanoparticles. One of the most intriguing results yielded from DNA mediated morphological growth of nanomaterials was the ability to rationally tune the absorption of Au nanorods from the NIR I window to the NIR II window.[84] It was observed that in the presence of A20, the L-LSPR peak was intensified and redshift to 856 nm. This is accompanied by the formation of dumbbell shape at the ends of the Au nanorods (Figure 3C). More interestingly, with the addition of phosphorothioate modified A20, the L-LSPR peak was found to be further turned to 1011 nm with four phosphorothioate to the DNA (Figure 4). This unique tuning to the NIR II window was attributed to the fact that with the increase of phosphorothioate modifications, there was larger intraparticle gaps with flattened ends to induce a red shift by a significant amount.
4.3. Cellular internalization:
One of the main challenges facing nanotechnology in biological applications has been to render nanoparticles not only soluble but also functional to detect, sense or target specific analytes in the biological matrix. As mentioned previously, most nanoparticle types ranging from semiconductor nanoparticles to lanthanide nanoparticles and gold nanoparticles are synthesized as hydrophobic nanoparticles due to the presence of oleic acid as capping ligands. While these syntheses yielded highly homogenous small nanoparticles, complex surface engineering has to be performed to render these nanoparticles soluble before it can be used as delivery agents or nanomedicine within the cells for various applications.[151]
To overcome the above limitation, Wang et al[80] reported that using DNA mediated morphological growth method, the synthesized AuNFs consist of high densities of DNA on its surface. Dark field microscopy of AuNF in CHO cells showed good distribution of nanoparticles within the cell (Figure 8B). By utilizing the light scattering properties of AuNF, there was strong evidence for the effective delivery of nanoparticles without the use of transfection agents. Additionally, the same study shows that the DNA tethered to the DNA-encoded AuNFs is more stable when treated with mercaptoethanol as compared to nanoparticles that were conjugated with thiolated DNA, probably because the DNA are embedded in between the nanoparticles during the growth, which prevents the DNA from dissociating from the nanoparticles. This particular finding is crucial in that biological environments contain many small thiolated molecules that can completely replace conjugated DNA from the nanoparticle surface but not the DNA that is present on a DNA-encoded nanoparticle. DNA ligand is the only ligand used in the synthesis of these DNA mediated nanoparticles, it is compelling that DNA functionalized nanoparticles resulted in the efficient cellular uptake of these shaped AuNF. This can be further supported by another independent work by Mirkin et al,[152] where high DNA density on the surface of the nanoparticles resulted in greater adsorption to surface proteins on the cell to induce better cellular uptake. Since DNA mediated nanoparticles result in a facile synthesis of nanoparticles of different shapes, there could also be different cellular uptake rates influenced by the shape of the nanoparticles.[153]
Yang and coworkers reported[87] that the silver nanoprisms derived from DNA mediated synthesis resulted in particles having good cell viability as compared to that of silver nanoparticles, while at the same time the silver nanoprisms exhibit better antibacterial properties. Due to the nature of DNA mediated nanomaterials, these particles have been demonstrated to be facile in synthesis to yield highly biocompatible properties and at the same time offers interesting biological application such as the ones observed in silver nanoparticles.
4.4. SERS properties:
SERS properties have also been demonstrated in these DNA-mediated nanoparticles. 4-methylbenenethiol (4-MBT) was used by the Lu group to demonstrate enhancement factors around 105 and 106 using DNA-mediated silver nanoparticles. The highest enhancement was reported to be with C10 on silver nanoparticles with good reproducibility. At the same time, truncated Ag nanoparticles with T10 could be used as hotspots for SERS enhancement.[85] In addition, SERS was also demonstrated in DNA mediated AuNRs. Enhancement factors between 104 and 106 was observed for different shapes such as rods, dumbbells, cracked octahedron and octahedron shapes using 4-MBT dye. Similar SERS enhancement was also reported by Yang and coworkers using crystal violet. It was observed that the SERS effects were stronger using AgNPr as compared to that of silver nanoparticles.[87] Furthermore, the NIR absorbing Pd@Au core-shell nanoparticles were used as SERS substrates for the same substrate with enhancement factors of up to ~107 when excited by higher wavelength lasers. The enhancement factors depended on the morphology of the nanoparticles tracing back to the sequence of DNA utilized in the synthesis (Figure 8C).[135]
5. Summary and Outlook:
This review summarizes recent advancements in the field of DNA-mediated growth of mono- and bi-metallic nanomaterials with controlled morphologies. Through the discovery of DNA codes, the use of DNA with different sequences to tune the morphologies has been demonstrated while using gold prisms, spheres, rods, silver and palladium nanocubes as seeds. As a result, different shapes and surface structures of nanomaterials were synthesized where DNA was demonstrated not only as a stabilizing capping ligand to the nanomaterials, but more importantly to systematically control the growth of nanomaterials. It has been found that the affinities of the nucleobases play a vital role in interacting and stabilizing different metal surfaces that consequently changes the metal deposition profiles in a sequence dependent matter. The versatility that this approach introduces to metal nanoparticle synthesis is extraordinary. Changing the DNA functionality by altering the sequences of DNA bases introduces novel design rules in the generating new morphologies of nanoparticles with exclusive properties. More interestingly, in a one-step reaction, these DNA directed nanoparticles retained bio-recognition capabilities on the surface. This is quite rare in gold and silver nanomaterials without post modification with DNA. The rational growth of nanoparticles has also yielded in a systematic tuning of optical properties such as LSPR from the visible range to the NIR II window. Several of the final morphologies exhibited excellent SERS properties owing to the unique shapes obtained via DNA-mediated synthesis. These interesting improvements and modifications to the properties of nanomaterials prompt further investigations into the role of DNA to rationally tune these properties. These facile routes to synthesize unique nanostructures have been proven to be reproducible and stable in aqueous media.
Many questions still remain in the field of DNA-mediated nanoparticle growth with different morphologies. While DNA interactions with metal ions and metal intercalators have been known,[96, 154, 155] there are only very few systematic studies on DNA interactions with nanoparticle surface. Even in the current literatures of the DNA interaction with bulk metal surface, the results are often inconsistent between different studies. To advance the field of using DNA to mediate the growth of nanomaterials, the detailed interaction of DNA with metal surfaces should be elucidated. Another area of interest would be to study the influence of different functional groups on the DNA in controlling the growth of nanomaterials. The ease of base modification allows one to remove or add certain functional groups onto the nucleobases enabling one to decouple functional parameters that influence the final morphologies. In addition, abasic analogues can establish the necessity of having purines and pyrimidines for the precise growth of nanoparticles. These studies could further help in the rational design of capping ligands that can predictably influence the morphology of nanomaterials and consequently their chemical and physical properties. Understanding the effects in these two areas will allow better control and design on mediating nanomaterial growth and its use as not only a stabilizing ligand but more importantly, a modular ligand that is capable of controlling the nanomaterial morphologies.
While the effect of different DNA sequences on the morphologies of Pd-Au nanoparticles have been elucidated, it would be interesting to see if the finding from this study can be applied to studying of other bimetallic nanoparticles, such as Au-Ag nanoparticles, given the similar lattice constants of Au and Ag and the knowledge we have gained on how DNA interacts with each of the metals individually. Additionally, almost all of the current studies have been focused on noble metal nanoparticles, it is important to explore extension of the approach to study more complicated systems that involve other metals and metal-oxide/semiconductor hybrid materials.[92] Since wide band gap semiconductors have relatively small optical cross-sections in the visible spectrum, these materials interact weakly with visible light. One way of enhancing this property is by integrating them with plasmonic metals either as an oxide core-metal shell or metal core-oxide shell[156–158] and the goal can be accomplished with DNA encoded synthesis.
To provide deeper insights into the mechanism of DNA-mediated nanoparticle formation, one can take advantage of in situ liquid cell to image growth of nanomaterials in real time under the transmission electron microscope (TEM).[159–161] This technique has allowed the extensive studies on the defect formation, particle growth rate,[162] oxidative etching,[163, 164] and Kirkendall effect.[165] Hence, to further advance the field, in situ TEM techniques may be employed to study the growth of NP in real time and to accurately observe the growth mechanism using both a wet cell and flow cell method. Using this tool, the TEM image and its facet changes at each stage of the nanoparticle growth can be accurately studied to fully understand what is happening at each stage of the nanoparticle growth. These studies are important as current mechanisms were proposed by using mercaptoethanol to stop the nucleation of nanoparticles and there could be inaccuracies in determining the exact growth of the nanomaterials. To study the exact growth, in situ TEM is current the best technique available to determine how the nanomaterial nucleates and further grows into the final shape. While DNA mediated growth of gold and silver nanoparticles has been reported, it is important to take extra precaution to avoid or minimize electron-beam induced reduction of metal ions in the solution that may contribute to some of the observations.
At the same time, advancements are being made to elucidate how shape and size of nanomaterials influence the cellular uptake in cells.[153, 166, 167] This is an important area of interest due to the emergence of nanomaterials in sensing, imaging and drug delivery. Besides the conventional studies to investigate the cytotoxicity of nanoparticles, another focus would be centered on understanding how shape influences the uptake of cells to maximize delivery into the cells as a delivery vehicle. These DNA mediated mono-metallic nanostructures can be further functionalized in the most facile manner with functional aptamers for biomedical applications. Another area of interest is the fate of the nanoparticle shape after cellular internalization and the extent to which shape related properties such as SPR can be utilized after the internalization event, which will be important for biomedical or bio-sensing applications.
Finally, most investigations have focused on homo-oligomeric DNA in order to elucidate effects of different nucleobases in controlling the morphology. It is important to build upon the progress and explore deeper into the effect of different sequence combinations on the morphologies of nanomaterials in order to be more analogs to biological genetic codes. The holy grail is to develop clear accurate design principles that governs DNA codes for abiological nanoparticle morphologies for all sequence combinations and across diverse ranges of metals in single or complex hybrid nanosystems. The realization of this goal would offer a myriad of new possibilities in the integration of these novel nanomaterials with healthcare platforms and offering engineering solutions to current challenges in nanoscale fabrication.
Acknowledgements:
We wish to thank the U.S. National Institutes of Health (GM124316 and MH110975) for financial support. N. S. R. S. would like to thank the Beckman Graduate Fellowship for financial support. We would also like to thank Dr. Tingjie Song and Yiming Wang for proof reading the manuscript and providing valuable input.
References:
- [1].Hu M, Chen J, Li Z-Y, Au L, Hartland GV, Li X, Marquez M, Xia Y, Chem. Soc. Rev 2006, 35, 1084–1094. [DOI] [PubMed] [Google Scholar]
- [2].Jain PK, Huang X, El-Sayed IH, El-Sayed MA, Acc. Chem. Res 2008, 41, 1578–1586. [DOI] [PubMed] [Google Scholar]
- [3].Lal S, Clare SE, Halas NJ, Acc. Chem. Res 2008, 41, 1842–1851. [DOI] [PubMed] [Google Scholar]
- [4].Tao Y, Li M, Ren J, Qu X, Chem. Soc. Rev 2015, 44, 8636–8663. [DOI] [PubMed] [Google Scholar]
- [5].Yu M, Zheng J, ACS Nano 2015, 9, 6655–6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kamat PV, J. Phys. Chem. B 2002, 106, 7729–7744. [Google Scholar]
- [7].Wang F, Han Y, Lim CS, Lu Y, Wang J, Xu J, Chen H, Zhang C, Hong M, Liu X, Nature 2010, 463, 1061–1065. [DOI] [PubMed] [Google Scholar]
- [8].Kauranen M, Zayats AV, Nat. Photon 2012, 6, 737–748. [Google Scholar]
- [9].Lewis LN, Chem. Rev 1993, 93, 2693–2730. [Google Scholar]
- [10].Haruta M, Catal. Today 1997, 36, 153–166. [Google Scholar]
- [11].Astruc D, Lu F, Aranzaes JR, Angew. Chem. Int. Ed 2005, 44, 7852–7872. [DOI] [PubMed] [Google Scholar]
- [12].Debe MK, Nature 2012, 486, 43–51. [DOI] [PubMed] [Google Scholar]
- [13].Mahmoud MA, Narayanan R, El-Sayed MA, Acc. Chem. Res 2013, 46, 1795–1805. [DOI] [PubMed] [Google Scholar]
- [14].Qian K, Sweeny BC, Johnston-Peck AC, Niu W, Graham JO, DuChene JS, Qiu J, Wang Y-C, Engelhard MH, Su D, Stach EA, Wei WD, J. Am. Chem. Soc 2014, 136, 9842–9845. [DOI] [PubMed] [Google Scholar]
- [15].Wei DW, Sweeny CB, Qiu J, DuChene SJ, Metallic Nanostructures for Catalytic Applications. In Metallic Nanostructures: From Controlled Synthesis to Applications, Xiong Y; Lu X, Eds. Springer International Publishing: Cham, 2015; pp 243–269. [Google Scholar]
- [16].Liu J, Lu Y, J. Am. Chem. Soc 2003, 125, 6642–6643. [DOI] [PubMed] [Google Scholar]
- [17].Rosi NL, Mirkin CA, Chem. Rev 2005, 105, 1547–1562. [DOI] [PubMed] [Google Scholar]
- [18].Willets KA, Duyne RPV, Annu. Rev. Phys. Chem 2007, 58, 267–297. [DOI] [PubMed] [Google Scholar]
- [19].Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, Van Duyne RP, Nat. Mater 2008, 7, 442–453. [DOI] [PubMed] [Google Scholar]
- [20].Shipway AN, Katz E, Willner I, ChemPhysChem 2000, 1, 18–52. [DOI] [PubMed] [Google Scholar]
- [21].Xia Y, Xiong Y, Lim B, Skrabalak SE, Angew. Chem. Int. Ed 2009, 48, 60–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kelly KL, Coronado E, Zhao LL, Schatz GC, J. Phys. Chem. B 2003, 107, 668–677. [Google Scholar]
- [23].Murphy CJ, Sau TK, Gole AM, Orendorff CJ, Gao J, Gou L, Hunyadi SE, Li T, J. Phys. Chem. B 2005, 109, 13857–13870. [DOI] [PubMed] [Google Scholar]
- [24].Narayanan R, El-Sayed MA, J. Phys. Chem. B 2005, 109, 12663–12676. [DOI] [PubMed] [Google Scholar]
- [25].Tao AR, Habas S, Yang P, Small 2008, 4, 310–325. [Google Scholar]
- [26].Laskar M, Zhong X, Li Z-Y, Skrabalak SE, ChemSusChem 2013, 6, 1959–1965. [DOI] [PubMed] [Google Scholar]
- [27].Xia Y, Xia X, Peng H-C, J. Am. Chem. Soc 2015, 137, 7947–7966. [DOI] [PubMed] [Google Scholar]
- [28].Wang Y, Xie S, Liu J, Park J, Huang CZ, Xia Y, Nano Lett 2013, 13, 2276–2281. [DOI] [PubMed] [Google Scholar]
- [29].Bhosale MA, Sasaki T, Bhanage BM, Powder Technol 2016, 291, 154–158. [Google Scholar]
- [30].Niu W, Zhang L, Xu G, ACS Nano 2010, 4, 1987–1996. [DOI] [PubMed] [Google Scholar]
- [31].Sardar R, Shumaker-Parry JS, J. Am. Chem. Soc 2011, 133, 8179–8190. [DOI] [PubMed] [Google Scholar]
- [32].Pastoriza-Santos I, Liz-Marzán LM, Adv. Funct. Mater 2009, 19, 679–688. [Google Scholar]
- [33].Wu B-H, Yang H-Y, Huang H-Q, Chen G-X, Zheng N-F, Chin. Chem. Lett 2013, 24, 457–462. [Google Scholar]
- [34].DeSantis CJ, Peverly AA, Peters DG, Skrabalak SE, Nano Lett 2011, 11, 2164–2168. [DOI] [PubMed] [Google Scholar]
- [35].DeSantis CJ, Sue AC, Bower MM, Skrabalak SE, ACS Nano 2012, 6, 2617–2628. [DOI] [PubMed] [Google Scholar]
- [36].Zhang J, Feng C, Deng Y, Liu L, Wu Y, Shen B, Zhong C, Hu W, Chem. Mater 2014, 26, 1213–1218. [Google Scholar]
- [37].Bower MM, DeSantis CJ, Skrabalak SE, J. Phys. Chem. C 2014, 118, 18762–18770. [Google Scholar]
- [38].Yu Y, Zhang Q, Xie J, Lee JY, Nat. Commun 2013, 4, 1454. [DOI] [PubMed] [Google Scholar]
- [39].DeSantis CJ, Sue AC, Radmilovic A, Liu H, Losovyj YB, Skrabalak SE, Nano Lett 2014, 14, 4145–4150. [DOI] [PubMed] [Google Scholar]
- [40].Ortiz N, Skrabalak SE, Angew. Chem. Int. Ed 2012, 51, 11757–11761. [DOI] [PubMed] [Google Scholar]
- [41].Ortiz N, Skrabalak SE, Langmuir 2014, 30, 6649–6659. [DOI] [PubMed] [Google Scholar]
- [42].Shao Y, Jin Y, Dong S, Chem. Commun 2004, 1104–1105. [DOI] [PubMed]
- [43].Kim F, Connor S, Song H, Kuykendall T, Yang P, Angew. Chem. Int. Ed 2004, 116, 3759–3763. [DOI] [PubMed] [Google Scholar]
- [44].Xie J, Lee JY, Wang DIC, J. Phys. Chem. C 2007, 111, 10226–10232. [Google Scholar]
- [45].Tan YN, Lee JY, Wang DIC, J. Am. Chem. Soc 2010, 132, 5677–5686. [DOI] [PubMed] [Google Scholar]
- [46].Jana NR, Gearheart L, Murphy CJ, J. Phys. Chem. B 2001, 105, 4065–4067. [Google Scholar]
- [47].Wiley BJ, Xiong Y, Li Z-Y, Yin Y, Xia Y, Nano Lett 2006, 6, 765–768. [DOI] [PubMed] [Google Scholar]
- [48].DuChene JS, Niu W, Abendroth JM, Sun Q, Zhao W, Huo F, Wei WD, Chem. Mater 2013, 25, 1392–1399. [Google Scholar]
- [49].Wang Y, Reddy Satyavolu NS, Lu Y, Curr. Opin. Colloid Interface Sci 2018, 38, 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Naik RR, Jones SE, Murray CJ, McAuliffe JC, Vaia RA, Stone MO, Adv. Funct. Mater 2004, 14, 25–30. [Google Scholar]
- [51].Chen C-L, Zhang P, Rosi NL, J. Am. Chem. Soc 2008, 130, 13555–13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chiu C-Y, Li Y, Ruan L, Ye X, Murray CB, Huang Y, Nat. Chem 2011, 3, 393–399. [DOI] [PubMed] [Google Scholar]
- [53].Dickerson MB, Sandhage KH, Naik RR, Chem. Rev 2008, 108, 4935–4978. [DOI] [PubMed] [Google Scholar]
- [54].Young M, Debbie W, Uchida M, Douglas T, Annu. Rev. Phytopathol 2008, 46, 361–384. [DOI] [PubMed] [Google Scholar]
- [55].Berti L, Burley GA, Nat. Nano 2008, 3, 81–87. [DOI] [PubMed] [Google Scholar]
- [56].Liu J, Phys. Chem. Chem. Phys 2012, 14, 10485–10496. [DOI] [PubMed] [Google Scholar]
- [57].Jones MR, Seeman NC, Mirkin CA, Science 2015, 347, 1260901. [DOI] [PubMed] [Google Scholar]
- [58].Laramy CR, O’Brien MN, Mirkin CA, Nat. Rev. Mater 2019.
- [59].Drummond TG, Hill MG, Barton JK, Nat. Biotech 2003, 21, 1192–1199. [DOI] [PubMed] [Google Scholar]
- [60].Torabi SF, Lu Y, Curr. Opin. Biotechnol 2014, 28, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tan LH, Xing H, Lu Y, Acc. Chem. Res 2014, 47, 1881–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chen Z, Liu C, Cao F, Ren J, Qu X, Chem. Soc. Rev 2018, 47, 4017–4072. [DOI] [PubMed] [Google Scholar]
- [63].Petty JT, Zheng J, Hud NV, Dickson RM, J. Am. Chem. Soc 2004, 126, 5207–5212. [DOI] [PubMed] [Google Scholar]
- [64].Vosch T, Antoku Y, Hsiang J-C, Richards CI, Gonzalez JI, Dickson RM, Proc. Natl. Acad. Sci. U.S.A 2007, 104, 12616–12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Han B, Wang E, Anal. Bioanal. Chem 2012, 402, 129–138. [DOI] [PubMed] [Google Scholar]
- [66].Richards CI, Choi S, Hsiang J-C, Antoku Y, Vosch T, Bongiorno A, Tzeng Y-L, Dickson RM, J. Am. Chem. Soc 2008, 130, 5038–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gwinn EG, O’Neill P, Guerrero AJ, Bouwmeester D, Fygenson DK, Adv. Mater 2008, 20, 279–283. [Google Scholar]
- [68].Liu J, TrAC, Trends Anal. Chem 2014, 58, 99–111. [Google Scholar]
- [69].Park SY, Lytton-Jean AKR, Lee B, Weigand S, Schatz GC, Mirkin CA, Nature 2008, 451, 553–556. [DOI] [PubMed] [Google Scholar]
- [70].Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ, Nature 1996, 382, 607–609. [DOI] [PubMed] [Google Scholar]
- [71].Chen Z, Chen H, Hu H, Yu M, Li F, Zhang Q, Zhou Z, Yi T, Huang C, J. Am. Chem. Soc 2008, 130, 3023–3029. [DOI] [PubMed] [Google Scholar]
- [72].Li L-L, Yin Q, Cheng J, Lu Y, Advanced Healthcare Materials 2012, 1, 567–572. [DOI] [PubMed] [Google Scholar]
- [73].Enustun BV, Turkevich J, J. Am. Chem. Soc 1963, 85, 3317–3328. [Google Scholar]
- [74].Frens G, Nature 1973, 241, 20–22. [Google Scholar]
- [75].Kimling J, Maier M, Okenve B, Kotaidis V, Ballot H, Plech A, J. Phys. Chem. B 2006, 110, 15700–15707. [DOI] [PubMed] [Google Scholar]
- [76].Wuithschick M, Birnbaum A, Witte S, Sztucki M, Vainio U, Pinna N, Rademann K, Emmerling F, Kraehnert R, Polte J, ACS Nano 2015, 9, 7052–7071. [DOI] [PubMed] [Google Scholar]
- [77].Cutler JI, Auyeung E, Mirkin CA, J. Am. Chem. Soc 2012, 134, 1376–1391. [DOI] [PubMed] [Google Scholar]
- [78].Daniel M-C, Astruc D, Chem. Rev 2004, 104, 293–346. [DOI] [PubMed] [Google Scholar]
- [79].Storhoff JJ, Elghanian R, Mirkin CA, Letsinger RL, Langmuir 2002, 18, 6666–6670. [Google Scholar]
- [80].Wang Z, Zhang J, Ekman JM, Kenis PJA, Lu Y, Nano Lett 2010, 10, 1886–1891. [DOI] [PubMed] [Google Scholar]
- [81].Millstone JE, Wei W, Jones MR, Yoo H, Mirkin CA, Nano Lett 2008, 8, 2526–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wang Z, Tang L, Tan LH, Li J, Lu Y, Angew. Chem. Int. Ed 2012, 51, 9078–9082. [DOI] [PubMed] [Google Scholar]
- [83].Huang X, El-Sayed IH, Qian W, El-Sayed MA, J. Am. Chem. Soc 2006, 128, 2115–2120. [DOI] [PubMed] [Google Scholar]
- [84].Song T, Tang L, Tan LH, Wang X, Satyavolu NSR, Xing H, Wang Z, Li J, Liang H, Lu Y, Angew. Chem. Int. Ed 2015, 54, 8114–8118. [DOI] [PubMed] [Google Scholar]
- [85].Wu J, Tan LH, Hwang K, Xing H, Wu P, Li W, Lu Y, J. Am. Chem. Soc 2014, 136, 15195–15202. [DOI] [PubMed] [Google Scholar]
- [86].Basu S, Jana S, Pande S, Pal T, J. Colloid Interface Sci 2008, 321, 288–293. [DOI] [PubMed] [Google Scholar]
- [87].Li J, Zhu Z, Liu F, Zhu B, Ma Y, Yan J, Lin B, Ke G, Liu R, Zhou L, Tu S, Yang C, Small 2016, 12, 5449–5487. [DOI] [PubMed] [Google Scholar]
- [88].Satyavolu NSR, Tan LH, Lu Y, J. Am. Chem. Soc 2016, 138, 16542–16548. [DOI] [PubMed] [Google Scholar]
- [89].Niu W, Zhang W, Firdoz S, Lu X, Chem. Mater 2014, 26, 2180–2186. [Google Scholar]
- [90].Satyavolu NSR, Pishevaresfahani N, Tan LH, Lu Y, Nano. Res 2018, 11, 4549–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Izatt RM, Christensen JJ, Rytting JH, Chem. Rev 1971, 71, 439–481. [DOI] [PubMed] [Google Scholar]
- [92].Lu C, Huang Z, Liu B, Liu Y, Ying Y, Liu J, Angew. Chem. Int. Ed 2017, 56, 6208–6212. [DOI] [PubMed] [Google Scholar]
- [93].Sigel H, Chem. Soc. Rev 1993, 22, 255–267. [Google Scholar]
- [94].Rodgers MT, Armentrout PB, J. Am. Chem. Soc 2000, 122, 8548–8558. [Google Scholar]
- [95].Lippert B, Coord. Chem. Rev 2000, 200–202, 487–516. [Google Scholar]
- [96].Anastassopoulou J, J. Mol. Struct 2003, 651–653, 19–26. [Google Scholar]
- [97].Clever GH, Shionoya M, Coord. Chem. Rev 2010, 254, 2391–2402. [Google Scholar]
- [98].Koo KM, Sina AAI, Carrascosa LG, Shiddiky MJA, Trau M, Anal. Methods 2015, 7, 7042–7054. [Google Scholar]
- [99].Demers LM, Östblom M, Zhang H, Jang N-H, Liedberg B, Mirkin CA, J. Am. Chem. Soc 2002, 124, 11248–11249. [DOI] [PubMed] [Google Scholar]
- [100].Östblom M, Liedberg B, Demers LM, Mirkin CA, J. Phys. Chem. B 2005, 109, 15150–15160. [DOI] [PubMed] [Google Scholar]
- [101].Piana S, Bilic A, J. Phys. Chem. B 2006, 110, 23467–23471. [DOI] [PubMed] [Google Scholar]
- [102].Gourishankar A, Shukla S, Ganesh KN, Sastry M, J. Am. Chem. Soc 2004, 126, 13186–13187. [DOI] [PubMed] [Google Scholar]
- [103].Kimura-Suda H, Petrovykh DY, Tarlov MJ, Whitman LJ, J. Am. Chem. Soc 2003, 125, 9014–9015. [DOI] [PubMed] [Google Scholar]
- [104].Tan LH, Yue Y, Satyavolu NSR, Ali AS, Wang Z, Wu Y, Lu Y, J. Am. Chem. Soc 2015, 137, 14456–14464. [DOI] [PubMed] [Google Scholar]
- [105].Jiang H, Materon EM, Sotomayor MDPT, Liu J, J. Colloid Interface Sci 2013, 411, 92–97. [DOI] [PubMed] [Google Scholar]
- [106].Yu L, Li N, Langmuir 2016, 32, 5510–5518. [DOI] [PubMed] [Google Scholar]
- [107].Liu B, Liu J, ACS Appl. Mater. Interfaces 2015, 7, 24833–24838. [DOI] [PubMed] [Google Scholar]
- [108].Liu B, Liu J, Langmuir 2015, 31, 371–377. [DOI] [PubMed] [Google Scholar]
- [109].Storhoff JJ, Elghanian R, Mucic RC, Mirkin CA, Letsinger RL, J. Am. Chem. Soc 1998, 120, 1959–1964. [Google Scholar]
- [110].Liu B, Liu J, J. Am. Chem. Soc 2017, 139, 9471–9474. [DOI] [PubMed] [Google Scholar]
- [111].Zhou W, Wang F, Ding J, Liu J, ACS Appl. Mater. Interfaces 2014, 6, 14795–14800. [DOI] [PubMed] [Google Scholar]
- [112].Phan AT, Mergny JL, Nucleic Acids Res 2002, 30, 4618–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Bochman ML, Paeschke K, Zakian VA, Nat. Rev. Genet 2012, 13, 770–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Benabou S, Avino A, Eritja R, Gonzalez C, Gargallo R, RSC Advances 2014, 4, 26956–26980. [Google Scholar]
- [115].Liu D, Cheng E, Yang Z, NPG Asia Mater 2011, 3, 109–114. [Google Scholar]
- [116].Nesterova IV, Nesterov EE, J. Am. Chem. Soc 2014, 136, 8843–8846. [DOI] [PubMed] [Google Scholar]
- [117].Aizen R, Golub E, Trifonov A, Shimron S, Niazov-Elkan A, Willner I, Small 2015, 11, 3654–3658. [DOI] [PubMed] [Google Scholar]
- [118].Tan LH, Doctoral Thesis, Studying the interface between DNA and inorganic nanoparticles to control shape and anisotropicity University of Illinois at Urbana-Champaign, 2015. [Google Scholar]
- [119].Xia Y, Gilroy KD, Peng H-C, Xia X, Angew. Chem. Int. Ed 2016, n/a-n/a.
- [120].Sun Y, Yin Y, Mayers BT, Herricks T, Xia Y, Chem. Mater 2002, 14, 4736–4745. [Google Scholar]
- [121].Gole A, Murphy CJ, Chem. Mater 2004, 16, 3633–3640. [Google Scholar]
- [122].Habas SE, Lee H, Radmilovic V, Somorjai GA, Yang P, Nat. Mater 2007, 6, 692–697. [DOI] [PubMed] [Google Scholar]
- [123].Quan Z, Wang Y, Fang J, Acc. Chem. Res 2013, 46, 191–202. [DOI] [PubMed] [Google Scholar]
- [124].Mel’nikov SM, Sergeyev VG, Yoshikawa K, J. Am. Chem. Soc 1995, 117, 2401–2408. [Google Scholar]
- [125].Porebski S, Bailey LG, Baum BR, Plant Molecular Biology Reporter 1997, 15, 8–15. [Google Scholar]
- [126].Dias R, Mel’nikov S, Lindman B, Miguel MG, Langmuir 2000, 16, 9577–9583. [Google Scholar]
- [127].Zhang X, Servos MR, Liu J, J. Am. Chem. Soc 2012, 134, 7266–7269. [DOI] [PubMed] [Google Scholar]
- [128].Zhao L, Ji X, Sun X, Li J, Yang W, Peng X, J. Phys. Chem. C 2009, 113, 16645–16651. [Google Scholar]
- [129].Rapino S, Zerbetto F, Langmuir 2005, 21, 2512–2518. [DOI] [PubMed] [Google Scholar]
- [130].Xia X, Xie S, Liu M, Peng H-C, Lu N, Wang J, Kim MJ, Xia Y, Proc. Natl. Acad. Sci. U.S.A 2013, 110, 6669–6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Gilroy KD, Ruditskiy A, Peng H-C, Qin D, Xia Y, Chem. Rev 2016. [DOI] [PubMed]
- [132].Njoki PN, Luo J, Kamundi MM, Lim S, Zhong C-J, Langmuir 2010, 26, 13622–13629. [DOI] [PubMed] [Google Scholar]
- [133].Thanh NTK, Maclean N, Mahiddine S, Chem. Rev 2014, 114, 7610–7630. [DOI] [PubMed] [Google Scholar]
- [134].Harada M, Kizaki S, Cryst. Growth Des 2016, 16, 1200–1212. [Google Scholar]
- [135].Satyavolu NSR, Pishevaresfahani N, Tan LH, Lu Y, Nano. Res 2018, 11, 4549–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Maxwell DJ, Taylor JR, Nie S, J. Am. Chem. Soc 2002, 124, 9606–9612. [DOI] [PubMed] [Google Scholar]
- [137].Liu J, Lu Y, Angew. Chem. Int. Ed 2006, 45, 90–94. [Google Scholar]
- [138].Tans SJ, Verschueren ARM, Dekker C, Nature 1998, 393, 49–52. [Google Scholar]
- [139].Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC, Clinical Pharmacology & Therapeutics 2008, 83, 761–769. [DOI] [PubMed] [Google Scholar]
- [140].Dreaden EC, Alkilany AM, Huang X, Murphy CJ, El-Sayed MA, Chem. Soc. Rev 2012, 41, 2740–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Poizot P, Laruelle S, Grugeon S, Dupont L, Tarascon JM, Nature 2000, 407, 496–499. [DOI] [PubMed] [Google Scholar]
- [142].Kamat PV, J. Phys. Chem. C 2008, 112, 18737–18753. [Google Scholar]
- [143].Zhang C, Macfarlane RJ, Young KL, Choi CHJ, Hao L, Auyeung E, Liu G, Zhou X, Mirkin CA, Nat. Mater 2013, 12, 741–746. [DOI] [PubMed] [Google Scholar]
- [144].Yang Y, Velmurugan B, Liu X, Xing B, Small 2013, 9, 2937–2944. [DOI] [PubMed] [Google Scholar]
- [145].Wang X, Zhuang J, Peng Q, Li Y, Nature 2005, 437, 121–124. [DOI] [PubMed] [Google Scholar]
- [146].Hurst SJ, Lytton-Jean AKR, Mirkin CA, Anal. Chem 2006, 78, 8313–8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Liu J, Lu Y, Nat. Protocols 2006, 1, 246–252. [DOI] [PubMed] [Google Scholar]
- [148].Liu B, Wu T, Huang Z, Liu Y, Liu J, Angew. Chem. Int. Ed 2019, 58, 2109–2113. [DOI] [PubMed] [Google Scholar]
- [149].Yang P, Zheng J, Xu Y, Zhang Q, Jiang L, Adv. Mater 2016, 28, 10508–10517. [DOI] [PubMed] [Google Scholar]
- [150].Si S, Liang W, Sun Y, Huang J, Ma W, Liang Z, Bao Q, Jiang L, Adv. Funct. Mater 2016, 26, 8137–8145. [Google Scholar]
- [151].Yu X, Hu L, He H, Zhang F, Wang M, Wei W, Xia Z, Anal. Chim. Acta 2019. [DOI] [PubMed]
- [152].Giljohann DA, Seferos DS, Patel PC, Millstone JE, Rosi NL, Mirkin CA, Nano Lett 2007, 7, 3818–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Chithrani BD, Ghazani AA, Chan WCW, Nano Lett 2006, 6, 662–668. [DOI] [PubMed] [Google Scholar]
- [154].Erkkila KE, Odom DT, Barton JK, Chem. Rev 1999, 99, 2777–2796. [DOI] [PubMed] [Google Scholar]
- [155].Pages BJ, Ang DL, Wright EP, Aldrich-Wright JR, Dalton Trans 2015, 44, 3505–3526. [DOI] [PubMed] [Google Scholar]
- [156].Linic S, Christopher P, Ingram DB, Nat. Mater 2011, 10, 911–921. [DOI] [PubMed] [Google Scholar]
- [157].Hou W, Cronin SB, Adv. Funct. Mater 2013, 23, 1612–1619. [Google Scholar]
- [158].Jiang R, Li B, Fang C, Wang J, Adv. Mater 2014, 26, 5274–5309. [DOI] [PubMed] [Google Scholar]
- [159].Parsons DF, Science 1974, 186, 407–414. [DOI] [PubMed] [Google Scholar]
- [160].Zheng H, Smith RK, Jun Y.-w., Kisielowski C, Dahmen U, Alivisatos AP, Science 2009, 324, 1309–1312. [DOI] [PubMed] [Google Scholar]
- [161].de Jonge N, Ross FM, Nat. Nano 2011, 6, 695–704. [DOI] [PubMed] [Google Scholar]
- [162].Woehl TJ, Park C, Evans JE, Arslan I, Ristenpart WD, Browning ND, Nano Lett 2014, 14, 373–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Jiang Y, Zhu G, Lin F, Zhang H, Jin C, Yuan J, Yang D, Zhang Z, Nano Lett 2014, 14, 3761–3765. [DOI] [PubMed] [Google Scholar]
- [164].Ye X, Jones MR, Frechette LB, Chen Q, Powers AS, Ercius P, Dunn G, Rotskoff GM, Nguyen SC, Adiga VP, Zettl A, Rabani E, Geissler PL, Alivisatos AP, Science 2016, 354, 874–877. [DOI] [PubMed] [Google Scholar]
- [165].Niu K-Y, Park J, Zheng H, Alivisatos AP, Nano Lett 2013, 13, 5715–5719. [DOI] [PubMed] [Google Scholar]
- [166].Chithrani BD, Chan WCW, Nano Lett 2007, 7, 1542–1550. [DOI] [PubMed] [Google Scholar]
- [167].Nel AE, Madler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M, Nat. Mater 2009, 8, 543–557. [DOI] [PubMed] [Google Scholar]