

Abstract
A method for the construction of boron-substituted quaternary carbons or diarylquaternary carbons by arylboration of highly substituted alkenylarenes is presented. A wide range of alkenes and arylbromides can participate in this reaction thus allowing for a diverse assortment of products to be prepared. In addition, a solvent dependent regiodivergent arylboration of 1,2-disubstituted alkenylarenes is presented, thus greatly increasing the scope of products that can be accessed.
Keywords: Nickel, Boron, Arylboration, Alkene, Cross Coupling
Graphical Abstract
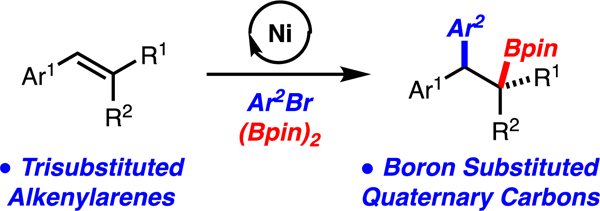
Alkene difunctionalization is a significant synthetic strategy as molecular complexity can be rapidly generated.1 Within this broad area, alkene carboboration has emerged as an effective strategy that allows for a diverse range of groups to be installed across the alkene by taking advantage of the synthetic versatility of the C-B bond. 2,3 Over the last five years, alkene arylboration methods have emerged with contributions from our lab4 and others.5 The arylboration of alkenylarenes is well developed with mono-, 1,2-di-, and 1,1-disubstituted alkenes (Scheme 1A).4,5 Notably, functionalization of trisubstituted alkenylarenes remains a challenge. We sought to develop an arylboration of these classes of substrates because the products would contain boron-substituted quaternary carbons or diarylquaternary carbons, motifs that are currently difficult to access (Scheme 1B). In particular, synthesis of boron-substituted quaternary carbons from widely available alkenes was seen as a key motivation for this study. Notable advances in the synthesis of boron substituted quaternary carbons from alkenes include conjugate addition/allylic substitution,6 hydroboration/diboration,7 conjunctive cross coupling,8 and aminoboration9,10 The majority of the known approaches utilize alkenes with strong activating group (e.g. substituted acrylates) or directing groups. The approach outlined herein is appealing as simple alkenylarenes are the starting materials and two bonds and two stereogenic centers are generated.
Scheme 1.
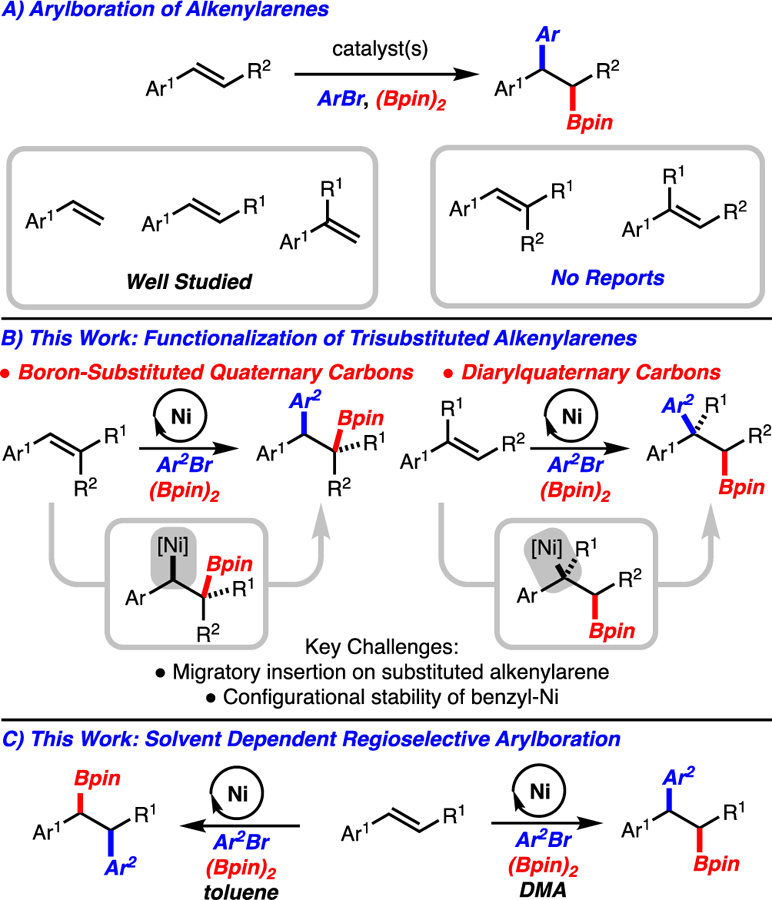
Arylboration of Alkenylarenes.
Early attempts from our group to develop arylboration reactions of trisubstituted alkenylarenes with Cu/Pd-catalysis were unsuccessful.4a-e However, our recent discovery of a Ni-catalyzed arylboration of unactivated alkenes,4f prompted us to explore Ni-catalysis in the context of highly substituted alkenylarenes. Herein, we disclose a method for the arylboration of a wide range of trisubstituted alkenylarenes that leads to the formation of challenging boron-substituted quaternary carbons or diarylquaternary carbons (Scheme 1B). In addition, we have uncovered a solvent dependent regiodivergent arylboration of 1,2-disubstiuted alkenylarenes that further increases the utility of this method (Scheme 1C).
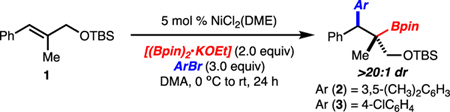
Arylboration of trisubstituted alkenylarenes under previously reported Ni-catalysis conditions resulted in low to moderate yield of the desired products due to incomplete conversion.4f Evaluation of reaction conditions ultimately led to the finding that use of DMA as solvent led to high yield of product (compare Table 1, entries 1 and 2). Unexpectedly, it was also found that premixing KOEt and (Bpin)2 prior to the addition of substrates and catalyst led to higher yields (compare Table 1, entries 1 and 3). Presumably premixing of KOEt and (Bpin)2 serves to sequester KOEt. It has been reported that alkoxide bases and Ni(II) salts can result in the formation of nanoparticles, which may deactivate the Ni-catalyst in this case.11 While 5 mol % NiCl2(DME) and 3.0 equiv aryl bromide were found to be optimal, lower catalyst loading or fewer equivalents of aryl bromide still resulted in useful yields (Table 1, entries 4 and 5). In addition, the reaction could be setup without the aid of glovebox techniques with little change in efficiency (Table 1, entries 6). Finally, it should be noted that the optimal conditions were particularly crucial for arylbromides bearing electron-withdrawing groups (compare Table 1, entries 7–9).
Table 1.
Change from Standard Conditions
 | |||
|---|---|---|---|
| entry | change from standard conditions | product | yield (%)[a] |
| 1 | no change | 2 | 98 |
| 2 | THF instead of DMA | 2 | 21 |
| 3 | no premixing of (Bpin)2 and KOEt | 2 | 79 |
| 4 | 1.5 equiv ArBr instead of 3.0 equiv ArBr | 2 | 65 |
| 5 | 2 mol % NiCl2 (DME) instead of 5 mol % NiCl2 (DME) | 2 | 75 |
| 6 | glovebox free reaction setup | 2 | 94 |
| 7 | no change | 3 | 65 |
| 8 | THF instead of DMA | 3 | 20 |
| 9 | no premixing of (Bpin)2 and KOEt | 3 | 29 |
Reactions performed on 0.2 mmol scale.
Yield determined by 1H NMR analysis of the unpurified reaction mixture with an internal standard.
Under the optimized conditions arylbromides bearing electron donating, electron withdrawing and sterically demanding substituents were well tolerated (Scheme 2). Various functional groups such as amines, alcohols, esters, and amides did not greatly impede the reaction. Finally, heterocycles could also be employed, albeit with slightly reduced yields. The primary limitation with respect to arylbromide scope is that strongly electron-withdrawing substituents such as CF3 resulted in low yields of product and formation of significant quantities of ArBpin.
Scheme 2.
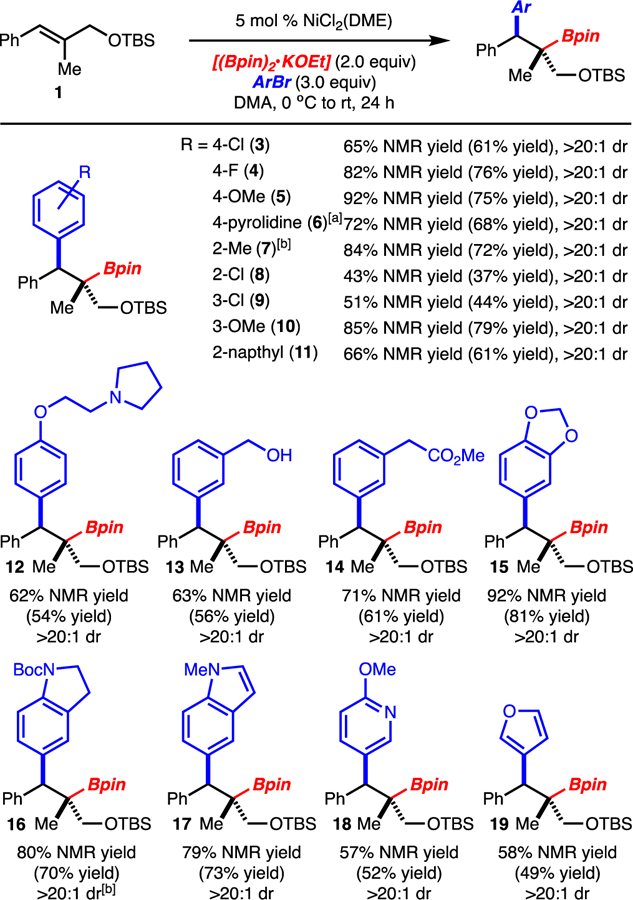
Reaction with Various Arylbromides. Yield of product after isolation. NMR yield determined by 1H NMR analysis of the unpurified reaction mixture with an internal standard. [a] Reaction with the 4-OMeAr analog of 1 used. [b] Products 7 and 16 were oxidized to the alcohol before isolation. Reactions performed on 0.4 mmol scale.
Regarding the scope of alkenes that can be used, several points are noteworthy (Scheme 3): 1) For the synthesis of boron-substituted quaternary carbons, the substitution pattern of the aryl group could be altered with various electron withdrawing, electron donating, sterically demanding substituents. 2) Both cyclic and acyclic substrates are suitable for this process. 3) The formation of 30 and 37 occurred with high levels of diastereoselectivity. In each case, the arylboration occurred from the least hindered face of the alkene. 3) For substrates with acyclic substituents, yield decreased with more sterically demanding substituents (compare 2 and 33). 4) The synthesis of diarylquaternary carbons can be achieved with both acyclic and cyclic substrates (products 38-44).
Scheme 3.

Arylboration of Trisubstituted Alkenes. See Scheme 2 for details.
The arylboration could also be performed on gram scale (Scheme 4). Furthermore, the C-B bond could be elaborated through oxidation (product 47) and Matteson homologation/oxidation (product 48). Alcohol 48 could be advanced to Glucocorticoid receptor modulators through standard sequences.12 Previously, these molecules were prepared by a non-stereoselective 6-step sequence. The route illustrated here required 3–4 operations, is stereoselective, and represents an orthogonal approach to the construction of biologically active molecules from alkenes.
Scheme 4.
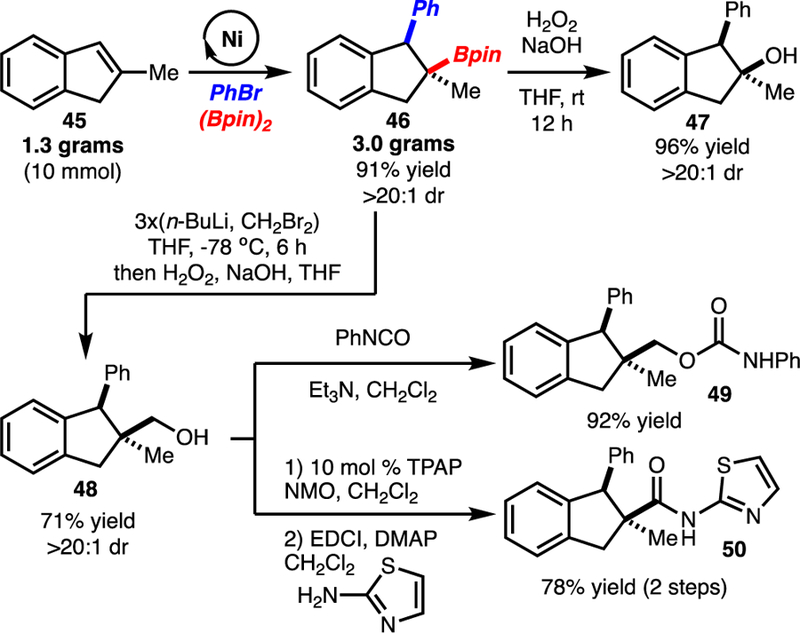
Gram Scale Reaction and Further Transformations.
In the course of these investigations, 1,2-disubstituted alkenylarenes were also probed. Under the standard conditions, 51 was generated in excellent selectivity (Table 2, entry 1). Interestingly, the observation was made that if toluene was used as a co-solvent, the formation of regioisomer 52 (>20:1 dr) was observed (Table 2, entry 2). Increasing the amount of toluene revealed that regioisomer 52 could be generated as the major product, albeit in low yield (Table 2, entry 7). Continued optimization led to the finding that higher reaction temperatures (60 °C vs. rt), use of KOMe (vs. KOEt), and excess alkene (relative to arylbromide) resulted in exclusive formation of 52 (Table 2, entry 12). Thus, through a simple change in solvent and minor modification to reaction conditions, the arylboration could be tuned to favor either regioisomer 51 or 52. Prior studies have demonstrated that product 51 can be prepared by a Cu/Pd-catalyzed arylboration.4,5 The Ni-catalyzed reaction reported herein offers an alternative that utilizes a simple catalyst. Methods for the synthesis of the regioisomeric product 52 do not exist. However, a recent report from Song, has demonstrated that styrene derivatives, but not 1,2-disubstituted alkenylarenes, can undergo Pd-catalyzed arylboration with aryldiazonium salts and (Bpin)2.5c
Table 2.
Regiodivergent Arylboration of 1,2-Disubstituted Alkenylarenes
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | solvent | temp (°C) | base | yield (%)[a] | 51 | : | 52 |
| 1 | DMA | rt | KOEt | 98 | >99 | : | 1 |
| 2 | toluene:DMA (1:1) | rt | KOEt | 98 | 93 | : | 1 |
| 3 | toluene:DMA (5:1) | rt | KOEt | 42 | 34 | : | 1 |
| 4 | toluene:DMA (10:1) | rt | KOEt | 28 | 14 | : | 1 |
| 5 | toluene:DMA (50:1) | rt | KOEt | 15 | 4.6 | : | 1 |
| 6 | toluene:DMA (160:1) | rt | KOEt | 17 | 2.3 | : | 1 |
| 7 | toluene | rt | KOEt | 15 | 1 | : | 9 |
| 8 | toluene | 60 | KOEt | 55 | 1 | : | 2.2 |
| 9 | toluene | 60 | KOMe | 42 | 1 | : | 5 |
| 10[b] | toluene | 60 | KOMe | 42 | 1 | 36 | |
| 11 [b],[c] | toluene | 60 | KOMe | 45 | 1 | 39 | |
| 12[c],[d] | toluene | 60 | KOMe | 58 | 1 | >99 | |
Reactions performed on 0.2 mmol scale.
Combined yield of 51 and 52 determined by GC using a calibrated internal standard.
ArBr (1.2 equiv).
NiCl2(DME) (15 mol%).
Alkene (2.0 equiv), ArBr, (1.0 equiv).
Under conditions outlined in Table 2 entries 1 and 12 both acyclic and cyclic 1,2-disubstituted alkenylarenes participated in the reactions with excellent regioselectivity and diastereoselectivity (products 51-60). While arylboration in DMA worked well with trisubstituted alkenes (Schemes 2–3), the conditions with toluene did not proceed, and starting material was recovered.
To account for the different regioselectivities observed in different solvents, two catalytic cycles are proposed (Scheme 6). In the case of reactions performed in DMA, and on the basis of prior studies, we propose that [Ni]-Bpin complex 62 is generated, which undergoes reaction with an alkene.4f,13 The regioselectivity of the migratory insertion occurs to generate benzyl-[Ni]-complex 63 regardless of alkene substitution pattern. The formation of product may occur through an oxidative addition with ArBr followed by reductive elimination via 64. In the case of reactions in toluene, it is proposed that migratory insertion occurs from Ni(II)ArBr complex 66.14 The regioselectivity is determined though formation of benzyl-[Ni]-complex 67. Transmetalation with (Bpin)2 and reductive elimination then occurs to generate product.
Scheme 6.
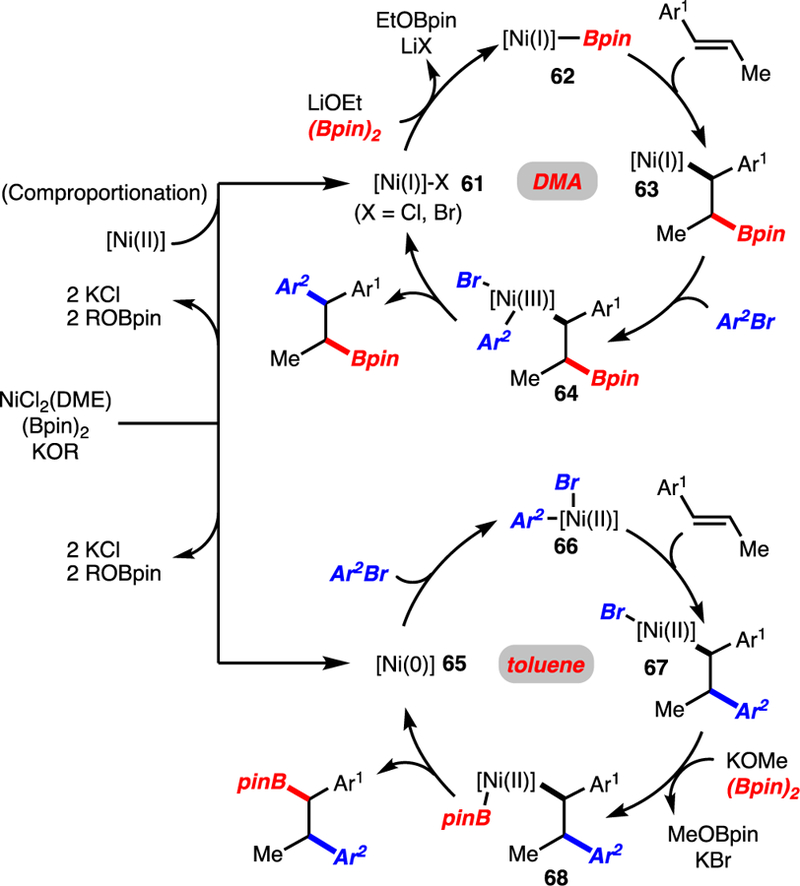
Putative Catalytic Cycles.
Support for the proposed catalytic cycles is illustrated in Scheme 7. It was found that addition of MeOH to the reaction mixture resulted in formation of protoboration adduct 69, thus suggesting that Bpin is incorporated prior to the aryl group.4f In addition, reaction of vinyl fluoride 70 led to the formation of vinyl boronic ester 71. Since direct cross coupling with vinyl fluorides is slow at ambient temperatures,15,16 it is suggested that the product formation occurs via 1,2-elimination of the corresponding alkyl-[Ni]-complex.17 With respect to the mechanistic proposal for the reaction in toluene, it was observed that reaction of α-methyl styrene (72) led to products resulting from a Heck-type reaction (74) and hydroarylation (75). The formation of these adducts can be rationalized with competitive reactions of the corresponding benzyl-[Ni]-complex via β-hydride elimination or protonation, respectively.18 At this stage, the role of solvent in determining the regioselectivity is not clear and is under investigation.
Scheme 7.
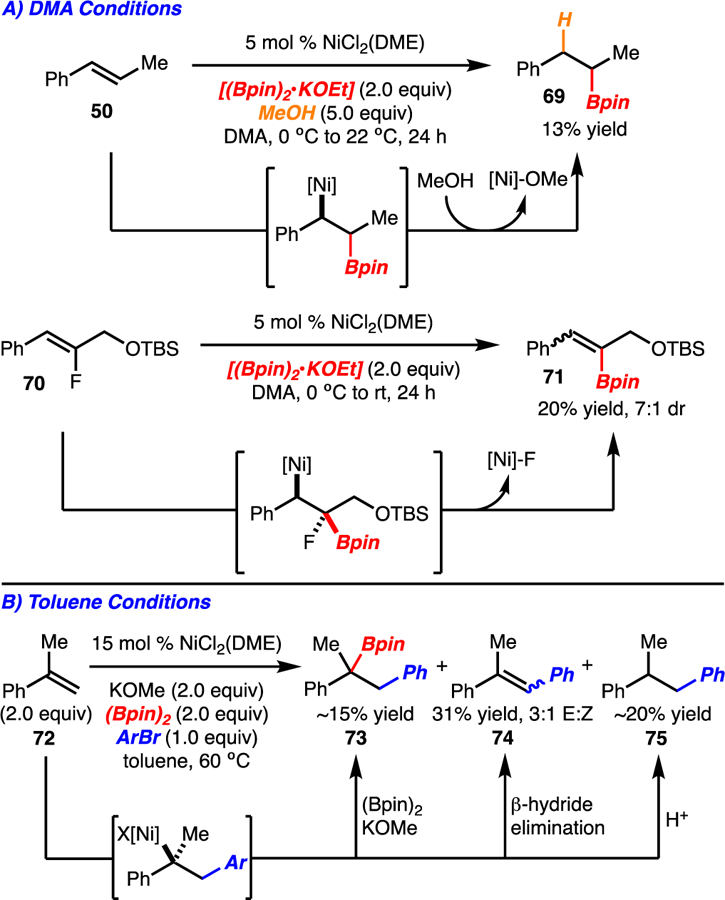
Mechanistic Experiments.
In summary, the synthesis of challenging boron-substituted quaternary carbons and diarylquaternary carbons has been achieved through an arylboration reaction. Furthermore, the arylboration regioselectivity of 1,2-disubstituted alkenylarenes can be tuned by altering the solvent, revealing a practical system in which diverse and versatile products can be accessed.19
Supplementary Material
Scheme 5.
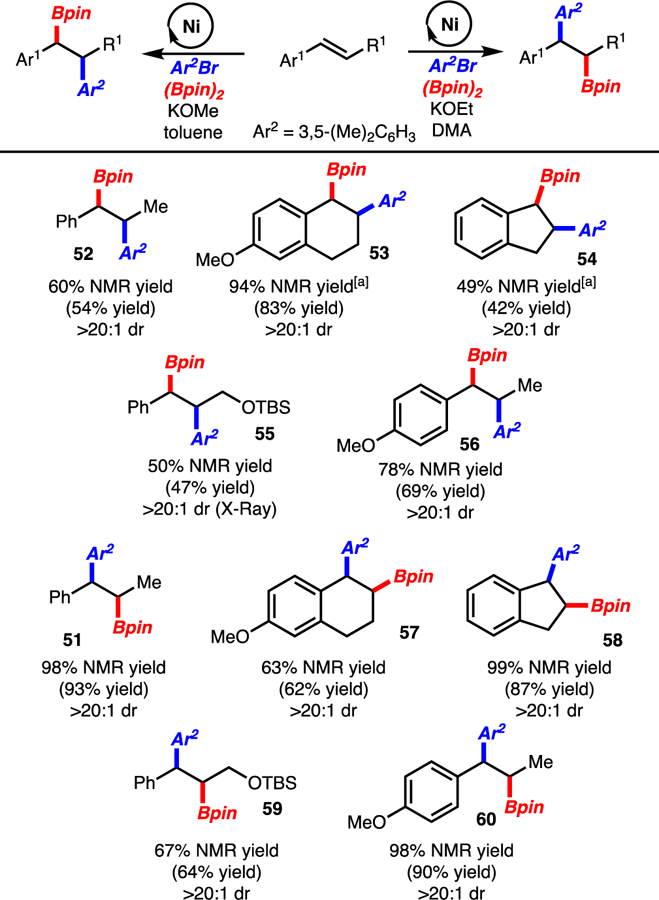
Regiodivergent Arylboration. See Table 2, entries 1 and 12 for conditions (0.4 mmol scale). Yield of product after isolation. NMR yield determined by 1H NMR analysis of the unpurified reaction mixture with an internal standard. [a] Reaction with 1.0 equiv alkene and 1.2 equiv ArBr.
Acknowledgements
We thank Indiana University and the National Institutes of Health (R01GM114443 and R35GM131755) for generous financial support. This project was partially funded by the Vice Provost for Research through the Research Equipment Fund.
References
- 1.a) Saini V, Stokes BJ, Sigman MS, Angew. Chem. Int. Ed 2013, 52, 11206 Angew. Chem. 2013, 125, 11414–11428 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Coombs JR, Morken JP, Angew. Chem. Int. Ed. Engl 2016, 55, 2636–2649. Angew. Chem. 2016, 128, 2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For reviews regarding carboboration, see: (a) Shimizu Y, Kanai M, Tetrahedron Lett 2014, 55, 3727. [Google Scholar]; (b) Semba K, Fujihara T, Terao J, Tsuji Y, Tetrahedron 2015, 71, 2183. [Google Scholar]; (c) Lazreg F, Nahra F, Cazin CSJ, Coord. Chem. Rev 2015, 293–294, 48. [Google Scholar]; (d) Neeve EC, Geier SJ, Mkhalid IAI, Westcott SA, Marder TB, Chem. Rev 2016, 116, 9091. [DOI] [PubMed] [Google Scholar]; (e) Hemming D, Fritzemeier R, Westcott SA, Santos WL, Steel PG, Chem. Soc. Rev 2018, 47, 7477. [DOI] [PubMed] [Google Scholar]
- 3.Sandford C, Aggarwal VK, Chem. Commun 2017, 53, 5481. [DOI] [PubMed] [Google Scholar]
- 4.a) Smith KB, Logan KM, You W, Brown MK, Chem. Eur. J 2014, 20, 12032; [DOI] [PubMed] [Google Scholar]; b) Logan KM, Smith KB, Brown MK, Angew. Chem. Int. Ed 2015, 54, 5228; Angew. Chem. 2015, 127, 5317; [DOI] [PubMed] [Google Scholar]; c) Logan KM, Brown MK, Angew. Chem. Int. Ed 2017, 56, 851; Angew. Chem. 2017, 129, 869; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Smith KB, Brown MK, J. Am. Chem. Soc 2017, 139, 7721; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Sardini SR, Brown MK, J. Am. Chem. Soc 2017, 139, 9823. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Bergman AM, Dorn SK, Smith KB, Logan KM, Angew. Chem. Int. Ed 2019, 58, 1719. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Logan KM, Sardini SR, White SD, Brown MK, J. Am. Chem. Soc 2017, 140, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Semba K, Nakao Y, J. Am. Chem. Soc 2014, 136, 7567. [DOI] [PubMed] [Google Scholar]; b) Semba K, Ohtagaki Y, Nakao Y, Org. Lett 2016, 18, 3956. [DOI] [PubMed] [Google Scholar]; c) Yang K, Song Q, Org. Lett 2016, 18, 5460. [DOI] [PubMed] [Google Scholar]; d) Chen B, Cao P, Yin X, Liao Y, Jiang L, Ye J, Wang M, Liao J ACS Catal 2017, 4, 2425. [Google Scholar]; e) Wang W, Ding C, Li Y, Li Y, Peng L, Yin G Angew. Chem. Int. Ed 2019, 58, 4612. [DOI] [PubMed] [Google Scholar]; f) Liu Z, Li X, Zeng T, Engle KM, ACS Catal 2019, 9, 3260. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) For a 1,1-arylboration, see: Nelson HM, Williams BD, Miro J, Toste FD J. Am. Chem. Soc 2015, 137, 3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Chen IH, Yin L, Itano W, Kanai M, Shibasaki M, J. Am. Chem. Soc 2009, 131, 11664; [DOI] [PubMed] [Google Scholar]; b) Chen IH, Yin L, Kanai M, Shibasaki M, Org. Lett 2010, 12, 4098; [DOI] [PubMed] [Google Scholar]; c) O’Brien JM, Lee K, Hoveyda AH, J. Am. Chem. Soc 2010, 132, 10630–10633; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Radomkit S, Hoveyda AH, Angew. Chem. Int. Ed 2014, 53, 3387; Angew.Chem. 2014, 126, 3455; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Feng X, Yun J, Chem. Eur. J 2010, 16, 13609. [DOI] [PubMed] [Google Scholar]; f) Guzman-Martinez AH Hoveyda, J. Am. Chem. Soc 2010, 132, 10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Blaisdell TP, Caya TC, Zhang L, Sanz-Marco A, Morken JP, J. Am. Chem. Soc 2014, 136, 9264. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kerchner HA, Montgomery J, Org. Lett 2016, 18, 5760; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Shoba VM, Thacker NC, Bochat AJ, Takacs JM, Angew. Chem. Int. Ed 2016, 55, 1465; Angew. Chem. 2016, 128, 1487; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chakrabarty S, Takacs JM, J. Am. Chem. Soc 2017, 139, 6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Silvi M, Sandford C, Aggarwal VK, J. Am. Chem. Soc 2017, 139, 5736; [DOI] [PubMed] [Google Scholar]; b) Kischkewitz M, Okamoto K, Muck-Lichtenfeld C, Studer A, Science 2017, 355, 936; [DOI] [PubMed] [Google Scholar]; c) Myhill JA, Zhang L, Lovinger GJ, Morken JP, Angew. Chem., Int. Ed 2018, 57, 12799; Angew. Chem. 2018, 130, 12981; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Tao Z, Robb KA, Panger JL, Denmark SE, J. Am. Chem. Soc 2018, 140, 15621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kato K, Hirano K, Miura M, J. Org. Chem 2017, 82, 10418. [DOI] [PubMed] [Google Scholar]
- 10.For selected examples of alternative approaches, see: a) Kato K, Hirano K, Miura M, J. Org. Chem 2017, 82, 10418. [DOI] [PubMed] [Google Scholar]; b) Atack TC, Cook SP, J. Am. Chem. Soc 2016, 138, 6139. [DOI] [PubMed] [Google Scholar]
- 11.Buslov I, Song F, Hu X Angew. Chem. Int. Ed 2016, 55, 12295–12299. Angew. Chem. 2016, 128, 12483. [DOI] [PubMed] [Google Scholar]
- 12.Duan J, Jiang B PCT Int. Appl 2007073503
- 13.Ni(I) may form through a comproportion pathway, as illustrated in Scheme 6. For a review regarding Ni(I), see: Lin C-Y, Power PP, Chem. Soc. Rev 2017, 46, 5347. [DOI] [PubMed] [Google Scholar]
- 14.Gao P, Chen L-A, Brown MK, J. Am. Chem. Soc 2018, 140, 10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X-W, Echavarren J, Zarate C, Martin R, J. Am. Chem. Soc 2015, 137, 12470. [DOI] [PubMed] [Google Scholar]
- 16.For β-fluoro elimination of an alkyl-Ni-complex, see: Zhou L, Zhu C, Bi P, Feng C, Chem. Sci 2019, 10, 1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.For a Cu-catalyzed variant, see: Sakaguchi H, Uetake Y, Ohashi M, Niwa T, Ogoshi S, Hosoya T, J. Am. Chem. Soc 2017, 139, 12855. [DOI] [PubMed] [Google Scholar]
- 18.The proton source may arise from the byproduct of β-hydride elimination.
- 19.During the preparation of this manuscript a Ni-catalyzed arylboration of vinylarenes was reported, see: Wang W, Ding C, Pang H, Yin G Org. Lett 10.1021/acs.orglett.9b01120. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.