

Abstract
Background:
Epigenetic abnormalities are manifold in all solid tumors and include changes in chromatin configuration and DNA methylation. We designed a phase I study to evaluate oral DNT methyltransferase inhibitor CC-486 with the histone deacetylase inhibitor romidepsin in advanced solid tumors with dose expansion to further evaluate pharmacodynamics and possible clinical benefit of the recommended phase II dose (RP2D).
Methods:
This was a phase I study with a 3+3 dose escalation design and an expansion phase for patients with virally mediated cancers. Disease control rate (DCR) was the primary outcome for the expansion cohort. Correlative studies included LINE-1 methylation and drug exposure in blood samples. Clinical Trial Registration: NCT01537744
Results:
Fourteen patients were enrolled in the dose escalation portion at three dose levels. Three patients experienced dose-limiting toxicities; the RP2D was CC-486 300mg orally daily days 1–14 and romidepsin 8mg/m2 days 8 and 15. Due to slow accrual into the expansion phase, the trial was closed after 4 patients enrolled. Common toxicities of the combination included nausea (83.3%), anorexia (72.2%), fatigue (61.1%), and constipation (55.6%). There were 12 patients evaluable for response, 5 with stable disease, 2 of which were > 4 cycles; there were no responses. CC-486 and romidepsin exposure were consistent with prior data. LINE-1 methylation on C1D8 was significantly reduced (mean: −6.23; 95% CI: −12.23, −0.24; p=0.04).
Conclusions:
While at the RP2D the combination of CC-486 and romidepsin was tolerable, no significant anti-cancer activity was observed. Significant demethylation in post-treatment ctDNA and biopsies provided proof of target acquisition.
Keywords: Phase I clinical trial, epigenetic therapy, DNA methyltransferase (DNMT) inhibitors, histone deacetylase (HDAC) inhibitors, CC-486, romidepsin
Precis:
Although the combination of CC-486 and romidepsin was safe and tolerable. While pharmacodynamic data confirmed on-target effects, no significant anti-cancer activity was observed. Further development in combination with other anti-neoplastic therapies may be feasible.
Alterations in the histone code regulating transcription can result in aberrant expression of genes leading to carcinogenesis. In particular, epigenetic silencing of tumor-suppressor genes through hypermethylation plays a key role in carcinogenesis 1, 2. While modifications of the primary sequence of DNA are unlikely to be reversible, epigenetic changes may be modulated by inhibitors targeting DNA methyltransferases (DNMT) and histone deacetylases (HDAC) leading to effective anti-neoplastic therapy. The DNMT inhibitors, azacitidine and decitabine, have been approved for the treatment of myelodysplastic syndrome; while HDAC inhibitors, such as romidepsin and panobinostat, are currently in clinical use in the treatment of cutaneous and peripheral T-cell lymphomas and multiple myeloma. However, in solid tumors, single agent therapy with either DNMT or HDAC inhibitors results in limited tumor response possibly secondary to a lower therapeutic index or limited bioavailability 3, 4.
In solid tumors, the combination of DNMT and HDAC inhibitors synergistically induces re-expression of tumor suppressor genes and inhibition of tumor growth in preclinical models 5–10. The combination results in reversal of hypermethylation which correlates with improved PFS in patients with colorectal cancer and objective, durable responses in patients with advanced non-small cell lung cancer 11, 12. Aberrant methylation of CpG islands in tumor suppressor gene promoter regions occurs more frequently in virally-mediated malignancies compared to their non-virally induced counterparts (reviewed in 13). Clinical responses to DNMT inhibition with the oral formulation of the DNMT inhibitor 5-azacitidine, CC-486, were observed in virally mediated cancers 14.
We hypothesized that combination therapy with DNMT and HDAC inhibitors would be effective in reversing abnormal gene DNA methylation and thus result in therapeutic benefit in patients with virally-mediated cancers. We designed a phase I study to determine the maximum tolerated dose (MTD) of the combination of CC-486 with romidepsin for patients with advanced solid tumors with an expansion cohort for patients with virally-mediated cancers.
Methods
Patients
This study was approved by the Johns Hopkins Institutional Review Board (NCT01537744). Written informed consent was obtained from all patients prior to performing study-related procedures in accordance with federal and institutional guidelines. Patients were eligible for this study if they had a histologically confirmed metastatic or unresectable solid tumor for the phase I dose escalation portion. Patients must have received at least one previous chemotherapy regimen for metastatic disease if standard therapies exist, have had measurable disease by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0, Eastern Cooperative Oncology Group performance status ≤ 1, and adequate hematologic, hepatic, and renal function. Exclusion criteria included chemotherapy, radiation therapy, or experimental drug therapy within 4 weeks of enrollment and any major comorbidity or intercurrent illness including active untreated brain metastases, active infection, symptomatic cardiac dysfunction, or advanced malignant hepatic tumors.
Once the study had identified the MTD of the combination, the clinical protocol was amended to limit the expansion cohort to patients with metastatic or unresectable virally-mediated cancer (such as HPV+ nasopharyngeal, cervical, or anal carcinoma) based on pre-clinical and clinical efficacy in this patient population 14.
Study design
This phase I dose escalation trial used a standard 3+3 phase I dose assessment schema with the endpoint of determining a safe and tolerable dose of CC-486 plus romidepsin, which was then used in the expansion component of the study. The primary endpoint of the expansion cohort of the trial was to determine the preliminary efficacy of CC-486 and romidepsin in subjects with virally-mediated cancers as determined by disease control rate (DCR; percent of patients with complete response + partial response + stable disease for ≥ 4 cycles) and to further characterize the toxicity of this regimen to ensure a dose limiting toxicity (DLT) rate of ≤30%. Secondary endpoints included the pharmacokinetics profile during treatment with the combination of CC-486 and romidepsin, changes in methylation on serial samples of circulating tumor DNA for pharmacodynamic assessment, and assessment of correlation between CC-486 and romidepsin exposure and pharmacodynamic effects.
CC-486 was administered orally on days 1–14 of each 28 day cycle. The CC-486 starting dose of 200mg daily was supported by previous studies in hematologic malignancies, which had identified the MTD of the single agent as 480mg daily for 7 days of each 28 day cycle 15. Four dose levels were planned (Table 1). Romidepsin was administered intravenously at a dose of 8mg/m2 on days 8 and 15 for dose levels 1–3 and was planned for days 8, 15, and 22 on dose level 4; this lower dose of romidepsin compared to the approved dose in lymphoma was selected due to concerns of overlapping toxicities and prior pharmacokinetic data indicating reasonable exposure at this dose. The selected doses and schedules were recommended by industry partners.
Table 1:
Planned Dose Levels
| Dose and Schedule (28 day cycles) | Number of subjects enrolled | |||
|---|---|---|---|---|
| Dose Level | 5-Azacitidine | Romidepsin | Phase I | Expansion |
| Level −1† | 100mg daily days 1–14 | 8mg/m2 days 8 and 15 | - | - |
| Level 1 | 200mg daily days 1–14 | 8mg/m2 days 8 and 15 | 3 | - |
| Level 2 | 300mg daily days 1–14 | 8mg/m2 days 8 and 15 | 8 | 4 |
| Level 3 | 300mg daily days 1–21 | 8mg/m2 days 8 and 15 | 3 | - |
| Level 4‡ | MTD daily days 1–21 | 8mg/m2 days 8, 15, and 22 | - | - |
Subjects would only be enrolled in Level −1 if >2 DLTs in Level 1
Level 4 was optional and based on discussions between the study investigator and Celgene was not opened to enrollment.
A DLT was defined as a study drug-related toxicity falling within pre-defined hematologic and non-hematologic parameters occurring during the DLT assessment window (days 1–28 of cycle 1). A hematologic DLT was defined as any of the following: grade 3 febrile neutropenia, grade 4 neutropenia lasting longer than 7 days, grade 4 thrombocytopenia, or grade 3 thrombocytopenia associated with clinically significant bleeding. A non-hematologic DLT was defined as any grade 3 or 4 non-hematologic toxicities related to study drug with the following exceptions: grade 3 nausea or vomiting if unresponsive to maximal medical therapy for 48 hours; grade 3 hyperglycemia, hypophosphatemia, hyponatremia, and hypocalcemia if unresponsive to medical therapy; or grade 3 or 4 diarrhea if unresponsive to maximal medical therapy for ≥ 48 hours.
Assessments
Toxicity was evaluated using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 4 at each intravenous infusion on the treatment schedule. Imaging assessments were performed for all patients at screening, at the end of every other cycle or as clinically indicated. Anti-tumor activity was assessed by investigators according to RECIST 1.1.
Pharmacodynamic analyses
Samples for pharmacokinetic analyses were obtained on Cycle 1 Day 1 for CC-486 and Cycle 1 Day 8 for romidepsin. CC-486 exposure was assessed as the active moiety, 5-azacitidine. Concentrations of 5-azacitidine and romidepsin were determined using a validated liquid chromatography-tandem mass spectrometry (LC/MS/MS) method16, 17. 5-azacitidine and romidepsin pharmacokinetic parameters were determined as previously described 17, 18.
Analysis of LINE-1 methylation
Plasma samples for LINE-1 methylation was obtained prior to starting treatment, as well as prior to romidepsin administration on days 8 and 15 of the study for Cycles 1 and 2, and at the beginning of each cycle, thereafter. Bisulfite converted DNA was obtained from plasma samples using Methylation on Beads technology 19. DNA was amplified using primers specific for Exon 1 of the L1H retrotransposon. Amplification was confirmed by gel electrophoresis followed by pyrosequencing on a Q24 (Qiagen) to determined methylation levels. LINE-1 methylation was analyzed as previously described 20. In brief, the first three CpG dinucleotides from the amplified sequence were used from each time-point and averaged to provide an average percent methylation for each sample. A positive control consisting of in-vitro methylated DNA, and a negative control consisting of DNA from a cell line (HCT116) with double knockout for DNMT1 and DNMT3a, were included with each sample batch (Zymo).
Statistical Considerations
Proportions are reported with exact 95% binomial confidence intervals. The event time distribution for overall survival (OS) was estimated with the method of Kaplan and Meier and confidence intervals were calculated using the method of Brookmeyer and Crowley21, 22. The Wilcoxon-Mann-Whitney test was used to compare mean changes in LINE-1 methylation (cycle 2 day 15 minus baseline) between patients with different clinical response categories. Within patient changes in LINE-1 methylation were assessed with paired t-tests. Pharmacokinetic parameters were summarized using descriptive statistics. Kruskal-Wallis tests were used to compare medians between the groups with respect to drug exposure, response, and toxicity. All P-values reported are two-sided, and the significance level set at 0.05 for all analyses. Statistical analyses were performed using SAS version 9.4 (SAS Institute Inc, Cary, North Carolina) and R version 3.4.4.
Results
Baseline patient demographics and disease characteristics.
Eighteen patients (14 in dose-escalation cohort, 4 in dose-expansion cohort) were enrolled in the study (Table 1 and Table 2) over three dose levels between May 13, 2103 and June 13, 2016. In the dose-escalation cohort, the median age of patients was 53 years (range 42 – 81). The most frequent primary tumor types were colorectal cancer (n = 5) and soft tissue sarcoma (n = 2). The median number of lines of prior treatment was 3 (range 1–8). DL2 was selected as the RP2D. An expansion cohort at the RP2D focused on virally-mediated tumors (including HPV+ nasopharyngeal, cervical, and anal cancers). Four patients were enrolled to the expansion cohort (cervical cancer (n = 3), anal cancer (n = 1)). The study was closed secondary to slow accrual. The median age in the expansion cohort was 64 years (range 42–69).
Table 2.
Patient Characteristics
| All | Dose Escalation | Expansion | |
|---|---|---|---|
| Number enrolled | 18 | 14 | 4 |
| Median Age, years (range) | 55 (42–81) | 53 (42–81) | 64 (42–69) |
| Gender, N (%) | |||
| - Female | 11 (61) | 7 (50) | 4 (100) |
| - Male | 7 (39) | 7 (50) | 0 |
| Performance Status, N (%) | |||
| - 0 | 8 (44) | 7 (50) | 1 (25) |
| - 1 | 7 (39) | 5 (36) | 1 (50) |
| - 2 | 3 (17) | 2 (14) | 1 (25) |
| Tumor Types, N (%) | |||
| - Colorectal | 5 (28) | 4 (36) | 0 |
| - Cervical | 3 (17) | 0 | 3 (75) |
| - Anal | 2 (11) | 1 (7) | 1 (25) |
| - Soft Tissue Sarcoma | 2 (11) | 2 (14) | 0− |
| - Esophageal | 1 (6) | 1 (7) | 0 |
| - HNSCC | 1 (6) | 1 (7) | 0 |
| - Lung | 1 (6) | 1 (7) | 0 |
| - Endometrial | 1 (6) | 1 (7) | 0 |
| - Pancreas | 1 (6) | 1 (7) | 0 |
| - Kidney | 1 (6) | 1 (7) | 0 |
| Prior lines of systemic therapy, N (%) | |||
| - 1 | 4 (22) | 2 (14) | 1 (50) |
| - 2 | 3 (17) | 2 (14) | 1 (25) |
| - 3 | 6 (28) | 4 (36) | 0 |
| - 4+ | 5 (33) | 5 (36) | 1 (25) |
| Prior radiation therapy, N (%) | |||
| - Yes | 12 (67) | 10 (71) | 1 (50) |
| - No | 6 (33) | 4 (29) | 2 (50) |
The median number of cycles received by the entire study population was 2 (range 1–6). Median duration of treatment on study was 56 days (range 13–181).
Adverse events
Nausea, anorexia, fatigue, and constipation were the most common adverse events (AEs) in this trial over the course of therapy (Table 3). Fifteen of 18 patients (83%) experienced any grade (G) of nausea with 1 (6%) experiencing ≥G3 nausea. Thirteen patients (72%) experienced anorexia with 2 (11%) reporting ≥G3 anorexia. Eleven patients (61%) reported fatigue with 4 (22%) reporting ≥G3 fatigue. Thirteen patients experienced ≥G3 hematologic toxicities [neutropenia (n = 5, 28%); anemia (n = 1, 6%); thrombocytopenia (n = 1, 6%)]. No grade 5 toxicities were observed.
Table 3.
Treatment Related Adverse Events
| Adverse Event | Level 1 (N=3) | Level 2 (N=12) | Level 3 (N=3) | Total (N=18) N (%) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cycle 1 | All Cycles | Cycle 1 | All Cycles | Cycle 1 | All Cycles | ||||||||
| Grade ≥3 | Grade 1/2 | Grade 3/4 | Total N (%) | Grade ≥3 | Grade 1/2 | Grade 3/4 | Total N (%) | Grade ≥3 | Grade 1/2 | Grade 3/4 | Total N (%) | ||
| Abdominal infection | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 (8) | 0 | 0 | 0 | 0 | 1 (6) |
| Anemia | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 2 (17) | 0 | 1 | 0 | 1 (33) | 3 (17) |
| Anorexia | 0 | 2 | 0 | 2 (67) | 0 | 7 | 1 | 8 (67) | 1 | 2 | 1 | 3 (100) | 13 (72) |
| Blurred vision | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 (8) | 0 | 0 | 0 | 0 | 1 (6) |
| Cheilitis | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 (8) | 0 | 0 | 0 | 0 | 1 (6) |
| Constipation | 0 | 2 | 0 | 2 (67) | 0 | 7 | 0 | 7 (58) | 0 | 1 | 0 | 1 (33) | 10 (56) |
| Diarrhea | 0 | 0 | 0 | 0 | 1 | 3 | 1 | 4 (33) | 0 | 0 | 0 | 0 | 4 (22) |
| Dizziness | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 (8) | 0 | 0 | 0 | 0 | 1 (6) |
| Dysgeusia | 0 | 1 | 0 | 1 (33) | 0 | 1 | 0 | 1 (8) | 0 | 1 | 0 | 1 (33) | 3 (17) |
| Dyspepsia | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 (8) | 0 | 1 | 0 | 1 (33) | 2 (11) |
| Edema | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 (8) | 0 | 0 | 0 | 0 | 1 (6) |
| Fatigue | 0 | 1 | 0 | 1 (33) | 1 | 6 | 2 | 8 (67) | 2 | 0 | 2 | 2 (67) | 11 (61) |
| Fever | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 2 (17) | 0 | 0 | 0 | 0 | 2 (11) |
| Flushing | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 (8) | 0 | 0 | 0 | 0 | 1 (6) |
| Headache | 0 | 1 | 0 | 1 (33) | 0 | 3 | 0 | 3 (25) | 0 | 1 | 0 | 1 (33) | 5 (28) |
| Hyperglycemia | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 (8) | 0 | 0 | 0 | 0 | 1 (6) |
| Hypomagnesemia | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 (8) | 0 | 0 | 0 | 0 | 1 (6) |
| Insomnia | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 (8) | 0 | 0 | 0 | 0 | 1 (6) |
| Lymphocyte count decreased | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 (8) | 0 | 0 | 0 | 0 | 1 (6) |
| Mucosal infection | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 (8) | 0 | 0 | 0 | 0 | 1 (6) |
| Myalgia | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 (8) | 0 | 0 | 0 | 0 | 1 (6) |
| Nausea | 0 | 3 | 0 | 3 (100) | 0 | 9 | 0 | 9 (75) | 1 | 2 | 1 | 3 (100) | 15 (83) |
| Neutrophil count decreased | 0 | 0 | 0 | 0 | 1 | 0 | 3 | 3 (25) | 0 | 0 | 2 | 2 (67) | 5 (28) |
| Platelet count decreased | 0 | 1 | 0 | 1 (33) | 0 | 3 | 0 | 3 (25) | 0 | 0 | 1 | 1 (33) | 5 (28) |
| Vomiting | 0 | 1 | 0 | 1 (33) | 0 | 2 | 0 | 2 (17) | 0 | 2 | 0 | 2 (67) | 5 (28) |
| Weight loss | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 2 (17) | 0 | 2 | 0 | 2 (67) | 4 (22) |
| White blood cell decreased | 0 | 0 | 0 | 0 | 1 | 0 | 2 | 2 (17) | 0 | 0 | 0 | 0 | 2 (11) |
Dose-limiting toxicities and selection of the RP2D
All 18 patients (14 in dose-escalation and 4 in dose-expansion cohorts) were evaluable for safety and toxicities of the combination. Three patients experienced at least 1 DLT (1 on DL2 and 2 on DL3). On DL2, one patient experienced a DLT of prolonged G3 fatigue. The DL2 cohort was expanded to 6 patients. Two patients on DL2 discontinued therapy before completing cycle 1 but did not meet DLT criteria. Thus, two additional patients were enrolled onto DL2 to allow 6 patients to complete cycle 1 prior to determining whether to escalate to the next dose level. On DL3, one patient experienced G4 thrombocytopenia and G3 fatigue lasting > 21 days; one patient experienced G3 anorexia and G3 fatigue. DL2 (CC-486 300mg daily days 1–14 and romidepsin 8mg/m2 days 8 and 15) was declared the MTD and selected as the RP2D used for the expansion cohort. No DLTs were observed in the four patients treated in the expansion cohort.
Response
Six patients discontinued treatment before response evaluation (4 withdrawals, 1 secondary to toxicity, 1 death secondary to disease progression,). Twelve patients (10 in dose-escalation and 2 in dose-expansion cohort) were evaluable for response by radiological assessment of target lesions using RECIST, v1.1. No complete or partial responses were observed. In the dose-escalation portion, progressive disease was seen in 6 patients. Best response of stable disease was observed in 4 patients, with one SD lasting >4 months. Of the four patients enrolled in the dose-expansion cohort, 2 were evaluable for response: one patient experienced SD >4 cycles and the other had progressive disease as best response. The median duration of study treatment for evaluable patients was 56 days (range 28–181, Figure 1). Of the 5 patients who achieved SD as best response, four ultimately discontinued due to disease progression; the fifth discontinued because of toxicity (diarrhea) without progression.
Figure 1:
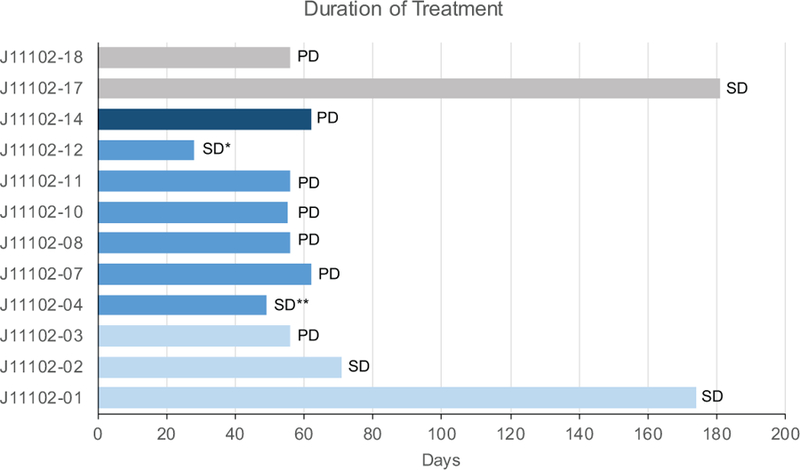
Time on study treatment (days) for patients evaluable for response. Light blue: dose level 1, medium blue: dose level 2, dark blue: dose level 3, gray: expansion cohort. Each bar is denoted with subject’s best response; SD: stable disease, PD: progressive disease. *denotes patient who discontinued treatment due to possible treatment-related toxicity after response evaluation, **denotes patient who discontinued due to clinical progression
Pharmacokinetics
CC-486 and romidepsin pharmacokinetic data were available for 17 (94%) and 16 (89%) patients, respectively (Table 4). Since CC-486 pharmacokinetics were obtained on Cycle 1 Day 1, there was no effect of romidepsin on the exposure with the data being presented as the active metabolite 5-azacitidine by dose. 5-azacitidine T max was observed at approximately 1.0 hour after CC-486 administration. There was no difference in dose-normalized 5-azacitidine exposure, apparent clearance, and apparent volume of distribution or half-life at 200 or 300 mg. 5-azacitidine dosing did not alter romidepsin exposure nor pharmacokinetic parameters.
Table 4.
Plasma pharmacokinetic parameters
| Cohort | CC-486 dose (mg) |
Romidepsin dose (mg/m2) |
Cmax (ng/mL) |
Tmax (h) |
AUCINF (ng*h/mL) |
t1/2 (h) |
Cl/F or Cl (L/h) |
V/F or V (L) |
|---|---|---|---|---|---|---|---|---|
| 5-azacitidine | ||||||||
| 1 | 200 | 8 | 324.0 ± 157.2 (3) |
1.00 (0.25 – 1.00; 3) |
537, 538 (2) |
0.55, 0.59 (2) |
372, 373 (2) |
293, 319 (2) |
| 2 and 3 | 300 | 8 | 225.7 ± 130.2 (14) |
1.08 (0.42 – 3.40; 14) |
523 ± 269 (10) |
0.62 ± 0.16 (10) |
922 ± 892 (10) |
731 ± 557 (10) |
| Romidepsin | ||||||||
| Any | Any | 8 | 455.1 ± 254.9 (16) |
4.00 (3.00–4.50; 16) |
1998 ± 1259 (16) |
13.4 ± 4.5 (16) |
10.7 ± 5.7 (16) |
15.7 ± 6.1 (16) |
Data are presented in the table as mean values ± SD (n). T max is presented as median (range; n). If n<3, the actual values are reported.
Abbreviations: AUCINF area under the plasma concentration-time curve to infinity; Cl systemic clearance; Cl/F apparent systemic clearance; Cmax, peak plasma concentration; Tmax, time to peak concentration; t1/2 half life; V volume of distribution; V/F apparent volume of distribution.
Pharmacodynamics
There were statistically significant correlations between the romidepsin total exposure [Area under the concentration versus time curve from time 0-infinity (AUCINF)] and nausea occurring during any cycle (p=0.03) and 5-azacitidine total exposure (AUCINF) and weight loss occurring during Cycle 1 (p=0.04) and any cycle (p=0.04). Otherwise, there were no correlations between worst grade of toxicity for the most common toxicities during cycle 1 or during treatment and 5-azacitidine or romidepsin exposure (p > 0.05). There were also no statistically significant correlations between the responses and 5-azacitidine or romidepsin exposure (p > 0.05).
Methylation status of free circulating tumor DNA was assessed from plasma samples obtained prior to start of cycle 1, on days 8 and 15 of cycle 1, as well as the first day of each subsequent cycle. Paired samples were available for 17 (94%) patients from C1D8 and C1D15 and 10 (56%) patients from C2D15. Mean LINE-1 methylation was decreased at each time point compared to baseline (Table 5), however the mean difference was only significant on C1D8 (−6.23, p=0.04) and C2D1 (−10.28, p=0.02). There was no association with dose level (Table 5 and Figure 2). Of the 12 patients with best response outcome, 9 had LINE-1 data at both baseline and C2D15. Patients with stable disease as best response had a trend towards a greater decrease in LINE-1 methylation by C2D15 than patients with progressive disease (Supplementary Table, p=0.14). Comparing the maximum de-methylation at any time prior to and including C2D15, the two best response groups appeared similar, p=0.46.
Table 5.
Mean difference in percent LINE-1 methylation compared to pre-treatment levels
| Timepoint | N | Mean difference (CI) | p-value |
|---|---|---|---|
| C1D8 | |||
| All dose levels | 17 | −6.23 (−12.23, −0.24) | 0.04 |
| DL1 | 3 | −6.67 (−24.84, 11.50) | 0.26 |
| DL2 | 11 | −4.82 (−13.06,3.43) | 0.22 |
| DL3 | 3 | −11.00 (−49.12, 27.12) | 0.34 |
| C1D15 | 17 | −5.76 (−13.31, 1.78) | 0.12 |
| C2D1 | 13 | −10.28 (−18.94, −1.63) | 0.02 |
| C2D15 | 10 | −9.03 (−19.99, 1.92) | 0.10 |
Mean difference reflects the change in percentage methylation between samples at the indicated timepoints.
Figure 2:
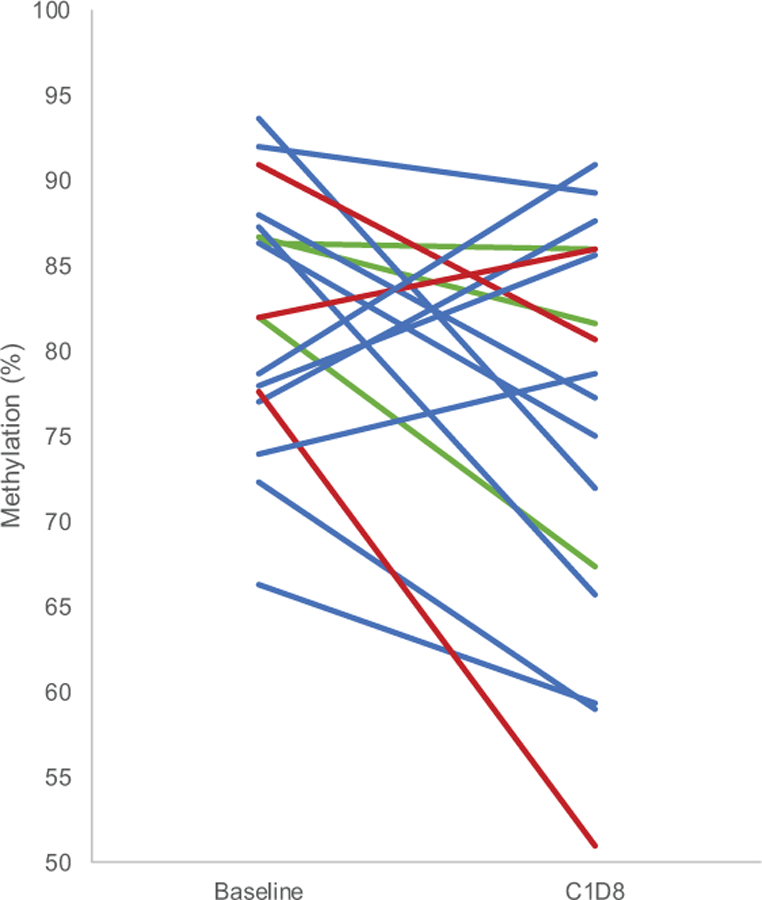
Change in LINE-1 methylation from baseline to C1D8. Paired plasma samples from baseline and C1D8 were evaluated for LINE-1 methylation. Each line represents paired samples and percent methylation for each sample is presented. The lines are color coded by dose level: dose level 1 (green lines), dose level 2 (blue lines), dose level 3 (red lines)
Discussion
Combination HDAC and DNMT inhibition have been previously tested in advanced solid tumor patients, beginning with phase I studies of subcutaneous 5-azacitidine with phenylbutyrate or valproic acid 23, 24. Both studies showed minimal efficacy, with the HDAC inhibitors utilized both suffering from unfavorable pharmacodynamics and therapeutic indices. A subsequent study of the class I specific HDAC inhibitor, entinostat, in combination with subcutaneous 5-azacitidine did show modest efficacy in heavily pre-treated non-small cell lung cancer (NSCLC) patients 12, resulting in that combination being moved forward in phase II studies in advanced NSCLC (NCT00387465), triple negative and hormone-resistant breast cancer 25, and colon cancer 11. These three studies failed to meet their primary endpoints, but for the NSCLC and breast cancer cohorts in particular, there was signal suggesting that epigenetic therapy could overcome resistance and/or increase sensitivity to multiple classes of agents including cytotoxic chemotherapy, hormonal therapy, and immunotherapy. Indeed, the strategy of using combination HDAC and DNMT inhibition to sensitize solid tumors to other classes of agents is presently the predominant therapeutic approach for these agents in ongoing studies (NCT02512172).
Both subcutaneous azacitidine and intravenous decitabine have short half-lives that may affect pharmacodynamic efficacy in solid tumors as well as compliance due to the required schedule of administration. Our trial was designed to find the maximum tolerated dose of the class 1 specific HDAC inhibitor romidepsin with an oral formulation of 5-azacitidine (CC-486), the latter of which had theoretical and practical advantages in terms of ease of administration and prolonged drug exposure to the DNMT inhibitor. Importantly, our trial was designed accepting the likelihood that this combination would likely need to move forward in combination with another class of agents as a therapeutic strategy, so the pharmacokinetic and pharmacodynamic analyses had particular importance. In this study, CC-486 and romidepsin exposure was consistent with previously reported data14, 17. Treatment with CC-486 did not alter romidepsin exposure, consistent with their independent routes of elimination. Exposure to the combination of CC-486 and romidepsin decreased LINE-1 methylation across time points, although the mean difference was only significant at Cycle 1 Day 8. The sample size was too small at individual dose levels to determine whether there was a dose-dependent effect.
A phase I study of CC-486 showed signs of potential preliminary efficacy in patients with nasopharyngeal cancer which has a known association with Epstein-Barr virus infection, with 7 of 8 patients treated with CC-486 monotherapy experiencing partial responses (3 patients) or stable disease (4 patients)14. Substantial preclinical data from multiple groups have demonstrated that DNMT inhibitors preferentially and consistently up-regulate viral response and host defense gene signaling pathways in multiple cancers 26–29. Accordingly, we chose to conduct an expansion cohort of a planned 15 patients in patients with virally-mediated cancers, hypothesizing that similar efficacy and modulation of viral response genes may be seen in these cancers. Target accrual was not reached due to competing studies of immune modulating agents (e.g PD-1/PD-L1 inhibitors) in the same patient populations. Only four patients were enrolled, with no statement regarding efficacy of the combination in virally-mediated tumors being possible.
Toxicities were as expected for this combination based on previous data regarding single agent adverse events. We were able to determine a recommended phase II dose for the combination, which could be useful in future studies of the combination. Multiple trials are ongoing of DNMT inhibitors and/or HDAC inhibitors with other classes of agents including cytotoxic chemotherapy, targeted agents, hormonal agents, and immunotherapy. With these approaches, there has been early demonstrated activity in reversing chemotherapy and hormonal therapy resistance. Indeed, the class I HDAC inhibitor, entinostat, has moved forward into phase III testing (NCT02115282) in combination with an aromatase inhibitor due to a survival benefit shown in a randomized phase II study, and responses being reported in immune-resistant tumors in combination with anti-PD-1 therapy30–33. It remains to be determined how these epigenetic modulators may best be utilized in advanced solid tumors and in combination with which classes of agents. Our trial defines the dose for the combination of romidepsin and oral azacitidine/CC-486 as another possible future regimen to be combined with other classes of agents.
Supplementary Material
Acknowledgements:
The project described was supported in part by the Analytical Pharmacology Core of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins (NIH grants 5P30CA006973 and 5UL1TR001079, and the Shared Instrument Grant (1S10RR026824)). Grant Number UL1 TR 001079 is from the National Center for Advancing Translational Sciences (NCATS) a component of the NIH, and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the Johns Hopkins ICTR, NCATS or NIH. Additional support was provided by Celgene.
Footnotes
Conflict of Interest: Dr. Rudek and Dr. Azad report research funding to the institution from Celgene. The remaining authors report no conflicts of interest related to the presented work.
Additional Information
Availability of data and material
Deidentified data will be made available by the authors on request.
References
- 1.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 2003;349: 2042–2054. [DOI] [PubMed] [Google Scholar]
- 2.Archer SY, Hodin RA. Histone acetylation and cancer. Curr Opin Genet Dev 1999;9: 171–174. [DOI] [PubMed] [Google Scholar]
- 3.Karpf AR, et al. Activation of the p53 DNA damage response pathway after inhibition of DNA methyltransferase by 5-aza-2’-deoxycytidine. Mol Pharmacol 2001;59: 751–757. [PubMed] [Google Scholar]
- 4.Palii SS, et al. DNA methylation inhibitor 5-Aza-2’-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol 2008;28: 752–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cameron EE, et al. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet 1999;21: 103–107. [DOI] [PubMed] [Google Scholar]
- 6.Belinsky SA, et al. Inhibition of DNA methylation and histone deacetylation prevents murine lung cancer. Cancer Res 2003;63: 7089–7093. [PubMed] [Google Scholar]
- 7.Gore SD, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res 2006;66: 6361–6369. [DOI] [PubMed] [Google Scholar]
- 8.Laird PW, et al. Suppression of intestinal neoplasia by DNA hypomethylation. Cell 1995;81: 197–205. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki H, et al. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet 2002;31: 141–149. [DOI] [PubMed] [Google Scholar]
- 10.Primeau M, et al. Synergistic antineoplastic action of DNA methylation inhibitor 5-AZA-2’-deoxycytidine and histone deacetylase inhibitor depsipeptide on human breast carcinoma cells. Int J Cancer 2003;103: 177–184. [DOI] [PubMed] [Google Scholar]
- 11.Azad NS, et al. Combination epigenetic therapy in metastatic colorectal cancer (mCRC) with subcutaneous 5-azacitidine and entinostat: a phase 2 consortium/stand up 2 cancer study. Oncotarget 2017;8: 35326–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Juergens RA, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov 2011;1: 598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li HP, et al. Epigenetic changes in virus-associated human cancers. Cell Res 2005;15: 262–271. [DOI] [PubMed] [Google Scholar]
- 14.Von Hoff DD, et al. Phase I Study of CC-486 Alone and in Combination with Carboplatin or nab-Paclitaxel in Patients with Relapsed or Refractory Solid Tumors. Clin Cancer Res 2018;24: 4072–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Manero G, et al. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J Clin Oncol 2011;29: 2521–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anders NM, et al. A robust and rapid liquid chromatography tandem mass spectrometric method for the quantitative analysis of 5-azacytidine. Biomed Chromatogr 2016;30: 494–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laille E, et al. Evaluation of CYP3A-mediated drug-drug interactions with romidepsin in patients with advanced cancer. J Clin Pharmacol 2015;55: 1378–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rudek MA, et al. Pharmacokinetics of 5-azacitidine administered with phenylbutyrate in patients with refractory solid tumors or hematologic malignancies. J Clin Oncol 2005;23: 3906–3911. [DOI] [PubMed] [Google Scholar]
- 19.Bailey VJ, et al. Single-tube analysis of DNA methylation with silica superparamagnetic beads. Clin Chem 2010;56: 1022–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang AS, et al. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res 2004;32: e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaplan EL, Meier P. Nonparametric-Estimation from Incomplete Observations. Journal of the American Statistical Association 1958;53: 457–481. [Google Scholar]
- 22.Brookmeyer R, Crowley J. A Confidence-Interval for the Median Survival-Time. Biometrics 1982;38: 29–41. [Google Scholar]
- 23.Lin J, et al. A phase I dose-finding study of 5-azacytidine in combination with sodium phenylbutyrate in patients with refractory solid tumors. Clin Cancer Res 2009;15: 6241–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braiteh F, et al. Phase I study of epigenetic modulation with 5-azacytidine and valproic acid in patients with advanced cancers. Clin Cancer Res 2008;14: 6296–6301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Connolly RM, et al. Combination Epigenetic Therapy in Advanced Breast Cancer with 5-Azacitidine and Entinostat: A Phase II National Cancer Institute/Stand Up to Cancer Study. Clin Cancer Res 2017;23: 2691–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daskalakis M, et al. Reactivation of endogenous retroviral elements via treatment with DNMT- and HDAC-inhibitors. Cell Cycle 2018;17: 811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stone ML, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci U S A 2017;114: E10981–E10990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiappinelli KB, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015;162: 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roulois D, et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015;162: 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gandhi L, et al. Efficacy and safety of entinostat (ENT) and pembrolizumab (PEMBRO) in patients with non-small cell lung cancer (NSCLC) previously treated with anti-PD-(L)1 therapy.. Journal of Clinical Oncology 2018;36: suppl abstr 9036. [Google Scholar]
- 31.Agarwala SS, et al. Efficacy and safety of entinostat (ENT) and pembrolizumab (PEMBRO) in patients with melanoma progressing on or after a PD-1/L1 blocking antibody. J Clin Oncol 2018;36: suppl abstr 9530. [Google Scholar]
- 32.Azad NS, et al. ENCORE 601: A phase 2 study of entinostat in combination with pembrolizumab in patients with microsatellite stable metastatic colorectal cancer. Journal of Clinical Oncology 2018;36: suppl abstr 3557. [Google Scholar]
- 33.Yardley DA, et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol 2013;31: 2128–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.