

Abstract
α-Amino-β-carboxymuconate-ϵ-semialdehyde decarboxylase (ACMSD) plays an important role in l-tryptophan degradation via the kynurenine pathway. ACMSD forms a homodimer and is functionally inactive as a monomer because its catalytic assembly requires an arginine residue from a neighboring subunit. However, how the oligomeric state and self-association of ACMSD are controlled in solution remains unexplored. Here, we demonstrate that ACMSD from Pseudomonas fluorescens can self-assemble into homodimer, tetramer, and higher-order structures. Using size-exclusion chromatography coupled with small-angle X-ray scattering (SEC-SAXS) analysis, we investigated the ACMSD tetramer structure, and fitting the SAXS data with X-ray crystal structures of the monomeric component, we could generate a pseudo-atomic structure of the tetramer. This analysis revealed a tetramer model of ACMSD as a head-on dimer of dimers. We observed that the tetramer is catalytically more active than the dimer and is in equilibrium with the monomer and dimer. Substituting a critical residue of the dimer–dimer interface, His-110, altered the tetramer dissociation profile by increasing the higher-order oligomer portion in solution without changing the X-ray crystal structure. ACMSD self-association was affected by pH, ionic strength, and other electrostatic interactions. Alignment of ACMSD sequences revealed that His-110 is highly conserved in a few bacteria that utilize nitrobenzoic acid as a sole source of carbon and energy, suggesting a dedicated functional role of ACMSD's self-assembly into the tetrameric and higher-order structures. These results indicate that the dynamic oligomerization status potentially regulates ACMSD activity and that SEC-SAXS coupled with X-ray crystallography is a powerful tool for studying protein self-association.
Keywords: NAD biosynthesis, small-angle X-ray scattering (SAXS), protein structure, protein folding, protein dynamics, decarboxylase, X-ray crystallography, metabolism, enzyme catalysis, α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase (ACMSD), amidohydrolase, protein self-association, SEC-SAXS, solution structure, quaternary structure, amino acid degradation
Introduction
The kynurenine pathway (KP)2 is a central metabolic route for degradation of the essential amino acid l-tryptophan. The KP produces metabolites that are neurologically active and affect neurotransmission and neuronal integrity (1–6). α-Amino-β-carboxymuconate-ϵ-semialdehyde (ACMS) decarboxylase (ACMSD), is a Zn-dependent decarboxylase of the KP (7–9). ACMSD removes a carboxylate group from ACMS to form 2-aminomuconate semialdehyde (2-AMS; Scheme 1). Both the substrate (ACMS) and the product (2-AMS) of the enzyme are unstable and nonenzymatically cyclize to form quinolinic acid (QA) and picolinic acid (PA), respectively (10, 11). QA, a side product of the KP, is an essential precursor to biosynthesize the coenzyme NAD+ (NADH) (6, 12–14). However, elevated QA levels cause abnormal glutamatergic activity leading to neuronal excitotoxicity and apoptosis (2, 5, 6). Injection of QA into healthy rats led to symptoms of Huntington disease (15). Therefore, QA production is tightly regulated in cells. The activity of ACMSD's downstream neighbor, α-aminomuconate-ϵ-semialdehyde dehydrogenase, competes with the nonenzymatic formation of PA, which has no well-established physiological roles (16). α-Aminomuconate-ϵ-semialdehyde dehydrogenase performs an unusual isomerization in addition to its dehydrogenation (17, 18), and its human version was recently established (19). In the bacterial 2-nitrobenzoic acid biodegradation pathway, which borrows from the core portion of the KP (20), the metabolites should be prevented from leaking to QA and PA because 2-nitrobenzoic acid is the primary source of carbon, nitrogen, and energy for those bacteria. Additionally, QA needed for NAD+ biosynthesis in bacteria is generated from a different pathway by quinolinate synthase from dihydroxyacetone phosphate and iminoaspartate (21). Therefore, the side product QA from l-tryptophan represents a metabolic waste in bacteria although it is essential for NAD+ biosynthesis in mammals.
Scheme 1.
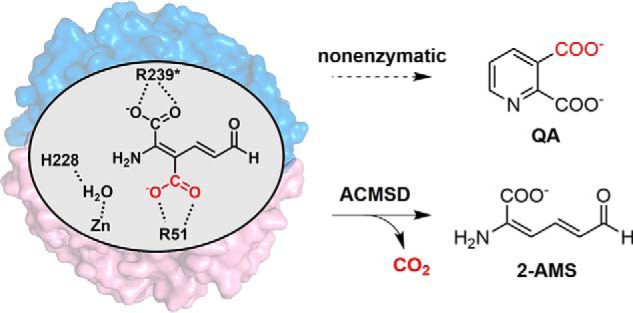
ACMSD requires dimerization for activity and competes with the nonenzymatic decay of its substrate to QA.
Enzymatic activity is generally regulated by several strategies to modulate metabolite concentrations: allosteric control, feedback inhibition, multiple forms of stable enzyme complexes, reversible covalent modification such as light-induced reduction (22), phosphorylation by a protein kinase, proteolytic activation, and controlling the amount of enzyme present. However, none of these strategies can be applied to (and they have not been studied for) the regulation of nonenzymatically derived side products of the KP, such as QA. ACMSD, as a critical enzyme in the KP (9), is directly linked to the QA levels and NAD+ homeostasis in the human body (6, 14). Inhibition of ACMSD activity was reported to increase the NAD+ levels in cellular and tissue systems and enhance mitochondrial function (6, 23).
A unique feature of ACMSD is that its activity is oligomeric state–dependent, with dimerization being required for function (24). As such, higher concentrations of ACMSD show greater specific activity. Structural analysis of ACMSD indicates that two arginine residues, Arg-51 and Arg-239* from the neighboring protomer in the monomer–monomer interface, are located in the active site for anchoring the substrate (Scheme 1), and mutation of either arginine eliminates the decarboxylase activity, providing a rationale for the requirement of dimeric or higher-order oligomers for catalytic activity (24). To better understand the oligomeric state distribution of ACMSD in solution and the protein self-association mechanism, size-exclusion chromatography (SEC)–coupled SAXS experiments were performed in this work to identify the macromolecular shape of oligomeric ACMSD. SEC separation, SAXS characterization, site-directed mutation, kinetic assays, and crystallographic studies were conducted to investigate the factors governing the protein self-association and oligomeric state distribution in solution and their impact on the catalytic activity of ACMSD.
Results
Identifying the oligomerization state of ACMSD by SEC-SAXS, revealing a previously unknown tetrameric form in solution
Fig. 1A shows a representative SEC chromatogram of ACMSD with a Superdex 200 column. The two major fractions were partially separated. Based on the molecular weight calibration curve (Fig. S1), these were presumably larger oligomer and dimer states of the protein, respectively. The monomeric form was not observed due to the retention time and high stock concentration of ACMSD applied to the column. The separation was improved by an analytical Superdex 200 column (Fig. S2A) and followed by SAXS characterizations (Table 1). A total of 500 images were recorded with 1 s of X-ray exposure for every 5 s during the elution. After data reduction and initial inspections, Guinier analysis was applied to each frame in order to obtain the overall dimension of the sample (i.e. the radius of gyration (Rg)) (25). Based on the flatness of the Rg values in the separation profile (Guinier Rg−I(0) plot; Fig. S2B), we selected two main fractions, which could be further processed for structural modeling. These were the major fractions at approximately image #360 and a minor fraction at approximately image #300 (Fig. S2B). The two fractions exhibited distinct scattering profiles, as shown in Fig. 1B. Porod volume estimations of the scattering profiles, assuming globular molecular shape (26), indicate molecular weights of major and minor fractions of 71.9 and 121 kDa (Fig. 1C), respectively. Given the calculated molecular weight of 39.7 kDa of ACMSD, the major and minor fractions are expected to be in the dimeric and trimeric states, respectively.
Figure 1.

Gel filtration chromatography and SEC-SAXS of ACMSD. A, SEC separation profile of ACMSD on a Superdex 200 column. B, SEC-SAXS profile of tetrameric (black) and dimeric (blue) states of ACMSD. C, pairwise distribution function, P(r), of the scattering profiles in B. D, Kratky plot of the tetrameric and dimeric samples. mAU, milli-absorbance units.
Table 1.
SAXS data collection and scattering-derived parameters
| ACMSD | ACMSD H110A | |
|---|---|---|
| Data collection | ||
| Beamline | SSRL BL4-2 | SSRL BL4-2 |
| Type of experiment | SEC-SAXS | SEC-SAXS |
| Beam defining slits size (mm) | 0.3 × 0.3 | 0.3 × 0.3 |
| Sample–detector distance (m) | 1.7 | 2.5 |
| Beam energy (keV) | 11.4 | 10.1 |
| Beam current (mA) | 500 | 500 |
| Exposure time per frame (s) | 1 | 1 |
| Frames per data set | 500 | 500 |
| Temperature (K) | 298 | 298 |
| Sample cell size (mm diameter) | 1.5 | 1.5 |
| Detector | Pilatus 3 X 1M | Pilatus 3 X 1M |
| SEC column | Superdex 200 Increase PC3.2/300 | Superdex 200 Increase PC3.2/300 |
| Sample volume (Concentration) | 35 μl (4 mg/ml) | 50 μl (4 mg/ml) |
| Flow rate (ml/min) | 0.05 | 0.05 |
| Guinier analysis | ||
| q × Rg limita | 1.3 | 1.3 |
| I(0) (cm−1) | 0.0612 ± 0.0011 | 0.0439 ± 0.0012 |
| Rg (Å) | 47.0 ± 1.06 | 52.2 ± 1.95 |
| P(r) and Porod volume estimation | ||
| Software | Primus/GNOM | Primus/GNOM |
| qmax (Å−1) | 0.30 | 0.15 |
| I(0) (cm−1), real space | 0.0610 ± 0.0010 | 0.0454 ± 0.0006 |
| Rg, real space (Å) | 48.28 ± 0.98 | 55.99 ± 0.84 |
| Dmax (Å) | 175 | 190 |
| Porod volume (Å3) | 195,000 | 238,000 |
| Expected mass (kDa) | 158.8 | 158.8 |
| Rigid-body modeling | ||
| Software | CORAL | CORAL |
| qmax (Å−1) | 0.3 | 0.2 |
| Number of runs | 20 | 20 |
| Ab initio modeling | ||
| Software (ab initio modeling) | DAMMIF | GASBOR |
| Software (averaging) | DAMAVER | DAMAVER |
| qmax (Å−1) | 0.18 | 0.15 |
| Number of runs | 20 | 20 |
| Normalized spatial discrepancy (NSD) | 0.763 ± 0.038 | 2.272 ± 0.242 |
aq = 4πsin(θ)/λ, where 2θ is the scattering angle.
Modeling the oligomerization state of ACMSD based on SAXS data
The eluate before the major, dimeric ACMSD peak in Fig. S2B (around image #300) provided stable Rg values over the chromatographic peak, indicating that overall dimension of the sample is uniform in the region. The linearity of the Guinier plot (Fig. S2C) of the higher oligomeric state sample (around image #300) indicated that it is not an aggregation artifact, and a single peak in the Kratky plot, which is often used for evaluation of sample flexibility and globularity, implies a well-folded sample (Fig. 1D). The estimated molecular weights by Porod volume suggested a trimeric state of ACMSD in solution; however, the crystal structures of ACMSD all show extensive salt bridges and hydrogen-bond interactions to form a dimer as the functional unit with no obvious trimer interface (8, 24, 27, 28). Therefore, additional macromolecular associations of ACMSD are expected to be multiples of the dimer (e.g. tetramer, hexamer, octamer, etc.). Interestingly, in our SEC-SAXS experiments, X-ray–induced aggregation actually decreased as the enzyme concentration increased, which is opposite to what is normally observed. Less aggregation may result from better-ordered structures of the enzyme in solution during concentration. Therefore, the aggregation portion eluted from the Superdex 200 column was tentatively considered as higher-order oligomers, especially because no precipitate was observed.
To further characterize the oligomeric state of ACMSD with the higher molecular weight, theoretical SAXS curves were generated based on given models derived from crystal structures for comparison with the experimental data. First, trimeric and dimeric models from crystal structures were tested to assess their agreement with the experimental SAXS data. The theoretical SAXS curves generated from trimeric and dimeric models did not faithfully reproduce the experimental data, especially in the middle q range (middle scattering angle) that is susceptible to the conformation of the sample (Fig. S3, A and B) (29). Next, two types of tetrameric models were constructed based on symmetry-generated neighboring dimers in crystal structures: a head-on, tandem model and a stacked, parallel model. Both forms were used to generate their theoretical SAXS profile, and comparison showed that the tandem dimer model provides the best recreation of the experimental data (Fig. S3, C and D).
Ab initio modeling was performed with the experimental SAXS curves without any template or predetermined model. For calculations, the shape of the scattering particle was selected as either prolate or oblate spheroids with various symmetries (P1, P2, P22, and P4). Density envelopes from oblate spheroids provided irregular shapes that are not amenable to encompassing ACMSD structures. In contrast, density envelopes from prolate models are similar to the tandem-dimeric tetramer models. The results showed that the models constructed with P4 symmetry provided additional envelope details that do not agree with the structural characters of ACMSD. However, those models made with P2 and P22 symmetry can contain the tandem-dimeric tetramer models with some excess density. Finally, the envelopes from models constructed with P1 symmetry encompass the tandem-dimeric tetramer models with little excess density generated by DAMMIF (Fig. 2A) (30). The excluded volume from a DAMAVER-processed model is 264,530 Å3, corresponding to an estimated molecular weight of 132.3 kDa, which can be assigned as tetrameric ACMSD (31). The normalized spatial discrepancy value is 0.763 ± 0.038.
Figure 2.

The ab initio structural model of ACMSD based on SEC-SAXS. A, the tetramer model generated by CORAL was docked into an ab initio envelope determined with DAMMIF using no symmetry constrains. B, back-calculated scattering curve (red line) of the CORAL model overlaid with scattering curve (black line).
Rigid-body modeling by CORAL was used as a complementary strategy to the ab initio model building (26, 32). Models were generated by moving and rotating given subunits (usually crystal structures) to fit experimental SAXS curves. The CORAL modeling also included possible positions of flexible regions, such as missing N- and C-terminal residues and flexible loops in crystal structures. First, the dimeric structure was set as the asymmetric unit to build the tetramer in silico using P2 symmetry. The crystallographic monomer–monomer interface was constrained during the modeling. The resulting models were clustered into three major groups with similar shapes by using DAMCLUST (26). Among those, a tandem-dimeric tetramer model was the most populated cluster. The major cluster also showed a lower averaged χ2 value of 0.30 compared with the other two clusters. In Fig. 2A, the tandem-dimeric tetramer model was fitted well with the envelope built by DAMMIF. As a secondary strategy, the two-tandem-dimeric tetramer model with P1 symmetry was used as the template. The processing included additional constraints for the monomer–monomer interface so that only the two dimers could move in relation to each other to fit the experimental SAXS data. As a result, the major cluster including tandem-dimeric tetramer models gave similar fits with a χ2 of 0.33. In contrast, any other ways to pack two dimers resulted in larger χ2 values (>0.5). The theoretical SAXS curve generated from the populated models fit the experimental data well (Fig. 2B). After establishing that ACMSD does indeed have a tetrameric conformation, the question arises of whether or not tetrameric ACMSD will exhibit any significantly different biological properties as compared with dimeric ACMSD.
ACMSD activity as a reporter of enzyme dynamics from oligomer to monomer
The requirement of dimerization for activity makes the specific activity of ACMSD an experimental handle that can be used to infer its oligomeric state (8, 28). ACMSD was initially stored at a relatively high concentration, 100 μm, to maximize tetramer structures. The enzyme was then diluted to 30 nm for assays, which should favor the monomer form at equilibrium (24). Measurement of the decreasing catalytic activity after dilution as a function of time reports on the dissociation of ACMSD from tetramer structures to the inactive monomer.
As shown in Fig. 3A, the time-dependent decrease of the specific activity of ACMSD upon dilution cannot be well-described by either single- or double-exponential functions. At 30 nm concentration, the specific activity of ACMSD drastically decreased (∼30%) in the first 10 min after dilution. The activity then decreased another ∼40% within 100 min. Finally, all activity was lost over the course of ∼12 h. Adequate representation of the decay of ACMSD activity after dilution requires three summed exponential functions with reciprocal relaxation times provided in Table 2. After the third decay stage, the ACMSD activity could not be rescued by reconcentration of the sample, indicating that the third decay phase is irreversible and hence represents the enzyme degrading or denaturing. The first two decay stages could be interpreted as the equilibria from tetramer to dimer and from dimer to monomer (Fig. 3B).
Figure 3.
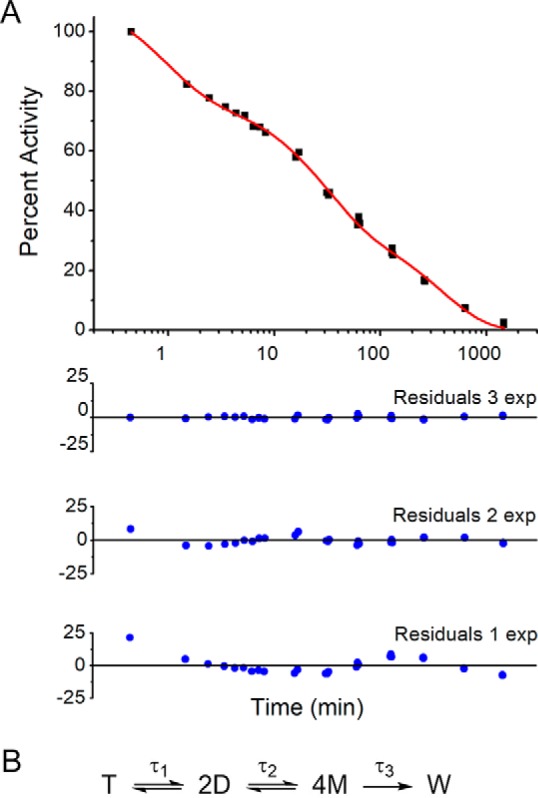
ACMSD dissociation as measured by specific activity. A, triple-exponential fitting of the decreasing specificity activity (black squares) with incubation time in the logarithmic scale. The fitting curves are shown as a red line. The residuals from fitting decreasing specificity activity data with triple-, double-, and single-exponential curves are shown as blue circles. B, proposed dissociation phases of ACMSD. T, D, M, and W, tetramer, dimer, monomer, and denatured protein, respectively.
Table 2.
The reciprocal relaxation times of each decay phase of wtACMSD and H110A
| Protein | τ1 | τ2 | τ3 |
|---|---|---|---|
| min | min | min | |
| ACMSD | 0.82 ± 0.25 | 30 ± 10 | 980 ± 170 |
| H110A | 14.5 ± 4.3 | 174 ± 38 | 4890 ± 410 |
| ACMSD with NaF | 1.55 ± 0.16 | 99 ± 10 | 7150 ± 410 |
| H110A with NaF | 2.98 ± 0.18 | 106 ± 20 | 5330 ± 420 |
Analysis of the dimer–dimer interface in the SAXS model of the tetramer
The structural models based on the SAXS data indicated that the quaternary structure of ACMSD is a tetramer of two head-on, tandem dimers. To better understand the model generated from SEC-SAXS data, crystal structures of ACMSD were reviewed to identify potential interacting residues for tetramer formation. In our previous studies, ACMSD and its variants were crystallized with two different space groups, C-centered monoclinic (C2) and primitive tetragonal (P42212) (24, 27, 28). The crystal packing of ACMSD in the tetragonal space group does not exhibit an interaction interface consistent with the above SAXS tetramer models (Fig. 2). However, the monoclinic space group packs with an interface similar to our SAXS-derived head-on, tandem dimer model (Fig. 4A). A perusal of the residues in the dimer–dimer interface led to the identification of residue His-110, which is involved in multiple interactions with Ser-17, Gln-19, and Glu-20 (Fig. 4, B and C).
Figure 4.

Tandem dimers from ACMSD crystal structures. A, surface structures of two dimers of ACMSD (PDB entry 2HBV). Monomers in each dimer are colored blue and pink. B, zoom-in image of the dimer–dimer interface; the interacting area is highlighted in yellow. C, residues involved in the dimer–dimer interaction networks. D, comparison of the dissociation profiles of wtACMSD (black squares) and the mutant H110A (blue squares) as measured by specific activity.
To investigate the role of the interaction network, His-110 was mutated to alanine to break any interactions. Deleting the side chain of His-110 had little effect on the steady-state kinetic parameters of ACMSD toward ACMS (Table S1); however the H110A variant did show altered dissociation behavior as measured by specific activity. As compared with WT ACMSD (wtACMSD), the dissociation profile of H110A still exhibits three decay phases. However, each phase takes substantially longer, increasing the time to reach equilibrium (Fig. 4D). In detail, the most significant change is that the first decay phase of H110A was elongated, and 90% activity was retained for nearly 10 min, whereas wtACMSD decreased to 60% activity over the same time period. The dissociation profile of H110A was fitted with three exponential functions, and the decay constants are compared with those of wtACMSD in Table 2. All of the decay constants of H110A are greater than wtACMSD, especially the first decay phase (18-fold), indicating elongation of the dissociation periods of the three phases. The H110A variant was anticipated to break the interaction networks in the dimer–dimer interface and was expected to show different dissociation behavior than wtACMSD. However, the observation of a seemingly more stable tetramer is opposite to our expectation. A more complicated mechanism for tetramer formation may need to be considered in ACMSD. Thus, we investigated other factors that may contribute to the oligomeric state distribution of the protein.
The effect of pH and ionic strength on ACMSD oligomeric state distribution
The influence of pH and ionic strength on the distribution of oligomeric states of ACMSD were investigated by SEC, which were performed on a Superdex 200 column. The buffer pH ranged from 6.0 to 9.0 with various buffer systems (see “Experimental procedures” for more details), which included 150 mm NaCl to maintain similar ionic strengths.
The separation profiles of wtACMSD and H110A are shown in Fig. 5 and Fig. S4. Three distinct fractions were observed, which represent different oligomeric states. Based on the molecular weight estimation from the SEC-SAXS results (Fig. 1), higher-order states (i.e. larger than tetramer) elute at a retention volume of 44 ml, tetramer at 54 ml, and dimer at 75 ml. Due to the relatively high concentration of ACMSD (100 μm) loaded to the column, no monomer peak was observed. At pH 7.0, 150 mm NaCl, wtACMSD was found mostly present as higher-order oligomers. For other pH values, the overall separation profiles are similar to each other and show a mixture of states, with the tetramer form being preferred (Fig. 5, A and B). The preference to form higher-order structures only at pH 7.0 may reflect an experimental pI value for ACMSD, whereas its theoretical pI value is 5.6. In contrast, the higher-order oligomer was the major fraction for H110A in the pH range between 6.0 and 7.5 (Fig. 5, C and D). The tetramer form became the predominant species only at pH 8.0 and above.
Figure 5.
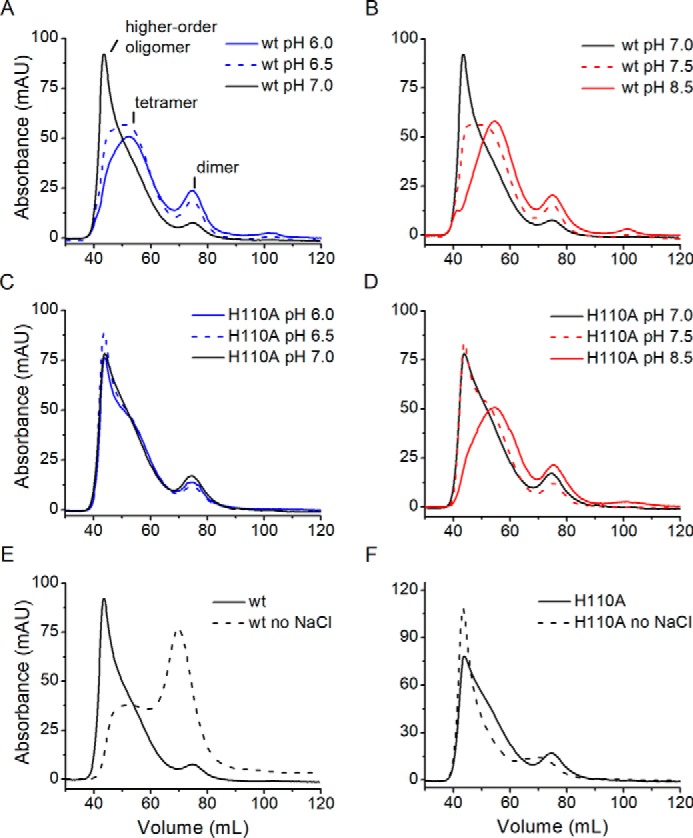
Effect of pH and ionic strength on ACMSD oligomeric state distribution. Representative separation profiles of wtACMSD (A and B) and H110A (C and D) under pH 6.0 (blue line), pH 6.5 (blue dashed line), pH 7.0 (black line), pH 7.5 (red dashed line), and pH 8.5 (red line). E and F, separation profiles of wtACMSD and H110A under pH 7.0 with (black line) and without (black dashed line) the addition of NaCl. mAU, milli-absorbance units.
Over the pH range from 6.0 to 9.0, increasing ionic strength, especially at low and neutral pH values, shifted the equilibrium composition to prefer higher-order states for wtACMSD (Fig. S4). These observations suggest that hydrophobic interactions are a significant contributor to oligomer formation. Moreover, the most pronounced effect on the separation profile of wtACMSD occurs at pH 7.0 (Fig. 5E). Under low ionic strength, wtACMSD was observed as 32.5 ± 1.6% tetramer and 65.1 ± 5.0% dimer, whereas at higher ionic strength, the tetrameric and higher-order oligomers accounted for nearly all (>90%) of the ACMSD observed (Table S2). In contrast, at pH 7.0, the majority (∼80%; Fig. 5F) of H110A was present as higher-order structures, even with low ionic strength. Also, despite very similar compositions at pH 7.0 with 150 mm NaCl, wtACMSD and the H110A variant showed opposite trends upon lowering the ionic strength. For wtACMSD, lowering the ionic strength led to dissociation, whereas for H110A, it led to increased subunit association to higher-order states. These data indicate that the His-110–involved interaction network may play a role in the distribution of tetramer and higher-order states, especially with respect to pH and ionic strength. It should also be noted that apo-ACMSD (the metal-free form) was unable to form any higher-order structure and was observed as exclusively in the dimeric form by gel filtration under the same conditions (Fig. S5).
SAXS modeling of H110A tetramer
The tetramer model of ACMSD H110A was also investigated by performing SEC-SAXS experiments at various pH values for comparison with the tetramer model of wtACMSD. Due to the presence of higher-order oligomers, no reasonable SAXS curves were obtained below pH 7.5. At pH 8.5, H110A showed consistent Rg values around the tetramer range. The Porod volume–estimated molecular weight was 148.8 kDa, suggesting the tetrameric state. After modeling with GASBOR, the populated envelope of H110A is a prolate shape capable of enclosing two-tandem-dimeric tetramer (Fig. 6), which is similar to the envelope of wtACMSD. These results suggest similar quaternary structures of wtACMSD and H110A even under different pH environments.
Figure 6.
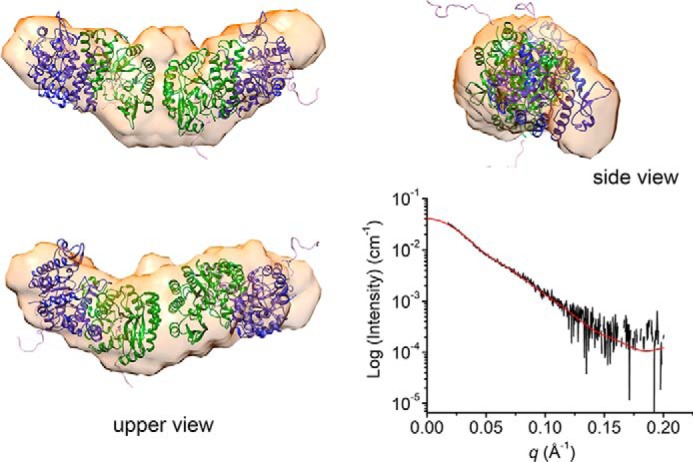
An ab initio model of H110A derived from the SAXS profile fitted with a CORAL model (most populated volume). Red line, back-calculated scattering curve of the CORAL model.
Effect of ionic strength on the dissociation of ACMSD
To test the effect of ionic strength on the dissociation profile, the dissociation assays of wtACMSD and H110A were also performed with increased ionic strength (from 5 to 39 mm) by adding 33.8 mm NaF. As shown in Table 2, the dissociation profile of ACMSD under higher ionic strength still required three exponential functions for fitting. The first and second decay constants of wtACMSD (τ1 and τ2) increased by 2-fold, and the third decay constants (τ3) increased ∼10-fold, suggesting that increasing the ionic strength (7.8-fold) slows down the transition from tetrameric to dimeric state and enhances the protein stability. In contrast, the first and second decay constants for H110A decreased about 4.9- and 1.6-fold (Table 2) upon increasing ionic strength. These values are more similar to those observed from wtACMSD. Overall, these results can be understood by recognizing that increasing ionic strength promotes tetramer formation for both wtACMSD and H110A. Therefore, the increased ionic strength is expected to extend wtACMSD dissociation by increasing the fraction of protein in tetramer and higher-order states at the start of the assay and decrease the relaxation times for H110A by decreasing the fraction of enzyme present in higher-order states.
Crystal structure differences of WT and H110A ACMSD proteins
Attempts to crystallize the H110A variant with reported crystal conditions for ACMSD were unsuccessful. However, we were able to crystallize both H110A and the WT protein under a new condition, and X-ray crystal structures were solved to resolutions of 2.77 and 3.15 Å, respectively (Table S3). The overall structures of wtACMSD and H110A are nearly identical to the published wtACMSD structures, with root mean square deviation values of 0.340 and 0.257 Å for all Cα atoms aligned in these structures, respectively. The wtACMSD shows a different space group (C2221), under the new crystallization conditions with three molecules per asymmetric unit, whereas H110A crystallizes in the tetragonal (P42212) space group with two molecules per asymmetric unit. After examining the neighboring units of wtACMSD, the packing manner is similar to monoclinic wtACMSD (PDB entry 2HBV) (Fig. 7A), and the potential tetramer-forming interface that includes His-110 is retained (Fig. 7B). However, the His-110–involved interacting network and tetramer-forming interface are not found in the H110A structures (Fig. 7C). The different packing behaviors under the same crystallization conditions between wtACMSD and H110A support an essential role for His-110 at the dimer–dimer interface.
Figure 7.
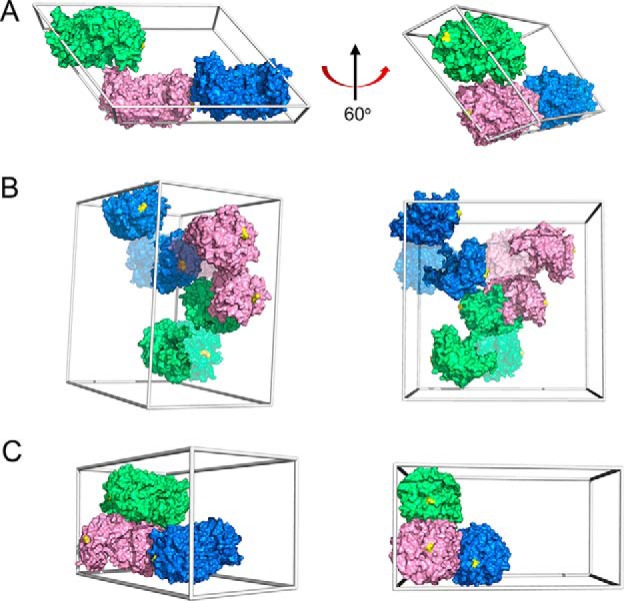
Crystal packing of wtACMSD and H110A. A, previously published wtACMSD crystallized in the monoclinic space group (C121, PDB entry 2HBV) (1). B, wtACMSD crystallizes under new conditions (see “Experimental procedures”) in the orthorhombic space group (C2221, PDB entry 6MGS). C, H110A under the same conditions as in B crystallized in the tetragonal space group (P42212, PDB entry 6MGT). Unit cells are shown as gray boxes, asymmetric units are marked by color, and chain B for wtACMSD in B is shown at 50% transparency for clarity. His-110 or Ala-110 is highlighted in yellow.
Sequence conservation of the dimer–dimer interface
Alignment of nonredundant sequences of ACMSD shows that His-110 is not conserved across all organisms. Histidine is the third most popular amino acid at the 110-position, with nine common residues observed (Fig. 8). In contrast, histidine is partially conserved in prokaryotes as the most abundant residue at position 110, with tyrosine, phenylalanine, and alanine as alternatives. Moreover, a few selected strains of prokaryotes exhibited relatively high conservation of His-110: Achromobacter, Ralstonia, Pseudomonas, Cupriavidus, Paraburkholderia, Herbaspirillum, Caballeronia, and Duganella (Fig. 8). Among the selected strains, in addition to Pseudomonas, several strains from Achromobacter (33), Ralstonia (34), and Cupriavidus (35) have been reported to utilize the nitrobenzoic acid (NBA) pathway as their sole source of carbon and energy, making the activity of ACMSD essential in those strains.
Figure 8.

The conservation of His-110–containing segment in ACMSD. The nonredundant sequences' BLAST of ACMSD was performed across all organisms, and the alignment results were shown by the Weblogo motif (top) (74), in prokaryotes (taxid 2; middle), and in selected strains (bottom) capable of degradating nitrobenzoic acids, including Achromobacter, Ralstonia, Pseudomonas, Cupriavidus, Paraburkholderia, Herbaspirillum, Caballeronia, and Duganella.
Discussion
Oligomeric state dictates the specific activity
ACMSD dissociation can be measured as a function of time by following the decrease in specific activity after dilution. Likewise, similar cases are known in other enzymes. For instance, dihydroorotate dehydrogenase A is also only active as a dimer by the interaction of two neighboring subunits (36). However, the loss of dehydrogenation activity upon dilution as a function of time can be fitted to a single decay phase with a reciprocal rate of ∼42 min. In contrast, ACMSD unexpectedly exhibited three decay phases during protein dissociation (Fig. 3). The monomeric form is catalytically inactive. The dimeric form of ACMSD is required for decarboxylation activity due to the two essential arginine residues in the active site, with one from the neighboring subunit. The dimer is formed by hydrogen bonding and salt-bridge networks at the monomer-monomer interface (24). The dissociation of a dimeric protein is only expected to take, at most, two phases, from dimer to monomer and protein degradation. Considering the given tetramer model in Fig. 2, one potential explanation is that in addition to dimerization being required for ACMSD activity, a tetrameric state may have higher specific activity than the dimer. In such a case, the first phase would represent maximally active tetramer structures disassociating to partially active dimers; the second phase would represent partially active dimers equilibrating with inactive monomers, with the eventual loss of all activity in the third phase as all of the enzyme degrades or denatures. Therefore, an important question arises as to why the tetrameric (or higher-order) form exhibits higher specific activity than the dimeric form.
A new insight for half-of-sites reactivity in homodimeric enzymes
Half-of-sites reactivity is a general phenomenon in enzymology reported in numerous homodimeric proteins (37–44). The homodimeric crystal structures of ACMSD invariably show one active site being well-ordered and another somewhat disordered, making ACMSD a candidate for half-of-sites reactivity (8, 24, 27, 28). However, the rationale for this phenomenon remains elusive (39, 44, 45). We revisited the 10 published crystal structures of ACMSD and mutants (8, 24, 27, 28) and compared each with the structures determined in this work to investigate the possible reason. Mutation of active-site residues of ACMSD did not affect the quaternary structures (24, 27). The averaged B-factor values were summarized in Table S4. In the four structures that refined at a relatively high resolution (1.65–2.00 Å), the averaged B-factor value of residues in one monomer (chain B) is ∼1.5-fold higher than the other monomer (chain A). Additionally, the poor density maps of residues in chain B with the resolution of ∼3 Å indicate that the residues in chain B are less ordered than chain A (28). The other six structures with a tetragonal space group have relatively low resolution (>2.40 Å). Although the overall B-factor values are high (>50 Å2), the averaged B-factor of chain B is ∼10 Å2 higher than that of chain A. Therefore, it is possible that in the dimeric state, only one of the active sites in ACMSD is well-ordered, and the well-ordered site performs more efficiently. We hypothesize that tetramer and higher-order forms of ACMSD may stabilize both active sites of each dimer, leading to higher specific activity in higher oligomeric states. This understanding may provide a novel insight into the half-of-sites reactivity because the enzymes would have room to increase their activity, if needed, by increasing the ratio of homotetramer under certain circumstances, so that the enzymes do not always operate at maximum capacity.
It should be noted that the concentrations used for in vitro studies may not be reflective of concentrations in the cell. However, the cellular environment is much more viscous and molecularly crowded than a dilute buffer solution. As such, effects that are only observable at relatively high protein concentration in vitro may occur at much lower concentrations in vivo.
Protein self-association and the quaternary structures of ACMSD in solution revealed by SEC-SAXS
The published ACMSD structures show two different space groups for packing, monoclinic and tetragonal, which may result from different packing during crystallization. Therefore, it is of interest to illustrate what the quaternary structure of ACMSD is in solution. Previous studies have shown that SAXS is an ideal tool to understand not only the molecular size but also the quaternary structures and shapes in the solution state, which is more similar to the physiological environment (32, 46, 47), especially combined with high-resolution structures from X-ray or cryo-EM (48). SAXS coupled with size-exclusion chromatography would increase the data quality by increasing the homogeneity of samples because the macromolecules might have various states in solution (49, 50). Fig. 2 shows the tetrameric form of ACMSD as two tandem, “head-on” dimers. Combined with published and new crystallography data, an interaction network involving His-110 is illustrated at the dimer–dimer interface to form a tetramer. Because of the long distance from the active site, mutating His-110 to alanine had little effect on the steady-state kinetic parameters of ACMSD (Table S1). However, the dissociation profile of H110A shows elongation of all three dissociation phases. As shown in Fig. 5, H110A presents a higher percentage of higher-order oligomers than wtACMSD. The higher-order portion would take more time to dissociate to dimer and monomer. Therefore, the three decay constants are increased compared with those in wtACMSD. There is no current rationale to preclude the elongation of tandem-dimeric tetramers of ACMSD with the same dimer–dimer interaction, resulting in higher-order oligomeric states (more than tetramer). Such a phenomenon would explain the observation of ACMSD species eluting before the tetramer during the SEC separation (Fig. S2A).
pH and ionic strength presenting major impact on the oligomeric state distribution
As might be expected, pH and ionic strength are both essential parameters that affect the oligomerization status of ACMSD, presumably by controlling protonation state of the charged residues of the subunit–subunit interface. In the case of ACMSD, the oligomeric state distribution of the native protein and the H110A mutant are entirely different under lower pH values but similar when the pH is above 8.0 (Fig. 5). Under higher pH values, the side chain of His-110 is deprotonated, and its interaction with surrounding residues becomes weaker, so deletion of His-110 becomes less important to the oligomeric state distribution under an alkaline environment. The similarity of the envelope shapes between H110A at pH 8.5 and wtACMSD at pH 7.0 suggests similar tetrameric structures between alkaline and neutral environment with or without mutating His-110. Another interaction mechanism may exist to maintain the tetramer state under alkaline conditions. In contrast, under lower pH values, there is a considerable tetramer portion of wtACMSD, whereas higher-order oligomers are the major portion of H110A. Therefore, deleting the side chain of His-110 results in a much larger portion of higher-order oligomeric states and less tetramer compared with wtACMSD. The results indicate that the protonated form of His-110, interacting with Ser-17, Gln-19, and Glu-20, is essential to maintain the tetrameric form of ACMSD below pH 7.
Several similar findings were reported recently (51–56). The voltage-dependent anion channel, the most abundant protein in the outer mitochondrial membrane, plays an essential biological role by exchanging ions and metabolites. Double electron–electron resonance spectroscopy and MS results indicate that the dimerization is pH-dependent primarily due to the protonation of Glu-73, which may be necessary for mitochondrial metabolic regulation and apoptosis as a response to cytosolic acidification during cellular stress (51). In another case, 2-Cys peroxiredoxins play a role in controlling the intracellular levels of H2O2 under oxidative stress conditions. Decreasing pH protonates His-113 to stabilize the doughnut-like decamer by interacting with Asp-76 (53), which is linked to the enzymatic activity and signaling (55). The dendritic cell specific protein DC-SIGN receptor recognizes glycosylated proteins and is involved in the early stages of numerous infectious diseases (54). Its extracellular domain exists in a monomer–tetramer equilibrium that is responsive to both pH and ionic strength. The nearby unique extended neck domain identified by SAXS is illustrated as a pH-sensor. In some instances, the stability of proteins is regulated with pH and ionic strength of the environment by changing their oligomeric states. The dimeric human small heat shock protein HSBP1 dissociates with decreasing pH, related to protonation of a histidine on the dimer interface (52). A nonnative secondary structure in equinatoxin II is induced by increasing ionic strength, decreasing its heat stability (56). Heterogeneous protein–protein interactions may be influenced by pH, which affects the charge states of residues on the interaction interface. ERp44 is a multifunctional chaperone in the early secretory pathway (57). Structural evidence indicates that the histidine-clustered region, located on the interface between ERp44 and its clients, involves a significant rearrangement to enlarge the positively charged regions for the clients binding at low pH compared with neutral pH. The pH-dependent association between ERp44 and its clients induced by the protonation of His and Cys, explains how ERp44 transports clients from acidic Golgi into the neutral endoplasmic reticulum.
We found that increasing the ionic strength of the buffer increased the fraction of ACMSD in the tetramer form between pH 6.0 and 7.5 (Fig. S4). Instead of an electrostatic interaction, hydrophobic interactions most likely explain the salt-induced oligomerization of ACMSD. In contrast, it is reported that the active monomer conformation of protein kinase CK2 is regulated by a self-polymerization to form inactive oligomers (58). The driving force of the oligomerization, in that case, is the electrostatic interaction from charged residues between two neighboring subunits. Increasing the ionic strength (0.5 m NaCl) disrupted the electrostatic interaction and resulted in the active conformation of CK2.
Moreover, the estimated molecular weight of tetramer fractions, based on the retention volume, is 550 kDa, ∼3-fold larger than the expected molecular weight of tetramer (158.8 kDa). The tetramer was modeled as a prolate-shaped, head-on dimer of dimers by SEC-SAXS. It is possible that the Superdex 200 column recognizes the tetrameric ACMSD as spherical molecules with the diameter of the long axis, which certainly are much larger molecules. Therefore, the tetrameric portion elutes earlier, resulting in an overestimated molecular weight.
Potential contribution of the quaternary structure of ACMSD in nitrobenzoic acid degradation
Nitrobenzoic acids are a relatively nascent environmental contaminant and resistant to biodegradation (59). Nevertheless, the broad applications of nitroaromatic compounds made them a prime candidate as a food source for bacteria that can tolerate and metabolize them (60). The tryptophan–kynurenine pathway, where ACMSD is located, was adapted to degrade NBA in bacteria such as Pseudomonas fluorescens strain KU-7 (61). Moreover, several strains from Achromobacter (33), Ralstonia (34), and Cupriavidus (35) were reported to utilize nitrobenzoate as the sole carbon and energy source, and they show relatively high conservation of His-110, implying higher probabilities for formation of more catalytically active tetramers. In other words, the biodegradation of NBA might require the higher specific activity of ACMSD that was partially regulated by the His-110–involved network, especially at lower pH, to guide the metabolic pathway to generate energy without side products in those bacteria. Considering that His-110 helps to stabilize the tetrameric form of ACMSD, especially at low pH and high ionic strength, it may be an adaptation for some bacteria that utilize NBA as their source of carbon, nitrogen, and energy.
Conclusion
In this study, the quaternary structure and its relation to activity in ACMSD were found to be surprisingly complex in solution. Although the dimer is the active unit, tetramer and higher-order structures show higher specific activity, and the distribution of oligomeric states is sensitive to the pH and ionic strength. A solution structure of a new ACMSD tetrameric state is described from an extensive SEC-SAXS study coupled with X-ray crystallography, and His-110 was found to be involved in regulating tetramer formation, especially at lower pH. The differential conservation of His-110 also provides potential new insight into the evolution of bacteria capable of efficiently degrading nitrobenzoic acids. These findings have broad implications in understanding the mechanism of protein self-association and the general half-site reactivity phenomenon in homodimeric enzymes.
Experimental procedures
Protein preparation
The expression and purification strategy of ACMSD (GenBankTM accession no. Q83V25) from P. fluorescens KU-7 was described in a previous publication (24). In brief, the recombinant Escherichia coli BL21 (DE3), harboring plasmid pET16b-ACMSD, was induced by 0.5 mm isopropyl β-d-thiogalactoside when the A600 reached 0.6. After cell disruption, the lysate was centrifuged to remove cell debris and loaded on a nickel-nitrilotriacetic acid column. Unbound proteins were washed by buffer containing 50 mm NaPi, 20 mm imidazole, and 300 mm NaCl, pH 8. Finally, ACMSD was eluted by the buffer with 100 mm imidazole and desalted with the buffer containing 25 mm HEPES, pH 7.0.
SAXS data collection and initial analysis
SEC-SAXS (online SEC coupled to SAXS) data for ACMSD were collected at the SSRL beamline 4-2 (62). Experimental details and structural parameters are summarized in Table 1. Briefly, the two-tandem analytical Superdex 200 columns were used for the SEC-SAXS experiments and equilibrated with the running buffer containing 25 mm HEPES, pH 7.0, 5 mm DTT. ACMSD (4 ml·min−1) was injected into the SEC column with a flow rate of 0.05 ml/min. A total of 500 images were recorded with 1-s exposure in every 5 s. The first 100 images prior to the column void volume were used for blank images. Data reduction and initial analysis were performed using SECPipe, a real-time data processing and initial analysis pipeline at SSRL beamline 4-2 (https://www-ssrl.slac.stanford.edu/smb-saxs/content/documentation-software-secpipe).3 Guinier analyses were performed on each SAXS profile of the eluted fractions using the program AUTORG (25), which is implemented in SECPipe. After assessing data quality (e.g. radiation damage of sample and cleanness of sample cell), SAXS profiles of interest were selected in the region having consistent Rg values in the Guinier Rg−I(0) plot (Fig. S2B).
SAXS modeling
Ab initio modeling was performed to predict solution structure of ACMSD using the program DAMMIF or GASBOR (63). Twenty independent ab initio envelopes were generated with respect to each symmetry operation (P1, P2, P22, or P4). The program DAMCLUST (26) was used to find and eliminate unrealistic outliers for further averaging by the program DAMAVER (31), as necessary. Overall, all of the generated envelopes with different symmetries give reasonable fits with the experimental SAXS curve.
Rigid-body refinement was utilized to build tetrameric models of ACMSD (20 independent runs) by using the program CORAL (64) with the crystal structure (PDB entry 2HBV) as individual initial models. The representative model was selected using the program DAMCLUST. The program CRYSOL was employed to generate the theoretical SAXS profile of the model for curve fitting with experimental data (65). UCSF Chimera (66) was used to manually fit the structural models with the ab initio envelope.
Site-directed mutagenesis for validation
The H110A mutant was constructed by the PCR overlap extension mutagenesis method. The plasmid pET16b containing acmsd was used as a template. The forward primer for H110A was 5′-GCTCTGGAGTTTGCGGCGGCGAATCCTCAGCGCATTAAG. The mutated plasmids were verified by DNA sequencing. The expression and purification strategy of the mutant was the same as for wtACMSD (24).
Kinetic assay of ACMSD
The substrate of ACMSD, ACMS, was generated by catalytic insertion of oxygen into 3-hydroxyanthranilic acid by 3-hydroxyanthranilic acid dioxygenase (7, 67). The enzymatic activity was measured by monitoring the decrease in absorbance of ACMS at 360 nm (ϵ360 = 47,500 cm−1 m−1), 320 nm (ϵ320 = 9600 cm−1 m−1), or 310 nm (ϵ310 = 5335 cm−1 m−1) at room temperature with an Agilent 8453 diode-array spectrophotometer.
Dissociation of ACMSD as measured by activity
ACMSD at a stock concentration of 100 μm was diluted in HEPES buffer to a final concentration of 30 nm. The specific activity of the diluted enzyme was then monitored as a function of time by taking aliquots at regular intervals and monitoring the decreasing absorbance of ACMS at 320 or 310 nm. The resulting dissociation profile was fitted with multiple exponential functions as follows,
where At is the percentage of specific activity at time x over the highest activity at the starting point, ti is the decay constant, and Ai is the amplitude on each decay phase i.
Analysis of the oligomeric states by size-exclusion chromatography
The calibration curve was obtained by using a standard proteins kit from Sigma-Aldrich (MWGF200-1KT), including β-amylase (200 kDa), alcohol dehydrogenase (150 kDa), albumin (66 kDa), carbonic anhydrase (29 kDa), and cytochrome c (12.4 kDa). Each protein was eluted from the HiLoad 16/600 Superdex 200 with the recommended buffer containing 50 mm Tris-HCl, pH 7.5, and 100 mm KCl and recorded as an elution volume (Ve). Blue dextran was used to determine the void volume (Vo). The molecular weight in log scale was plotted as a function of Ve/Vo.
The oligomers of ACMSD were separated by using a Superdex 200 column under various buffers, including sodium phosphate (pH 6.0–6.5), HEPES (pH 7.0–8.0), Tris (pH 8.5), and CHES (pH 9.0), with and without the addition of 150 mm NaCl. The separation profiles were obtained with an ÄKTApurifier. The separation profile of wtACMSD at pH 8.5 was analyzed with UNICORN version 5.11 (Fig. 5B). The three eluted portions, higher-order oligomers, tetramer, and dimer were separated by drop lines, and the area under their respective curves was integrated with the unit ml·mAU. The percentages of each portion were calculated based the integrated area and listed in Table S2. Then the same procedure was used to analyze the separation profiles under other conditions mentioned in the legends to Fig. 5 and Fig. S4. The results were summarized in Table S2.
Crystallization of WT and H110A ACMSD proteins
ACMSD H110A was not able to be crystallized with the reported conditions. After screening, a new condition containing 8% (v/v) TacsimateTM, pH 6.0, and 20% (w/v) PEG 3350 was identified, which was able to produce crystals for both H110A and wtACMSD. The X-ray diffraction data sets were collected, processed, and scaled by HKL-2000 (68) at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory. The structures of both H110A and wtACMSD were solved by molecular replacement using wtACMSD (PDB entry 2HBV) as the template and refined with Phenix version 1.10.1 (69) and Coot version 0.8.3 (70). The wtACMSD data set was processed with a diffraction anisotropy server (71) before modeling due to its anisotropic character. Structural packing figures were drawn with PyMOL 1.8.6.0 (72).
Author contributions
Y. Y., I. D., T. M., and I. R. data curation; Y. Y., I. D., and T. M. formal analysis; Y. Y., I. D., T. M., and A. L. validation; Y. Y., I. D., T. M., I. R., and A. L. investigation; Y. Y., I. D., T. M., and A. L. visualization; Y. Y. writing-original draft; I. D. and A. L. project administration; I. D., T. M., I. R., and A. L. writing-review and editing; T. M. software; T. M. methodology; A. L. conceptualization; A. L. supervision; A. L. funding acquisition.
Supplementary Material
Acknowledgments
X-ray synchrotron data were collected at the beamlines BL4-2 and BL9-2 of the Stanford Synchrotron Radiation Lightsource (SSRL) under user program 5B14, SLAC National Accelerator Laboratory. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the Department of Energy Office of Biological and Environmental Research and by NIGMS, National Institutes of Health, Grant P41GM103393.
The work was supported in part by National Institutes of Health Grant R01GM108988 and National Science Foundation Grant CHE-1808637 (to A. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1–S4 and Figs. S1–S5.
The atomic coordinates and structure factors (codes 6MGS and 6MGT) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The SAXS data of WT and H110A ACMSD described here were deposited in the SASBDB (73) with accession codes SASDFN5 and SASDFM5, respectively.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- KP
- kynurenine pathway
- ACMS
- 2-amino-3-carboxymuconate-6-semialdehyde
- ACMSD
- α-amino-β-carboxymuconate-ϵ-semialdehyde decarboxylase
- QA
- quinolinic acid
- PA
- picolinic acid
- SEC
- size-exclusion chromatography
- SAXS
- small-angle X-ray scattering
- NBA
- nitrobenzoic acid
- 2-AMS
- 2-aminomuconate semialdehyde
- wtACMSD
- wild-type ACMSD
- PDB
- Protein Data Bank
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid.
References
- 1. Stone T. W., and Darlington L. G. (2002) Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug. Discov. 1, 609–620 10.1038/nrd870 [DOI] [PubMed] [Google Scholar]
- 2. Schwarcz R. (2004) The kynurenine pathway of tryptophan degradation as a drug target. Curr. Opin. Pharmacol. 4, 12–17 10.1016/j.coph.2003.10.006 [DOI] [PubMed] [Google Scholar]
- 3. Schwarcz R., Bruno J. P., Muchowski P. J., and Wu H.-Q. (2012) Kynurenines in the mammalian brain: when physiology meets pathology. Nat. Rev. Neurosci. 13, 465–477 10.1038/nrn3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vécsei L., Szalárdy L., Fülöp F., and Toldi J. (2013) Kynurenines in the CNS: recent advances and new questions. Nat. Rev. Drug. Discov. 12, 64–82 10.1038/nrd3793 [DOI] [PubMed] [Google Scholar]
- 5. Cervenka I., Agudelo L. Z., and Ruas J. L. (2017) Kynurenines: tryptophan's metabolites in exercise, inflammation, and mental health. Science 357, eaaf9794 10.1126/science.aaf9794 [DOI] [PubMed] [Google Scholar]
- 6. Katsyuba E., Mottis A., Zietak M., De Franco F., van der Velpen V., Gariani K., Ryu D., Cialabrini L., Matilainen O., and Liscio P., Giacchè N., Stokar-Regenscheit N., Legouis D., de Seigneux S., Ivanisevic J., et al. (2018) De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 563, 354–359 10.1038/s41586-018-0645-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li T., Iwaki H., Fu R., Hasegawa Y., Zhang H., and Liu A. (2006) α-Amino-β-carboxymuconic-ϵ-semialdehyde decarboxylase (ACMSD) is a new member of the amidohydrolase superfamily. Biochemistry 45, 6628–6634 10.1021/bi060108c [DOI] [PubMed] [Google Scholar]
- 8. Huo L., Liu F., Iwaki H., Li T., Hasegawa Y., and Liu A. (2015) Human α-amino-β-carboxymuconate-ϵ-semialdehyde decarboxylase (ACMSD): a structural and mechanistic unveiling. Proteins 83, 178–187 10.1002/prot.24722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li T., Walker A. L., Iwaki H., Hasegawa Y., and Liu A. (2005) Kinetic and spectroscopic characterization of ACMSD from Pseudomonas fluorescens reveals a pentacoordinate mononuclear metallocofactor. J. Am. Chem. Soc. 127, 12282–12290 10.1021/ja0532234 [DOI] [PubMed] [Google Scholar]
- 10. Li T., Ma J. K., Hosler J. P., Davidson V. L., and Liu A. (2007) Detection of transient intermediates in the metal-dependent nonoxidative decarboxylation catalyzed by α-amino-β-carboxymuconate-ϵ-semialdehyde decarboxylase. J. Am. Chem. Soc. 129, 9278–9279 10.1021/ja073648l [DOI] [PubMed] [Google Scholar]
- 11. Colabroy K. L., and Begley T. P. (2005) The pyridine ring of NAD is formed by a nonenzymatic pericyclic reaction. J. Am. Chem. Soc. 127, 840–841 10.1021/ja0446395 [DOI] [PubMed] [Google Scholar]
- 12. Zhang H., Ryu D., Wu Y., Gariani K., Wang X., Luan P., D'Amico D., Ropelle E. R., Lutolf M. P., Aebersold R., Schoonjans K., Menzies K. J., and Auwerx J. (2016) NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443 10.1126/science.aaf2693 [DOI] [PubMed] [Google Scholar]
- 13. Hershberger K. A., Martin A. S., and Hirschey M. D. (2017) Role of NAD+ and mitochondrial sirtuins in cardiac and renal diseases. Nat. Rev. Nephrol. 13, 213–225 10.1038/nrneph.2017.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoshino J. (2019) ACMSD: a novel target for modulating NAD+ homeostasis. Trends Endocrinol. Metab. 30, 229–232 10.1016/j.tem.2019.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beal M. F., Kowall N. W., Ellison D. W., Mazurek M. F., Swartz K. J., and Martin J. B. (1986) Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature 321, 168–171 10.1038/321168a0 [DOI] [PubMed] [Google Scholar]
- 16. Davis I., and Liu A. (2015) What is the tryptophan kynurenine pathway and why is it important to neurotherapeutics? Expert Rev. Neurother. 15, 719–721 10.1586/14737175.2015.1049999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huo L., Davis I., Liu F., Andi B., Esaki S., Iwaki H., Hasegawa Y., Orville A. M., and Liu A. (2015) Crystallographic and spectroscopic snapshots reveal a dehydrogenase in action. Nat. Commun. 6, 5935 10.1038/ncomms6935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang Y., Davis I., Ha U., Wang Y., Shin I., and Liu A. (2016) A pitcher-and-catcher mechanism drives endogenous substrate isomerization by a dehydrogenase in kynurenine metabolism. J. Biol. Chem. 291, 26252–26261 10.1074/jbc.M116.759712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davis I., Yang Y., Wherritt D., and Liu A. (2018) Reassignment of the human aldehyde dehydrogenase ALDH8A1 (ALDH12) to the kynurenine pathway in tryptophan catabolism. J. Biol. Chem. 293, 9594–9603 10.1074/jbc.RA118.003320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Muraki T., Taki M., Hasegawa Y., Iwaki H., and Lau P. C. (2003) Prokaryotic homologs of the eukaryotic 3-hydroxyanthranilate 3,4-dioxygenase and 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase in the 2-nitrobenzoate degradation pathway of Pseudomonas fluorescens strain KU-7. Appl. Environ. Microbiol. 69, 1564–1572 10.1128/AEM.69.3.1564-1572.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chandler J. L., and Gholson R. K. (1972) Studies on the de novo biosynthesis of NAD in Escherichia coli. II. Quantitative method for isolating quinolinic acid from biological materials. Anal. Biochem. 48, 529–535 10.1016/0003-2697(72)90108-X [DOI] [PubMed] [Google Scholar]
- 22. Dai S., Schwendtmayer C., Schürmann P., Ramaswamy S., and Eklund H. (2000) Redox signaling in chloroplasts: cleavage of disulfides by an iron-sulfur cluster. Science 287, 655–658 10.1126/science.287.5453.655 [DOI] [PubMed] [Google Scholar]
- 23. Pellicciari R., Liscio P., Giacchè N., De Franco F., Carotti A., Robertson J., Cialabrini L., Katsyuba E., Raffaelli N., and Auwerx J. (2018) α-Amino-β-carboxymuconate-ϵ-semialdehyde decarboxylase (ACMSD) inhibitors as novel modulators of de novo nicotinamide adenine dinucleotide (NAD+) biosynthesis. J. Med. Chem. 61, 745–759 10.1021/acs.jmedchem.7b01254 [DOI] [PubMed] [Google Scholar]
- 24. Huo L., Davis I., Chen L., and Liu A. (2013) The power of two: arginine 51 and arginine 239* from a neighboring subunit are essential for catalysis in α-amino-β-carboxymuconate-ϵ-semialdyhyde decarboxylase. J. Biol. Chem. 288, 30862–30871 10.1074/jbc.M113.496869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Petoukhov M. V., Konarev P. V., Kikhney A. G., and Svergun D. I. (2007) ATSAS 2.1–towards automated and web-supported small-angle scattering data analysis. J. Appl. Crystallogr. 40, s223–s228 10.1107/S0021889807002853 [DOI] [Google Scholar]
- 26. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 10.1107/S0021889812007662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huo L., Fielding A. J., Chen Y., Li T., Iwaki H., Hosler J. P., Chen L., Hasegawa Y., Que L. Jr., and Liu A. (2012) Evidence for a dual role of an active site histidine in α-amino-β-carboxymuconate-ϵ-semialdehyde decarboxylase. Biochemistry 51, 5811–5821 10.1021/bi300635b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martynowski D., Eyobo Y., Li T., Yang K., Liu A., and Zhang H. (2006) Crystal structure of α-amino-β-carboxymuconate-ϵ-semialdehyde decarboxylase: insight into the active site and catalytic mechanism of a novel decarboxylation reaction. Biochemistry 45, 10412–10421 10.1021/bi060903q [DOI] [PubMed] [Google Scholar]
- 29. Putnam C. D., Hammel M., Hura G. L., and Tainer J. A. (2007) X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 40, 191–285 10.1017/S0033583507004635 [DOI] [PubMed] [Google Scholar]
- 30. Franke D., and Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 10.1107/S0021889809000338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 10.1107/S0021889803000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Korasick D. A., and Tanner J. J. (2018) Determination of protein oligomeric structure from small-angle X-ray scattering. Protein Sci. 27, 814–824 10.1002/pro.3376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ruiz-Herrera J., Amezcua-Ortega R., and Trujillo A. (1968) Purification and properties of a disulfide reductase obtained from Achromobacter starkeyi. J. Biol. Chem. 243, 4083–4088 [PubMed] [Google Scholar]
- 34. Samanta S. K., Bhushan B., Chauhan A., and Jain R. K. (2000) Chemotaxis of a Ralstonia sp. SJ98 toward different nitroaromatic compounds and their degradation. Biochem. Biophys. Res. Commun. 269, 117–123 10.1006/bbrc.2000.2204 [DOI] [PubMed] [Google Scholar]
- 35. Basu S., Pal Chowdhury P., Deb S., and Dutta T. K. (2016) Degradation pathways of 2- and 4-nitrobenzoates in Cupriavidus sp. strain ST-14 and construction of a recombinant strain, ST-14::3NBA, capable of degrading 3-nitrobenzoate. Appl. Environ. Microbiol. 82, 4253–4263 10.1128/AEM.00739-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ottosen M. B., Björnberg O., Nørager S., Larsen S., Palfey B. A., and Jensen K. F. (2002) The dimeric dihydroorotate dehydrogenase A from Lactococcus lactis dissociates reversibly into inactive monomers. Protein Sci. 11, 2575–2583 10.1110/ps.0220302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sjöberg B. M., Karlsson M., and Jörnvall H. (1987) Half-site reactivity of the tyrosyl radical of ribonucleotide reductase from Escherichia coli. J. Biol. Chem. 262, 9736–9743 [PubMed] [Google Scholar]
- 38. Shi J., Dertouzos J., Gafni A., Steel D., and Palfey B. A. (2006) Single-molecule kinetics reveals signatures of half-sites reactivity in dihydroorotate dehydrogenase A catalysis. Proc. Natl. Acad. Sci. U.S.A. 103, 5775–5780 10.1073/pnas.0510482103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sinha S. C., and Sprang S. R. (2006) Structures, mechanism, regulation and evolution of class III nucleotidyl cyclases. Rev. Physiol. Biochem. Pharmacol. 157, 105–140 [DOI] [PubMed] [Google Scholar]
- 40. Mochalkin I., Miller J. R., Evdokimov A., Lightle S., Yan C., Stover C. K., and Waldrop G. L. (2008) Structural evidence for substrate-induced synergism and half-sites reactivity in biotin carboxylase. Protein Sci. 17, 1706–1718 10.1110/ps.035584.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tsai A.-L., and Kulmacz R. J. (2010) Prostaglandin H synthase: resolved and unresolved mechanistic issues. Arch. Biochem. Biophys. 493, 103–124 10.1016/j.abb.2009.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu Q., Chai J., Moche M., Guy J., Lindqvist Y., and Shanklin J. (2015) Half-of-the-sites reactivity of the castor Δ9–18:o-acyl carrier protein desaturase. Plant Physiol. 169, 432–441 10.1104/pp.15.00622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Orlando B. J., and Malkowski M. G. (2016) Substrate-selective inhibition of cyclooxygeanse-2 by fenamic acid derivatives is dependent on peroxide tone. J. Biol. Chem. 291, 15069–15081 10.1074/jbc.M116.725713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee A. L., and Sapienza P. J. (2018) Thermodynamic and NMR assessment of ligand cooperativity and intersubunit communication in symmetric dimers: application to thymidylate synthase. Front. Mol. Biosci. 5, 47 10.3389/fmolb.2018.00047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wielgus-Kutrowska B., Grycuk T., and Bzowska A. (2018) Part-of-the-sites binding and reactivity in the homooligomeric enzymes–facts and artifacts. Arch. Biochem. Biophys. 642, 31–45 10.1016/j.abb.2018.01.011 [DOI] [PubMed] [Google Scholar]
- 46. Svergun D. I., and Koch M. H. (2003) Small-angle scattering studies of biological macromolecules in solution. Rep. Prog. Phys. 66, 1735–1782 10.1088/0034-4885/66/10/R05 [DOI] [Google Scholar]
- 47. Korasick D. A., Končitíková R., Kopečná M., Hájková E., Vigouroux A., Moréra S., Becker D. F., Šebela M., Tanner J. J., and Kopečný D. (2019) Structural and biochemical characterization of aldehyde dehydrogenase 12, the last enzyme of proline catabolism in plants. J. Mol. Biol. 431, 576–592 10.1016/j.jmb.2018.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ho M.-C., Ménétret J.-F., Tsuruta H., and Allen K. N. (2009) The origin of the electrostatic perturbation in acetoacetate decarboxylase. Nature 459, 393–397 10.1038/nature07938 [DOI] [PubMed] [Google Scholar]
- 49. Mathew E., Mirza A., and Menhart N. (2004) Liquid-chromatography-coupled SAXS for accurate sizing of aggregating proteins. J. Synchrotron Radiat. 11, 314–318 10.1107/S0909049504014086 [DOI] [PubMed] [Google Scholar]
- 50. David G., and Pérez J. (2009) Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. J. Appl. Crystallogr. 42, 892–900 10.1107/S0021889809029288 [DOI] [Google Scholar]
- 51. Bergdoll L. A., Lerch M. T., Patrick J. W., Belardo K., Altenbach C., Bisignano P., Laganowsky A., Grabe M., Hubbell W. L., and Abramson J. (2018) Protonation state of glutamate 73 regulates the formation of a specific dimeric association of mVDAC1. Proc. Natl. Acad. Sci. U.S.A. 115, E172–E179 10.1073/pnas.1715464115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Clouser A. F., and Klevit R. E. (2017) pH-dependent structural modulation is conserved in the human small heat shock protein HSBP1. Cell Stress Chaperones 22, 569–575 10.1007/s12192-017-0783-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Morais M. A., Giuseppe P. O., Souza T. A., Alegria T. G., Oliveira M. A., Netto L. E., and Murakami M. T. (2015) How pH modulates the dimer-decamer interconversion of 2-Cys peroxiredoxins from the Prx1 subfamily. J. Biol. Chem. 290, 8582–8590 10.1074/jbc.M114.619205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tabarani G., Thépaut M., Stroebel D., Ebel C., Vivès C., Vachette P., Durand D., and Fieschi F. (2009) DC-SIGN neck domain is a pH-sensor controlling oligomerization: SAXS and hydrodynamic studies of extracellular domain. J. Biol. Chem. 284, 21229–21240 10.1074/jbc.M109.021204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wood Z. A., Poole L. B., Hantgan R. R., and Karplus P. A. (2002) Dimers to doughnuts: redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry 41, 5493–5504 10.1021/bi012173m [DOI] [PubMed] [Google Scholar]
- 56. Ulrih N. P., Anderluh G., Macek P., and Chalikian T. V. (2004) Salt-induced oligomerization of partially folded intermediates of equinatoxin II. Biochemistry 43, 9536–9545 10.1021/bi049616h [DOI] [PubMed] [Google Scholar]
- 57. Watanabe S., Harayama M., Kanemura S., Sitia R., and Inaba K. (2017) Structural basis of pH-dependent client binding by ERp44, a key regulator of protein secretion at the ER–Golgi interface. Proc. Natl. Acad. Sci. U.S.A. 114, E3224–E3232 10.1073/pnas.1621426114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lolli G., Naressi D., Sarno S., and Battistutta R. (2017) Characterization of the oligomeric states of the CK2 α2β2 holoenzyme in solution. Biochem. J. 474, 2405–2416 10.1042/BCJ20170189 [DOI] [PubMed] [Google Scholar]
- 59. Symons Z. C., and Bruce N. C. (2006) Bacterial pathways for degradation of nitroaromatics. Nat. Prod. Rep. 23, 845–850 10.1039/b502796a [DOI] [PubMed] [Google Scholar]
- 60. Ju K.-S., and Parales R. E. (2010) Nitroaromatic compounds, from synthesis to biodegradation. Microbiol. Mol. Biol. Rev. 74, 250–272 10.1128/MMBR.00006-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hasegawa Y., Muraki T., Tokuyama T., Iwaki H., Tatsuno M., and Lau P. C. (2000) A novel degradative pathway of 2-nitrobenzoate via 3-hydroxyanthranilate in Pseudomonas fluorescens strain KU-7. FEMS Microbiol. Lett. 190, 185–190 10.1111/j.1574-6968.2000.tb09284.x [DOI] [PubMed] [Google Scholar]
- 62. Smolsky I. L., Liu P., Niebuhr M., Ito K., Weiss T. M., and Tsuruta H. (2007) Biological small-angle X-ray scattering facility at the Stanford Synchrotron Radiation Laboratory. J. Appl. Crystallogr. 40, s453–s458 10.1107/S0021889807009624 [DOI] [Google Scholar]
- 63. Svergun D. I., Petoukhov M. V., and Koch M. H. (2001) Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80, 2946–2953 10.1016/S0006-3495(01)76260-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Petoukhov M. V., and Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 10.1529/biophysj.105.064154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Svergun D., Barberato C., and Koch M. H. (1995) CRYSOL-a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 10.1107/S0021889895007047 [DOI] [Google Scholar]
- 66. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 67. Yang Y., Liu F., and Liu A. (2018) Adapting to oxygen: 3-hydroxyanthrinilate 3,4-dioxygenase employs loop dynamics to accommodate two substrates with disparate polarities. J. Biol. Chem. 293, 10415–10424 10.1074/jbc.RA118.002698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 69. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 71. Strong M., Sawaya M. R., Wang S., Phillips M., Cascio D., and Eisenberg D. (2006) Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 103, 8060–8065 10.1073/pnas.0602606103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. DeLano W. L. (2014) The PyMOL Molecular Graphics System, version 1.7.0.0, Schrödinger, LLC, New York [Google Scholar]
- 73. Valentini E., Kikhney A. G., Previtali G., Jeffries C. M., and Svergun D. I. (2015) SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res. 43, D357–D363 10.1093/nar/gku1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Crooks G. E., Hon G., Chandonia J.-M., and Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.