

Abstract
Cardiolipin (CL) is the signature phospholipid of mitochondrial membranes. Although it has long been known that CL plays an important role in mitochondrial bioenergetics, recent evidence in the yeast model indicates that CL is also essential for intermediary metabolism. To gain insight into the function of CL in energy metabolism in mammalian cells, here we analyzed the metabolic flux of [U-13C]glucose in a mouse C2C12 myoblast cell line, TAZ-KO, which is CL-deficient because of CRISPR/Cas9-mediated knockout of the CL-remodeling enzyme tafazzin (TAZ). TAZ-KO cells exhibited decreased flux of [U-13C]glucose to [13C]acetyl-CoA and M2 and M4 isotopomers of tricarboxylic acid (TCA) cycle intermediates. The activity of pyruvate carboxylase, the predominant enzyme for anaplerotic replenishing of the TCA cycle, was elevated in TAZ-KO cells, which also exhibited increased sensitivity to the pyruvate carboxylase inhibitor phenylacetate. We attributed a decreased carbon flux from glucose to acetyl-CoA in the TAZ-KO cells to a ∼50% decrease in pyruvate dehydrogenase (PDH) activity, which was observed in both TAZ-KO cells and cardiac tissue from TAZ-KO mice. Protein–lipid overlay experiments revealed that PDH binds to CL, and supplementing digitonin-solubilized TAZ-KO mitochondria with CL restored PDH activity to WT levels. Mitochondria from TAZ-KO cells exhibited an increase in phosphorylated PDH, levels of which were reduced in the presence of supplemented CL. These findings indicate that CL is required for optimal PDH activation, generation of acetyl-CoA, and TCA cycle function, findings that link the key mitochondrial lipid CL to TCA cycle function and energy metabolism.
Keywords: cardiolipin, pyruvate dehydrogenase complex (PDC), pyruvate carboxylase (PC), tricarboxylic acid cycle (TCA cycle) (Krebs cycle), mitochondria
Introduction
The phospholipid cardiolipin (CL)8 is the signature lipid of mitochondrial membranes. It interacts with a wide variety of mitochondrial proteins (1) and has multiple roles in mitochondrial biogenesis as well as other cellular functions (2–4). Loss of CL leads to deficiencies in respiratory function, mitochondrial membrane potential, and ATP synthesis (1, 5–7). CL that is synthesized de novo has predominantly saturated fatty acids (8). However, the lipid is then remodeled via phospholipase-mediated deacylation to form monolysocardiolipin (MLCL), which is subsequently acylated by the enzyme tafazzin to synthesize CL with predominantly unsaturated fatty acids (9, 10).
Abnormal CL composition is associated with several human disorders, including diabetes, Alzheimer's disease, and Parkinson's disease (11–13). However, Barth syndrome (BTHS) is the only disorder identified to date that is caused by altered CL metabolism (14, 15). BTHS is a severe X-linked disorder characterized by dilated cardiomyopathy, skeletal myopathy, neutropenia, exercise intolerance, lactic acidosis, and sudden death from arrhythmia (16–18). The underlying cause of BTHS is mutations in the tafazzin (TAZ) gene, which codes for the transacylase that remodels CL (10, 19). As a result, BTHS patients exhibit an aberrant CL profile characterized by decreased total CL, increased MLCL, and abnormal CL acylation patterns (20). Tetralinoleoyl-CL, the most prevalent CL species in mitochondria of the human heart and skeletal muscle cells, is virtually absent from BTHS cells (21).
Although the clinical phenotypes of BTHS point to mitochondrial bioenergetic defects, the molecular basis whereby CL deficiency leads to the pathology is not understood. Several studies suggest that metabolic dysregulation is a key pathological mechanism in BTHS. A clear distinction has been shown in plasma metabolite profiles from BTHS patients compared with control patients of similar age (22). For example, 3-methylglutaconic aciduria is commonly observed in BTHS patients, sometimes accompanied by increased levels of lactic acid (23–26). 3-Methylglutaconic aciduria occurs as a result of increased excretion of 3-methylglutaconic and 3-methylglutaric acid, both of which are products of leucine catabolism (27). In addition, the organic acid 2-ethylhydracrylic acid has been reported in the urine of BTHS patients, indicating altered isoleucine metabolism (28, 29). Furthermore, the levels of skeletal muscle phosphodiesters, intermediates of membrane phospholipid breakdown, were elevated in BTHS patients, indicative of a damaged sarcolemma, atrophied muscle, and loss of fiber (30, 31).
Although the specific mechanisms leading to the metabolic changes in BTHS are unknown, they suggest that CL, long known to be required for oxidative phosphorylation and bioenergetics (2, 3), is also required for optimal intermediary metabolism. This functional role is supported by studies in yeast strongly suggesting that CL deficiency leads to defects in the TCA cycle (32, 33). Raja et al. (32) showed that yeast cells that cannot synthesize CL (crd1Δ) exhibit decreased levels of acetyl-CoA. Interestingly, although expression of the acetyl-CoA biosynthetic enzyme pyruvate dehydrogenase (PDH) is up-regulated in the yeast mutant, PDH activity is not increased, suggesting that optimal PDH catalytic activity may require CL. The PDH complex catalyzes the oxidative decarboxylation of pyruvate to acetyl-CoA by three catalytic activities: PDH (E1), dihydrolipoamide acetyltransferase (E2), and dihydrolipoamide dehydrogenase (E3) (34, 35). PDH kinases (PDKs) (36, 37) and PDH phosphatases (PDPs) (38) are also components of mammalian PDH complexes. Flux of carbon through the PDH complex is tightly regulated by the phosphorylation state of the enzyme, which is controlled by PDKs and PDPs in response to altered metabolic conditions (35, 39). Importantly, phosphorylation by PDKs of three sites in the E1α subunit leads to inactivation of the PDH complex; activity is restored by PDP-mediated dephosphorylation (40–42). A second finding linking CL to the TCA cycle was that of Patil et al. (33), who reported that Fe-S biogenesis is perturbed in CL-deficient yeast cells. This is reflected in decreased activity of Fe-S–requiring enzymes, including the TCA cycle enzymes aconitase and succinate dehydrogenase (SDH). Consistent with perturbation of the TCA cycle, a recent report showed that anaplerotic pathways are required to ameliorate TCA cycle dysfunction in yeast cells (43). These discoveries, linking CL and TCA cycle function in yeast, raise the possibility that CL may play an essential role in metabolic regulation associated with BTHS pathology. Indeed, CL has been reported to be required for the stability and optimal enzymatic activity of SDH (44), and, accordingly, decreased protein levels and activity of SDH were also observed in cardiac tissue from BTHS mice (45).
To gain insight into the role of CL in energy metabolism, we analyzed the metabolic flux of [U-13C]glucose in a tafazzin knockout mouse cell line, TAZ-KO (46). In this study, we demonstrate that the TCA cycle is perturbed in tafazzin-deficient cells, which exhibit decreased flux of glucose carbon to acetyl-CoA, TCA cycle metabolites, and related amino acids. We further show that CL activates PDH, supporting the hypothesis that decreased flux of carbon from glucose to acetyl-CoA in CL-deficient cells results from decreased PDH activity. To our knowledge, this is the first demonstration that the mitochondrial lipid CL is required for PDH activation, acetyl-CoA synthesis, and TCA cycle function in mammalian cells.
Results
Decreased flux of carbon from glucose to acetyl-CoA in TAZ-KO cells
To investigate metabolic changes resulting from tafazzin deficiency, TAZ-KO and isogenic WT cells were cultured in medium containing [U-13C]glucose, and glycolytic and TCA pathway metabolites in cell extracts were analyzed by LC-MS and GC-MS. [U-13C]glucose was metabolized quickly to glycolytic intermediates during fermentation in both WT and TAZ-KO cells. However, the relative percentage of [13C]acetyl-CoA was sharply reduced in TAZ-KO cells (Fig. 1A). Consistent with decreased flux of [U-13C]glucose to [13C]acetyl-CoA, which enters the TCA cycle by condensation with oxaloacetate (OAA) to form citrate, the total percentages of [13C]citrate/isocitrate, [13C]malate, [13C]succinate, [13C]glutamate, and [13C]aspartate were decreased in TAZ-KO cells relative to the WT (Fig. 1B).
Figure 1.

Flux of [U-13C]glucose in TAZ-KO cells relative to WT. A and B, the percentage of 13C-labeled metabolites was measured after 1-h incubation with [U-13C]glucose. Metabolites from glycolysis (A) and the TCA cycle (B) were analyzed by LC-MS and GC-MS. Data shown are mean ± S.D. (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
M2, M3, and M4 isotopomers were the most abundant TCA cycle metabolites detected. M2 metabolites are generated in the first round of the TCA cycle. Condensation of M2 [13C]acetyl-CoA with M2 [13C]OAA produces M4 isotopomers in the second round of the cycle. Consistent with decreased M2 [13C]acetyl-CoA in TAZ-KO cells, M2 and M4 [13C]citrate/isocitrate, [13C]glutamate, [13C]succinate, [13C]malate, [13C]fumarate, and [13C]aspartate were decreased in mutant cells relative to WT (Fig. 2).
Figure 2.

Mass-isotopomer distribution (MID) of M2 and M4 metabolites. MID of M2 (top panel) and M4 (bottom panel) isotopomers was determined by LC-MS and GC-MS after 1-h incubation with [U-13C]glucose. Data shown are mean ± S.D. (n = 3). *, p < 0.05; **, p < 0.01; ***p < 0.001.
Increased PC activity in TAZ-KO cells
PC is an anaplerotic enzyme that replenishes TCA cycle intermediates by catalyzing the conversion of pyruvate to OAA (47). Elevated PC activity (Fig. 3A) and increased levels of PC protein (Fig. 3B) were observed in TAZ-KO cells. In the presence of phenylacetic acid, an inhibitor of PC (48), growth of TAZ-KO cells was decreased, whereas growth of WT cells was not affected (Fig. 3C). These data suggest that PC is required to replenish the TCA cycle in TAZ-KO cells.
Figure 3.

Increased PC activity in TAZ-KO cells. A, PC activity was assayed in isolated mitochondria as described under “Experimental procedures.” Data shown are mean ± S.D. (n = 3). B, levels of PC from cell extracts were determined by Western blot analysis with anti-PC antibody. 50 μg of total protein from each sample was loaded onto an SDS gel under reducing conditions, and actin was used as a loading control. C, cells were treated with 4 mm phenylacetic acid (PAA, a PC inhibitor, pH adjusted to 7.4). Cell viability was measured by MTT viability assay as described under “Experimental procedures.”
As all three carbons of [13C]pyruvate generated from [U-13C]glucose are 13C-labeled, PC converts M3 [13C]pyruvate to M3 [13C]OAA. The reversibility of TCA cycle enzymes, including SDH, fumarase, and malate dehydrogenase (MDH), enables interconversion between succinate, fumarate, malate, and OAA (49). Interestingly, M3 [13C]malate and [13C]aspartate levels did not differ between WT and TAZ-KO cells, suggesting that these metabolites are replenished through the PC pathway (Fig. 4). However, M3 [13C]succinate was decreased by 74%, whereas M3 [13C]fumarate accumulated markedly in TAZ-KO cells, in contrast to WT cells. This finding suggests that conversion of fumarate to succinate by SDH was decreased in TAZ-KO cells. In agreement with this, mutant mitochondria exhibited a 40% decrease in SDH activity compared with the control (Fig. 5A). By comparison, activity of the TCA cycle enzyme MDH was not affected in mutant mitochondria (Fig. 5B). Taken together, these experiments indicate that carbon flux from glucose to TCA cycle intermediates is reduced in TAZ-KO cells. This defect is partially compensated by up-regulation of PC, but compensation is limited by decreased SDH activity in the mutant.
Figure 4.

MID of M3 metabolites. MID of M3 isotopomers was determined by LC-MS and GC-MS after 1-h incubation with [U-13C]glucose. Data shown are mean ± S.D. (n = 3). *, p < 0.05; ***, p < 0.001.
Figure 5.
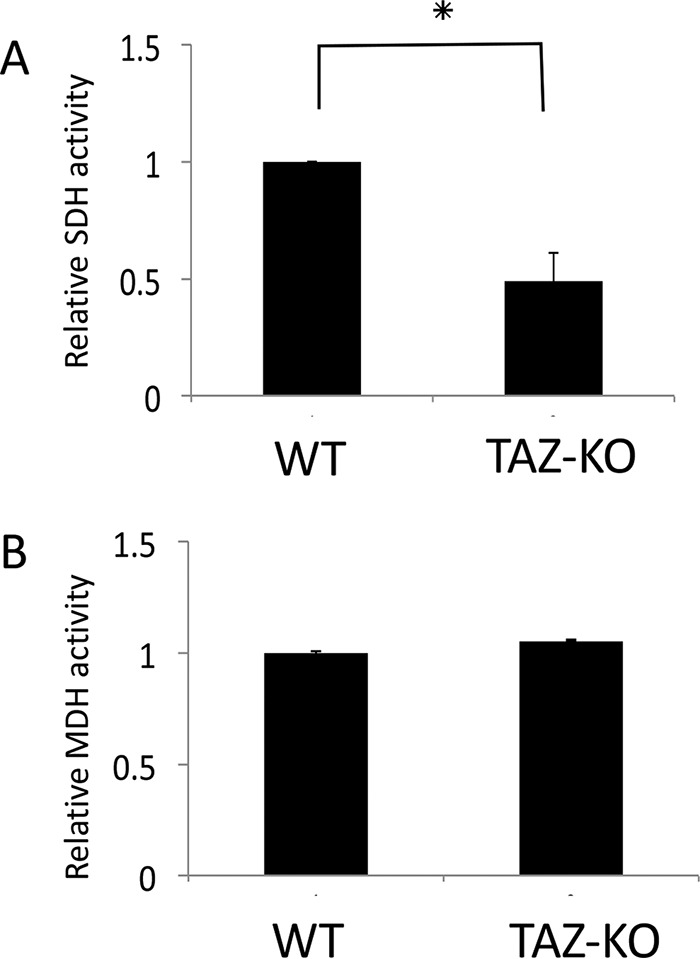
TAZ-KO cells exhibit decreased SDH activity. A and B, the activity of SDH (A) and MDH (B) was assayed in isolated mitochondria as described under “Experimental procedures.” Data shown are mean ± S.D. (n = 3). *, p < 0.05.
PDH activity is decreased in tafazzin-deficient cells
To investigate the mechanism underlying diminished carbon flux from glucose to acetyl-CoA in mutant cells, we explored the possibility that PDH-mediated conversion of pyruvate to acetyl-CoA was defective. As glucose-derived pyruvate is the primary source of carbon for the generation of acetyl-CoA by PDH, decreased [13C]acetyl-CoA in TAZ-KO cells suggested that PDH activity was decreased in the mutant. To test this possibility, PDH was assayed in mitochondrial extracts from TAZ-KO and WT cells. PDH activity was decreased by about 50% in TAZ-KO mitochondria (Fig. 6A), whereas the protein levels of PDH-E1α (the catalytic subunit of the PDH complex) were not decreased (Fig. 6B), suggesting that the PDH defect resulted from decreased catalytic activity of the enzyme. The deficiency in PDH activity was also apparent in cardiac tissue isolated from TAZ-KO mice (Fig. 6C), suggesting that our observations in myoblast cultures remain valid in a fully developed muscular organ such as the heart.
Figure 6.

Decreased activity of PDH in tafazzin-deficient cells. A, PDH activity was assayed in isolated mitochondria as described under “Experimental procedures.” Data shown are mean ± S.D. (n = 3). *, p < 0.05. B, levels of PDH-E1α in mitochondrial extracts were determined by Western blot analysis with anti-PDH-E1α antibody. 50 μg of total protein from each sample was loaded onto an SDS gel under reducing conditions, and the mitochondrial protein Ndufb6 was used as a loading control. C, PDH activity was assayed in heart tissue extracted from WT and TAZ-KO mice. Each column represents a separate mouse. Data shown are mean ± S.D. (n = 2). D, WT and TAZ-KO cells were seeded in DMEM (without glucose, pyruvate, or glutamine/1% FCS), followed by 3-h incubation in Dulbecco's PBS (Life Technologies) supplemented with 100 μm [1-14C]pyruvate, and the PDH flux assay was performed in the presence or absence of 5 mm DCA. PDH activity was assayed by measuring the release of 14CO2 from [1-14C]pyruvate as described under “Experimental procedures.” Data shown are mean ± S.D. (n = 3). *, p < 0.05.
We further tested the in vivo activity of PDH in TAZ-KO cells by assaying the production of [14C]CO2 from [14C]pyruvate. In agreement with the in vitro assay, TAZ-KO cells exhibited an ∼50% decrease in PDH flux. When treated with the PDH kinase inhibitor dichloroacetate (DCA), which activates PDH flux, [14C]CO2 production in TAZ-KO cells was restored to WT levels (Fig. 6D), suggesting that the reduced PDH flux in TAZ-KO cells is due to active inhibition rather than impaired capacity.
Cardiolipin activates PDH
To investigate the possibility that PDH is activated by CL, levels of which are reduced in TAZ-KO cells, digitonin-solubilized mitochondria from TAZ-KO and WT cells were incubated with exogenous CL. As seen in Fig. 7A, CL restored the activity of PDH in mutant mitochondria to WT levels. In agreement with this finding, incubation of CL with purified PDH complex resulted in an increase in enzymatic activity (Fig. 7B). Binding of CL to the PDH complex was assessed by an in vitro protein lipid overlay assay. Both CL and phosphatidic acid bound to the PDH complex (Fig. 7C), but activation of PDH was specific for CL (Fig. 7B). MLCL, which accumulates in tafazzin-deficient cells (46), did not stimulate activity (data not shown).
Figure 7.

CL binds to PDH and increases its activity. A, digitonin-solubilized mitochondria from WT and TAZ-KO cells were treated with or without CL. PDH activity was assayed as described under “Experimental procedures.” B, purified PDH complex (Sigma) was incubated with exogenous CL or phosphatidic acid (PA). PDH activity was assayed as described under “Experimental procedures”. Data shown are mean ± S.D. (n = 3). *, p < 0.05. C, the indicated lipids CL, phosphatidylethanolamine (PE), phosphatidylcholine (PC), phosphatidylinositol 4,5-bisphosphate (PIP2), phosphatidylinositol 3,4,5-trisphosphate (PIP3), and phosphatidic acid were serially diluted and spotted onto a nitrocellulose membrane, which was incubated overnight in buffer containing 10 μg of PDH complex. Interactions were detected by immunoblotting with an antibody against the PDH complex.
As discussed above, PDH is regulated by phosphorylation, which decreases the catalytic activity (40). We therefore addressed the possibility that CL activates PDH by facilitating a decrease in phosphorylation of the enzyme. Consistent with decreased PDH activity, mitochondria isolated from TAZ-KO cells exhibited an increase in phosphorylated PDH (Fig. 8A). When digitonin-solubilized TAZ-KO mitochondria were incubated with exogenous CL, phosphorylation of PDH was reduced (Fig. 8B).
Figure 8.

CL decreases levels of phosphorylated PDH. Phosphorylated PDH was identified by Western blot analysis with an antibody against the phosphorylated enzyme (Phos-PDH). The mitochondrial protein NDUFB6 was included as a loading control. For DCA-treated samples, 5 mm DCA was added to cultured WT cells for 24 h before mitochondrial isolation. A, levels of phosphorylated PDH in mitochondrial protein from WT and TAZ-KO cells. B, levels of phosphorylated PDH in digitonin-solubilized mitochondria from WT and TAZ-KO cells incubated for 2 h on ice with or without CL. Bottom panels, the signal intensities of protein bands and surrounding background were scanned and quantified using ImageJ. The background-subtracted value for each protein band was normalized to that of NDUFB6 and quantified relative to the DCA-treated sample.
Taken together, these experiments suggest that CL links mitochondrial lipid biosynthesis with energy metabolism by mediating a decrease in PDH phosphorylation and enhancing PDH activity. Deficient PDH activity in CL-deficient cells results in decreased synthesis of acetyl-CoA with concomitant perturbation of the TCA cycle.
Discussion
In this study, we show for the first time that CL is required for optimal activity of the acetyl-CoA biosynthetic enzyme PDH and TCA cycle function. CL-deficient TAZ-KO cells exhibited decreased PDH activity (Fig. 6), increased levels of the phosphorylated, inactive enzyme (Fig. 8A), and decreased flux of glucose to acetyl-CoA and TCA cycle intermediates (Figs. 1 and 2). A requirement for anaplerosis was demonstrated in the mutant by up-regulation of PC and sensitivity to PC inhibition (Figs. 3 and 4). These findings identify a regulatory link between the mitochondrial membrane lipid CL and the reactions of energy metabolism in the mitochondrial matrix.
Our findings suggest that the mechanism whereby CL regulates PDH is that of facilitating a decrease in PDH phosphorylation. In agreement with this, supplementation of digitonin-solubilized mitochondria with exogenous CL led to a decrease in phosphorylated PDH. Dephosphorylation of PDH is facilitated by Ca2+-dependent binding of PDP to the L2 domain of the E2 subunit dihydrolipoyl acetyltransferase (50, 51). This suggests the possibility that CL, which has a high affinity for Ca2+ (52), facilitates binding of PDP to the E2 subunit, increasing dephosphorylation of the enzyme. This mechanism would be consistent with an early report showing that the PDH complex binds tightly to the mitochondrial inner membrane (53). Our findings do not rule out the possibility that CL may inhibit PDK activity, resulting in decreased phosphorylation of the enzyme. Experiments to test these possible mechanisms are in progress.
An increase in the protein level and cellular activity of PC was observed in TAZ-KO cells (Fig. 3, A and B). The importance of PC in TAZ-KO cells is reflected in the delayed growth of these cells in the presence of the PC inhibitor phenylacetic acid (Fig. 3C). A previous study reported that increased PC is necessary for continued mitochondrial metabolism and cellular anabolism in SDH mutant human cells (54). These findings suggest that TCA cycle perturbation in TAZ-KO cells is anaplerotically rescued by PC.
The decrease in SDH activity in TAZ-KO cells (Fig. 5A) is in agreement with the finding of heart-specific SDH deficiency in the BTHS mouse model (45). We have shown previously that yeast crd1Δ cells, which lack CL, exhibit decreased activity of SDH and other enzymes requiring Fe-S cofactors because of defective Fe-S biosynthesis (33). Decreased SDH activity in TAZ-KO cells may thus result from a deficiency in Fe-S cofactors.
This study may have important implications for BTHS. Defects in PDH activity and perturbation of the TCA cycle are expected to lead to a reduction in ATP production and energy deficits, causing cardiomyopathy and heart failure (55, 56). Increased expression of PC may be essential to compensate for energy deficits resulting from decreased acetyl-CoA synthesis. PDH deficiency further results in increased generation of lactic acid, which may contribute to the observed lactic acidosis and organic aciduria. Importantly, BTHS is characterized by a wide disparity in symptoms, even among patients with identical tafazzin mutations, suggesting that physiological modifiers of tafazzin deficiency contribute to the clinical presentation of the disease (57). We speculate that PDH, PC, and SDH are likely candidates for physiological modifiers of BTHS, as defects in these enzymes may exacerbate the effects of tafazzin deficiency. In conclusion, this study identifies a multifunctional role of CL in cellular metabolism and may shed light on the wide disparities in clinical phenotypes observed in BTHS patients.
Experimental procedures
CRISPR/Cas9 off-target analysis of TAZ-KO cells
The TAZ-KO cell line used in this study was generated previously using CRISPR/Cas9 targeted against exon 3 of the tafazzin (Taz) gene in C2C12 mouse myoblasts (46). The top 10 predicted off-target sites were identified using the CCTop CRISPR/Cas9 target predictor tool (https://crispr.cos.uni-heidelberg.de/)9 based on the Mus musculus GRCm38 reference genome and the gRNA sequence (5′-TCCTAAAACTCCGCCACATC-3′) utilized to create the TAZ-KO cell line (46, 63). Next-generation whole-genome sequencing was performed on WT and TAZ-KO cells, and a differential variant analysis was run to identify deviations between the WT and TAZ-KO genomes (GENEWIZ Biotechnology, South Plainfield, NJ). The readout from the differential variant analysis lists all genomic loci (also mapped to the M. musculus GRCm38 reference genome) at which the sequence of the TAZ-KO cell line differs from that of the WT cell line. The genomic coordinates of each of the 10 predicted off-target sites as well as the on-target Taz locus were manually compared against the differential variant analysis data. This analysis indicated that none of the predicted off-target sites were altered in the TAZ-KO cell line relative to isogenic WT cells (Table S1).
Cell lines and growth conditions
WT and tafazzin-knockout C2C12 cells were used as described previously (46). Cells were grown in DMEM (Gibco) containing 10% FBS (Hyclone), 2 mm glutamine (Gibco), penicillin (100 units/ml), and streptomycin (100 μg/ml) (Invitrogen) at 37 °C in a humidified incubator with 5% CO2. For flux analysis, 1 g/liter [U-13C]glucose was mixed in glucose- and serum-free medium. Cells were incubated in medium containing [U-13C]glucose for 1 h, collected, and stored at −80 °C.
Mouse heart tissue preparation
Mouse protocols were approved by the Institutional Animal Care and Use Committee of the New York University School of Medicine. Mice (C57BL/6) were housed under temperature-controlled conditions and a 12-h light/dark cycle with free access to drinking water and food. The TAZ-KO mouse model was developed in the C57BL/6 background by Dr. D. Strathdee of the Cancer Research UK Beatson Institute and is available upon request. TAZ-KO mice and WT mice were euthanized to harvest their hearts. The hearts were dissected, and the tissue pieces were stored at −80 °C before measurement of PDH activity.
Cell proliferation assay
3000 cells were suspended in 100 μl of growth medium and seeded into 96-well plates. Viable cells were measured in triplicate using an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Fisher) assay after 3, 24, and 48 h. In brief, 10 μl of 5 mg/ml MTT was added to each well. As a negative control, MTT was applied to wells lacking cells. The plate was incubated for 4 h at 37 °C. The medium was carefully removed, and 150 μl of DMSO was added to dissolve the MTT product. The plate was covered with foil and incubated for 10 min at 37 °C. Samples from each well were mixed with a pipette, and absorbance was read at 570 nm.
Immunoblotting
Cell or mitochondrial extract corresponding to 50 μg of protein was analyzed using a 10% SDS-PAGE gel. Immunoblotting was performed using primary antibodies to PDH-E1 (1:1000, Santa Cruz Biotechnology, sc-377092, lot D1615), phos-PDH (1:1000, Millipore, AP1062, lot 2923467), PC (1:1000, Santa Cruz Biotechnology, sc-271862, lot G1514), NDUFB6 (1:5000), and actin (1:3000) (Santa Cruz Biotechnology). Corresponding secondary antibodies conjugated to horseradish peroxidase were used. Immunoreactivity was visualized using ECL substrate (Thermo). To determine the effect of CL on PDH phosphorylation, aliquots of mitochondria (400 μg of protein) were suspended in 40 ml of 50 mm NaCl and 50 mm imidazole HCl (pH 7.0). 2 μl of CL (bovine heart, Sigma) from a stock solution containing 20 mg/ml in 5% digitonin and 50% ethanol and 1 ml of digitonin (10% in water) was added to obtain a CL/protein ratio of 1:10 (g/g) and a digitonin/protein ratio of 0.5 (g/g). After 2-h incubation on ice, levels of phosphorylated PDH were determined by Western blot analysis with antibody against phos-PDH (Millipore).
Mitochondrial extraction
Cells were cultured in 150-mm dishes until they reached 100% confluency, collected, and centrifuged at 720 × g for 5 min. Cell pellets were washed with cold PBS and suspended in mitochondrial isolation buffer (280 mm sucrose, 0.25 mm EDTA, and 20 mm Tris-HCl (pH 7.2)) and manually homogenized with a glass homogenizer. Cell debris was removed by centrifugation at 720 × g for 5 min. Mitochondria were subsequently collected by centrifugation at 14,812 × g for 10 min. Protein concentration was determined using a detergent compatible protein assay kit (Bio-Rad).
Metabolomic flux analysis
Metabolomic flux analysis was carried out at the University of Michigan Metabolomics Core Services as follows.
Central carbon metabolism sample preparation
Cell culture plates were removed from −80 °C storage and maintained on wet ice throughout the processing steps. To each 6-cm plate, 0.5 ml of a mixture of methanol, chloroform, and water (8:1:1) was added. Plates were gently agitated and then scraped to release cells, which were transferred to a microtube, vortexed, and incubated at 4 °C for 10 min to complete metabolite extraction. Samples were vortexed a second time and then centrifuged at 22,000 × g for 10 min at 4 °C. After centrifugation, 100 μl of the extraction solvent was transferred to an autosampler vial for LC-MS analysis. 10 μl of each sample was removed and pooled in a separate autosampler vial for quality control analyses. The remaining supernatant from each sample was transferred to an autosampler vial and dried. To each sample, 50 μl of a 20 mg/ml solution of methoxyamine hydrochloride in pyridine was added. Samples were vortexed briefly, incubated at 37 °C for 90 min, removed from heating, and allowed to cool to room temperature. 50 μl of N-methyl-N-tert-butyldimethylsilyltrifluoroacetamide) with 1% tertbutyldimethylchloro silane (Regis Technologies) was added, and samples were incubated at 70 °C for 1 h. A series of calibration standards was prepared along with samples for quality control of instrument performance and chromatography for LC and GC analysis.
LC-MS analysis
LC-MS analysis was performed on an Agilent system consisting of a 1260 ultra-high performance liquid chromatography module coupled with a 6520 Quadrupole-TOF mass spectrometer (Agilent Technologies). Metabolites were separated on a 150 × 1 mm Luna NH2 hydrophilic interaction chromatography column (Phenomenex) using 10 mm ammonium acetate in water, adjusted to pH 9.9 with ammonium hydroxide as mobile phase A and acetonitrile as mobile phase B. The flow rate was 0.075 ml/min, and the gradient was linear (20% to 100% A) over 15 min, followed by isocratic elution at 100% A for 5 min. The system was returned to starting conditions (20% A) in 0.1 min and held there for 10 min to allow for column re-equilibration before injecting another sample. The mass spectrometer was operated in electrospray ionization mode according to conditions published previously (58).
GC-MS analysis
GC-MS analysis was performed on an Agilent 69890N GC-5975 MS detector with the following parameters. A 1-μl sample was injected splitlessly on an HP-5MS 15m column (Agilent Technologies, Santa Clara, CA) with a He gas flow rate of 1.4 ml/min. The GC oven initial temperature was 60 °C, was increased at 10 °C per minute to 300 °C, and held at 300 °C for 5 min. The inlet temperature was 250 °C, and the MS source and quad temperatures were 230 °C and 150 °C, respectively.
Data analysis
Metabolites were identified by matching the retention time and mass to authentic standards. For LC-MS, metabolites were identified using Agilent's Profinder Isotopologue extraction program, which does automatic isotope subtraction based on the expected isotope distribution for the molecular formula provided. For GC-MS data, metabolites were identified by matching the retention time and mass (0.1-Da resolution) to authentic standards. Isotope peak areas were integrated using MassHunter Quantitative Analysis vB.07.00 (Agilent Technologies). Peak areas were corrected for natural isotope abundance (using a software package written in-house based on the method of Fernandez et al. (58)), and the residual isotope signal was reported.
Protein lipid overlay assay
Protein lipid overlay assays were performed according to the protocol of Dowler et al. (59) with modification. In brief, lyophilized lipids (tetraoleoyl-CL, phosphatidylethanolamine, phosphatidylcholine, phosphatidylinositol 4,5-bisphosphate, phosphatidylinositol 3,4,5-trisphosphate, and phosphatidic acid) were dissolved in a 1:1 solution of methanol and chloroform to make 1 mm stocks. Lipids were diluted to five different concentrations (500, 250, 125, 62.5, and 31.25 μm) in a 2:1:0.8 solution of methanol:chloroform:water. Then 1 μl of lipid sample from each dilution was spotted on a nitrocellulose membrane. After drying for 1 h at room temperature, membranes were incubated in blocking buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl2, 0.1% Tween 20, and 2 mg/ml BSA) with gentle rocking for 1 h at room temperature and then incubated overnight at 4 °C with gentle rocking in 10 ml of blocking buffer containing 25 μg of purified PDH complex (Sigma). PDH bound to lipids on the membrane was detected by immunoblotting using primary antibody (Assaypro) against the PDH complex (1:1000) and secondary antibody conjugated to horseradish peroxidase (1:3000).
Enzyme assays
In vivo assay of PDH activity
Pyruvate oxidation was determined by measuring the release of 14CO2 from [1-14C]pyruvate as described previously (60). Cells were seeded in glass vials at ∼100 μg/vial in DMEM (without glucose, pyruvate, or glutamine/1% FCS). The following day, cells were washed twice with Dulbecco's PBS prior to 3-h incubation in Dulbecco's PBS (Life Technologies) supplemented with 100 μm [1-14C]pyruvate (specific activity, 0.1 μCi/ml; Perkin Elmer Life Sciences). A center well containing 2 m NaOH was placed to trap CO2. After 1 h of shaking at 37 °C, medium was acidified with 2.6 m perchloric acid at a final concentration of 0.4 m to stop the reaction. After 3 h of trapping, the 14CO2 collected in the center well was counted by liquid scintillation. Pyruvate oxidation flux was determined by the amount of pyruvate oxidized to CO2 normalized to protein content.
In vitro assay of PDH activity
Mitochondrial PDH activity was measured spectrophotometrically by determining the reduction of NAD+ to NADH, coupled to the reduction of a reporter dye to yield a colored reaction product with an increase in absorbance at 450 nm at 37 °C (Biovision). PDH enzyme activity was expressed in units per milligram of total protein. To determine the effect of phospholipids on PDH activity, aliquots of mitochondria (400 μg of protein) were suspended in 40 ml of 50 mm NaCl and 50 mm imidazole HCl (pH 7.0). 2 μl of CL (from bovine heart, Sigma), phosphatidic acid (Avanti), or MLCL (Avanti) from a stock solution containing 20 mg/ml in 5% digitonin and 50% ethanol and 1 ml of digitonin (10% in water) were added to obtain a phospholipid/protein ratio of 1:10 (g/g) and a digitonin/protein ratio of 0.5 (g/g). After 2-h incubation on ice, 10 ml of digitonin (10% stock solution in water) was added to obtain a digitonin/protein ratio of 3:1 (g/g), which was sufficient for quantitative solubilization. The samples were used directly in PDH activity assays. To determine the effect of phospholipids on purified PDH (Sigma) activity, 2 ml of CL or phosphatidic acid from a stock solution containing 20 mg/ml in 5% digitonin and 50% ethanol was incubated with purified PDH complex for 10 min at room temperature.
MDH activity
MDH activity was measured as oxidation of NADH, determined by a decrease of absorbance at 340 nm. The assays were performed in buffer containing 100 mm KPi, 0.1 mm NADH, and 0.2 mm oxaloacetate. After stabilization, Triton X-100 solubilized mitochondria were added, and the absorbance was measured at 340 nm.
PC activity
PC activity was measured spectrophotometrically by monitoring the formation of CoA at 412 nm as modified from the protocol of Payne and Morris (61). The assay was performed in buffer containing 1 m Tris-HCl (pH 8), 0.5 m NaHCO3, 0.1 m MgCl2, 1 mm acetyl-CoA, 0.1 m pyruvate, 0.1 m ATP, 0.0039 g of 5,5′-dithiobis-(2-nitrobenzoic acid), and 1000 units/ml citrate synthase. After stabilization, the reaction was started by adding Triton X-100–solubilized mitochondria.
SDH activity
SDH activity was measured as the velocity of 2,6-dichlorophenolindophenol (DCPIP) reduction (62), corresponding to a decrease in absorbance at 600 nm. Assays were performed in buffer containing 1 g/liter BSA, 80 mm potassium phosphate, 2 mm EDTA, 10 mm succinate, 0.2 mm ATP, 0.3 mm potassium cyanide, 80 μm DCPIP, 1 μm antimycin-A, 50 μm decylubiquinone, and 3 μm rotenone. After preincubation with the reaction mixture, DCPIP was added, and absorbance was measured at 600 nm.
Author contributions
Y. L., W. L., C. A. R., and M. L. G. conceptualization; Y. L. data curation; Y. L. validation; Y. L., W. L., V. R., S. D., W. Y., C. A. R., M. S., R. H. H., and M. L. G. investigation; Y. L., W. L., V. R., S. D., M. W. S., R. H. H., and M. L. G. methodology; Y. L. writing-original draft; Y. L., M. W. S., and M. L. G. writing-review and editing; M. W. S. and M. L. G. formal analysis; M. L. G. resources; M. L. G. supervision; M. L. G. funding acquisition; M. L. G. project administration.
Supplementary Material
Acknowledgments
We thank Avanti Polar Lipids, Inc. for the generous gift of MLCL. The University of Michigan Metabolomics Core Services is supported by grant U24 DK097153 from the National Institutes of Health Common Funds Project to the University of Michigan.
This work was supported by National Institutes of Health Grant HL 117880 (to M. L. G.) and the Barth Syndrome Foundation (to M. L. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.
- CL
- cardiolipin
- MLCL
- monolysocardiolipin
- BTHS
- Barth syndrome
- TCA
- tricarboxylic acid
- PDH
- pyruvate dehydrogenase
- PDK
- pyruvate dehydrogenase kinase
- PDP
- pyruvate dehydrogenase phosphatase
- SDH
- succinate dehydrogenase
- TAZ
- tafazzin
- OAA
- oxaloacetate
- PC
- pyruvate carboxylase
- MDH
- malate dehydrogenase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- DCPIP
- 2,6-dichlorophenolindophenol
- MID
- mass-isotopomer distribution
- DCA
- dichloroacetate.
References
- 1. Planas-Iglesias J., Dwarakanath H., Mohammadyani D., Yanamala N., Kagan V. E., and Klein-Seetharaman J. (2015) Cardiolipin interactions with proteins. Biophys. J. 109, 1282–1294 10.1016/j.bpj.2015.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ren M., Phoon C. K., and Schlame M. (2014) Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 55, 1–16 10.1016/j.plipres.2014.04.001 [DOI] [PubMed] [Google Scholar]
- 3. Paradies G., Paradies V., De Benedictis V., Ruggiero F. M., and Petrosillo G. (2014) Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 1837, 408–417 10.1016/j.bbabio.2013.10.006 [DOI] [PubMed] [Google Scholar]
- 4. Houtkooper R. H., and Vaz F. M. (2008) Cardiolipin, the heart of mitochondrial metabolism. Cell Mol. Life Sci. 65, 2493–2506 10.1007/s00018-008-8030-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Joshi A. S., Zhou J., Gohil V. M., Chen S., and Greenberg M. L. (2009) Cellular functions of cardiolipin in yeast. Biochim. Biophys. Acta 1793, 212–218 10.1016/j.bbamcr.2008.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang F., Ryan M. T., Schlame M., Zhao M., Gu Z., Klingenberg M., Pfanner N., and Greenberg M. L. (2000) Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J. Biol. Chem. 275, 22387–22394 10.1074/jbc.M909868199 [DOI] [PubMed] [Google Scholar]
- 7. Claypool S. M., Oktay Y., Boontheung P., Loo J. A., and Koehler C. M. (2008) Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 182, 937–950 10.1083/jcb.200801152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hatch G. M. (1994) Cardiolipin biosynthesis in the isolated heart. Biochem. J. 297, 201–208 10.1042/bj2970201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beranek A., Rechberger G., Knauer H., Wolinski H., Kohlwein S. D., and Leber R. (2009) Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J. Biol. Chem. 284, 11572–11578 10.1074/jbc.M805511200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu Y., Kelley R. I., Blanck T. J., and Schlame M. (2003) Remodeling of cardiolipin by phospholipid transacylation. J. Biol. Chem. 278, 51380–51385 10.1074/jbc.M307382200 [DOI] [PubMed] [Google Scholar]
- 11. Monteiro-Cardoso V. F., Oliveira M. M., Melo T., Domingues M. R., Moreira P. I., Ferreiro E., Peixoto F., and Videira R. A. (2015) Cardiolipin profile changes are associated to the early synaptic mitochondrial dysfunction in Alzheimer's disease. J. Alzheimers Dis. 43, 1375–1392 [DOI] [PubMed] [Google Scholar]
- 12. He Q., and Han X. (2014) Cardiolipin remodeling in diabetic heart. Chem. Phys. Lipids 179, 75–81 10.1016/j.chemphyslip.2013.10.007 [DOI] [PubMed] [Google Scholar]
- 13. Tyurina Y. Y., Winnica D. E., Kapralova V. I., Kapralov A. A., Tyurin V. A., and Kagan V. E. (2013) LC/MS characterization of rotenone induced cardiolipin oxidation in human lymphocytes: implications for mitochondrial dysfunction associated with Parkinson's disease. Mol. Nutr. Food Res. 57, 1410–1422 10.1002/mnfr.201200801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Adès L. C., Gedeon A. K., Wilson M. J., Latham M., Partington M. W., Mulley J. C., Nelson J., Lui K., and Sillence D. O. (1993) Barth syndrome: clinical features and confirmation of gene localisation to distal Xq28. Am. J. Med. Genet. 45, 327–334 10.1002/ajmg.1320450309 [DOI] [PubMed] [Google Scholar]
- 15. Bolhuis P. A., Hensels G. W., Hulsebos T. J., Baas F., and Barth P. G. (1991) Mapping of the locus for X-linked cardioskeletal myopathy with neutropenia and abnormal mitochondria (Barth syndrome) to Xq28. Am. J. Hum. Genet. 48, 481–485 [PMC free article] [PubMed] [Google Scholar]
- 16. Clarke S. L., Bowron A., Gonzalez I. L., Groves S. J., Newbury-Ecob R., Clayton N., Martin R. P., Tsai-Goodman B., Garratt V., Ashworth M., Bowen V. M., McCurdy K. R., Damin M. K., Spencer C. T., Toth M. J., et al. (2013) Barth syndrome. Orphanet J. Rare Dis. 8, 23 10.1186/1750-1172-8-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Christodoulou J., McInnes R. R., Jay V., Wilson G., Becker L. E., Lehotay D. C., Platt B. A., Bridge P. J., Robinson B. H., and Clarke J. T. (1994) Barth syndrome: clinical observations and genetic linkage studies. Am. J. Med. Genet. 50, 255–264 10.1002/ajmg.1320500309 [DOI] [PubMed] [Google Scholar]
- 18. Barth P. G., Wanders R. J., and Vreken P. (1999) X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): MIM 302060. J. Pediatr. 135, 273–276 10.1016/S0022-3476(99)70118-6 [DOI] [PubMed] [Google Scholar]
- 19. Bione S., D'Adamo P., Maestrini E., Gedeon A. K., Bolhuis P. A., and Toniolo D. (1996) A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 12, 385–389 10.1038/ng0496-385 [DOI] [PubMed] [Google Scholar]
- 20. Hauff K. D., and Hatch G. M. (2006) Cardiolipin metabolism and Barth syndrome. Prog. Lipid Res. 45, 91–101 10.1016/j.plipres.2005.12.001 [DOI] [PubMed] [Google Scholar]
- 21. Schlame M., Towbin J. A., Heerdt P. M., Jehle R., DiMauro S., and Blanck T. J. (2002) Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann. Neurol. 51, 634–637 10.1002/ana.10176 [DOI] [PubMed] [Google Scholar]
- 22. Sandlers Y., Mercier K., Pathmasiri W., Carlson J., McRitchie S., Sumner S., and Vernon H. J. (2016) Metabolomics reveals new mechanisms for pathogenesis in Barth syndrome and introduces novel roles for cardiolipin in cellular function. PLoS ONE 11, e0151802 10.1371/journal.pone.0151802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gibson K. M., Elpeleg O. N., Jakobs C., Costeff H., and Kelley R. I. (1993) Multiple syndromes of 3-methylglutaconic aciduria. Pediatr. Neurol. 9, 120–123 10.1016/0887-8994(93)90046-F [DOI] [PubMed] [Google Scholar]
- 24. Schmidt M. R., Birkebaek N., Gonzalez I., and Sunde L. (2004) Barth syndrome without 3-methylglutaconic aciduria. Acta Paediatr. 93, 419–421 10.1111/j.1651-2227.2004.tb02974.x [DOI] [PubMed] [Google Scholar]
- 25. Marziliano N., Mannarino S., Nespoli L., Diegoli M., Pasotti M., Malattia C., Grasso M., Pilotto A., Porcu E., Raisaro A., Raineri C., Dore R., Maggio P. P., Brega A., and Arbustini E. (2007) Barth syndrome associated with compound hemizygosity and heterozygosity of the TAZ and LDB3 genes. Am. J. Med. Genet. A 143A, 907–915 10.1002/ajmg.a.31653 [DOI] [PubMed] [Google Scholar]
- 26. Kelley R. I., Cheatham J. P., Clark B. J., Nigro M. A., Powell B. R., Sherwood G. W., Sladky J. T., and Swisher W. P. (1991) X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methylglutaconic aciduria. J. Pediatr. 119, 738–747 10.1016/S0022-3476(05)80289-6 [DOI] [PubMed] [Google Scholar]
- 27. Wysocki S. J., and Hähnel R. (1976) 3-Hydroxy-3-methylglutaric aciduria: 3-hydroxy-3-methylglutaryl-coenzyme A lyase levels in leucocytes. Clin. Chim. Acta 73, 373–375 10.1016/0009-8981(76)90186-8 [DOI] [PubMed] [Google Scholar]
- 28. Mamer O. A., Tjoa S. S., Scriver C. R., and Klassen G. A. (1976) Demonstration of a new mammalian isoleucine catabolic pathway yielding an Rseries of metabolites. Biochem. J. 160, 417–426 10.1042/bj1600417a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Korman S. H., Andresen B. S., Zeharia A., Gutman A., Boneh A., and Pitt J. J. (2005) 2-Ethylhydracrylic aciduria in short/branched-chain acyl-CoA dehydrogenase deficiency: application to diagnosis and implications for the R-pathway of isoleucine oxidation. Clin. Chem. 51, 610–617 10.1373/clinchem.2004.043265 [DOI] [PubMed] [Google Scholar]
- 30. Lanza I. R., and Nair K. S. (2010) Mitochondrial function as a determinant of life span. Pflugers Arch. 459, 277–289 10.1007/s00424-009-0724-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bashir A., Bohnert K. L., Reeds D. N., Peterson L. R., Bittel A. J., de Las Fuentes L., Pacak C. A., Byrne B. J., and Cade W. T. (2017) Impaired cardiac and skeletal muscle bioenergetics in children, adolescents, and young adults with Barth syndrome. Physiol. Rep. 5, e13130 10.14814/phy2.13130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Raja V., Joshi A. S., Li G., Maddipati K. R., and Greenberg M. L. (2017) Loss of cardiolipin leads to perturbation of acetyl-CoA synthesis. J. Biol. Chem. 292, 1092–1102 10.1074/jbc.M116.753624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patil V. A., Fox J. L., Gohil V. M., Winge D. R., and Greenberg M. L. (2013) Loss of cardiolipin leads to perturbation of mitochondrial and cellular iron homeostasis. J. Biol. Chem. 288, 1696–1705 10.1074/jbc.M112.428938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reed L. J. (2001) A trail of research from lipoic acid to α-keto acid dehydrogenase complexes. J. Biol. Chem. 276, 38329–38336 10.1074/jbc.R100026200 [DOI] [PubMed] [Google Scholar]
- 35. Patel M. S., and Roche T. E. (1990) Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 4, 3224–3233 10.1096/fasebj.4.14.2227213 [DOI] [PubMed] [Google Scholar]
- 36. Gudi R., Bowker-Kinley M. M., Kedishvili N. Y., Zhao Y., and Popov K. M. (1995) Diversity of the pyruvate dehydrogenase kinase gene family in humans. J. Biol. Chem. 270, 28989–28994 10.1074/jbc.270.48.28989 [DOI] [PubMed] [Google Scholar]
- 37. Roche T. E., Baker J. C., Yan X., Hiromasa Y., Gong X., Peng T., Dong J., Turkan A., and Kasten S. A. (2001) Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog. Nucleic Acid Res. Mol. Biol. 70, 33–75 10.1016/S0079-6603(01)70013-X [DOI] [PubMed] [Google Scholar]
- 38. Harris R. A., Bowker-Kinley M. M., Huang B., and Wu P. (2002) Regulation of the activity of the pyruvate dehydrogenase complex. Adv. Enzyme Regul. 42, 249–259 10.1016/S0065-2571(01)00061-9 [DOI] [PubMed] [Google Scholar]
- 39. Korotchkina L. G., Sidhu S., and Patel M. S. (2006) Characterization of testis-specific isoenzyme of human pyruvate dehydrogenase. J. Biol. Chem. 281, 9688–9696 10.1074/jbc.M511481200 [DOI] [PubMed] [Google Scholar]
- 40. Korotchkina L. G., and Patel M. S. (1995) Mutagenesis studies of the phosphorylation sites of recombinant human pyruvate dehydrogenase: site-specific regulation. J. Biol. Chem. 270, 14297–14304 10.1074/jbc.270.24.14297 [DOI] [PubMed] [Google Scholar]
- 41. Kolobova E., Tuganova A., Boulatnikov I., and Popov K. M. (2001) Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem. J. 358, 69–77 10.1042/0264-6021:3580069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Korotchkina L. G., and Patel M. S. (2001) Probing the mechanism of inactivation of human pyruvate dehydrogenase by phosphorylation of three sites. J. Biol. Chem. 276, 5731–5738 10.1074/jbc.M007558200 [DOI] [PubMed] [Google Scholar]
- 43. Raja V., Salsaa M., Joshi A. S., Li Y., van Roermund C. W. T., Saadat N., Lazcano P., Schmidtke M., Hüttemann M., Gupta S. V., Wanders R. J. A., and Greenberg M. L. (2019) Cardiolipin-deficient cells depend on anaplerotic pathways to ameliorate defective TCA cycle function. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 1864, 654–661 10.1016/j.bbalip.2019.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schwall C. T., Greenwood V. L., and Alder N. N. (2012) The stability and activity of respiratory complex II is cardiolipin-dependent. Biochim. Biophys. Acta 1817, 1588–1596 10.1016/j.bbabio.2012.04.015 [DOI] [PubMed] [Google Scholar]
- 45. Dudek J., Cheng I. F., Chowdhury A., Wozny K., Balleininger M., Reinhold R., Grunau S., Callegari S., Toischer K., Wanders R. J., Hasenfuß G., Brügger B., Guan K., and Rehling P. (2016) Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Mol. Med. 8, 139–154 10.15252/emmm.201505644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lou W., Reynolds C. A., Li Y., Liu J., Hüttemann M., Schlame M., Stevenson D., Strathdee D., and Greenberg M. L. (2018) Loss of tafazzin results in decreased myoblast differentiation in C2C12 cells: myoblast model of Barth syndrome and cardiolipin deficiency. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1863, 857–865 10.1016/j.bbalip.2018.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jitrapakdee S., St Maurice M., Rayment I., Cleland W. W., Wallace J. C., and Attwood P. V. (2008) Structure, mechanism and regulation of pyruvate carboxylase. Biochem. J. 413, 369–387 10.1042/BJ20080709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee J. H., Jung I. R., Choi S. E., Lee S. M., Lee S. J., Han S. J., Kim H. J., Kim D. J., Lee K. W., and Kang Y. (2014) Toxicity generated through inhibition of pyruvate carboxylase and carnitine palmitoyl transferase-1 is similar to high glucose/palmitate-induced glucolipotoxicity in INS-1 β cells. Mol. Cell Endocrinol. 383, 48–59 10.1016/j.mce.2013.12.002 [DOI] [PubMed] [Google Scholar]
- 49. Dalziel K., and Londesborough J. C. (1968) The mechanisms of reductive carboxylation reactions: carbon dioxide or bicarbonate as substrate of nicotinamide-adenine dinucleotide phosphate-linked isocitrate dehydrogenase and malic enzyme. Biochem. J. 110, 223–230 10.1042/bj1100223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shan C., Kang H. B., Elf S., Xie J., Gu T. L., Aguiar M., Lonning S., Hitosugi T., Chung T. W., Arellano M., Khoury H. J., Shin D. M., Khuri F. R., Boggon T. J., and Fan J. (2014) Tyr-94 phosphorylation inhibits pyruvate dehydrogenase phosphatase 1 and promotes tumor growth. J. Biol. Chem. 289, 21413–21422 10.1074/jbc.M114.581124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen G., Wang L., Liu S., Chuang C., and Roche T. E. (1996) Activated function of the pyruvate dehydrogenase phosphatase through Ca2+-facilitated binding to the inner lipoyl domain of the dihydrolipoyl acetyltransferase. J. Biol. Chem. 271, 28064–28070 10.1074/jbc.271.45.28064 [DOI] [PubMed] [Google Scholar]
- 52. Brenza J. M., Neagle C. E., and Sokolove P. M. (1985) Interaction of Ca2+ with cardiolipin-containing liposomes and its inhibition by adriamycin. Biochem. Pharmacol. 34, 4291–4298 10.1016/0006-2952(85)90287-4 [DOI] [PubMed] [Google Scholar]
- 53. Phelps A., and Lindsay J. G. (1986) Mammalian pyruvate dehydrogenase complex binds tightly to the mitochondrial inner membrane. Biochem. Soc. Transactions 14, 893–893 10.1042/bst0140893 [DOI] [Google Scholar]
- 54. Lussey-Lepoutre C., Hollinshead K. E., Ludwig C., Menara M., Morin A., Castro-Vega L. J., Parker S. J., Janin M., Martinelli C., Ottolenghi C., Metallo C., Gimenez-Roqueplo A. P., Favier J., and Tennant D. A. (2015) Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat. Commun. 6, 8784 10.1038/ncomms9784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Neubauer S. (2007) The failing heart: an engine out of fuel. N. Engl. J. Med. 356, 1140–1151 10.1056/NEJMra063052 [DOI] [PubMed] [Google Scholar]
- 56. Ingwall J. S. (2009) Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 81, 412–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Johnston J., Kelley R. I., Feigenbaum A., Cox G. F., Iyer G. S., Funanage V. L., and Proujansky R. (1997) Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am. J. Hum. Genet. 61, 1053–1058 10.1086/301604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fernandez C. A., Des Rosiers C., Previs S. F., David F., and Brunengraber H. (1996) Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J. Mass. Spectrom. 31, 255–262 10.1002/(SICI)1096-9888(199603)31:3<255::AID-JMS290>3.0.CO;2-3 [DOI] [PubMed] [Google Scholar]
- 59. Dowler S., Kular G., and Alessi D. R. (2002) Protein lipid overlay assay. Sci. STKE 2002, pl6 [DOI] [PubMed] [Google Scholar]
- 60. Held N. M., Kuipers E. N., van Weeghel M., van Klinken J. B., Denis S. W., Lombès M., Wanders R. J., Vaz F. M., Rensen P. C. N., Verhoeven A. J., Boon M. R., and Houtkooper R. H. (2018) Pyruvate dehydrogenase complex plays a central role in brown adipocyte energy expenditure and fuel utilization during short-term β-adrenergic activation. Sci. Rep. 8, 9562 10.1038/s41598-018-27875-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Payne J., and Morris J. G. (1969) Pyruvate carboxylase in Rhodopseudomonas spheroides. J. Gen. Microbiol. 59, 97–101 10.1099/00221287-59-1-97 [DOI] [PubMed] [Google Scholar]
- 62. Rustin P., Chretien D., Bourgeron T., Gérard B., Rötig A., Saudubray J. M., and Munnich A. (1994) Biochemical and molecular investigations in respiratory chain deficiencies. Clin. Chim. Acta 228, 35–51 10.1016/0009-8981(94)90055-8 [DOI] [PubMed] [Google Scholar]
- 63. Stemmer M., Thumberger T., Del Sol Keyer M., Wittbrodt J., and Mateo J.L. (2015) CCTop: an intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS ONE 10, e0124633 10.1371/journal.pone.0124633 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.