

Abstract
Human telomerase maintains genome stability by adding telomeric repeats to the ends of linear chromosomes. Although previous studies have revealed profound insights into telomerase functions, the low cellular abundance of functional telomerase and the difficulties in quantifying its activity leave its thermodynamic and kinetic properties only partially characterized. Employing a stable cell line overexpressing both the human telomerase RNA component and the N-terminally biotinylated human telomerase reverse transcriptase and using a newly developed method to count individual extension products, we demonstrate here that human telomerase holoenzymes contain fast- and slow-acting catalytic sites. Surprisingly, both active sites became inactive after two consecutive rounds of catalysis, named single-run catalysis. The fast active sites turned off ∼40-fold quicker than the slow ones and exhibited higher affinities to DNA substrates. In a dimeric enzyme, the two active sites work in tandem, with the faster site functioning before the slower one, and in the monomeric enzyme, the active sites also perform single-run catalysis. Interestingly, inactive enzymes could be reactivated by intracellular telomerase-activating factors (iTAFs) from multiple cell types. We conclude that the single-run catalysis and the iTAF-triggered reactivation serve as an unprecedented control circuit for dynamic regulation of telomerase. They endow native telomerase holoenzymes with the ability to match their total number of active sites to the number of telomeres they extend. We propose that the exquisite kinetic control of telomerase activity may play important roles in both cell division and cell aging.
Keywords: enzyme kinetics, thermodynamics, cellular regulation, cancer biology, cellular senescence, catalysis-dependent control, ddTRAP, hTERT, single-run catalysis, telomerase reactivation
Introduction
Telomeres refer to terminal sequences of linear chromosomes in eukaryotic cells (1). Their catalytic extension provides an evolutionarily conserved solution to the “end replication” problem. To maintain proper telomeric length, eukaryotic cells utilize telomerase to catalyze addition of telomeric repeats following an intrinsic RNA template. In human cells, telomerase adds hexameric repeats, (TTAGGG)n, to telomeres (2). During a cell cycle, a telomerase enzyme is recruited to a transiently-uncapped telomere before it can function in a controlled fashion (3). It is believed that the telomerase preferentially acts on shorter telomeres (4–8). Every telomere should be acted on to maintain proper telomere length equilibrium; otherwise, some telomeres would become shorter over time, leading to cellular senescence (9). How telomerase-expressing cells regulate telomere length in a global scale remains unclear.
Telomerase plays a critical role in human diseases, in particular cancers and other age-related diseases. With down-regulated telomerase activity, differentiating cells reach a critical state after a certain number of cell divisions so that cell cycle will be arrested and cells will enter “senescence.” How a cell senses changes in telomere length before senescence remains a mystery. Approximately 85–90% of human cancers exhibit elevated telomerase activity (1, 10–18). The importance of telomerase activity to cancer cells and other proliferative stem-like cells has been well demonstrated. Chemical inhibitors and activators of human telomerase are being explored for cancer treatment and anti-aging therapies, respectively (19–21). These two aspects make the structural and functional studies of human telomerase and telomere maintenance a key area in cell biology and cancer biology (22–30).
In human cells, the telomerase holoenzymes are heterogeneous and contain both monomers and dimers (31). A dimeric holoenzyme contains two copies of hTERT5 (telomerase reverse transcriptase), dyskerin and hTR (telomerase RNA component), as well as other factors. It has an apparent mass of ∼700 kDa (12, 32, 33). A subclass of telomerase holoenzyme was found to be monomeric and contained one copy of hTERT, hTR, and TCAB1, and two copies of NHP2, dyskerin, NOP10, and GAR1 as recently resolved by cryoEM (34), which is of similar mass (∼600 kDa) as a dimer and difficult to separate from dimers by biochemical procedures. Each monomer has one active site. By reconstitution in vitro, every hTERT/hTR pair forms a minimally active enzyme that has one active site (35). A proliferating human cell may have 50–100 copies of functional telomerase (9, 36). It remains unclear how free telomerase complexes are physically recruited to recognize the telomeres when chromosomal 3′-overhangs become uncapped in S-phase; it is not known how a telomerase adds telomeric repeats in a processive fashion or finishes its reaction when it stochastically falls off its product (9, 37–41). Energetically, telomerase uses the chemical energy from dNTP hydrolysis to catalyze its RNA-guided DNA synthesis. For every six-nucleotide repeat, perhaps following a Boltzmann distribution among states with different numbers of nucleotides added, the DNA–RNA hybrids are presumably most stable when all six positions are paired, explaining the main peaks for every repeat addition observed by gel-based activity assays. For processive extension, an energetically costly step is expected to melt the DNA–RNA hybrid to translocate the substrate by six nucleotides or fall off from the product sporadically to terminate the reaction. The kinetics and the mechanistic programs for the processive addition of telomeric repeats have yet to be elucidated.
Prior studies of human telomerase holoenzyme have revealed important insights (12, 42, 43), but the dynamic control of its catalytic activity, the structural relationship between hTR and hTERT, and the interactions between a telomerase holoenzyme and its substrates remain incompletely understood. Semi-quantitative analyses by gel-based direct or TRAP assays have derived incomplete kinetic and thermodynamic properties of telomerase, partially because low abundance of telomerase has significantly limited quantitative analysis. In this study, we will use quantitative analysis to reveal an unexpected kinetic property of human telomerase.
Results
In single-turnover settings, human telomerase becomes inactive after two sequential runs of catalysis
To analyze telomerase activity quantitatively, we took advantage of telomerase's ability to stably bind triple telomeric repeats (TTAGGG)3 (R3) at the GGG position but to quickly fall off when it adds three nucleotides (3-nt) to the substrate and reaches the TTA position in vitro (Fig. 1A) (32). Each addition of TTA is thus half a telomeric repeat and constitutes a single-turnover condition for analyzing the catalytic activity. A single-step pulldown using R3 as ligands enriched active enzymes from cell lysates (Fig. S1A), which contained both monomers and dimers (31). When studying the macroscopic and thermodynamic behavior of telomerase, we were not able to computationally separate the monomers from the dimers as was done during cryoEM analysis or as in single molecule enzymology (34, 44). Instead, we used two sequential pulldowns to separate the dimers from the monomers because a monomer contains only one active site and would not be able to bind to fresh DNA substrates due to enzyme shutoff observed in Fig. 1B. A conventional TRAP assay, using lysates from 50, 500 and 5000 cells as a control, showed that a single-step pulldown sequestered ∼25% of the total activity (Fig. S1A), which is not surprising because only enzyme molecules with their active sites accessible to substrates were pulled down. In a paper by Cohen et al. (32, 45), antibody-based pulldown was used to sequester telomerase, which apparently allowed enzymes more time to empty their active sites and enriched higher activity than our R3-based pulldown. Furthermore, during our R3 pulldown, the enzyme went through one reaction cycle and a significant fraction shut off (see Fig. 1B).
Figure 1.

Catalysis-dependent loss of telomerase activity. A, schematic representation of two repetitive cycles of affinity-based pulldown and elution from R3-telomeric substrates to purify endogenous telomerase from H1299 cells. Fresh beads and oligonucleotides were used for the second round. dATP and dTTP were inactivated by apyrase hydrolase enzyme in order for eluted telomerase to bind again to fresh beads. Apyrase was not used in other assays. B, gel-based TRAP assay of samples from the double-affinity pulldown. Samples were loaded according to cell equivalence. Cell equivalence is labeled separately (50, 500, 5000, etc.). The bottom bands (IC) represent the internal PCR control, iTAS. H1299 control samples on the far left represent total activity from cell lysate. C, schematic representation of the extension assay for tethered telomerase through the biotinylated hTERT. Extension products were separated from the beads (and the enzymes) and quantified using the ddTRAP assay after every reaction cycle. D, ddTRAP assays for the tethered telomerase. Aliquots of enzymes with a cell equivalency of 250 were used for individual reactions. Ext 1 is the first reaction. The enzymes were washed twice before the second reaction (Ext 2). Two more washes before the third reaction (Ext 3). Ext 1* denotes the control sample with the delayed extension reaction (after being kept for 4 h at room temperature before substrates were presented to it). Error bars, S.D. (n = 3).
Using the total telomerase from 50 to 5000 cells as controls, we estimated that the gel-based TRAP assay is insensitive to changes of telomerase activity by less than 2–5-fold (data in Fig. 1B and Fig. S1A), consistent with past findings (46). The direct assays based on radioactivity suffered from similar insensitivity and nonlinearity in quantifying enzymes from 5,000 or fewer cells. For example, please refer to Ref. 45. To increase the amount of telomerase for experiments, we generated a stable cell line overexpressing both hTR and N-terminally biotinylated hTERT (Fig. S2A). When compared with parental H1299 cells and stable H1299 cells overexpressing hTERT alone by the gel-based TRAP assay, the engineered cells had ∼20-fold more activity (Fig. S3A) and will be called the super-H1299. The biotinylation was accomplished by mammalian intracellular biotinylation machinery (47). When incubated with streptavidin-coated beads, biotinylated telomerase bound to the beads, but not the endogenous enzyme (Fig. S2, C versus D), suggesting that the recombinant hTERT, ∼10 kDa heavier than the endogenous one in SDS-PAGE, was indeed biotinylated. Biotinylation provided an effective way to selectively enrich recombinant telomerase, allowing separation of the recombinant hTERT from the endogenous hTERT as well as possible contaminating proteins (Fig. S3, B and C).
We used a two-step pulldown procedure to enrich selectively endogenous telomerase holoenzymes (Fig. 1A). Between the two steps, apyrase was introduced to remove dATP/dTTP from the eluate (elution 1) of the first pulldown (Fig. S4). This was the only step in which we used apyrase, which showed no effect on the stable telomerase-binding to fresh R3 beads at the GGG positions. With even distribution of all activity in a total sample volume in each step, we used a proportional aliquot of the samples in each step to represent the equivalent number of cells in the starting material, which is called cell equivalency. An aliquot of the sample loaded to the substrate (R3) beads, equivalent to the enzymes from 5000 parental H1299 cells (or called 5000 cell equivalency), was analyzed as the first input in Fig. 1B. Cell-equivalent samples from other fractions were analyzed in parallel. A small amount of activity (<10%) showed in the flow-through (FT in Fig. 1B). After two washing steps, the eluted fraction (elution 1) showed strong activity (∼25% of the starting input activity; Fig. 1B). Based on the test results in Fig. S4, 0.1 unit of apyrase was introduced to hydrolyze dTTP and dATP in elution 1. After apyrase treatment, elution 1 became input 2*, which was mixed with fresh R3 beads, washed, and eluted (Fig. 1B). Apyrase had no effect on the TRAP assay (elution 1 versus input 2* in Fig. 1B) and was removed during the wash step. Surprisingly, although based on the recovery ratio of elution 1 versus the first input in Fig. 1B, we expected to recover 25% or more of the telomerase activity in input 2* in the second eluate, literally no activity was detected in all subsequent fractions (FT2, wash 1 and 2, and elution 2). Because of negative data and to highlight this point, 100 times more materials, 500,000 cell equivalents of FT2, wash 1 and 2, and elution 2 from the second pulldown were analyzed in a gel-based TRAP assay (Fig. 1B), clearly showing that after binding to fresh R3 substrates and performing two rounds of 3-nucleotide extension, telomerase became inactive (Fig. 1, A and B).
The surprising results in Fig. 1B suggested the possibility that the telomerase holoenzyme, once separated from the other components of the cell lysates, shuts itself off after two rounds of short 3-nucleotide single-turnover extension reactions. If this holds true for telomerase holoenzymes from cell lysates, telomerase should shut off after two rounds of processive extension reactions that extend more than 3 nt. Although it is unclear to what extent the fall-off of telomerase from the TTA position contributes to the release of telomerase from processively extended substrates inside cells, the single-turnover mechanism uncovered by two runs of a 3-nt extension reaction does come from active enzymes and thus probably reflects a fundamental property. We next tested it under different reaction conditions when telomerase underwent processive extension reactions.
To test our prediction, we examined the telomerase shutoff (inactivation) without apyrase treatment or other potential limitations of the short single-turnover 3-nt extension in Fig. 1B. We tethered biotinylated telomerase from super-H1299 cells to streptavidin-coated beads and used a more quantitative droplet digital TRAP assay (ddTRAP) to count individual extension products (48). ddTRAP is accurate to one extension product and thus is much more sensitive than both the direct primer-extension methods and the conventional gel-based TRAP assay. It is linear in a broad dynamic range of the extension products being detected in our experiments. On the beads, streptavidin molecules were at least 50 nm apart such that the telomerase holoenzymes tethered on the bead surface could not physically interact with each other. The ddTRAP assay counted individual extension products in an all-or-none fashion, only registering the frequency of successful catalytic interactions between the enzymes and the DNA substrates (TS primers) without measuring the length of the products. Within experimental concentration ranges, the results of ddTRAP are linearly proportional to those of the conventional gel-based TRAP assay that does count the length of products from processive reactions (49). The ddTRAP is more precise in quantification and fast for high-throughput analysis, and thus suitable for our analysis.
When analyzed in a time-lapsed fashion, the tethered recombinant telomerase holoenzyme was stable for at least 4–5 h at room temperature (Figs. 1C and 2, A and B), suitable for the kinetic analysis. After each run of processive extension by the tethered enzymes, longer than the half-turn addition of three nucleotides in Fig. 1A, the products were separated from the enzymes and quantified by ddTRAP (Fig. 1D). No apyrase was needed in this process. The enzymes were washed thoroughly before the second run of reaction (Ext 2 in Fig. 1D), and the products were 60–70% of those from the first run (Ext 1). During the third run (Ext 3), the yield dropped to <10% of those from the first run with the same amount of tethered enzyme. As a control, the same aliquot of enzyme preparation, which went through the same mechanical manipulations for three rounds of buffer changes containing all components except the TS primers (DNA substrates) in the first two rounds, was able to generate nearly the same amount of products (Ext 1* versus Ext 1 in Fig. 1D) as the first round (Ext 1) after the TS primers were added to the third round of reaction mixture. This indicated that the waiting time and the manipulations through tethering and washing caused no substantial loss of activity in our assays. The loss of activity is thus use (catalysis)-dependent.
Figure 2.

Activity loss of telomerase was not due to enzyme instability, protein degradation, product inhibition, or loss of hTR from complex. A, partially purified telomerase (at different stages indicated by colored bars) retained most activity over 3 days at 4 °C as quantified by ddTRAP assay. B, partially purified telomerase enzyme over 5 h at room temperature (RT), as quantified by ddTRAP activity assay. C, Western blotting of hTERT showing telomerase stability throughout the full cycle of the extension assay of the tethered recombinant telomerase (∼5 h at room temperature) from super-H1299 cells. D, no product inhibition of telomerase activity. After one extension reaction for 120 min, the enzyme was separated from the reaction mixture containing the extension products. Fresh telomerase (equal amount) was added to the same reaction mixture. After another 120 min of extension, the products (from two tandem reactions) were quantified with ddTRAP activity assay (right), roughly twice of the control (left) from the first reaction. E, schematic representation of modified tethered telomerase extension assay. In this assay, ddPCR was used to quantify the hTR content in the products to determine whether hTR dissociated from the telomerase complex. F, ddPCR quantification of hTR during multiple rounds of extension reactions (Ext 1–3). More than 95% of hTR molecules remained bound to the tethered enzymes. Enzyme aliquots with cell equivalency of 250 were used for experiments showed in A, B, and D.
The loss of telomerase activity after two single-turnover runs of catalysis (Fig. 1B) might have resulted from multiple possibilities. To name a few, telomerase might be degraded or lost during the apyrase treatment; its RNA component (hTR) might dissociate from the complex; one or more of its intrinsic accessory factors might fall off after catalysis; or the products from its extension reaction might cause inhibition even in the presence of a saturating concentration of substrates through either active-site occupation (competitive inhibition) or allosteric effects (noncompetitive inhibition). We tested all of these possibilities.
First, partially purified telomerase using streptavidin beads or other methods was stable in maintaining enzyme activity for a few days at 4 °C (Fig. 2A and Fig. S5, A and C) or for 5–6 h at room temperature (Fig. 2B) with a limited loss of activity by 5–15%. The endogenous telomerase directly from cell lysates, which also shut off after two runs of extensions, lost a bit more activity (35%) over a period of 5 h (Fig. S1B), but such a loss was much lower than the observed >90% use-dependent loss. To maintain good stability of enzyme activity in our analysis, we thus preferentially used partially purified telomerase.
Second, Western blotting of recombinant hTERT found that the tethered enzymes after two rounds of extension reactions (Fig. 1D) suffered no obvious hTERT degradation (Fig. 2C). The same was found true for endogenous hTERT selectively enriched onto antibody-bound protein A/G beads.
Third, RT-qPCR analysis of the hTR content found that the “elution 2” fraction in Fig. 1B had ∼25% of the hTR in the elution 1 (Fig. S6), but zero activity in even 100× more cell-equivalent materials (Fig. 1B). The elution 1 fraction contained a major portion of inactive dimers and monomers and a small portion of active dimers (in E1 state; see Fig. 4), which accounted for the 25% hTR in elution 2 (mostly inactive dimers in E2 state; see Fig. 4) compared with that of elution 1 as measured by RT-qPCR.
Figure 4.

Two distinct active sites in human telomerase. A, time course for endogenous telomerase before one-step pulldown purification as in Fig. 1B. Telomerase cartoons indicate three populations within the sample. Yellow star indicates a “once used” enzyme. The enzyme without a star represents either a “pristine” E0 dimer or an M1 monomer. The red dashed lines represent the amounts of products if the active sites remained continuously active. B, time course of endogenous telomerase after one-step pulldown purification (elution 1). The telomerase cartoons indicated a “once-used” E1 dimer and slow M1 monomer, respectively. The red dashed line represents the amounts of products if the active sites remained continuously active. C, Michaelis-Menten plot for fast-acting components of the endogenous telomerase in A. Fitting of the data found Km ∼10 nm. D, Michaelis-Menten plot for endogenous telomerase used in B. Fitting of the data found Km ∼28 nm. E, time course for ddTRAP assay of partially purified endogenous telomerase activity in fractions from glycerol gradient. Error bars, S.D. (n = 3). The red dashed lines represent the amounts of products if the active sites remained continuously active. F, time course for ddTRAP assay of partially purified recombinant telomerase in glycerol gradient fractions. Error bars, S.D. (n = 3). The dashed line represents the amounts of products if the active sites remained continuously active. Aliquots of enzymes with cell equivalency of 50 were used in A, B, E, and F for better performance in ddPCR. G, parallel Markov models for monomeric telomerase by switching fast- (fM1) and slow-acting (sM1) monomers into inactive monomers (M2) and three different kinetic models for a telomerase dimer: Sequential with delay, Sequential and Parallel. H, “fast” and “slow” kinetic constants (k1 and k2) from data in E and F after two-exponential fittings.
Fourth, using the recombinant telomerase containing N-terminally biotinylated hTERT (Fig. S3A and Fig. 1C), we started one reaction with a batch of fresh enzyme, and after 2 h, we separated the enzymes from the products. When fresh telomerase was added into the reaction mixture that contained the products from the first round of catalysis, it produced similar amounts of products (the total product amount doubled as in Fig. 2D). The presence of products does not inhibit the activity of fresh telomerase holoenzymes.
Fifth, when ddPCR was used to detect reverse-transcripts of hTR extracted from the tethered biotinylated enzymes (Fig. 2F), almost all hTR was retained with the tethered enzymes, suggesting that the telomerase holoenzyme did not fall apart after multiple rounds of extension reactions.
Last, when the holoenzymes after multiple manipulations were blotted for dyskerin, NHP2, and NOP10, all three major accessory factors of the active enzymes were still retained (Fig. S5D), suggesting that the key protein factors were still present because the enzyme did not fall apart, although we could not rule out the possibility that during the catalytic reaction an unknown factor might fall off from the holoenzyme and lead to inactivation. These data revealed the integrity of a telomerase RNP complex by retaining the core components of hTERT, hTERC, and key protein factors (Fig. 2F and Fig. S5D). Together, these experiments demonstrated that the use-dependent loss of telomerase activity was not caused by chromatographic manipulations, apyrase treatment, instability, or degradation of the holoenzyme, hTR dissociation, release of key accessory factors, or product inhibition.
Use-dependent loss of activity is not uncommon for enzymes. Conventional “single-turnover” enzymes are good examples. With saturating substrates, a normal enzyme with the demonstrated stability of telomerase (Fig. 2A) should catalyze a reaction continuously with a linear increase of products over a long period of time (indicated by red dashed lines in Fig. 4, A, B, E, and F). A single-turnover enzyme, however, is self-limiting by turning itself off after one round of catalysis. In a similar fashion, human telomerase shuts off after two rounds of single-turnover processive extension, if it follows the sample principles of the single-turnover reaction as in Fig. 1B. At the molecular level, there are three potential explanations for the single-run catalysis. One is that each enzyme, e.g. monomeric telomerase, has only one active site, which has the same affinity to the substrates and is able to count the rounds of catalysis and shut off after exactly two rounds. As a thermodynamic system, such counting in an exact fashion is improbable. The second possibility is that there are two types of monomeric enzymes, such as monomeric telomerase holoenzymes made of the core factors and different auxiliary factors and named fM1 and sM1, whose active sites differ. The fM1 functions mainly in the first round of fast catalysis, and the sM1, a good fraction of which does not bind to the TS primers in the first round, works in the second round. Third, there are two types of active sites in a dimeric enzyme, both of which shut off after two separate rounds of reaction. The dimeric enzymes may co-exist with fM1 or both fM1 and sM1, especially in consideration of the mixture of dimers and monomers seen by cryoEM (34). With the currently available techniques, it remains difficult to separate fM1, sM1, or the different states (E0 and E1) of dimers for our studies.
Human telomerase holoenzyme from super-H1299 cells is heterogeneous in composition
In the literature, the telomerase holoenzymes in human cells are found heterogeneous, containing both dimers and monomers (32, 50–52). The minimally-active hTERT/hTR complex is monomeric with only one active site and a simple kinetic behavior (35); however, without other protein components of the holoenzyme (Fig. S5D), it may not have real physiology. Enzymes attached to slides or exposed to or near air–water interfaces were found to be a mixture of both dimers and monomers. It is unclear whether such operations might have changed the ratio of dimers to monomers (31). The acutely assembled monomeric enzymes classified and analyzed by cryoEM showed a complex of ∼590 kDa (34). To evaluate our recombinant enzymes, we used a continuous glycerol gradient to separate active enzymes from cell lysates. Thyroglobulin (∼669 kDa), a molecular weight marker, was used as a benchmark in our gradient (Fig. 3A, fractions F7/F8). Western blotting found that the fractionated enzymes from the super-H1299 cells were distributed near the bottom of the density gradient (F8–F11), suggesting that the holoenzymes are heterogeneous in size. A good fraction of the enzymes was apparently heavier than a 669-kDa globular protein (Fig. 3, B and C). Here, we did not take consideration of variations in partial specific volumes, frictional resistance, and solution viscosity and density for individual proteins because they are difficult to measure for each complex individually. Instead, we used a molecular weight marker. Similarly, size-exclusion chromatography of the glycerol-gradient fractions in a Superose 6 column found that a major fraction of the telomerase had a retention volume of 11–19 ml, equivalent to globular proteins in a broad range of ∼0.3–0.9 MDa (Fig. 3D). The gel-based TRAP assay showed that the active human telomerase holoenzymes may be larger or smaller than the thyroglobulin (669 kDa) in size. Because 700-kDa dimers and 590-kDa monomers were not separated well biochemically, the gel-filtration profile of active enzymes isolated from the super-H1299 cells indicated heterogeneous composition containing both monomers and dimers as reported before (31, 34, 53).
Figure 3.

Recombinant telomerase complexes from super-H1299 cells have a major fraction of dimers. Molecular weight marker thyroglobulin (∼669 kDa) was run on a 10–30% glycerol gradient in parallel with recombinant telomerase. A, Coomassie-stained SDS-PAGE assay of thyroglobulin in all 11 gradient fractions (top F1 to bottom F11). Red box marks the fractions where telomerase activity was detected. B, gel-based TRAP assay on gradient fractions. Lysates of super-H1299 fractionated in the glycerol gradient were assayed. Majority of the activity was found in fractions 8–11. C, Western blotting (anti-hTERT) of gradient fractions shows that the recombinant hTERT (red arrow) is primarily in F8–F11. The holoenzyme is thus slightly heavier than 669 kDa. D, recombinant telomerase run through a Superose 6 column. ddTRAP assay of the eluted fractions is plotted against fraction number (1 ml each). Red box marks the major activity containing fractions (S12–S16). Cell equivalency for each fraction was 50 due to the need for better droplet separation in ddPCR. Thyroglobulin run on the same column eluted with its peak in fractions S14–S15, suggesting that the recombinant telomerase holoenzyme complex be heavier than 669 kDa.
Time-lapse experiments reveal two distinct active sites for human telomerase
Partially fractionated telomerase holoenzymes were sufficiently stable for measurements in a period of 3–6 h (Fig. 2A). The same was found to be true for telomerase enriched in glycerol-gradient fractions of cell lysates (Fig. 3B), the fractions eluted from single-step pulldown (Fig. 1B), and the fractions of the biotinylated enzymes eluted from the monomeric avidin beads (Fig. 3D). High stability of the active telomerase in all these preparations made it feasible to conduct the time-lapse experiments under different conditions.
We asked the question whether two active sites in a dimeric telomerase holoenzyme or in two separate monomeric enzymes were active at the same time and, if both were active, whether they would both undergo catalysis-dependent loss of activity. We first characterized the basic thermodynamic and kinetic properties of the active sites. Because two different monomers may show kinetics similar to dimers with two different active sites, our analysis will use dimeric enzymes as examples, and the principles then apply to the two different monomers. A dimeric enzyme with two types of active sites might be in three distinct conformational states: the pristine enzyme (E0) with both active sites functional, the once-used enzyme (E1), with one site functional and the other nonfunctional, and the exhausted enzyme (E2) with no activity. A monomeric enzyme has one active site in either the active (M1) or the exhausted state (M2). Quantitative kinetic analysis would be needed to characterize enzymes distributed among these states (49).
With saturating concentrations of substrates (200 nm), catalytic activity of telomerase from H1299 cell lysates or partially purified in a continuous glycerol gradient was compared with the enzymes eluted from (TTAGGG)3-conjugated beads (as the elution 1 in Fig. 1B). The same amount (cell equivalency) of enzymes was used for extension reaction, which extended the DNA substrates (TS primers), but was stopped at different time points by heat inactivation to halt the reactions and dissociate the products (Fig. 4, A and B). The released products were quantified by ddTRAP and normalized against the measured products at the time point of 90 min. Without exception, the enzymes from different batches of biochemical preparations all exhibited a fast and a slow kinetic component (Fig. 4, A–D; Fig. S7). The fast one was saturated after ∼5–10 min. The slow one slowly increased without saturation at 90 min, but with a longer incubation, it reached saturation after ∼300 min (Fig. 4, A, B, E, and F, and Fig. S7). The kinetic difference between the two components is striking (Fig. 4, A, and E). Both components deviate significantly from what is expected from enzymes that are continuously active (red dashed lines based on the initial reaction rates in Fig. 4 and Fig. S7). In accord with the use-dependent loss of activity, the two saturating kinetic components in product accumulation suggest that there are two different types of active sites that both become inactive after catalysis. As depicted above, the two types of active sites may come from two distinct monomeric holoenzymes or be contained within one dimeric complex. Because of experimental limits in kinetic analysis, we parsimoniously assigned that the fast monomers have a similar kinetic constant as the fast dimeric site (in E0) and the slow monomers as the slower dimeric site in E1.
Compared with the two kinetic components of telomerase from cell lysates, the telomerase from the one-step single-turnover affinity purification using the R3 beads exhibited only the slow component (Fig. 4B). Because the single-turnover affinity purification would remove all monomeric enzymes (fast or slow) due to their turn-off after one round of a 3-nt extension (Figs. 1B and 4B), the leftover active enzymes are dominated by dimers (Fig. 4B), which exhibit the same kinetic constant as the slow-component in the mixed enzymes from cell lysates (Fig. 4, B versus F). Comparing this result with those in Figs. 1B and 4, A and E, we deduced that active telomerase enzymes (Fig. 4A) with the fast kinetic component, either dimers in the E0 state or fast monomers (fM1), and those with the slow kinetic component in sM1 monomers or starting E1 state were no longer active following the first single turnover addition of three nucleotides, such that the telomerase activity in the elution 1 fraction (Fig. 4B) represents the activity of telomerase enzymes with slow-acting active sites (E1 coming from the E0 → E1 conversion). The two kinetic components were different in time domain such that two exponential components were needed to fit the data (red solid lines in Fig. 4, E and F). As compared in Fig. S7, one exponential component is clearly insufficient to fit the data.
We next evaluated the initial rates of product accumulation by varying the DNA-substrate concentration while keeping the dNTPs saturated, under which the binding of the DNA substrate became rate-limiting. The average initial rate, which was represented by the slope within the first 5 min and extrapolated to near t = 0 min (the boundary condition), was obtained for the fast component as illustrated by a red dashed line in Fig. 4A. The average initial rate in the first 10 min was obtained for the slow-acting component as shown in Fig. 4B. We normalized the initial reaction rates against the maximal rates at 200 nm substrates and plotted them against substrate concentration (Fig. 4, C and D, respectively). Without an accurate way to determine the absolute quantity of active telomerase, we instead used the same aliquots of enzymes to set up the extension reaction. The reactions in triplicates were stopped at different time points by heat inactivation. Usually each aliquot of enzymes in the extension reaction was equivalent to telomerase from ∼500 to 5000 cells, making the number of reaction products in the right range for ddTRAP assays. Because we measured the initial rates with a trace amount of enzymes, which has cell equivalency of ∼10–1000, depending on the amount of telomerase activity recovered by specific purification procedure, and a small fraction of product accumulation, a Michaels-Menten equation (V0/Vmax = 1/(1 + [S]/Km)) could be used to describe the normalized initial rate as a function of the DNA substrate, yielding Km = 10 and 28 nm for the fast-acting and slow-acting components, respectively. These are fairly close to those measured previously using different dansyl substrates (54–56). The difference in Km values argued against the hypothesis that the same active site counts two rounds of reaction before shutting itself off. It further confirmed that the fast- and slow-acting components co-exist in the heterogeneous holoenzymes. Because of the slow off-rate (koff <1/72,000 s−1; at the GGG position) (54), Km ∼ koff/kon could provide an upper-limit for kon of ∼1.4 × 103 m−1 s−1, suggesting that the reaction of the holoenzymes would be slow if the telomere concentration is <100 nm in cells and no other factors are present to significantly accelerate its on-rate.
Two active sites in a dimer bind substrates in a sequential manner and act in tandem
Because our one-step R3 binding can separate the dimeric enzymes in the E1 state, we next ask whether the two active sites in a dimer function in parallel or in sequence. The kinetic differences between the slow and fast components could be explained by different Markov models: a two-step sequential model in a dimer, a parallel model in a dimer, or two different monomers (fM1 and sM1), and a sequential model with a delay in a dimer (Fig. 4G). Here, the parallel model accounted for either two different types of monomeric enzymes or one type of dimeric enzyme harboring two distinct active sites. Using MATLAB programs to derive numerical solutions to each model (supporting information; Fig. 4, E and F, red lines), we estimated two kinetic constants from these models, ∼0.4 min−1 (60%) and 0.01 min−1 (40%) for the fast and the slow components, respectively (Fig. 4H). All Markov models fitted the time-lapse data reasonably well. The sequential model with a delay worked slightly better than the others.
To experimentally test the sequential or parallel actions of the two active sites in the dimeric enzymes, we examined whether the two sites interfere with each other during the DNA substrate-binding step, i.e. whether a DNA substrate's binding to the first site affects the binding of a second DNA substrate to the other active site. The parallel model of one dimer or two different monomers would support simultaneous binding of two substrates to two active sites (Fig. 4G). The different monomers must act in parallel. In contrast, a sequential model of a dimer would indicate that the enzyme extends one DNA substrate at a time and only after the first site finishes its catalysis can the second site perform its reaction.
We asked whether the dimers may act as in a sequential model. We used the tethered recombinant telomerase holoenzymes, which were physically separated from each other by at least 50 nm on the surfaces of streptavidin-coated beads so that, on average, two neighboring enzyme complexes were physically independent of each other (Fig. 5A). Biotinylated telomerase on the streptavidin-coated beads was first incubated with the TS primers (DNA substrates), which would be stabilized at the GGG-position and stay bound when there was no catalysis (32). The saturating concentration of TS primers would fill in all accessible active sites (sM1, fM1, E1, and E0). After the first round of extension, the products were separated from the tethered enzymes within ∼20 min, which was much shorter than 20 h (inverse of the off-rate), such that only a very small fraction (∼(1 −exp(−20/1200)) = 1.6%) of the bound substrates in the slow-acting sites (sM1, or in either E1 or E0) that were not catalyzing would dissociate. During this round, monomeric fM1 would all become inactive; all E0 dimers would become E1, and a good fraction (exp(−20/109) = 82%) of slow-acting monomeric sM1 or dimeric E1 would remain bound with the products (occupied). If both active sites of E0 were bound with the substrates, then at the end of the first round, most of the leftover active sites (∼exp(−20/1200) = 98.3%) would remain occupied by the DNA substrates during the experimental interval when reaction products were removed. If the assumption of simultaneous binding holds, the products from the second round of reaction in the absence of fresh DNA primers (−TS) would be expected to be more than exp(−90/1200) = 93% of those from the parallel reaction done in the presence of fresh primers.
Figure 5.

Sequential binding of two active sites to substrates suggests a sequential kinetic model for dimeric enzymes. A, schematic representation of tethered telomerase being tested for whether two active sites are saturated by substrates at the same time. B, ddTRAP assay on samples obtained from experiments as designed in A. Telomerase extension products were collected, and equal amounts were used for the ddTRAP assay. Error bars, S.D. (n = 3). C, ddTRAP assay of tethered telomerase performed at varying times for extension 1 only. Blue, gray, and black bars represent 30, 90, and 120 min, respectively. Extensions 2 and 3 were all performed for 2 h. Ctl** samples were time-delayed samples that were left at room temperature for 5 h before starting the extension reaction for a duration of 30, 90, or 120 min. Error bars, S.D. (n = 3). Enzyme aliquots with cell equivalency of 250 were used for B and C.
We next tested whether the above prediction is true by conducting a second round of reactions in the presence and absence of fresh TS primers in parallel. After the first round of reactions, the enzymes on the beads were split into two equal aliquots, and we performed the reactions in an extension mixture with or without fresh TS primers (Fig. 5, A and B, CTL + TS and −TS). If all slow-acting active sites were saturated during the first round of substrate binding, >96% (i.e. exp(−20/1200)) of bound substrates would not have enough time to dissociate during the fast separation of tethered enzymes from free TS primers and products, and the second round of extension reaction from the bound primers should produce approximately the same amount of products, regardless of whether or not fresh TS primers were added. However, our data showed the opposite. In the second round of reactions, the tethered enzymes with no fresh substrates (“−TS”) exhibited a significant drop (∼75% less; 16% versus 60%) in extension products, when compared with the control sample (Fig. 5B, extension 2). The significant reduction in product yield during the second round must have come from the lack of bound substrates in a majority of the slow-acting active sites of the dimeric enzymes (E0) when the first round of reaction ended. The residual 16% (extension 2 (−TS)) could come from slow-acting active sites in dimers (E1) or slow monomers (sM1) that were in the middle of their reactions or from a small fraction of active sites (<6% based on extension 3 (+TS) in Fig. 5B) that completed their reactions but did not release their products quick enough.
Our observation was not caused by the degradation of the bound TS primers because we used DNase/RNase-free solutions and nuclease inhibitors in our buffers, and the amount of DNA substrates overwhelmed the active sites of the telomerase by at least 9 orders of magnitude. Thus, the only sound explanation is that a significant fraction of slow active sites (44/60 = 75% of those measured from extension 2 (+TS) were equivalent to 60–16% = 44% of total active sites in extension 1) was inaccessible to the substrates during the first round of reaction, and thus were empty when the first round finished. Based on the data in Fig. 5B and the ratio of total slow and fast active sites in Fig. 4B, we estimated that at the beginning of the first round of reaction, the enzyme mixture contained ∼44% E0 dimers, 36% fM1 monomers, and 20% (sM1 monomers + E1 dimers), which could provide an upper-bound estimate for the hTR content of ∼28% in the elution 2 fraction in Fig. S6 by assuming a partial recovery in the R3-pulldown experiments (Fig. 1B). In an E0 dimer, TS primer binding to its fast-acting site negatively impacts on the binding of a second primer to the slow active site. This represents a strong negative cooperativity between the two active sites in the E0 dimer.
Because of no physical contact between neighboring telomerase complexes on the streptavidin-coated beads, the strong negative cooperativity is only possible when the two types of active sites are within a dimer, not from two separate enzymes (Figs. 5, A and B, and 4G). Furthermore, negative cooperativity in substrate binding suggests that a significant fraction (∼44%; above-specified) of E0 dimers exist in the enzyme mixture from cell lysates even though the new intermediate-resolution cryoEM structure reveals a monomer. The negative cooperativity also explains the observation that the two active sites exhibit different affinities to the same DNA substrates (Fig. 4, C and D).
As depicted in Fig. 4G, the dimers in cell lysates belong to three subgroups: E0, E1, and E2. In the first round of extension, the pristine enzymes (E0) were converted to E1 (Fig. 1A), and a fraction (∼63% = exp(−90/109), based on 0.00915 s−1 in Fig. 4H) of E1 changed into E2 (Figs. 1A, 4A, and 5A). In the second round, the leftover E1 enzymes became inactive E2, explaining why the flow-through fraction, the washed ones, or the elution 2 in Fig. 1B had no activity. A sequential model (with or without a delay) for the dimers is provided in the supporting Experimental procedures and can explain the kinetic property (Fig. 4G). Between the two monomeric forms, our limited resolution in recognizing only two kinetic constants (Fig. 4, A, E, and H) led to a parsimonious assignment that the slow sM1 enzymes behave the same as the E1 and contribute partially to the products detected in extension 2 (−TS), whereas the fast fM1 enzymes (∼36% in the mixture; above estimated) turn into inactive M2 (Fig. 5B). If we split the 20% (sM1 and E1) evenly between the two, the ratio of monomers (36% + 10%) versus dimers (44% + 10%) (∼45:55) estimated here is close to what was derived by single molecule imaging (31).
To further test the sequential model for dimeric telomerase, we varied the incubation time for the first round of extension by the tethered enzymes. For this series of experiments, the same amounts of enzymes were allowed to extend the TS primers for 30, 90, or 120 min in the first extension reaction, respectively. Afterwards, the enzymes were separated from the products and subjected to an equal extension time (120 min) for the second and the third round. Based on its time constant, the fast-acting sites became completely inactive after ∼30 min (round 1). The fixed amount of time in the three rounds within the regime of a sequential model predicted that the total products from three rounds would be similar for three groups. Group 1 (30-min extension in round 1, Fig. 5C) had the lowest activity in the first round, but greater activity in both the second and third rounds (Fig. 5C) as compared with group 2 (90 min extension 1) and group 3 (120 min extension 1) samples. The enzymes in group 3, due to sufficient time during the first two rounds, showed almost no activity in the third round because of catalysis-dependent shutoff. The time-dependent catalysis of the telomerase complex followed the prediction from the models in Fig. 4G with substantial amounts of dimeric holoenzymes. Because of the processive reaction in each active site, we named the catalysis-dependent shutoff of human telomerase “single-run catalysis,” instead of single-turnover (Fig. 4G).
A significant fraction (∼44%; see above) of functional enzymes in the H1299 cell lysates were dimeric E0. Our analysis accounted for the existence of monomeric enzymes, which followed single-exponential kinetics (Fig. 4G). Combination of the sequential model for the dimeric holoenzymes with the parallel model for two monomeric forms was thus sufficient to fit all experimental data well (Fig. 4 and Fig. S7). It is thus not necessary to biochemically purify the telomerase monomers from the dimers for us to deduce the fundamental property of the enzyme. A simple assumption of the fM1 and sM1 following the kinetics for fast- and slow-acting sites was sufficient for reaching the biophysical understanding (Fig. 4H).
iTAFs turn on inactive telomerases
Allosterically, the inactive enzymes probably reside in a stable conformation that does not switch back to the active state due to an energy barrier or due to missing accessory factors that might have dissociated during the extension reaction. There are three potential destinies for inactive telomerase inside a cell: being recycled into the active form, being degraded, or being stored in cells as a reserve. Telomerase is known to be active in dividing cells in every cell cycle. This led us to the question whether cells have a capacity to turn on the inactive telomerase.
We first tested cell lysates of a telomerase-negative cell line, BJ fibroblasts, and used ddTRAP to measure the fraction of reactivated enzymes (Fig. 6A). Tethered enzymes on streptavidin-coated beads were made fully inactive after three rounds of extension reactions (in E2 or M2 states in Fig. 6B). Afterwards, the beads with inactive enzymes were mixed with the BJ cell lysates and the extension reaction mixture for 2 h before the reaction products were counted using ddTRAP. To our surprise, the cell lysates from the telomerase-negative BJ cells were able to reactivate a significant fraction of inactive enzymes (as exemplified in Fig. 6B). As a negative control, the BJ cell lysates had no activity (Fig. 6B). These results clearly demonstrated that the BJ cells contain iTAFs that can reactivate the inactive holoenzymes (E2 and M2). Similar iTAF activity was detected in other types of telomerase-negative proliferating cells, such as SAOS-2 and SKLU-1 (Fig. 6F).
Figure 6.

iTAFs reactivate inactive telomerase holoenzymes. A, schematic representation of catalytically exhausted tethered telomerase enzymes (E2 or M2) mixed with telomerase-negative cell lysate (of BJ cells) to test enzyme reactivation. B, telomerase reactivation assay performed using ddTRAP. Error bars, S.D. (n = 3). Inactive telomerase treated with the BJ cell lysate showed a 7-fold increase in activity compared with the background readout from control enzymes. BJ cell lysate shows no telomerase activity. C and D, time courses for the tethered telomerase before and after treatment with the iTAFs (from BJ cells). The solid lines were fitted with the sequential model with a short delay. After reactivation, the slow-acting enzyme is dominating (75%). Error bars, S.D. (n = 3). The dashed lines represent the amount of products if the active sites remained continuously active. Every set of experiments in B and C started with enzyme aliquots of cell equivalency of 1 million. E, iTAF fractions were heat-inactivated or treated with the DNase before being incubated with the inactive tethered telomerase. The background activity was from the tethered E2 enzymes with residual activity. Other samples were normalized against the control. Error bars, S.D. (n = 3). F, cell lysates of SAOS-2 and SKLU-1 contain iTAF activity. These are two alternative lengthening telomere (ALT) cell lines that have no telomerase activity (green bars of zero height).
When partially purified telomerase holoenzymes were incubated with iTAFs, time-lapse experiments showed that iTAF activation increased the amount of the fast active sites (E0 or fM1) by a minor fraction (∼25%; 500 increased to ∼620 in Fig. 6, C and D), but augmented the slow active sites (E1 or sM1) more significantly (∼480%; 500 increased to 2400) (Fig. 6, C versus D). The control telomerase (Fig. 6C) had roughly equal amounts of fast and slow sites. Under the current experimental conditions, the (re)activated enzymes were seemingly dominated by slow active sites (E1 or sM1), suggesting that the iTAFs preferentially switched the inactive enzymes into the E1 or sM1 states. Furthermore, the fast active sites in the reactivated enzymes displayed slightly faster kinetics, which remains comparable with that of the native enzymes (Fig. 6C versus Fig. 4A). These data further demonstrate that the two kinetic components reflect the intrinsic properties of the native, functional telomerase holoenzymes (Fig. S7).
The iTAFs are proteinaceous components. We were able to separate iTAFs from the telomerase fractions in a glycerol gradient. The iTAFs heated in a boiling temperature lost their activity in reactivating the inactive enzymes, whereas DNase treatment had almost no effect on iTAF activity (Fig. 6E). When the cell lysates were fractionated and eluted in a Superdex 200 column, the iTAFs were found to be equivalent to an ∼150-kDa globular protein (Fig. S8). Further experiments are needed to identify the iTAFs and characterize their function.
Discussion
Kinetic model for catalysis-dependent inactivation and iTAF-mediated reactivation
Our results show that in the telomerase mixture of both dimers and monomers inside a human cell, a dimeric holoenzyme contains both fast and slow active sites and two isoforms of monomeric enzymes may harbor similar fast and slow active sites, respectively (Fig. 4, A–D). The two types of active sites exhibit different affinities for the DNA substrates. Negative cooperativity between them within a dimeric enzyme makes the slow site inaccessible to substrates when the fast site is in catalysis. A dimeric E0 enzyme may undergo a symmetry breakdown when one of its active sites binds a DNA substrate, and its two sites act in tandem. The monomers do not have negative interactions between them (Fig. 5B). More importantly, both types of active sites perform “single-run catalysis” (Fig. 7A), and iTAFs can turn the inactive enzymes (E2 or M2) into different active states (Fig. 7B). Our results can be incorporated into a kinetic model in Fig. 7A, where one active site goes through one round of processive extension reaction and becomes inactive after it falls off the product (Fig. 1, A and B). The inactive site can be turned back on (step IV; recycle) by iTAFs. The switching of the enzymes between active and inactive states is further diagrammed in Fig. 7B. Both the endogenous telomerase holoenzymes and the recombinant ones exhibited similar kinetic property under different conditions. We therefore propose that the use-dependent loss of activity and the iTAF-dependent gain of activity represent fundamental properties of human telomerase holoenzymes and provide an intrinsic on-off control of their activity in human cells.
Figure 7.
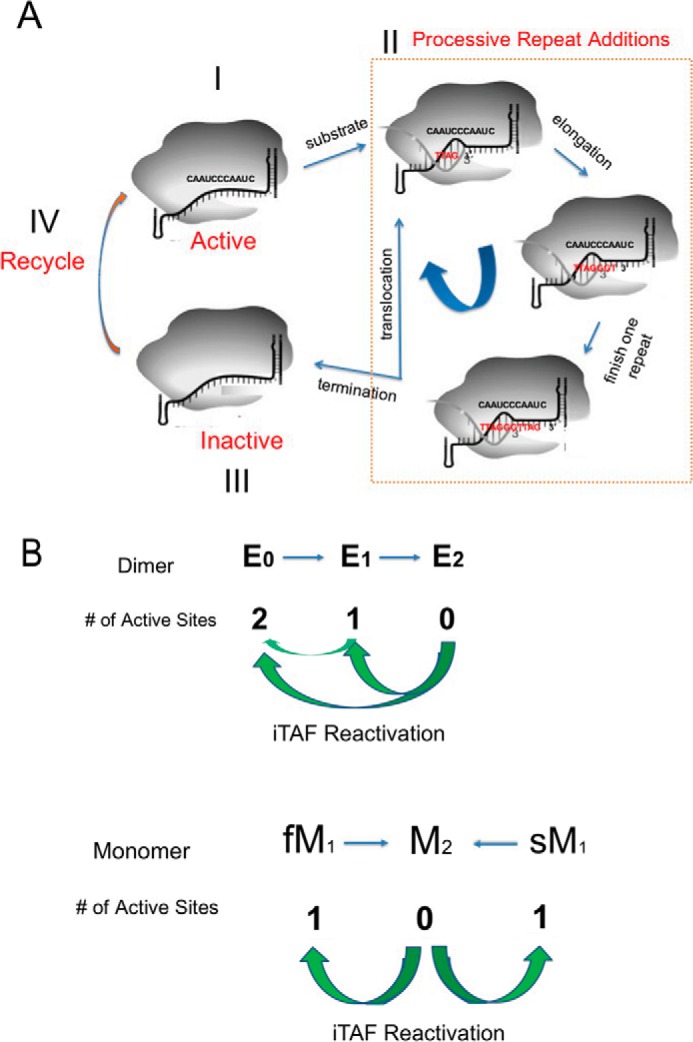
Kinetic on-off control of human telomerase holoenzyme. A, each active site undergoes catalysis-dependent shutoff. During the processive catalytic reaction, after each translocation of the newly added repeat the enzyme has a frequency in falling off the substrate, which results in the shutdown of the active site. The iTAFs can reactivate the inactivated enzyme. The dashed line box denotes the processive catalysis of the telomerase activity. B, sequential model for the two active sites of the human telomerase holoenzyme and the parallel action of two different monomeric enzymes. Newly assembly dimeric enzymes (E0) have two active sites. E1 has its fast-acting site shut off, and E2 has no activity. M1 has one active site and M2 none. The iTAFs can switch the inactive sites into active states through E2 → E1 or E2 → E0 with E2 → E1 seemingly dominating the reactivation or through M2 → M1. E1 → E0 is shown as well.
The sequential action of the two sites in a dimeric telomerase (Fig. 7B) suggests that a dimeric enzyme does not act on two telomeres, say those of sister chromosomes, at the same time (43, 50, 57). Two monomers can act on two telomeres simultaneously. Our in vitro assays of the slow-acting sites suggest that the in vivo enzymes might be able to use both slow and fast active sites within the average duration of S-phases of human cell cycles (6–8 h). For quick-dividing cells, the S-phase might be <30 min such that only the fast active sites (E0 and fM1) are suitable for fast chromosomal replication. Under such conditions, iTAFs for turning more telomerases into E0 or fM1 states or extra copies of mature telomerase holoenzymes would be needed to avoid critically short telomeres. A combination of fast and slow active sites in dimers and monomers could be utilized to match the time duration needed for telomerase to finish its catalysis with the duration of the S-phases so that all telomerase activity can be maximally utilized. Although speculative, this might be one of the reasons why the telomerase holoenzymes have two types of active sites with contrastingly different kinetics. This mechanism further indicates a possible role of the iTAFs in regulating the S/G2 transition.
Single-run catalysis of human telomerase
When counting the extension products by ddTRAP with single digit accuracy, we could avoid complications from the average length of extension products, the differences in catalysis rates, and the possible delay between the moment the fast site in a dimer is turned off and the time point when the slow site becomes accessible to a new substrate (Fig. 4G). Using the substrate pulldown to select only active enzymes in the single-turnover setting (Fig. 1, A and B), we avoided the often observed mismatching between a larger amount of enzymes (or hTERT) and a smaller number of extension products in the enzyme assays (34), which indicates a significant fraction of inactive enzymes. The similar distribution of the products of different lengths in the input 1 and that in the elution 1 (E1 or slow M1) in Fig. 1B from the endogenous telomerase suggests that the processive additions of repeats to substrates are similar between the slow and the fast active sites (Fig. 7A). The main differences between the fast and slow active sites are thus due to other factors, such as accessibility to substrates, initiation of the successful extension reaction, pause duration after the complete extension of each repeat, etc. Singe molecule enzymology would be suitable for determining in detail the differences between these two active sites (44, 58, 59), even though the slow sites might be difficult to study, the dimeric enzymes might fall apart in vitro, and the enzymes remain heterogeneous in stoichiometry and conformation.
The single-run catalysis may not be limited to human telomerase (60, 61) because monomeric telomerase enzymes in other organisms may follow similar kinetics. It is tempting to speculate a similar mechanism in yeast and Tetrahymena even though they vary in TERT and TR size and in complex stoichiometry. The single-run catalysis also occurs to other RNP complexes, for example, CRISPR/cas9 (62), suggesting a more universal mechanism among RNP enzymes. A generic mechanism of catalysis-dependent turnoff and iTAF-dependent turn-on of the telomerase holoenzymes in other species, if true, would reveal a more general role of the single-run catalysis in achieving tight regulation of telomerase activity.
iTAFs differ from other proteinaceous factors that regulate telomerase activity
The iTAFs are proteinaceous factors that can be fractionated. They are the first group of factors that directly control the recycling of human telomerase, and they are mechanistically different from the recruitment factors in the shelterin complex, the hTR-binding factors, and the factors involved in the assembly of nascent active enzymes (63). It is still too early to say whether they function at the same location and/or time as the recruitment and/or activation of the telomerase happens next to the shelterin complex (63, 64), and we are not able to explain why iTAFs are available in telomerase-free cells. Future study will be needed to identify the iTAFs as we did previously (see Ref. 65) and to verify their functions in both telomerase-positive and -negative cells.
With the slow kinetics and the stability of the telomerase holoenzymes within 5–8 h at room temperature, it is not feasible to determine how many times each telomerase can be turned off and turned back on before its physical breakdown. The high stability of a holoenzyme in the nuclear environment may allow multiple off-on cycles to occur before it becomes defective and is marked for degradation. More broadly, whether the iTAFs function in controlling the cell cycle and what other functions they might perform in cell proliferation and cell aging are fascinating questions for the future.
Single-run catalysis as a built-in brake for human telomerase
Similar to single-turnover enzymes, catalysis-dependent inactivation of human telomerase offers an exquisite regulatory mechanism. There are at least two layers of control. The first is that the fast-acting sites are quickly exhausted, ∼40 times faster than the slow sites. The second is that the iTAFs may be regulated through different pathways.
The single-run catalysis explains the need for telomere extension to happen in focused areas in the nucleus. With each active site functioning once on one telomere in a co-replicational manner in the S-phase, ∼92 active sites are needed, which is equivalent to 46 copies of pristine dimeric telomerase holoenzymes in the E0 state to satisfy the need of extending all telomeres once. If these enzymes are randomly distributed in a nucleus of ∼5 μm in diameter, the average concentration of the active sites is ∼0.1–0.2 nm, 2 orders of magnitude lower than the measured Km values for these sites (Fig. 4, C and D). To solve this mass-action problem, the telomerase molecules may be concentrated in a small area (< 0.5 μm) such that their local concentration would be ∼200 nm, sufficient to support productive collisions between telomerase active sites and individual telomeres. Similarly, the telomeres of different chromosomes may be looped from individual chromosomal territories into the telomerase-concentrated regions, which could be the telomere processing centers, so that the local concentration of the substrates (uncapped telomeres) can approach ∼200 nm, which would be sufficient for ensuring a sizeable volume of extension reactions within a short period of time.
In conclusion, the native human telomerase has two kinetically distinct types of catalytic sites that manifest varying affinities for the telomeric substrates. The two sites in dimeric enzymes show negative cooperativity and act in tandem by sequentially becoming accessible to the DNA substrates. The active sites in monomeric enzymes function independently. Both types of active sites function as a single-run enzyme because they undergo catalysis-dependent shutoff. Inactive enzymes can be reactivated by iTAFs from certain cells. Identification of the iTAFs and their roles in regulating telomerase activity await future studies.
Experimental procedures
Cell culture and cell lines
All cell lines (H1299, H1299 +hTR +hTERT, BJ fibroblast, SKLU, and SAOS-2) were cultured at 37 °C in 5% CO2 in 4:1 Dulbecco's modified Eagle's medium containing 10% calf serum (HyClone, Logan, UT). All cell lines were obtained from ATCC. Cells used for preparing the active telomerases were either H1299 lung adenocarcinoma cells or super-H1299 overexpressing recombinant hTR and N-terminally biotinylated hTERT.
Generation of stable super-H1299 cell line overexpressing recombinant hTERT and hTR
Biotin-tagged hTERT carried in pBabe-hygro retroviral vector was first used to transfect the transient packaging line PhoenixE. The virus supernatant was then used to infect the stable amphotropic packaging line PA317. PA317 was then selected with hygromycin and the resulted hygromycin-resistant cells produced a stable virus that was then used to infect the parental cell line H1299. The infected H1299 cells were selected with hygromycin. For hTR, the pSSI 7661 lentiviral vector together with two helper plasmids, psPAX2 and pMD2G, were used to transfect 293 packaging cells. The virus supernatant was used to infect H1299 cells that already overexpress biotin-tagged hTERT. The infected H1299 cells were further selected with blasticidin.
RT-droplet digital PCR and RT-qPCR measurements for hTR
RNA purification from samples was performed with RNAzol® RT (MRC, Inc.) kits according to the manufacturer's protocol. For hTR analyses, we used 50 ng of purified total RNA in each RT reaction (iSCRIPT Select Advance cDNA synthesis kit from Bio-Rad) to generate cDNAs, diluted 1:4 and used within 48 h of production in ddPCR and qPCR measures. For long-term storage, all cDNAs were diluted 1:4 before use and stored at −80 °C. For both ddPCR (Supermix for Probes (No dUTP) no. 1863024) and qPCR (TaqMan Master no. 0435286001), we employed the same set of primers and probe: forward primer, cgaggttcaggcctttca, and reverse primer, ccacagctcagggaatcg (UPL Probe no. 4 (Roche Applied Science catalog no. 04683633001)).
Gel-based TRAP and ddTRAP assay
Quantitation of telomerase enzyme activity was performed using a modified telomeric repeat amplification protocol (TRAP). Briefly, cells were lysed, diluted, and added to the telomerase extension reaction mixture followed by heat inactivation of telomerase. An aliquot of the extension products was amplified in a droplet digital PCR for 40 cycles, and fluorescence was measured, and droplets were read and counted on the droplet reader (QX200, Bio-Rad). Afterward, data were processed and telomerase extension products per unit were determined. Gel-based TRAP assay was performed as described before in Herbert et al. (46). Detailed procedures and analysis for ddTRAP can be found in Ludlow et al. (49).
Western blot analysis
SDS-PAGE analysis and Western blotting detection of protein components were performed based on published procedures (66). Briefly, protein samples were mixed with 2× Laemmli buffer and boiled for 5 min at 95 °C. Anti-hTERT (Y182, ab32020 from Abcam) rabbit mAb was used for primary detection prior to secondary detection. Blots were imaged with Bio-Rad Chemidoc XRS+ Molecular Imager and quantified with Bio-Rad Image Lab software.
Purification of recombinant telomerase from Super H1299 (H1299 + hTR + hTERT)
Partial purification of recombinant telomerase was done in four steps. All steps were performed in a manner as not to contaminate the sample with RNase. Nuclease-free water and buffers were employed for every step to ensure stability of telomerase. 200–500 million super-H1299 cells were pooled together and lysed in a nuclease-free 1.5% CHAPS buffer: 10 mm Tris-HCl, pH 7.5, 1 mm MgCl2, 1 mm EGTA, pH 8.0, 5 mm β-mercaptoethanol, 10% ultrapure glycerol, 0.01 mm phenylmethylsulfonyl fluoride, Ribolock RNase inhibitor 40 units/ml (Thermo Fisher Scientific catalog no. EO0381). The lysate was cleared by centrifugation and then fractionated in a continuous 10–30% glycerol gradient. The active fractions were pooled together and incubated with monomeric avidin beads (Pierce catalog no. 20228) for 2 h at 4 °C. After the beads were washed with buffer A (0.150 m sodium phosphate buffer, pH 7.0, 0.1% Triton X-100, 400 mm NaCl), the active enzymes were eluted with buffer A containing d-biotin (2 mm final). The active fractions were then pooled together and diluted to lower the salt concentrations (50 mm NaCl final) prior to being incubated with SPFF beads (GE Healthcare catalog no. 17072910) for 2 h at 4 °C. Samples were eluted in a salt gradient with 0.2–1.0 m NaCl in buffer B (50 mm sodium phosphate buffer, pH 7.0, and 0.1% Triton X-100).
Affinity purification of endogenous human telomerase from H1299 cells (one-step) and double-affinity purification (two-step R3 pulldown)
5′-Biotinylated telomeric repeats ((TTAGGG)3; R3) were loaded to streptavidin-coated Dynabeads. After complete wash to remove free R3, the R3-conjugated beads were used for single-step pulldown of human telomerase holoenzymes as described before (32). However, no antibody to telomerase was used to pull down telomerase from lysates. Instead, we employed the whole-cell lysate for subsequent steps to analyze all telomerase molecules. 0.1 mm dATP/dTTP was used to elute the bound enzymes from the beads. Apyrase treatment was introduced to remove the nucleotides when two such pulldown steps were performed in tandem. Apyrase was added (0.1 units) to treat elution samples from the first-step pulldown on ice for 30 min. The elution samples were then reloaded to a new set of beads as a new input for a second round of affinity pulldown.
Tethered telomerase extension assay
Purified recombinant telomerase enzymes were incubated with streptavidin-coated magnetic Dynabeads (MyOne T1, Invitrogen) for 2 h at 4 °C. Beads were used to present the purified biotinylated telomerase holoenzymes for multiple rounds of extension reactions at room temperature (200 nm TS substrate and 50 μm dNTPs mix). Based on the surface area of the beads and the total number of telomerase enzymes used in our experiments, the average distance between the neighboring streptavidin molecules would be at least 50 nm. A magnet was used to achieve quick separation of the enzymes and the reaction products in less than 10 s. Extension reactions were carried out at intervals indicated in the main text and figure legends. Products were analyzed via ddPCR to quantify telomerase extension molecules. The same products were also analyzed for RNA content to determine hTR levels via ddPCR.
Purification of iTAFs
Partial purification of iTAFs from cell lysates followed a four-step procedure. After cell lysis, the cleared lysates were fractionated in a continuous glycerol gradient. The fractions containing iTAF activity were pooled and concentrated before being run in a Superdex 200 gel-filtration column. The active fractions from size-exclusion were loaded into a Mono S column or Sulfopropyl sepharose fast flow (SPFF) for further purification. The active fractions were tested for telomerase reactivation via ddTRAP and subjected to further kinetic analysis. Similar to our previous study (65), mass spectrometry and proteomic analysis of the active fractions was performed to identify candidate proteins for iTAFs.
Author contributions
M. E. S. and Q.-X. J. data curation; M. E. S. and Q.-X. J. formal analysis; M. E. S. and Q.-X. J. validation; M. E. S., A. C., G. P. Y., and Q.-X. J. investigation; M. E. S., A. C., G. P. Y., A. T. L., W. E. W., and Q.-X. J. methodology; M. E. S., A. C., G. P. Y., A. T. L., J. W. S., W. E. W., and Q.-X. J. writing-review and editing; J. W. S., W. E. W., and Q.-X. J. supervision; J. W. S. and Q.-X. J. funding acquisition; W. E. W. and Q.-X. J. conceptualization; Q.-X. J. resources; Q.-X. J. software; Q.-X. J. visualization; Q.-X. J. writing-original draft; Q.-X. J. project administration.
Supplementary Material
Acknowledgments
We are indebted to members of the Jiang laboratory and those of the Shay/Wright laboratory. Three members of the Ph.D. thesis committee for M. E. S., Drs. Hamid Mirazei, Robert Eberhart, and Michael Cho, provided insightful advice and comments. Drs Sixue Chen and Jin Koh in the Proteomics Core at the Interdisciplinary Center of Biotechnology Research (ICBR) of University of Florida offered technical support. Some experiments reported here were performed in a laboratory constructed with support from National Institutes of Health Grant C06RR30414.
This work was supported by Cancer Prevention and Research Institute of Texas Grant RP120474 (to Q.-X. J.) and in part by National Institutes of Health Grants R01GM111367 and R01GM093271 (to Q.-X. J.), Cystic Fibrosis Foundation Grant JIANG15G0 (to Q.-X. J.), Welch Foundation Grant I-1684 (to Q.-X. J.), and startup funds from the University of Florida (to Q.-X. J.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S8 and supporting Experimental procedures.
- hTERT
- human telomerase reverse transcriptase
- TRAP
- telomerase repeat amplification protocol
- ddTRAP
- droplet digital telomerase repeat amplification protocol
- hTR
- human telomerase RNA
- RT-qPCR
- reverse transcriptase q-PCR
- TS
- telomerase substrate
- nt
- nucleotide
- Ext
- extension
- RNP
- ribonucleoprotein
- dansyl
- 5-dimethylaminonaphthalene-1-sulfonyl.
References
- 1. Blackburn E. H. (2010) Telomeres and telomerase: the means to the end (Nobel lecture). Angew. Chem. Int. Ed. Engl. 49, 7405–7421 10.1002/anie.201002387 [DOI] [PubMed] [Google Scholar]
- 2. Blackburn E. H. (2000) Telomeres and telomerase. Keio J. Med. 49, 59–65 10.2302/kjm.49.59 [DOI] [PubMed] [Google Scholar]
- 3. de Lange T. (2009) How telomeres solve the end-protection problem. Science 326, 948–952 10.1126/science.1170633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ouellette M. M., Liao M., Herbert B. S., Johnson M., Holt S. E., Liss H. S., Shay J. W., and Wright W. E. (2000) Subsenescent telomere lengths in fibroblasts immortalized by limiting amounts of telomerase. J. Biol. Chem. 275, 10072–10076 10.1074/jbc.275.14.10072 [DOI] [PubMed] [Google Scholar]
- 5. Hemann M. T., Strong M. A., Hao L. Y., and Greider C. W. (2001) The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 107, 67–77 10.1016/S0092-8674(01)00504-9 [DOI] [PubMed] [Google Scholar]
- 6. Marcand S., Gilson E., and Shore D. (1997) A protein-counting mechanism for telomere length regulation in yeast. Science 275, 986–990 10.1126/science.275.5302.986 [DOI] [PubMed] [Google Scholar]
- 7. Steinert S., Shay J. W., and Wright W. E. (2000) Transient expression of human telomerase extends the life span of normal human fibroblasts. Biochem. Biophys. Res. Commun. 273, 1095–1098 10.1006/bbrc.2000.3080 [DOI] [PubMed] [Google Scholar]
- 8. Zhu L., Hathcock K. S., Hande P., Lansdorp P. M., Seldin M. F., and Hodes R. J. (1998) Telomere length regulation in mice is linked to a novel chromosome locus. Proc. Natl. Acad. Sci. U.S.A. 95, 8648–8653 10.1073/pnas.95.15.8648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao Y., Sfeir A. J., Zou Y., Buseman C. M., Chow T. T., Shay J. W., and Wright W. E. (2009) Telomere extension occurs at most chromosome ends and is uncoupled from fill-in in human cancer cells. Cell 138, 463–475 10.1016/j.cell.2009.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Palm W., and de Lange T. (2008) How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334 10.1146/annurev.genet.41.110306.130350 [DOI] [PubMed] [Google Scholar]
- 11. de Lange T. (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19, 2100–2110 10.1101/gad.1346005 [DOI] [PubMed] [Google Scholar]
- 12. Blackburn E. H., and Collins K. (2011) Telomerase: an RNP enzyme synthesizes DNA. Cold Spring Harb. Perspect. Biol. 3, a003558 10.1101/cshperspect.a003558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsu M., McEachern M. J., Dandjinou A. T., Tzfati Y., Orr E., Blackburn E. H., and Lue N. F. (2007) Telomerase core components protect Candida telomeres from aberrant overhang accumulation. Proc. Natl. Acad. Sci. U.S.A. 104, 11682–11687 10.1073/pnas.0700327104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blackburn E. H., Greider C. W., and Szostak J. W. (2006) Telomeres and telomerase: the path from maize, Tetrahymena, and yeast to human cancer and aging. Nat. Med. 12, 1133–1138 10.1038/nm1006-1133 [DOI] [PubMed] [Google Scholar]
- 15. Greider C. W. (2006) Telomerase RNA levels limit the telomere length equilibrium. Cold Spring Harb. Symp. Quant. Biol. 71, 225–229 10.1101/sqb.2006.71.063 [DOI] [PubMed] [Google Scholar]
- 16. Smogorzewska A., and de Lange T. (2004) Regulation of telomerase by telomeric proteins. Annu. Rev. Biochem. 73, 177–208 10.1146/annurev.biochem.73.071403.160049 [DOI] [PubMed] [Google Scholar]
- 17. Cech T. R. (2004) Beginning to understand the end of the chromosome. Cell 116, 273–279 10.1016/S0092-8674(04)00038-8 [DOI] [PubMed] [Google Scholar]
- 18. Lei M., Podell E. R., Baumann P., and Cech T. R. (2003) DNA self-recognition in the structure of Pot1 bound to telomeric single-stranded DNA. Nature 426, 198–203 10.1038/nature02092 [DOI] [PubMed] [Google Scholar]
- 19. Townsley D. M., Dumitriu B., Liu D., Biancotto A., Weinstein B., Chen C., Hardy N., Mihalek A. D., Lingala S., Kim Y. J., Yao J., Jones E., Gochuico B. R., Heller T., Wu C. O., et al. (2016) Danazol treatment for telomere diseases. N. Engl. J. Med. 374, 1922–1931 10.1056/NEJMoa1515319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marian C. O., Cho S. K., McEllin B. M., Maher E. A., Hatanpaa K. J., Madden C. J., Mickey B. E., Wright W. E., Shay J. W., and Bachoo R. M. (2010) The telomerase antagonist, imetelstat, efficiently targets glioblastoma tumor-initiating cells leading to decreased proliferation and tumor growth. Clin. Cancer Res. 16, 154–163 10.1158/1078-0432.CCR-09-2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mender I., Gryaznov S., Dikmen Z. G., Wright W. E., and Shay J. W. (2015) Induction of telomere dysfunction mediated by the telomerase substrate precursor 6-thio-2′-deoxyguanosine. Cancer Discov. 5, 82–95 10.1158/2159-8290.CD-14-0609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morrish T. A., and Greider C. W. (2009) Short telomeres initiate telomere recombination in primary and tumor cells. PLoS Genet. 5, e1000357 10.1371/journal.pgen.1000357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Corey D. R. (2009) Telomeres and telomerase: from discovery to clinical trials. Chem. Biol. 16, 1219–1223 10.1016/j.chembiol.2009.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Armanios M., Alder J. K., Parry E. M., Karim B., Strong M. A., and Greider C. W. (2009) Short telomeres are sufficient to cause the degenerative defects associated with aging. Am. J. Hum. Genet. 85, 823–832 10.1016/j.ajhg.2009.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boukamp P., and Mirancea N. (2007) Telomeres rather than telomerase a key target for anti-cancer therapy? Exp. Dermatol. 16, 71–79 10.1111/j.1600-0625.2006.00517.x [DOI] [PubMed] [Google Scholar]
- 26. Blackburn E. H. (2005) Telomerase and cancer: Kirk A. Landon–AACR prize for basic cancer research lecture. Mol. Cancer Res. 3, 477–482 10.1158/1541-7786.MCR-05-0147 [DOI] [PubMed] [Google Scholar]
- 27. Folini M., Venturini L., Cimino-Reale G., and Zaffaroni N. (2011) Telomeres as targets for anticancer therapies. Expert Opin. Ther. Targets 15, 579–593 10.1517/14728222.2011.556621 [DOI] [PubMed] [Google Scholar]
- 28. Montanaro L. (2010) Dyskerin and cancer: more than telomerase. The defect in mRNA translation helps in explaining how a proliferative defect leads to cancer. J. Pathol. 222, 345–349 10.1002/path.2777 [DOI] [PubMed] [Google Scholar]
- 29. Trahan C., and Dragon F. (2009) Dyskeratosis congenita mutations in the H/ACA domain of human telomerase RNA affect its assembly into a pre-RNP. RNA 15, 235–243 10.1261/rna.1354009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Venteicher A. S., Abreu E. B., Meng Z., McCann K. E., Terns R. M., Veenstra T. D., Terns M. P., and Artandi S. E. (2009) A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323, 644–648 10.1126/science.1165357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu R. A., Dagdas Y. S., Yilmaz S. T., Yildiz A., and Collins K. (2015) Single-molecule imaging of telomerase reverse transcriptase in human telomerase holoenzyme and minimal RNP complexes. Elife 4, 2015 10.7554/eLife.08363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cohen S. B., Graham M. E., Lovrecz G. O., Bache N., Robinson P. J., and Reddel R. R. (2007) Protein composition of catalytically active human telomerase from immortal cells. Science 315, 1850–1853 10.1126/science.1138596 [DOI] [PubMed] [Google Scholar]
- 33. Venteicher A. S., Meng Z., Mason P. J., Veenstra T. D., and Artandi S. E. (2008) Identification of ATPases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell 132, 945–957 10.1016/j.cell.2008.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nguyen T. H. D., Tam J., Wu R. A., Greber B. J., Toso D., Nogales E., and Collins K. (2018) Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature 557, 190–195 10.1038/s41586-018-0062-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alves D., Li H., Codrington R., Orte A., Ren X., Klenerman D., and Balasubramanian S. (2008) Single-molecule analysis of human telomerase monomer. Nat. Chem. Biol. 4, 287–289 10.1038/nchembio.82 [DOI] [PubMed] [Google Scholar]
- 36. Mozdy A. D., and Cech T. R. (2006) Low abundance of telomerase in yeast: implications for telomerase haploinsufficiency. RNA 12, 1721–1737 10.1261/rna.134706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blackburn E. H. (2000) Telomere states and cell fates. Nature 408, 53–56 10.1038/35040500 [DOI] [PubMed] [Google Scholar]
- 38. Blackburn E. H. (2001) Switching and signaling at the telomere. Cell 106, 661–673 10.1016/S0092-8674(01)00492-5 [DOI] [PubMed] [Google Scholar]
- 39. Bianchi A., and Shore D. (2008) How telomerase reaches its end: mechanism of telomerase regulation by the telomeric complex. Mol. Cell 31, 153–165 10.1016/j.molcel.2008.06.013 [DOI] [PubMed] [Google Scholar]
- 40. Shore D., and Bianchi A. (2009) Telomere length regulation: coupling DNA end processing to feedback regulation of telomerase. EMBO J. 28, 2309–2322 10.1038/emboj.2009.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Förstemann K., and Lingner J. (2005) Telomerase limits the extent of base pairing between template RNA and telomeric DNA. EMBO Rep. 6, 361–366 10.1038/sj.embor.7400374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jiang J., Miracco E. J., Hong K., Eckert B., Chan H., Cash D. D., Min B., Zhou Z. H., Collins K., and Feigon J. (2013) The architecture of Tetrahymena telomerase holoenzyme. Nature 496, 187–192 10.1038/nature12062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sauerwald A., Sandin S., Cristofari G., Scheres S. H., Lingner J., and Rhodes D. (2013) Structure of active dimeric human telomerase. Nat. Struct. Mol. Biol. 20, 454–460 10.1038/nsmb.2530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Parks J. W., and Stone M. D. (2017) Single-molecule studies of telomeres and telomerase. Annu. Rev. Biophys. 46, 357–377 10.1146/annurev-biophys-062215-011256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cohen S. B., and Reddel R. R. (2008) A sensitive direct human telomerase activity assay. Nat. Methods 5, 355–360 10.1038/nmeth.f.209 [DOI] [PubMed] [Google Scholar]
- 46. Herbert B. S., Hochreiter A. E., Wright W. E., and Shay J. W. (2006) Nonradioactive detection of telomerase activity using the telomeric repeat amplification protocol. Nat. Protoc. 1, 1583–1590 10.1038/nprot.2006.239 [DOI] [PubMed] [Google Scholar]
- 47. Maine G. N., Li H., Zaidi I. W., Basrur V., Elenitoba-Johnson K. S., and Burstein E. (2010) A bimolecular affinity purification method under denaturing conditions for rapid isolation of a ubiquitinated protein for mass spectrometry analysis. Nat. Protoc. 5, 1447–1459 10.1038/nprot.2010.109 [DOI] [PubMed] [Google Scholar]
- 48. Skvortsov D. A., Zvereva M. E., Shpanchenko O. V., and Dontsova O. A. (2011) Assays for detection of telomerase activity. Acta Naturae 3, 48–68 [PMC free article] [PubMed] [Google Scholar]
- 49. Ludlow A. T., Robin J. D., Sayed M., Litterst C. M., Shelton D. N., Shay J. W., and Wright W. E. (2014) Quantitative telomerase enzyme activity determination using droplet digital PCR with single cell resolution. Nucleic Acids Res. 42, e104 10.1093/nar/gku439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wenz C., Enenkel B., Amacker M., Kelleher C., Damm K., and Lingner J. (2001) Human telomerase contains two cooperating telomerase RNA molecules. EMBO J. 20, 3526–3534 10.1093/emboj/20.13.3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Beattie T. L., Zhou W., Robinson M. O., and Harrington L. (1998) Reconstitution of human telomerase activity in vitro. Curr. Biol. 8, 177–180 10.1016/S0960-9822(98)70067-3 [DOI] [PubMed] [Google Scholar]
- 52. Gardano L., Holland L., Oulton R., Le Bihan T., and Harrington L. (2012) Native gel electrophoresis of human telomerase distinguishes active complexes with or without dyskerin. Nucleic Acids Res. 40, e36 10.1093/nar/gkr1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang J., and Hwang T. C. (2017) Electrostatic tuning of the pre- and post-hydrolytic open states in CFTR. J. Gen. Physiol. 149, 355–372 10.1085/jgp.201611664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wallweber G., Gryaznov S., Pongracz K., and Pruzan R. (2003) Interaction of human telomerase with its primer substrate. Biochemistry 42, 589–600 10.1021/bi026914a [DOI] [PubMed] [Google Scholar]
- 55. Pongracz K., Li S., Herbert B. S., Pruzan R., Wunder E., Chin A., Piatyszek M., Shay J., and Gryaznov S. M. (2003) Novel short oligonucleotide conjugates as inhibitors of human telomerase. Nucleosides Nucleotides Nucleic Acids 22, 1627–1629 10.1081/NCN-120023085 [DOI] [PubMed] [Google Scholar]
- 56. Jurczyluk J., Nouwens A. S., Holien J. K., Adams T. E., Lovrecz G. O., Parker M. W., Cohen S. B., and Bryan T. M. (2011) Direct involvement of the TEN domain at the active site of human telomerase. Nucleic Acids Res. 39, 1774–1788 10.1093/nar/gkq1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sandin S., and Rhodes D. (2014) Telomerase structure. Curr. Opin. Struct. Biol. 25, 104–110 10.1016/j.sbi.2014.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Holmstrom E. D., and Nesbitt D. J. (2014) Single-molecule fluorescence resonance energy transfer studies of the human telomerase RNA pseudoknot: temperature-/urea-dependent folding kinetics and thermodynamics. J. Phys. Chem. B 118, 3853–3863 10.1021/jp501893c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hengesbach M., Kim N. K., Feigon J., and Stone M. D. (2012) Single-molecule FRET reveals the folding dynamics of the human telomerase RNA pseudoknot domain. Angew. Chem. Int. Ed. Engl. 51, 5876–5879 10.1002/anie.201200526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Prescott J., and Blackburn E. H. (1997) Functionally interacting telomerase RNAs in the yeast telomerase complex. Genes Dev. 11, 2790–2800 10.1101/gad.11.21.2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wu R. A., Upton H. E., Vogan J. M., and Collins K. (2017) Telomerase mechanism of telomere synthesis. Annu. Rev. Biochem. 86, 439–460 10.1146/annurev-biochem-061516-045019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sternberg S. H., Redding S., Jinek M., Greene E. C., and Doudna J. A. (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 10.1038/nature13011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Armstrong C. A., and Tomita K. (2017) Fundamental mechanisms of telomerase action in yeasts and mammals: understanding telomeres and telomerase in cancer cells. Open Biol. 7, 160338 10.1098/rsob.160338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Armstrong C. A., Pearson S. R., Amelina H., Moiseeva V., and Tomita K. (2014) Telomerase activation after recruitment in fission yeast. Curr. Biol. 24, 2006–2011 10.1016/j.cub.2014.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yadav G. P., Zheng H., Yang Q., Douma L. G., Bloom L. B., and Jiang Q.-X. (2018) Secretory granule protein chromogranin B (CHGB) forms an anion channel in membrane. Life Sci. Alliance 1, e201800139 10.26508/lsa.201800139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jiang Q. X., Thrower E. C., Chester D. W., Ehrlich B. E., and Sigworth F. J. (2002) Three-dimensional structure of the type 1 inositol 1,4,5-trisphosphate receptor at 24 A resolution. EMBO J. 21, 3575–3581 10.1093/emboj/cdf380 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.