

Abstract
DHX9/RNA helicase A (RHA) is a host RNA helicase that participates in many critical steps of the HIV-1 life cycle. It co-assembles with the viral RNA genome into the capsid core. Virions deficient in RHA are less infectious as a result of reduced reverse transcription efficiency, demonstrating that the virion-associated RHA promotes reverse transcription before the virion gains access to the new host's RHA. Here, we quantified reverse-transcription intermediates in HIV-1–infected T cells to clarify the mechanism by which RHA enhances HIV-1 reverse transcription efficiency. Consistently, purified recombinant human RHA promoted reverse transcription efficiency under in vitro conditions that mimic the early reverse transcription steps prior to capsid core uncoating. We did not observe RHA-mediated structural remodeling of the tRNALys3–viral RNA-annealed complex. RHA did not enhance the DNA synthesis rate until incorporation of the first few nucleotides, suggesting that RHA participates primarily in the elongation phase of reverse transcription. Pre–steady-state and steady-state kinetic studies revealed that RHA has little impact on the kinetics of single-nucleotide incorporation. Primer extension assays performed in the presence of trap dsDNA disclosed that RHA enhances the processivity of HIV-1 reverse transcriptase (RT). The biochemical assays used here effectively reflected and explained the low RT activity in HIV-1 virions produced from RHA-depleted cells. Moreover, RT activity in our assays indicated that RHA in HIV-1 virions is required for the efficient catalysis of (−)cDNA synthesis during viral infection before capsid uncoating. Our study identifies RHA as a processivity factor of HIV-1 RT.
Keywords: human immunodeficiency virus (HIV), reverse transcription, processivity, viral replication, RNA helicase, host-pathogen interaction, DNA synthesis, DHX9/RHA, lymphocyte infection, Primer, primer extension, Template
Introduction
Human immunodeficiency virus, type 1 (HIV-1)5 reverse transcribes its single-stranded RNA genome into dsDNA that integrates into the host chromosome, establishing provirus that engenders life-long infection. HIV-1 hijacks many host factors to aid in its replication. Proteomic studies of HIV-1 virions have identified more than 200 host factors incorporated into the viral core (1). Among the co-assembled RNA-binding proteins, DHX9/RNA helicase A (RHA) is recruited at one-to-one stoichiometry to viral genomic RNA (2). Although the down-regulation of host endogenous RHA exerts little impact on HIV-1 genome dimerization and packaging (3), the progeny virions depleted of RHA are less infectious, primarily because of a decrease in reverse transcription efficiency/activity. These results indicate that the incorporation of RHA enhances the activity of virion reverse-transcription ribonucleoprotein (RNP) complex in subsequently infected cells (3–6).
The initiation of retroviral reverse transcription is a highly regulated process that occurs within intact viral capsid (CA) cores (7, 8). It is primed by the annealing of the 3′-terminal nucleotides (nt) of human tRNALys3 onto the complementary sequences in the viral genome known as the primer-binding site (PBS). Reverse transcriptase (RT) synthesizes minus-strand DNA until it reaches the 5′-end of genomic RNA and yields a ∼200-nt-long cDNA product named (−)strong-stop DNA (or (−)ssDNA), which is then strand-transferred to the 3′-end of viral genomic RNA, forming an RNA–cDNA hybrid by base pairing at the R (repeat) region. The 3′-terminal bases of the (−)ssDNA are used to prime RT for the synthesis of the full-length (∼9000 nt) viral (−)cDNA (9). In vitro reverse-transcription assays showed that synthesis of (−)ssDNA proceeds in two steps: an initiation phase occurring within the first several nucleotides and an extension phase (+181 nt) (10–13). During the initiation phase, RT synthesizes DNA in a distributive manner, resulting in slow, measurable incorporation of the first 5–6 nucleotides. During the extension phase, RT functions in a processive manner, and DNA synthesis is much faster (11, 14). Viral nucleocapsid (NC) proteins participate in reverse transcription by promoting tRNALys3 annealing and stimulating specific cDNA synthesis primed by tRNALys3 (15, 16). Several cellular factors found in virus particles are reported to impact RT activity, but their roles remain controversial (17).
Similar to other DEAH-box helicases, RHA unwinds both RNA and DNA duplexes and remodels RNP complexes, which facilitates complex reactions such as splicing and recombination (18, 19). RHA contains two tandem dsRNA-binding domains (dsRBDs) at the N terminus. It unwinds DNA and RNA in a 3′ to 5′ direction using the energy from the hydrolysis of dNTPs or NTPs (20–22), using two RecA like domains (I and II) and a helical core domain (23–25). RHA selectively binds structured domains of the 5′-UTR of many retroviruses and facilitates viral RNA metabolism (4, 19, 26, 27).
As one of the beneficial host factors recruited into HIV-1 virions, RHA significantly enhances virion infectivity (3). We have recently reported that RHA assembles with HIV-1 genomic RNA through a direct interaction between the dsRBDs of RHA and a 5′-UTR segment in proximity to the reverse transcription initiation site (5). These results posit RHA influences tRNALys3 annealing and/or subsequent reverse transcription.
Previous studies agree that RHA enhances HIV-1 reverse transcription, but the mechanism remains controversial. Endogenous reverse-transcription assays of replication-competent HIV-1 identified virions depleted of RHA maintained tRNALys3 annealing to the template RNA, although these virions experienced significantly reduced (−)cDNA synthesis (3). Conversely, endogenous reverse-transcription assays using protease-deficient virions identified that virions depleted of RHA were deficient in promoting tRNALys3 annealing to viral RNA, positing RHA interferes with unprocessed Gag chaperone activity that promotes annealing (6).
Here, we report the quantification of the reverse-transcription intermediates in HIV-1 infected T cells. The data support the results of the endogenous reverse-transcription assays, which conclude that RHA promotes reverse transcription. The attenuated infectivity of virions produced from RHA-depleted cells was attributable to diminished RT products, which can be rescued by expressing exogenous RHA in producer cells. Next, we established an in vitro primer extension assay by utilizing physiological stoichiometry between HIV-1 template RNAs and various recombinant protein components (RT, RHA, and NC) to evaluate the role of virion-associated RHA in (−)ssDNA and (−)cDNA synthesis. We measured the impact of RHA on RT kinetics using pre–steady-state and steady-state studies. The results show that RHA does not affect tRNALys3 annealing or reverse transcription initiation. It acts as a processive factor that enhances the processivity of RT. In summary, our data demonstrate that RHA promotes HIV-1 virion infectivity by accelerating reverse transcription mainly in the elongation step. Virion-associated RHA significantly increases the processivity of HIV-1 RT and therefore the rate of (−)ssDNA and (−)cDNA synthesis.
Results
HIV-1 virions produced in cells depleted of RHA are deficient in synthesizing the early product of reverse transcription
A virion-associated host factor, RHA is recruited into HIV-1 virions in a stoichiometry proportional to genomic RNA (2). Previously we reported that virions produced in RHA-depleted cells are poorly infectious (4), and the decrease in infectivity is directly proportional to the decrease in cell-endogenous RHA (5). Here, we used qPCR to quantify the early and late reverse transcription products in lymphocytes newly infected with RHA-deficient virions. RHA-specific siRNA down-regulated cell-endogenous RHA by >80% compared with nontargeting control siRNA (NT) (Fig. 1A). The exogenous expression of FLAG-tagged RHA rescued RHA levels. Cell endogenous Gag production was significantly reduced by RHA down-regulation. The diminished Gag production was similar in magnitude between cell-associated protein and cell-free virions (Fig. 1, A and B). Virions generated in three independent experiments were used to infect MT4 target cells. Given the significant decline in Gag production by RHA down-regulation, the volume of equivalent Gag (200 ng) from the RHA-deficient cells was 7-fold greater than the NT control and exogenous FLAG-tagged RHA rescue virions.
Figure 1.

Virions produced in RHA–down-regulated cells have diminished HIV early reverse-transcription intermediates in MT4-infected cells. HEK293 cells were transfected with siRNA targeting DHX9 or NT for 8 h, followed by transfection of FLAG-RHA that is resistant to the siRNA. After overnight incubation, the cultures were transfected with HIV molecular clone pNL4–3. 24 h later, the cell lysates and cell-free medium were collected for immunoblotting or Gag ELISA. Equivalent Gag containing cell-free medium was used for infection of MT4 cells. A, representative immunoblot of cell lysates (20 μg) with antiserum to RHA, FLAG, Gag, or GAPDH. The antiserum detected the specific proteins on the immunoblots, as shown relative to the prestained protein standards. B, virion production from the HIV producer cells was measured by Gag ELISA on cell-free medium. The volume of medium corresponding to 200 ng of Gag was used for the MT4 infections. The data obtained from three independent experiments (blue, experiment 1; orange, experiment 2; gray, experiment 3) are summarized on the scatter plot showing standard deviation and significance, as determined by Student's t test (**, p ≤ 0.01). C, diagram to show the placement of primers (red thin lines) and PCR amplicons (thick red lines) on the minus strand DNA (dashed line) to measure strong stop DNA (early RT product) and the completed dsDNA intermediate (gray boxes and thick black lines) to measure the late RT product in MT4-infected cells. D, DNA was extracted from the infected MT4 cells and subjected to the qPCR using primer pairs in C. DNA copies were determined relative to standard curves. Statistically significant differences were measured by Student's t test: *** p ≤ 0.001; ** p ≤ 0.01. WB, Western blotting.
Cellular DNA from newly infected MT4 cells was isolated and analyzed by qPCR using primer pairs to amplify the early and late products of reverse transcription (Fig. 1C). As shown in Fig. 1D, the amount of early RT product, (−)ssDNA, in MT4 cells was significantly diminished for virions depleted of RHA (p < 0.001). The 80% reduction in RT activity directly corresponds to the depletion of endogenous RHA levels in the virion-producer cells. The significant reduction in early RT activity carried forward, because the late RT product was also diminished by 80%. The amount of early and late RT products was completely rescued in virions produced in cells expressing exogenous FLAG-RHA. These results indicate that the cell-endogenous RHA in producer cells plays an important role in the early replication steps of progeny virions before the reverse transcription RNP has access to RHA in the newly infected lymphocytes. More specifically, virion-associated RHA is necessary for efficient reverse transcription of the RNA template.
RHA does not remodel the reverse transcription initiation RNA complex
Recombinant full-length RHA was expressed using a baculovirus system and purified (Fig. 2A). Its RNA helicase activity was confirmed by unwinding an RNA–RNA duplex with a 3′-overhang (6). A 3′-FTSC–labeled 34-nt RNA was preannealed to a 78-nt unlabeled RNA (Fig. 2B). In the presence of ATP, RHA unwound the complex and released the FTSC-labeled 34-nt RNA over a time range of 0–20 min. In the absence of ATP, no unwinding was detected because no free FTSC-labeled 34-nt RNA was observed at 20 min, demonstrating that the recombinant RHA unwinding activity is NTP-dependent (Fig. 2C).
Figure 2.
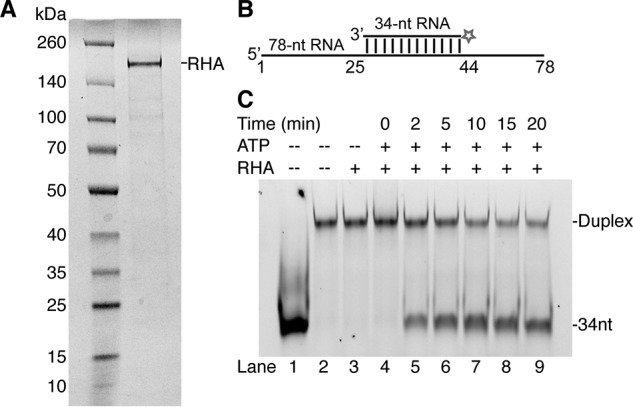
Recombinant RHA purified from insect cells exhibits RNA unwinding activity. A, a representative SDS-PAGE showing molecular mass markers (left lane) and recombinant RHA isolated from insect cells with a purity of ∼85% (right lane). The gel was visualized using Coomassie Blue staining. B, diagram of the RNA duplex used in the helicase assay. The duplex was formed by an unlabeled 78-nt RNA annealed to a complementary 34-nt 3′-FTSC–labeled RNA, which forms a 22-nt 3′-overhang. C, recombinant RHA unwinds RNA duplex. The reaction mixtures were collected and analyzed on a native PAGE at the indicated time points. In the presence of RHA (900 nm) and ATP (333 nm), the FTSC-labeled RNA duplex (42 nm) disassociated, and the free FTSC–34-nt RNA band increased over time (lanes 4–9). In the absence of ATP, the FTSC-labeled RNA duplex remained intact (lane 3). Free 34-nt RNA (lane 1) and annealed 34–78-nt RNA (lane 2) were run as controls.
The helicase activity of RHA is capable of unwinding RNA structures, as well as remodeling RNA complexes (21). It was previously suggested that RHA facilitated Gag in promoting formation of a slow migrating tRNALys3–5′-UTR complex structure that initiated reverse transcription efficiently (6). Here, gel electrophoresis under native conditions was employed to evaluate the RNA complexes annealed in the presence of NC and RHA. The PBS segment (nt 125–223) within the 5′-UTR with an additional two nonnative G-C pairs at the 3′ and 5′ end was used for better resolution of gel electrophoresis. The proteins were removed from the RNA complex prior to running the native agarose gel. No difference was observed between the complex of bands formed in the presence or absence of RHA (Fig. 3A), suggesting that either RHA did not remodel the RNA–RNA complex or it remodeled the annealed RNA complex into an alternative structure that migrated at the same rate.
Figure 3.

Recombinant HIV-1 RT catalyzes reverse transcription and is augmented by recombinant RHA. The synthesis of (−)ssDNA was primed by a 5′-Cy3–labeled tRNALys3 annealed to the HIV-1 5′-UTR template, and reactions were collected and analyzed at the indicated time points. The RNA size marker is labeled with M at the top of the gels. A, visualization of the annealed RNA complexes on a native agarose gel. PBS segment and tRNALys3 (1 μm) were annealed by NC (30 μm) in the absence and presence of RHA (1 μm) at 37 °C. B, diagram of the 5′-Cy3–labeled tRNALys3 annealed to the HIV-1 5′-UTR RNA template. C, comparison of the tRNALys3 primer extension products, synthesized in the absence and presence of RHA (1 μm) and separated by 8% urea–PAGE. Representative gels are shown.
Recombinant RHA accelerates the synthesis of (−)ssDNA by RT
Because the recombinant RHA does not seem to affect the tRNALys3 annealing, we next examined its impact on (−)ssDNA synthesis using an in vitro RT-primer extension assay. A 5′-Cy3–labeled tRNALys3 was annealed to the 5′-UTR (nt 1–344) of the HIV-1 genomic RNA in the presence of NC protein (Fig. 3B). NC was added at a ratio of one NC per 15 nt RNA, which is predicted to be close to the ratio within virions without inhibiting reverse transcription (15, 28). The 5′-UTR was predimerized to mimic the RNA conformation that directs genome packaging in the late phase of replication (29, 30). HIV-1 RT and dNTPs were added to the mixture to initiate RT-primer extension. This process generated a 181-nt DNA product covalently linked to the 3′-end of tRNALys3, mimicking the (−)ssDNA produced during early infection (Fig. 3C). At later time points, a band higher than the full-length product was observed, which was likely produced by self-priming in the absence of an acceptor for strand transfer (31). The ratio of RHA to 5′-UTR RNA was slightly greater than 1:1, because we were not able to ensure that all the purified recombinant RHA molecules were enzymatically active. Two major differences were observed between reactions containing or lacking RHA. First, the RT pausing was less frequent in reactions containing RHA, as shown by the pause product bands at the early time points (0–6 min) (Fig. 3C). This suggests that RHA is likely to remove RNA structures and/or NC protein on the template that hinder RT processivity. Second, the RHA-containing reactions exhibited an acceleration in the formation of the full-length product (257 nt). In the absence of RHA, RT completed the (−)ssDNA synthesis in an interval between 4 and 6 min, whereas in the presence of RHA, RT synthesized a full-length product between 2 and 4 min (Fig. 3C). Linear regression of the full-length product amount against time was employed to calculate how soon the full-length product started to form under our experimental conditions. Based on three independent assays, we estimated that in the absence of RHA, the formation of completed product occurred at 4.0 ± 0.3 min, whereas in the presence of RHA, the process was significantly accelerated, and the full-length product was generated as early as 2.1 ± 0.1 min (p < 0.001). RHA seems only to accelerate the rate of RT extension but not increase the yield of full-length products, because no significant differences of full-length (and self-priming) products between the no-RHA and RHA groups were observed after 10 min.
RHA acts downstream of reverse transcription initiation
Synthesis of (−)ssDNA consists of a slow initiation step and the subsequent rapid elongation step (10–13). To examine the impact of RHA on the initiation step of reverse transcription, we replaced dATP by ddATP in the primer extension assay. Because of the lack of a 3′-hydroxyl group, incorporation of ddATP at the sixth nucleotide position, after the tRNALys3 primer, terminated the reverse transcription reaction and yielded a tRNALys3 +6-nt product, referred to as +6 product. In the absence and presence of RHA, similar amounts of the +6 products were formed at each time point examined (Fig. 4A). Quantification of the +6 products showed no significant difference in the presence or absence of RHA (Fig. 4B). These data demonstrate that RHA does not aid reverse transcription at the initiation step. RHA neither remodels the primer–template (P/T) complex (Fig. 3A) nor accelerates the incorporation of the first couple of nucleotides. Combining with the results shown in Fig. 3C, these data show that RHA promotes RT activity in the elongation step of (−)ssDNA synthesis.
Figure 4.
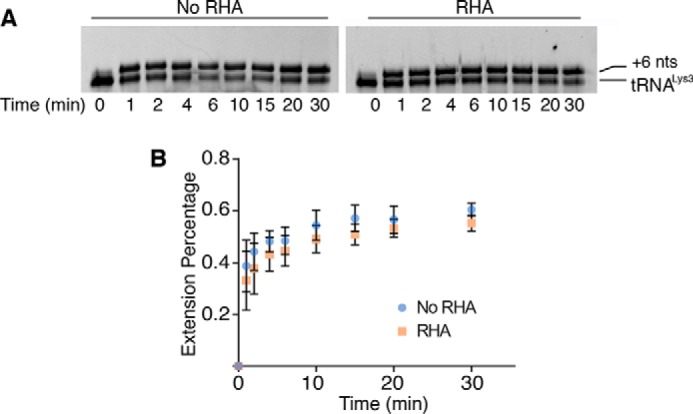
Initiation of reverse transcription is not augmented by the presence of RHA. Reactions contained tRNALys3 annealed to the PBS segment (333 nm) and ddATP (333 μm) instead of dATP, which terminates the RT reaction at the +6 nt position, and aliquots were collected and analyzed at the indicated time points. A, comparison of the +6 tRNALys3 primer extension products, synthesized in the absence and presence of RHA (1 μm), and separated by 8% urea–PAGE. Representative gels are shown. B, the amount of the +6 tRNALys3 product was quantified and normalized to total tRNALys3 to calculate the primer extension percentage. The average results of three independent experiments are shown with standard deviation (±).
RHA resolves significant structural barriers to reverse transcription created by guanine residues in the 3′-UTR
During the first strand transfer, the (−)ssDNA anneals with the 3′-UTR to continue to synthesize (−)cDNA. The viral CA core uncoating occurs after the first-strand transfer (8, 32), suggesting that the virion-associated RHA may have an impact on (−)cDNA synthesis. We next examined whether RHA could improve the rate of (−)cDNA synthesis by using the 3′-UTR as a template in a primer extension assay. The U3 region of the 3′-UTR contains stretches of guanosines (nt 8995–9023) that are predicted to fold into G-quadruplexes (Fig. 5A). These stable RNA structures would hinder RT progression and cause strong pauses (33). RHA is likely to resolve such stable RNA structures in the 3′-UTR because it has been previously reported to be able to unwind G-quadruplexes (21). The 3′-UTR RNA template and a Cy3-labeled DNA primer (19 nt) were annealed in the presence of K+ to promote the formation of G-quadruplexes, and the RT-primer extension assay was then carried out (Fig. 5A). RHA significantly enhanced RT's efficiency on the 3′-UTR template (Fig. 5B), shown by the reduction in time it took to generate the full-length product (222 nt) from 5.4 ± 1.0 to 2.0 ± 0.3 min (p < 0.001). The predicted G-quadruplex structures are ∼130 nt upstream of the primer annealing site. As expected, a strong pause product at ∼150 nt accumulated when the template RNA was prepared in buffer containing K+ (Fig. 5B). Such pause products were not detected when the K+ was replaced by Li+, an ion that disrupts formation of G-quadruplex structures (Fig. 5C) (34). Taken together, Figs. 3–5 show that RHA can enhance the rate of RT extension on RNA templates that are mimicking two different stages of reverse transcription.
Figure 5.
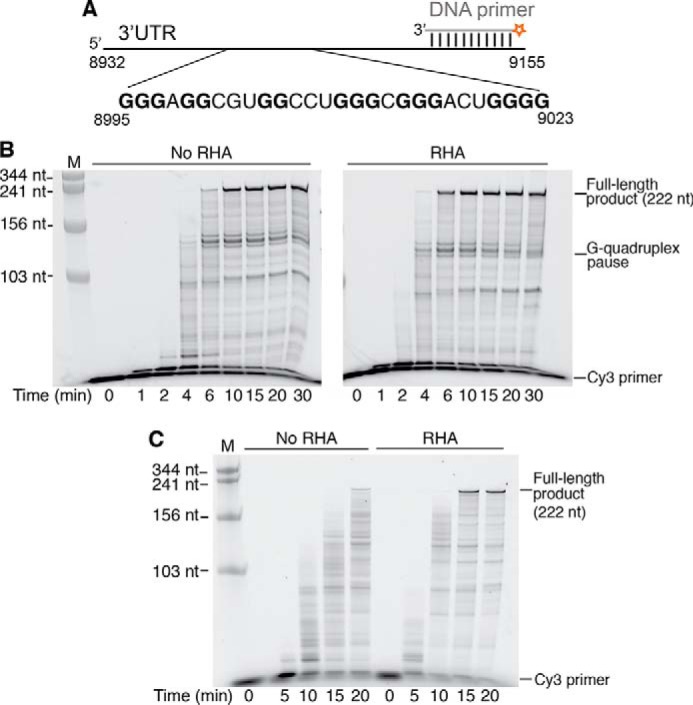
The efficiency of reverse transcription on the 3′-UTR template RNA is increased in the presence of RHA. Synthesis of the (−)cDNA was carried out using a 5′-Cy3–labeled DNA as the primer annealed to the 3′-UTR template (333 nm), and reactions were collected and analyzed at the indicated time points. The RNA size marker is labeled with M on the top of the gels. A, diagram of the 5′-Cy3–labeled DNA primer annealed to the RNA template. Guanosines (bold letters) in the sequence 8995–9023 are predicted to form G-quadruplexes. B, the 3′-UTR RNA template was preincubated in the presence of K+ to promote G-quadruplex formation. Comparison of the primer extension products, synthesized in the absence and presence of RHA (1 μm), and separated by 8% urea–PAGE. Representative gels are shown. C, primer extension assay carried out with the 3′-UTR template (333 nm) preincubated in Li+ buffer that diminishes G-quadruplex formation. The strong RT pause bands caused by the putative G-quadruplex in B were not observed.
RHA enhances the processivity of RT on the RNA template
Next, experiments were set out to investigate how RHA facilitates RT to accelerate (−)cDNA synthesis on an RNA template. It is possible that RHA activates the catalytic activity of RT by increasing RT's binding affinity to the P/T (Kd,RNA), reducing the dissociation rate of RT from P/T prior to nucleotide incorporation, or accelerating the single-nucleotide turnover. Another possibility is that RHA acts as a processivity factor that clears roadblocks such as RNA structures or proteins bound on the template to enhance RT's processivity.
The Kd,RNA of RT in the presence and absence of RHA was determined under pre–steady-state conditions using rapid quench-flow analysis (Fig. 6A). For these assays, 50 nm RT was incubated (with or without equimolar RHA) with increasing concentrations of the 3′-UTR–primer complex. The extension products at 250 ms were plotted against increasing primer–template concentrations and fit to a quadratic function (35, 36). The Kd,RNA in the absence and presence of RHA were 51.2 ± 1.6 and 45.00 ± 1.3 nm, respectively (Fig. 6A).
Figure 6.

RHA does not affect RT's binding and kinetic properties of single-nucleotide incorporation. A, determination of RNA template binding affinity (Kd,RNA) of RT in the absence and presence of RHA under pre–steady-state conditions. Active-site titration of RT in the presence and absence of RHA was carried out by preincubating RT (50 nm) in the absence (■) and presence of RHA (50 nm) (▴) with varying concentrations of P/T complex (40, 80, 160, 320, and 640 nm) followed by the addition of dTTP (100 μm) and MgCl2 (2.5 mm). B, determination of RT's dissociation rate (koff) from the P/T complex before nucleotide incorporation. Under steady-state conditions, 30 nm RT was incubated with 30 nm P/T in the presence and absence of RHA. The solutions were then mixed with dsDNA trap at varying time points and initiated with dTTP in which reactions were run for 10 s. RT's dissociation rate (koff) was determined by fitting to a single exponential decay equation. C, comparison of observed catalytic rate (kobs) of single-nucleotide incorporation under steady-state conditions. Reactions contained 5′-Cy3–labeled DNA primer annealed to the 3′-UTR (333 nm) and only dTTP (333 μm), which terminates the RT reaction at the +1 nt position. The kobs was obtained by the quantification of the DNA primer extension products and nonlinear square fitting of the data. The average results of three independent experiments are shown with standard deviation (±).
The dissociation of RT from the P/T before catalysis
| (Eq. 1) |
measured by adding 0.3 μg of dsDNA to trap the free enzyme prior to nucleotide incorporation (11). Dissociation of RT from an RNA/DNA P/T was slow: koff was measured as 0.0007 and 0.001 s−1 when RHA was absent and present, respectively (Fig. 6B).
The impact of RHA on the incorporation of one nucleotide by RT was then evaluated under steady-state conditions to determine whether RHA affected the catalytic rate of nucleotide incorporation. The observed catalytic rate (kobs) for single-nucleotide incorporation
| (Eq. 2) |
was calculated according to [P + 1] = A(1 − e−kobs·t) under saturating substrate conditions (11). In the absence and presence of RHA, the kobs values for single-nucleotide incorporation were 0.07 ± 0.003 and 0.06 ± 0.003 s−1, respectively (Fig. 6C). These data demonstrate that RHA does not alter the observed catalytic rate of RT under steady-state kinetics.
After demonstrating that RHA does not affect the kinetics of single-nucleotide incorporation, the effect of RHA on RT's processivity was investigated. Processivity is the probability that polymerase incorporates the next nucleotide in a single binding event without falling off the P/T. The processivity of HIV-1 RT in the presence of RHA was evaluated by introducing a dsDNA trap in the primer extension system. The dsDNA trap sequesters disassociated RT and prevents it from reassociating with the P/T. To gain higher resolution of primer extension bands in the early time points, 20% sequencing gels were used to resolve the primer extension products. As shown in Fig. 7A, the extension products were of much larger size in the RHA-containing lanes than that of the no RHA lanes. For example, at 20 s, the no RHA lane extended ∼8 nt (Fig. 7A, lane 2), whereas the RHA-containing lane extended more than 15 nt (Fig. 7A, lane 2′). These data recapitulate RHA's enhancement of reverse transcription elongation.
Figure 7.

RHA enhances the processivity of RT. A, single turnover processivity assay sof RT using dsDNA trap in the absence (lane 1–9) and presence (lane 1′–9′) of RHA. RT (400 nm) was incubated with P/T (200 nm) or NC (3.2 μm) in the absence and presence of RHA (400 nm). The reactions were initiated with dNTPs (100 μm), MgCl2 (5 mm), and dsDNA trap (0.3 μg). The reactions were carried out for 0, 20, 40, 60, 90, 120, 180, 240, and 300 s and loaded to lanes 1–9 and 1′-9′. Primer extension products were resolved in 20% urea–PAGE. B, the +1 products were quantified and fit into the single exponential decay equation. C, the +5 products were quantified and fit into the single exponential decay equation. D, the +6 products were quantified. The no RHA data were fit into the double exponential decay equation, and the RHA data were fit into the single exponential decay equation.
Each extension band represents the sum of nucleotide incorporation at its template position and the decay of the product because of subsequent nucleotide incorporation. The decay rate reflects the probability of RT dissociating from P/T and thus the processivity. Several template positions were selected to quantify the rate of product decay, including +1, +5, and +6 products. The product decay rate was quantified by fitting the product concentration to a double exponential equation (37, 38). In some cases, the largest accumulation of products occurred at the earliest time point measured. Thus, these data were fit into an exponential decay equation.
As shown in Fig. 7, the decay rate of +1 product was 0.034 ± 0.004 and 0.020 ± 0.003 s−1 with and without RHA, respectively, indicating that RHA enhances the processivity of RT at +1 position. A 5-fold processivity enhancement was observed in the +5 position, because the decay rate of +5 product was 0.035 ± 0.002 and 0.0077 ± 0.0020 s−1 with and without RHA, respectively (Fig. 7C). On the other hand, the decay rate of +6 product is unaffected by RHA, 0.013 ± 0.003 and 0.014 s−1 with and without RHA, respectively (Fig. 7D). These data indicate that RHA's processive enhancement is a dynamic process that depends on nucleotide position.
In summary, our results demonstrate that RHA positively regulates the processivity during the elongation phase of reverse transcription. RNA secondary/tertiary structures and proteins bound to the RNA template typically hinder RT scanning and lead to dissociation of RT from the template (15, 33, 39). Hence, RHA most likely removes the physical roadblocks for RT, such as RNA structures, NC, and/or other proteins coating the RNA and thereby improves RT's processivity (Fig. 8).
Figure 8.
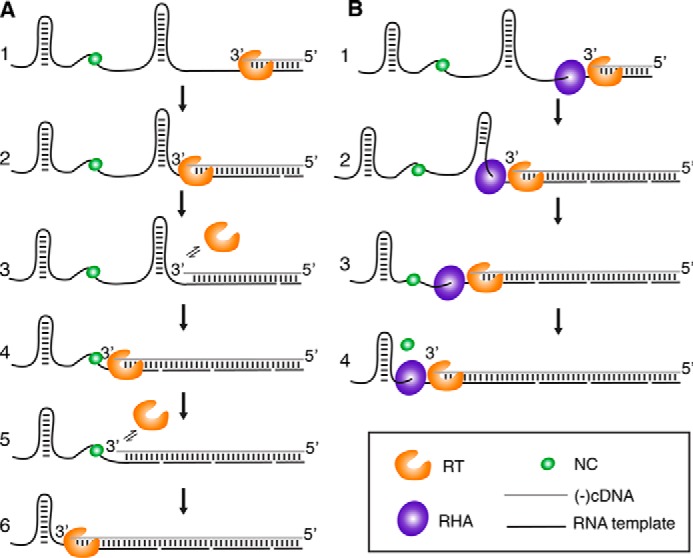
Model of how RHA is enhancing RT's reverse transcription efficiency. A, in the absence of RHA, RT (orange) falls off the template on residues involved in RNA secondary structures and protein-bound regions. These RT pauses result in early terminated cDNA products (referred as pause products in Fig. 3C). B, RHA (purple) unwinds RNA secondary structures in a 3′ to 5′ manner and subsequently remove proteins, such as NC (green), on the template. RT remains on the RNA template as reverse transcription progresses. As a result, the processivity of RT is enhanced, and the extension efficiency is increased.
Our in vitro results recapitulate the 1:1 stoichiometry between the genomic RNA template and host RNA helicase in virions (2). Notably, the amount of reverse transcript synthesized in newly infected lymphocytes prior to uncoating diminished in direct proportion to the depletion of RHA from producer cells. We conjecture RHA-deficient virions provide a viable strategy to generate live-attenuated HIV vaccine.
Discussion
RHA is recruited during biogenesis of virion RNPs within the late phase of viral replication. The incorporated RHA enhances the rate at which genomic RNA is reverse transcribed into DNA. Hence, RHA serves a critically important function during the early phase of viral replication prior to CA uncoating. Reverse transcription efficiency of virions produced from RHA-deficient cells was attenuated and can be rescued by exogenously expressed RHA (Fig. 1). These data are in agreement with Roy et al. (3), whose work showed that RHA enhances the endogenous RT activity of virions. Although it is possible that RHA may antagonize replication restriction factors in virions, here we demonstrate that under optimized in vitro conditions that involve NC, RHA directly promotes reverse transcription in the elongation phase on the RNA template. These findings are different from another study where NC was removed from the viral RNA prior to primer extension (6). Thus, strictly controlling the stoichiometry of viral and host components in the in vitro assays is likely the key to obtain primer extension data that are consistent with data from infected cells. Our in vitro assays recapitulate RHA's impact on virion reverse transcription observable in infected lymphocytes and allow us to evaluate the role of template RNA structure quantitatively.
Although the MT4 cells used in our infection assays maintained a normal RHA expression level, the new host's RHA was not able to rescue the (−)cDNA synthesis in RHA-deficient virions (Fig. 1D). These data demonstrate that RHA promotes reverse transcription in the early stage of (−)cDNA synthesis before the CA core disassociation, which is initiated after the first strand transfer (8, 32). Thus, two in vitro P/T systems that recapitulate the (−)cDNA synthesis prior to or during CA core disassociation were employed to investigate the impact of RHA in reverse transcription. The tRNALys3–5′-UTR P/T represents the synthesis of the (−)ssDNA, where RT enters a slow initiation phase with low processivity and switches to a rapid elongation phase after incorporating the first several nucleotides (10–13). The other P/T (DNA:3′-UTR) represents the synthesis of (−)cDNA after the first strand transfer, where RT directly enters the rapid elongation phase (11, 14). RHA facilitates RT activity by reducing the time necessary to produce full-length products in both P/T combinations. The rate enhancement by RHA is pronounced when the template RNA bears stable RNA structures such as G-quadruplexes (Fig. 5). Thus, data demonstrate that RHA promotes reverse transcription efficiency after initiating on the tRNALys3 primer and during the elongation phase, most likely by unwinding the secondary/tertiary structures of the RNA template (Fig. 8).
RHA has been previously reported to facilitate Gag in promoting tRNALys3 annealing to HIV-1 RNA. Xing et al. (6) showed that tRNALys3 and HIV-1 viral RNA (nt 1–386) formed three complexes, and the population distribution of each complex was sensitive to annealing conditions. In the presence of Gag, RHA requires ATP to promote formation of a slow-migrating complex, which exhibited a higher efficiency of reverse transcription initiation in protease-deficient virions. The same group later reported SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension) reactivities of the annealed complex extracted from WT and protease-deficient cells and showed that Gag is less efficient than NC in promoting tRNALys3 annealing (40). Reverse transcription occurs after Gag is proteolytically processed into matrix, CA, and NC. Hence, we examined whether RHA remodels the RNA complex annealed by NC, which is considered biologically relevant. No significant differences were observed between the RNA complexes annealed by NC in the absence and presence of RHA (Fig. 3A). Although we cannot exclude the possibility that the gel resolution was not high enough to detect subtle structural differences or that the remodeled PBS–tRNALys3 complex annealed by NC and RHA happened to migrate at the same rate as the complex annealed solely by NC, no significant difference in the rate of the early product (+6) formation was observed. Collectively, these results demonstrate that RHA does not participate in remodeling the PBS–tRNALys3 complex in a way that enhances initiation.
The dsRBDs of RHA mediate recruitment of RHA into HIV-1 virions through specific interactions with HIV-1 RNA elements near the reverse transcription initiation site (3, 5). Alanine substitution of surface exposed lysine residues in either of the two dsRBDs significantly diminish affinity for retrovirus 5′-UTR RNA (41). It is unclear whether other helicases could also promote reverse transcription, but the dsRBDs of RHA ensure specific co-assembly of RHA into HIV-1 virions. This specific interaction is utilized until the helicase activity is initiated, after which the interaction becomes transient. In addition, the nucleotides recognized by the dsRBDs are likely to undergo large structural rearrangements upon tRNALys3 annealing, which could lead to the dissociation of the RHA from the RNA, allowing RHA to processively unwind RNA template.
During the early stage of HIV-1 infection, RHA participates in reverse transcription by promoting the rate of RT elongation on the RNA template. The helicase activity is likely required to unwind RNA secondary/tertiary structures to enhance the processivity of RT during elongation. Because RHA does not affect RT's incorporation of one nucleotide under steady-state and pre–steady-state conditions, the rate enhancement by RHA must occur after initiation. During early infection, RT copies the 5′-UTR of the genome, which is a highly structured RNA that binds NC specifically and nonspecifically (28–30, 42, 43). RHA likely unwinds the structured RNA and removes NC from the template, allowing for efficient production of (−)ssDNA (Fig. 8).
Polymerases collaboratively couple with helicases and rely on their NTP hydrolysis in both prokaryotic and eukaryotic DNA replication (44, 45). Some RNA and DNA viruses encode helicases that modulate RNA structures during replication (46, 47). For example, the nonstructural protein 3 of flaviviruses, belonging to the helicase superfamily 2, like RHA, assists RNA-dependent RNA polymerase in viral nucleic acid synthesis (48, 49). The nonstructural protein 13 of coronaviruses and 2CATPase in human enterovirus, belonging to superfamily 1 and 3 helicases, respectively, are a group of helicases that utilize the energy from ATP hydrolysis to unwind nucleic acid structures during synthesis of the viral genome and the sub-genomic mRNAs (50, 51). Some RNA viruses, such as influenza A virus, tomato bushy stunt virus, and Flock House virus, do not code their own helicases but recruit host helicases to enhance genome replication (47, 52–54). It seems retroviruses employ a similar strategy, because they hijack host helicases in the nucleus and cytoplasm for efficient gene expression (55). Because the access to host helicases is limited within an intact viral core, it is thus necessary to recruit host helicase(s) during packaging to facilitate the early stage of reverse transcription. The existence of an ATP-dependent RNA helicase DDX1-like protein in simian immunodeficiency virus is probably for this reason (56). Similarly, RHA co-assembles with the HIV-1 RNP complex during packaging (5), and here we show that it enhances RT's processivity and increases reverse transcription efficiency on the RNA template during the elongation stage.
Disrupting the timing and localization of CA core uncoating is detrimental to viral infectivity (57), because it affects multiple replication steps including the nuclear translocation of the preintegration complex, such as proviral DNA integration and host responses (58–61). CA core uncoating is a highly regulated process that initiates after the first strand transfer of reverse transcription, because inhibiting the strand transfer step by inactivating the RNase H activity led to a delayed uncoating in the cytoplasm (8, 32). In the absence of RHA, the synthesis of (−)ssDNA is inefficient, which may delay the first strand transfer and the subsequent uncoating. RT is sensitive to RNA secondary/tertiary structures, which results in pauses of reverse transcription. Although this could allow time for recombination during strand transfer (62, 63), the optimal timing of generating the accurately sized product in an efficient manner is essential. The coordination between RHA-promoting reverse transcription and subsequent early phase replication events should be investigated for a comprehensive understanding of RHA's role during HIV-1 replication.
Experimental procedures
Plasmids
The expression plasmid expressing siRNA-resistant RHA was generated by two rounds of site-directed mutagenesis on pcDNA3.1 FLAG-RHA with the primer sets (Table S1), which introduced silent codon changes: isoleucine 903, histidine 904, arginine 905, asparagine 906, arginine 928, glycine 930, glycine 931, and glutamic acid 933. Plasmid sequences were verified by Sanger sequencing (Sequencing Core, Ohio State University). The plasmids used as templates for in vitro transcription were generated by amplifying the corresponding region in HIV-1 pNL4–3 (National Institutes of Health AIDS Reagent Program) and fusing it to a pUC19 vector. Plasmid sequences were verified by Sanger sequencing (DNA Core, University of Missouri, Columbia, MO). The HIV-1 RT plasmid was a kind gift from Dr. Stefan Sarafianos. The p66 and p51 of HIV-1 subtype B (GenBankTM accession no. K03455) were cloned in the pETDuet-1 vector (Novagen). The RT protein was expressed in BL21(DE3)-pLysS (Invitrogen) and purified as described elsewhere (64).
Cell lines, transfection, and virus propagation
HEK293 cells were maintained in Eagle's minimum essential medium (ATCC) containing 10% fetal bovine serum (Gibco) and 1× antibiotic–antimycotic (Gibco) and seeded in 6-well plates for transfection (1 × 106/well). The cells were transfected using Lipofectamine 2000 (Invitrogen) with either siRNAs targeting the RHA ORF or scrambled NT siRNAs (19). 8 h post-transfection, the cells were co-transfected with or without FLAG-RHA resistant to the anti-RHA siRNA (19). The cells were cultured for 16 h and co-transfected with 1 μg of the pNL4–3 molecular clone using 3 μl/μg plasmid XtremeGene (Roche) in OptiMEM (250 μl). 24 h post-transfection, the cells and cell-free medium were collected and lysed in ice-cold lysis buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 2 mm EDTA, 1% Nonidet P-40). Soluble proteins were collected in a centrifuge at 12,000 rpm for 1 min. Equivalent volumes of cell lysate were subjected to Western blotting with the following primary antibodies: anti-RHA, anti-FLAG, anti-HIV-1 Gag, and anti-GAPDH, and the protein–antibody complexes were detected by enhanced chemiluminescence (Amersham Biosciences). Cell-free medium was subjected to Gag p24 ELISA (XpressBio catalog no. XB-1000).
Infection and quantification of HIV-1 reverse-transcription intermediates
MT4 lymphocytes (5 × 105 cells in 0.5 ml of RPMI medium) were seeded in 12-well plates and spinoculated for 2 h with equivalent virus (200 ng of Gag p24) in cell-free medium. The negative control samples were heated to 100 °C for 5 min, cooled to room temperature, centrifuged at 12,000 rpm for 1 min, and treated with DNase I for 1 h. The MT4 cells were washed twice in RPMI medium and cultured for another 2 h; then cells were collected and washed twice with phosphate-buffered saline, and DNA was extracted using QIAamp DNA blood mini kit (Qiagen). 600 ng of cellular DNA was evaluated in qPCRs with HIV-1–specific primer sets that detect early and late products of HIV-1 reverse transcription, as described (65). The abundance of copies was determined relative to standard curves generated on gene-specific PCR amplicons minus negative control, heat-inactivated virus infection. The values are the averages ± standard deviation of three biological replicate experiments.
In vitro RNA transcription
PCR products served as templates for in vitro T7 transcriptions. The corresponding plasmid template was amplified using primers (Table S1) for the 5′-UTR (nt 1–344) and 3′-UTR (nt 8932–9155) primer extension templates. Trial transcription reactions using varying concentrations of MgCl2 and NTPs were performed to determine optimal conditions prior to large-scale RNA transcription. Remaining components included transcription buffer (40 mm Tris-HCl, pH 8.0, 5 mm DTT, 10 mm spermidine, 0.01% (v/v) Triton X-100), and double-distilled H2O to a total volume of 8–10 ml. The transcription reactions were carried out at 37 °C for 3 h, quenched with 25 mm EDTA and 1 m urea, mixed with 10% (v/v) of 10% glycerol, and run on 6% denaturing acrylamide gels at 20 W overnight. RNA was visualized by UV shadowing, extracted from the gel through electroelution (Elutrap, Whatman), and washed in Amicon ultracentrifugal filters.
The Cy3-labeled tRNALys3 primer was made by transcribing tRNALys3 in a NTP mixture containing 1:10 ratio of GTP to GMP and regular ATP, UTP, and CTP. To label it with a 5′-Cy3 fluorescence tag, 18 nmol of GMP-primed tRNALys3 was lyophilized and resuspended in 75 μl of reaction buffer (10 mm phosphate buffer, pH 7.2, 10 mm EDTA, 150 mm NaCl). The resuspended tRNALys3 solution was then added to a tube containing 12.5 mg of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide, immediately followed by adding 50 μl of 0.25 m ethylenediamine buffer (dissolved in 0.1 m imidazole, pH 6.0). The reaction was incubated overnight at 37 °C. The RNA sample was then washed using Amicon ultracentrifugal filters with three bed volumes of RNA reaction buffer, concentrated to 50 μm, and 200-μl aliquots were mixed with 160 μl 0.5 mm Cy3-NHS (Lumiprobe) in 10% DMSO and 40 μl of 1 m PBS (pH 8). The solution was placed at 37 °C for 4 h. The RNA was separated from the unincorporated Cy3-NHS dye using a NAP5 desalting column (GE Healthcare).
Purification of RHA and HIV-1 RT
Human DHX9/RHA was cloned with a His-tag and a mocr tag appended to the N terminus, and recombinant protein was expressed in the High Five insect cell line. The cells were cultured, induced, and collected. The pellet was resuspended in lysis buffer (20 mm Hepes, pH 7.5, 300 mm KCl, 10 mm NaCl, 1 mm MgCl2, 5 mm imidazole, 150 mm urea, 10% glycerol, 1 mm tris(2-carboxyethyl)phosphine, 1× protease inhibitor mixture (Sigma). The cells were lysed in a French press, and the soluble fraction was collected and filtered through a 0.22-μm filter for His-tag affinity chromatography (cOmplete, Roche). The HIS-mocr-RHA protein was eluted with lysis buffer containing 50 mm imidazole, concentrated, and buffer-exchanged into storage buffer (20 mm Hepes, pH 7.5, 300 mm KCl, 10 mm NaCl, 1 mm MgCl2, 10% glycerol, 1 mm tris(2-carboxyethyl)phosphine) using centrifugal filters (Pall). The purity was assessed using 4–20% ExpressPlusTM PAGE gels (GenScript), and the bands were visualized using Coomassie Blue staining.
Recombinant RHA helicase assay
A 3′-FTSC–labeled 34-nt RNA was annealed to a 78-nt unlabeled RNA by boiling and then slow cooling the RNAs to room temperature in the presence of 140 mm KCl, 10 mm NaCl, 1 mm MgCl2, 10 mm Tris-HCl, pH 7.5. The annealed RNAs were then mixed to a final reaction concentration of 42 nm with 333 nm ATP, 1.67 μm unlabeled 34-nt RNA, 100 mm KCl, 3.8 mm NaCl, 2.5 mm MgCl2, 3.8 mm Tris-HCl, pH 7.5. The reaction was initiated with 2.3 μg of RHA (900 nm) and carried out at 37 °C for the allotted time: 0, 2, 5, 10, 15, and 20 min. The reactions were quenched with 3 μl of 100 mg/ml SDS and 3 μl of 370 mm EDTA with 20 μg of proteinase K (Gold Biotechnology) and incubated at 37 °C for 5 min. The reactions were then mixed with phenol-chloroform at a 1:2 (v/v) ratio to remove RHA. The samples were then centrifuged for 3 min at 21,100 × g, and the aqueous phase of the solution was collected. Glycerol was added to the extracted RNA samples to a final concentration of 7%, and they were loaded onto a 14% native PAGE at room temperature with 1× Tris–boric acid. The gels were imaged using the Typhoon FLA 9000 biomolecular imager.
Annealing PBS segment and tRNALys3 with NC
The PBS segment and tRNALys3 were annealed by mixing 1 μm of each RNA, 140 mm KCl, 10 mm NaCl, 1 mm MgCl2, 10 mm Tris-HCl (pH 7.5), 30 μm NC and in the absence and presence of 1 μm RHA. After mixing, the samples were incubated at 37 °C for 1 h. The samples were treated with 20 μg of proteinase K (Gold Biotechnology) and incubated at 37 °C for 20 min. The reaction was quenched with 3 μl of 100 mg/ml of SDS, and the samples were phenol-extracted. The samples were mixed with 2 μl of 50% glycerol, loaded onto a 2.5% native agarose gel (1× Tris–boric acid) containing ethidium bromide, and run at 150 V for 1 h.
Primer extension assay
The 5′-UTR was annealed to the Cy3-tRNALys3 primer by mixing 2 μm of each RNA and incubating at 37 °C for 30 min in the presence of 140 mm KCl, 10 mm NaCl, 1 mm MgCl2, 10 mm Tris-HCl (pH 7.5), 55 μm NC, and 1.3 units/μl of RNase inhibitor (Roche Diagnostics). The annealed complex was then mixed in the presence and absence of 1 μm RHA to a final reaction concentration of 333 nm annealed complex, 0.33 mm dNTPs, 2 mm MgCl2, 70 mm NaCl, and 20 mm Hepes, pH 7.5. To initiate reverse transcription, 3 μm of HIV-1 RT was added to the reaction mixtures and placed in 37 °C incubator for the allotted time: 0, 1, 2, 4, 6, 10, 15, 20, and 30 min. To quench the reaction and remove NC from the template, 3 μl of 370 mm EDTA with 20 μg of proteinase K (Gold Biotechnology) were added to the mixtures and incubated for another 5 min at 37 °C. The reaction mixtures were then mixed with 100 mg/ml of SDS and boiled for 5 min at 95 °C. Proteins were removed from the reaction mixtures by phenol-chloroform extraction at a 1:2 (v/v) ratio. The samples were then centrifuged for 3 min at 21,100 × g, and the aqueous phase of the solution was collected and mixed with sample loading buffer (90% formamide v/v, 50 mm EDTA, bromphenol blue, and xylene cyanol). The samples were then boiled for 5 min and loaded onto a 10% urea–PAGE at 35 mAmps. The gels were imaged using the Typhoon FLA 9000 biomolecular imager. The +6 primer extension assays were carried out by replacing dATP by ddATP to terminate the primer extension at the +6 nt position. The +1 primer extension assays were carried out by replacing the dNTP mixture with only dTTP to terminate the primer extension at the +1 position. The samples were run on a 20% denaturing gel at 5 mAmps. Nonlinear square fitting using the BoxLucas function (see details under “Results”) was performed for kobs calculation (Origin 7).
Determination of primer–template binding affinity (Kd,RNA)
The primer–template binding affinity was determined using pre–steady-state kinetics conditions in which 50 nm RT was incubated with increasing concentrations of the 3′-UTR–primer (10, 20, 40, 80, and 160 nm) annealed complex in 25 mm Hepes (pH 7.8) buffer containing 80 mm NaCl, in the presence and absence of 50 nm RHA. The reactions were initiated by rapidly mixing a solution containing 2.5 mm MgCl2 and 100 μm dTTP in a rapid quench-flow instrument (RQF-3, KinTek Corp., Austin, TX). The reactions were allowed to proceed for 0.25 s and quenched by the addition of 10-fold excess EDTA. The samples were lyophilized overnight and resuspended in 34 μl of water. The samples were mixed with gel loading dye (95% formamide, 50 mm EDTA, and bromphenol blue), and the products were resolved on a 20% polyacrylamide, 8 m urea gel. The products were visualized on the Typhoon FLA 9000 biomolecular imager, and the product bands were quantitated by ImageQuant TL 1.2 (GE Healthcare). The Kd,RNA was determined by plotting the amount of extended primer against the concentration of the P/T. The data points were fit to a quadratic function as described previously (35). The quadratic equation used to fit the data is.
| (Eq. 3) |
Determination of dissociation rate (Koff)
The dissociation rate of RT was determined using steady-state kinetics conditions in which 30 nm RT was incubated at 37 °C with 30 nm 3′-UTR–primer annealed complex in 25 mm Hepes (pH 7.8) buffer containing 80 mm NaCl, in the absence or presence of 30 nm RHA. Reaction mixtures were then incubated with 300 nm dsDNA at varying time points (0, 5, 20, 40, 80, 160, and 320 s). The reactions were initiated by rapidly mixing the solutions with 5 mm MgCl2 and 100 μm dTTP in a rapid quench-flow instrument (RQF-3, KinTek Corp., Austin, TX). The reactions were allowed to proceed for 10 s and quenched by the addition of 50 mm EDTA. The samples were boiled for 5 min and mixed with gel loading dye at a 1:1 ratio (95% formamide, 50 mm EDTA, and bromphenol blue), and the products were resolved on a 20% polyacrylamide, 8 m urea gel. The products were visualized on the Typhoon FLA 9000 biomolecular imager, and the product bands were quantitated by ImageQuant TL 1.2 (GE Healthcare). The data were fit to a single exponential decay equation to determine Koff (11).
Single-turnover processivity assay
The 3′-UTR–primer was annealed by incubating 2 μm of T/P with 32 μm of NC protein at 37 °C for 30 min. The T/P was then mixed to a final concentration of 200 nm with 400 nm RT (with or without equimolar RHA). The reactions were then initiated with 100 μm dNTPs, 5 mm MgCl2, and 0.3 μg of dsDNA trap and ran at varying time points (0.01, 20, 40, 60, 90, 120, 180, 240, and 300 s). The reactions were quenched with 500 mm EDTA, pH 8.0, and then the samples were boiled for 5 min and mixed with gel loading dye at a 1:1 ratio (95% formamide, 50 mm EDTA, and bromphenol blue), and the products were resolved on a 20% polyacrylamide, 8 m urea gel. The products were visualized on the Typhoon FLA 9000 biomolecular imager, and the product bands were quantitated by ImageQuant TL 1.2 (GE Healthcare). The processivity index was obtained by fitting the intensities of the +6 product without RHA bands to a double exponential equation:
| (Eq. 4) |
where P is the amount of 25-mer, A is the amplitude, k1 is the rate of product appearance, k2 is the rate of decay, and C is a constant (37, 38). However, all of the other values were fit to a single exponential decay equation.
| (Eq. 5) |
Author contributions
S. B., Z. S., K. B.-L., and X. H. conceptualization; S. B., G. S., K. W., and B. T. data curation; S. B., G. S., and K. B.-L. formal analysis; S. B. investigation; S. B., G. S., C. B., Z. S., I. B., K. S., and K. B.-L. methodology; S. B. and X. H. writing-original draft; S. B., G. S., K. B.-L., and X. H. writing-review and editing; Z. S., W.C. B., and X. H. resources; K. B.-L. and X. H. funding acquisition; X. H. supervision; X. H. project administration.
Supplementary Material
Acknowledgments
We appreciate Donald Burke-Agüero for kindly allowing us to use the KinTek rapid quench flow instrument. We thank Anna Davis in the Singh laboratory for training and helping us run the sequencing gels.
This work was supported by National Institutes of Health Grant R01AI150460 (reassigned from R01GM119932; to X. H. and K.-B.-L.) and R01CA108882 (to K. B.-L). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1.
- HIV-1
- human immunodeficiency virus, type 1
- RHA
- RNA helicase A
- RNP
- reverse-transcription ribonucleoprotein
- RT
- reverse transcriptase
- CA
- capsid
- nt
- nucleotide(s)
- PBS
- primer-binding site
- NC
- nucleocapsid
- dsRBD
- dsRNA-binding domain
- qPCR
- quantitative PCR
- NT siRNA
- nontargeting siRNA
- P/T
- primer–template
- ddATP
- dideoxyATP
- FTSC
- fluorescein-5-thiosemicarbazide.
References
- 1. Santos S., Obukhov Y., Nekhai S., Bukrinsky M., and Iordanskiy S. (2012) Virus-producing cells determine the host protein profiles of HIV-1 virion cores. Retrovirology 9, 65 10.1186/1742-4690-9-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sharma A., and Boris-Lawrie K. (2012) Determination of host RNA helicases activity in viral replication. Methods Enzymol. 511, 405–435 10.1016/B978-0-12-396546-2.00019-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roy B. B., Hu J., Guo X., Russell R. S., Guo F., Kleiman L., and Liang C. (2006) Association of RNA helicase a with human immunodeficiency virus type 1 particles. J. Biol. Chem. 281, 12625–12635 10.1074/jbc.M510596200 [DOI] [PubMed] [Google Scholar]
- 4. Bolinger C., Sharma A., Singh D., Yu L., and Boris-Lawrie K. (2010) RNA helicase A modulates translation of HIV-1 and infectivity of progeny virions. Nucleic Acids Res. 38, 1686–1696 10.1093/nar/gkp1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boeras I., Song Z., Moran A., Franklin J., Brown W. C., Johnson M., Boris-Lawrie K., and Heng X. (2016) DHX9/RHA binding to the PBS-segment of the genomic RNA during HIV-1 assembly bolsters virion infectivity. J. Mol. Biol. 428, 2418–2429 10.1016/j.jmb.2016.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xing L., Liang C., and Kleiman L. (2011) Coordinate roles of Gag and RNA helicase A in promoting the annealing of tRNALys3 to HIV-1 RNA. J. Virol. 85, 1847–1860 10.1128/JVI.02010-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coffin J. M., Hughes S. H., and Varmus H. E. (1997) Retroviruses, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 8. Cosnefroy O., Murray P. J., and Bishop K. N. (2016) HIV-1 capsid uncoating initiates after the first strand transfer of reverse transcription. Retrovirology. 13, 58 10.1186/s12977-016-0292-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gilboa E., Mitra S. W., Goff S., and Baltimore D. (1979) A detailed model of reverse transcription and tests of crucial aspects. Cell 18, 93–100 10.1016/0092-8674(79)90357-X [DOI] [PubMed] [Google Scholar]
- 10. Isel C., Lanchy J.-M., Le Grice S. F., Ehresmann C., Ehresmann B., and Marquet R. (1996) Specific initiation and switch to elongation of human immunodeficiency virus type 1 reverse transcription require the post-transcriptional modifications of primer tRNA3Lys. EMBO J. 15, 917–924 10.1002/j.1460-2075.1996.tb00426.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lanchy J.-M., Ehresmann C., Le Grice S. F., Ehresmann B., and Marquet R. (1996) Binding and kinetic properties of HIV-1 reverse transcriptase markedly differ during initiation and elongation of reverse transcription. EMBO J. 15, 7178–7187 10.1002/j.1460-2075.1996.tb01109.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lanchy J.-M., Keith G., Le Grice S. F., Ehresmann B., Ehresmann C., and Marquet R. (1998) Contacts between reverse transcriptase and the primer strand govern the transition from initiation to elongation of HIV-1 reverse transcription. J. Biol. Chem. 273, 24425–24432 10.1074/jbc.273.38.24425 [DOI] [PubMed] [Google Scholar]
- 13. Arts E. J., Stetor S. R., Li X., Rausch J. W., Howard K. J., Ehresmann B., North T. W., Wöhrl B. M., Goody R. S., Wainberg M. A., and Le Grice S. F. (1996) Initiation of (−) strand DNA synthesis from tRNA(3Lys) on lentiviral RNAs: implications of specific HIV-1 RNA–tRNA(3Lys) interactions inhibiting primer utilization by retroviral reverse transcriptases. Proc. Natl. Acad. Sci. U.S.A. 93, 10063–10068 10.1073/pnas.93.19.10063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lanchy J.-M., Isel C., Ehresmann C., Marquet R., and Ehresmann B. (1996) Structural and functional evidence that initiation and elongation of HIV-1 reverse transcription are distinct processes. Biochimie 78, 1087–1096 10.1016/S0300-9084(97)86734-X [DOI] [PubMed] [Google Scholar]
- 15. Li X., Quan Y., Arts E. J., Li Z., Preston B. D., de Rocquigny H., Roques B. P., Darlix J.-L., Kleiman L., Parniak M. A., and Wainberg M. A. (1996) Human immunodeficiency virus Type 1 nucleocapsid protein (NCp7) directs specific initiation of minus-strand DNA synthesis primed by human tRNA(Lys3) in vitro: studies of viral RNA molecules mutated in regions that flank the primer binding site. J. Virol. 70, 4996–5004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo F., Saadatmand J., Niu M., and Kleiman L. (2009) Roles of Gag and NCp7 in facilitating tRNA(Lys3) annealing to viral RNA in human immunodeficiency virus type 1. J. Virol. 83, 8099–8107 10.1128/JVI.00488-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Warren K., Warrilow D., Meredith L., and Harrich D. (2009) Reverse transcriptase and cellular factors: regulators of HIV-1 reverse transcription. Viruses 1, 873–894 10.3390/v1030873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robb G. B., and Rana T. M. (2007) RNA helicase A interacts with RISC in human cells and functions in RISC loading. Mol. Cell 26, 523–537 10.1016/j.molcel.2007.04.016 [DOI] [PubMed] [Google Scholar]
- 19. Hartman T. R., Qian S., Bolinger C., Fernandez S., Schoenberg D. R., and Boris-Lawrie K. (2006) RNA helicase A is necessary for translation of selected messenger RNAs. Nat. Struct. Mol. Biol. 13, 509–516 10.1038/nsmb1092 [DOI] [PubMed] [Google Scholar]
- 20. Koh H. R., Xing L., Kleiman L., and Myong S. (2014) Repetitive RNA unwinding by RNA helicase A facilitates RNA annealing. Nucleic Acids Res. 42, 8556–8564 10.1093/nar/gku523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chakraborty P., and Grosse F. (2011) Human DHX9 helicase preferentially unwinds RNA-containing displacement loops (R-loops) and G-quadruplexes. DNA Repair 10, 654–665 10.1016/j.dnarep.2011.04.013 [DOI] [PubMed] [Google Scholar]
- 22. Lee C. G., and Hurwitz J. (1992) A new RNA helicase isolated from HeLa cells that catalytically: translocates in the 3′ to 5′ direction. J. Biol. Chem. 267, 4398–4407 [PubMed] [Google Scholar]
- 23. Schütz P., Wahlberg E., Karlberg T., Hammarström M., Collins R., Flores A., and Schüler H. (2010) Crystal structure of human RNA helicase A (DHX9): Structural basis for unselective nucleotide base binding in a DEAD-box variant protein. J. Mol. Biol. 400, 768–782 10.1016/j.jmb.2010.05.046 [DOI] [PubMed] [Google Scholar]
- 24. Zhang S., and Grosse F. (1997) Domain structure of human nuclear DNA helicase II (RNA helicase A). J. Biol. Chem. 272, 11487–11494 10.1074/jbc.272.17.11487 [DOI] [PubMed] [Google Scholar]
- 25. Fuller-Pace F. V. (2006) DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 34, 4206–4215 10.1093/nar/gkl460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Butsch M., and Boris-Lawrie K. (2000) Translation is not required to generate virion precursor RNA in human immunodeficiency virus type 1–infected T cells. J. Virol. 74, 11531–11537 10.1128/JVI.74.24.11531-11537.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bolinger C., Yilmaz A., Hartman T. R., Kovacic M. B., Fernandez S., Ye J., Forget M., Green P. L., and Boris-Lawrie K. (2007) RNA helicase A interacts with divergent lymphotropic retroviruses and promotes translation of human T-cell leukemia virus type 1. Nucleic Acids Res. 35, 2629–2642 10.1093/nar/gkm124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Karpel R. L., Henderson L. E., and Oroszlan S. (1987) Interactions of retroviral structural proteins with single-stranded nucleic acids. J. Biol. Chem. 262, 4961–4967 [PubMed] [Google Scholar]
- 29. Lu K., Heng X., Garyu L., Monti S., Garcia E. L., Kharytonchyk S., Dorjsuren B., Kulandaivel G., Jones S., Hiremath A., Divakaruni S. S., LaCotti C., Barton S., Tummillo D., Hosic A., et al. (2011) NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science 334, 242–245 10.1126/science.1210460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heng X., Kharytonchyk S., Garcia E. L., Lu K., Divakaruni S. S., LaCotti C., Edme K., Telesnitsky A., and Summers M. F. (2012) Identification of a minimal region of the HIV-1 5′-leader required for RNA dimerization, NC binding, and packaging. J. Mol. Biol. 417, 224–239 10.1016/j.jmb.2012.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Driscoll M. D., and Hughes S. H. (2000) Human immunodeficiency virus type 1 nucleocapsid protein can prevent self-priming of minus-strand strong stop DNA by promoting the annealing of short oligonucleotides to hairpin sequences. J. Virol. 74, 8785–8792 10.1128/JVI.74.19.8785-8792.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mamede J. I., Cianci G. C., Anderson M. R., and Hope T. J. (2017) Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc. Natl. Acad. Sci. U.S.A. 114, E7169–E7178 10.1073/pnas.1706245114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Piekna-Przybylska D., Sullivan M. A., Sharma G., and Bambara R. A. (2014) U3 region in the HIV-1 genome adopts a G-quadruplex structure in its RNA and DNA sequence. Biochemistry 53, 2581–2593 10.1021/bi4016692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Venczel E. A., and Sen D. (1993) Parallel and antiparallel G-DNA structures from a complex telomeric sequence. Biochemistry 32, 6220–6228 10.1021/bi00075a015 [DOI] [PubMed] [Google Scholar]
- 35. Singh K., Marchand B., Rai D. K., Sharma B., Michailidis E., Ryan E. M., Matzek K. B., Leslie M. D., Hagedorn A. N., Li Z., Norden P. R., Hachiya A., Parniak M. A., Xu H. T., Wainberg M. A., et al. (2012) Biochemical mechanism of HIV-1 resistance to rilpivirine. J. Biol. Chem. 287, 38110–38123 10.1074/jbc.M112.398180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kati W. M., Johnson K. A., Jerva L. F., and Anderson K. S. (1992) Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 267, 25988–25997 [PubMed] [Google Scholar]
- 37. Ndongwe T. P., Adedeji A. O., Michailidis E., Ong Y. T., Hachiya A., Marchand B., Ryan E. M., Rai D. K., Kirby K. A., Whatley A. S., Burke D. H., Johnson M., Ding S., Zheng Y.-M., Liu S.-L., et al. (2012) Biochemical, inhibition and inhibitor resistance studies of xenotropic murine leukemia virus-related virus reverse transcriptase. Nucleic Acids Res. 40, 345–359 10.1093/nar/gkr694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Patel S. S., Wong I., and Johnson K. A. (1991) Pre–steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry 30, 511–525 10.1021/bi00216a029 [DOI] [PubMed] [Google Scholar]
- 39. Wang X., Ao Z., Chen L., Kobinger G., Peng J., and Yao X. (2012) The cellular antiviral protein APOBEC3G interacts with HIV-1 reverse transcriptase and inhibits its function during viral replication. J. Virol. 86, 3777–3786 10.1128/JVI.06594-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Seif E., Niu M., and Kleiman L. (2015) In virio SHAPE analysis of tRNALys3 annealing to HIV-1 genomic RNA in wild type and protease-deficient virus. Retrovirology 12, 40 10.1186/s12977-015-0171-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ranji A., Shkriabai N., Kvaratskhelia M., Musier-Forsyth K., and Boris-Lawrie K. (2011) Features of double-stranded RNA-binding domains of RNA helicase A are necessary for selective recognition and translation of complex mRNAs. J. Biol. Chem. 286, 5328–5337 10.1074/jbc.M110.176339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Keane S. C., Heng X., Lu K., Kharytonchyk S., Ramakrishnan V., Carter G., Barton S., Hosic A., Florwick A., Santos J., Bolden N. C., McCowin S., Case D. A., Johnson B. A., Salemi M., et al. (2015) Structure of the HIV-1 RNA packaging signal HHS public access. Science 348, 917–921 10.1126/science.aaa9266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kutluay S. B., Zang T., Blanco-Melo D., Powell C., Jannain D., Errando M., and Bieniasz P. D. (2014) Global changes in the RNA binding specificity of HIV-1 Gag regulate virion genesis. Cell 159, 1096–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Langston L. D., and O'Donnell M. (2006) DNA replication: keep moving and don't mind the gap. Mol. Cell 23, 155–160 10.1016/j.molcel.2006.05.034 [DOI] [PubMed] [Google Scholar]
- 45. Lohman T. M., Tomko E. J., and Wu C. G. (2008) Non-hexameric DNA helicases and translocases: mechanisms and regulation. Nat. Rev. Mol. Cell Biol. 9, 391–401 10.1038/nrm2394 [DOI] [PubMed] [Google Scholar]
- 46. Kadaré G., and Haenni A. (1997) Virus-encoded RNA helicases. J. Virol. 71, 2583–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ranji A., and Boris-Lawrie K. (2010) RNA helicases. RNA Biol. 7, 775–787 10.4161/rna.7.6.14249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matusan A. E., Pryor M. J., Davidson A. D., and Wright P. J. (2001) Mutagenesis of the Dengue virus type 2 NS3 protein within and outside helicase motifs: effects on enzyme activity and virus replication. J. Virol. 75, 9633–9643 10.1128/JVI.75.20.9633-9643.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lam A. M., and Frick D. N. (2006) Hepatitis C virus subgenomic replicon requires an active NS3 RNA helicase. J. Virol. 80, 404–411 10.1128/JVI.80.1.404-411.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tanner J. A., Watt R. M., Chai Y.-B., Lu L.-Y., Lin M. C., Peiris J. S., Poon L. L., Kung H.-F., and Huang J.-D. (2003) The severe acute respiratory syndrome (SARS) coronavirus NTPasefhelicase belongs to a distinct class of 5′ to 3′ viral helicases. J. Biol. Chem. 278, 39578–39582 10.1074/jbc.C300328200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xia H., Wang P., Wang G.-C., Yang J., Sun X., Wu W., Qiu Y., Shu T., Zhao X., Yin L., Qin C.-F., Hu Y., and Zhou X. (2015) Human enterovirus nonstructural protein 2CATPase functions as both an RNA helicase and ATP-independent RNA chaperone. PLOS Pathog. 11, e1005067 10.1371/journal.ppat.1005067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kovalev N., Barajas D., and Nagy P. D. (2012) Similar roles for yeast Dbp2 and Arabidopsis RH20 DEAD-box RNA helicases to Ded1 helicase in tombusvirus plus-strand synthesis. Virology 432, 470–484 10.1016/j.virol.2012.06.030 [DOI] [PubMed] [Google Scholar]
- 53. Kovalev N., Pogany J., and Nagy P. D. (2012) A co-opted DEAD-box RNA helicase enhances tombusvirus plus-strand synthesis. PLoS Pathog. 8, e1002537 10.1371/journal.ppat.1002537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lin L., Li Y., Pyo H.-M., Lu X., Raman S. N., Liu Q., Brown E. G., and Zhou Y. (2012) Identification of RNA helicase A as a cellular factor that interacts with influenza A virus NS1 protein and its role in the virus life cycle. J. Virol. 86, 1942–1954 10.1128/JVI.06362-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Singh G., Heng X., and Boris-Lawrie K. (2018) Cellular RNA helicases support early and late events in retroviral replication. In Retrovirus–Cell Interactions (Parent L. J., ed) 1st Ed., pp. 253–271, Academic Press, Orlando, FL [Google Scholar]
- 56. Stephenson S. T., Bostik P., Song B., Rajan D., Bhimani S., Rehulka P., Mayne A. E., and Ansari A. A. (2010) Distinct host cell proteins incorporated by SIV replicating in CD4+ T cells from natural disease resistant versus non-natural disease susceptible hosts. Retrovirology 7, 107 10.1186/1742-4690-7-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Forshey B. M., von Schwedler U., Sundquist W. I., and Aiken C. (2002) Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76, 5667–5677 10.1128/JVI.76.11.5667-5677.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schaller T., Ocwieja K. E., Rasaiyaah J., Price A. J., Brady T. L., Roth S. L., Hué S., Fletcher A. J., Lee K., KewalRamani V. N., Noursadeghi M., Jenner R. G., James L. C., Bushman F. D., and Towers G. J. (2011) HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 7, e1002439 10.1371/journal.ppat.1002439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Matreyek K. A., and Engelman A. (2011) The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J. Virol. 85, 7818–7827 10.1128/JVI.00325-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lelek M., Casartelli N., Pellin D., Rizzi E., Souque P., Severgnini M., Di Serio C., Fricke T., Diaz-Griffero F., Zimmer C., Charneau P., and Di Nunzio F. (2015) Chromatin organization at the nuclear pore favours HIV replication. Nat. Commun. 6, 6483 10.1038/ncomms7483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lahaye X., and Manel N. (2015) Viral and cellular mechanisms of the innate immune sensing of HIV. Curr. Opin. Virol. 11, 55–62 10.1016/j.coviro.2015.01.013 [DOI] [PubMed] [Google Scholar]
- 62. Derebail S. S., and DeStefano J. J. (2004) Mechanistic analysis of pause site-dependent and -independent recombinogenic strand transfer from structurally diverse regions of the HIV genome. J. Biol. Chem. 279, 47446–47454 10.1074/jbc.M408927200 [DOI] [PubMed] [Google Scholar]
- 63. Lanciault C., and Champoux J. J. (2006) Pausing during reverse transcription increases the rate of retroviral recombination. J. Virol. 80, 2483–2494 10.1128/JVI.80.5.2483-2494.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Michailidis E., Marchand B., Kodama E. N., Singh K., Matsuoka M., Kirby K. A., Ryan E. M., Sawani A. M., Nagy E., Ashida N., Mitsuya H., Parniak M. A., and Sarafianos S. G. (2009) Mechanism of inhibition of HIV-1 reverse transcriptase by 4′-ethynyl-2-fluoro-2′-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J. Biol. Chem. 284, 35681–35691 10.1074/jbc.M109.036616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mbisa J. L., Delviks-Frankenberry K. A., Thomas J. A., Gorelick R. J., and Pathak V. K. (2009) Real-time PCR analysis of HIV-1 replication post-entry events. Methods Mol. Biol. 485, 55–72 10.1007/978-1-59745-170-3_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.