

Abstract
The synthesis of bio‐based and biodegradable plastics is a hot topic in research due to growing environmental problems caused by omnipresent plastics. As a result, polylactide, which has been known for years, has seen a tremendous increase in industrial production. Nevertheless, the manufacturing process using the toxic catalyst Sn(Oct)2 is very critical. As an alternative, five zinc acetate complexes have been synthesized with Schiff base‐like ligands that exhibit high activity in the ring‐opening polymerization of non‐purified lactide. The systems bear different side arms in the ligand scaffold. The influence of these substituents has been analyzed. For a detailed description of the catalytic activities, the rate constants k app and k p were determined using in‐situ Raman spectroscopy at a temperature of 150 °C. The polymers produced have molar masses of up to 71 000 g mol−1 and are therefore suitable for a variety of applications. Toxicity measurements carried out for these complexes proved the nontoxicity of the systems.
Keywords: polylactide, catalysis, biobased materials, sustainable chemistry, ring-opening polymerization
1. Introduction
The rising littering of our planet with plastics and the increasing scarcity of crude oil pose new challenges for society.1 In addition to recycling systems and natural materials, bio‐based and biodegradable plastics are a good alternative.2 A plastic that meets both criteria is polylactide.3 Sugarcane, sugar beets or maize serve as raw material source. After a fermentation process of the material, the lactic acid is obtained, which is esterified in a subsequent condensation reaction to the cyclic dimer, being the monomer unit lactide. By a controlled ring‐opening polymerization, the corresponding polymer polylactide is then synthesized.4 The controlled ring‐opening succeeds with the aid of suitable catalysts.5 From an economic point of view, some requirements are placed on the catalyst. In addition to cost‐effective production, high activities, low concentrations of use and robustness against air and moisture are in the foreground. In addition, the turnover must be ensured at temperatures beyond 130 °C and colourless polymers are to be obtained.6 The tin octanoate (Sn(Oct)2) (Oct=OCO(CH2)6CH3) fulfils these properties and is therefore currently the most widely used catalyst in the industrial production of PLA.7 Since the catalyst is not removed after melt polymerization, it remains in the polymer and it is assumed that the tin(II) compound accumulates during the compost degradation of polylactide. For a long time, the toxicity of this tin compound has been known, so a replacement for Sn(Oct)2 is strongly advised to keep the bioplastic PLA “green” even if the catalyst remains in the polymer matrix.8 Zinc‐based catalyst systems are therefore an excellent alternative. Thus, Coates et al. developed various zinc complexes with β‐diiminates as ligand.9 Zinc aminophenolates from Ma et al.,10 Mehrkhodavandi et al.,11 Tolman et al.12 showed high activities and stereoselectivities. Hayes and Wheaton et al. developed zinc complexes bearing phosphinimines13 and Schulz et al.14 zinc ketoiminate and β‐diketiminate complexes. Different zinc alkoxides with trispyrazolyl‐ and trisindazolylborate ligands have been designed and tested by Chisholm et al.15 Zinc complexes containing OOO‐tridentate bis(phenolate) or tris(pyrazolyl) methane ligands have been applied successfully in the ROP of lactide by Mountford et al.16 In 2016 Williams et al. presented dinuclear zinc systems, which reached the highest activity in the area of zinc catalysts up to now.17 While the above‐mentioned systems have been tested mainly in solution and with purified lactide, the activity of the catalyst with non‐purified lactide, low catalyst concentrations and high temperatures is an important criterion for industrial use. Along the way, Davidson et al. developed titanium, zirconium and hafnium aminophenolate complexes for the polymerization in melt.18 Jones et al.19 recently presented zinc aminophenolate complexes that showed high activity in melt using singly recrystallized lactide. At a ratio of [LA]:[I]:[BnOH]=10 000 : 1 : 100 and a temperature of 180 °C a conversion of 90 % as well as controlled molar masses have been reached.20 Another attractive class of ligands in this context are guanidines.21 As neutral donors they form stable and robust complexes in combination with zinc.22 In the past, several hybrid and bisguanidines with N,N donors have been reported to be good catalysts in the field of non‐purified lactide polymerization. In recent years, zinc hybrid guanidines with neutral N,O donors have come into the focus as they have significantly higher activity and produce molar masses up to 86 000 g mol−1 under industrially relevant conditions.23 Recently, iron guanidine complexes have been published, which show higher activities than pure Sn(Oct)2 using non‐purified rac‐LA at 150 °C.24
However, the search for easily accessible catalyst systems for the ROP of lactide goes on. At this point we report zinc systems containing Schiff base‐like ligand scaffolds. Their synthesis succeeds starting from commercially available substances and cost‐effectively in just one step. Various complexes were tested under industrial conditions and their activity was recorded in situ using Raman spectroscopy. An investigation of the mechanistic ring‐opening was carried out by means of MALDI‐ToF measurements.
2. Results and Discussion
2.1. Synthesis
The Zn(II) complexes were obtained by a condensation reaction between Zn(OAc)2 ⋅ 2H2O and the tridentate Schiff base‐like ligands in ethanol (HL1, HL3, and HL5) or methanol (HL2 and HL4). The tridentate ligands were synthesized by a facile condensation reaction as described previously.25 The synthesis and numbering scheme is given in Scheme 1. The acetate anion is acting as base for the deprotonation of the ligand. The coordination compounds were obtained as white, crystalline powder and their purity was confirmed by means of elemental analysis, mass spectrometry, TGA, and IR spectroscopy.
Scheme 1.
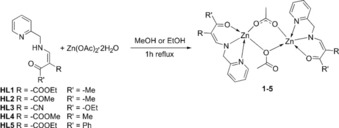
General synthetic procedure for the synthesis of the Zn(II) complexes described in this work.
2.2. X‐Ray Structure Analysis
Crystals suitable for X‐ray structure analysis were obtained for 1 by liquid‐liquid diffusion of a methanol solution of the ligand and an aqueous solution of Zn(OAc)2 ⋅ 2H2O, and for 5 from the mother liquor. The crystallographic data were collected at 133 K and are summarized in Table S1. Complex 1 crystallized in the triclinic space group P‐1, 5 in the monoclinic space group P21/c. Both complexes crystallized as dimers, with each metal centre coordinated by one tridentate ligand and two acetate anions bridging the Zn(II) centres. One anion is coordinating with only one of the two oxygen atoms, while for the other both are coordinating. The asymmetric units of both complexes are depicted in Figure 1. The bond lengths of the first coordination sphere are given in Table S2. The Zn−Npy bond lengths are slightly longer (2.15 Å in average for 1, 2.14 Å for 5) than the other bond lengths of the first coordination sphere of the Zn(II) atoms (average values: Zn−Nax 2.03 Å [1], 2.04 Å [5]; Zn−Oax 2.06 Å [1], 2.05 Å [5]; Zn−O53 2.03 Å [1], 2.04 Å [5]; Zn−O51 2.03 Å [1], 2.01 Å [5]; Zn−O52 1.98 Å [1], 1.97 Å [5]). The assignment of a single or double bond in the acetate anions is clear for the ion in which only one oxygen is bridging the Zn(II) centres (C53−1.311(5) Å/1.307(2) Å and C53−O54 1.222(5) Å/1.221(2) Å for 1 and 5, respectively), whereas for the other acetate ion the delocalization of the negative charge over both oxygen atoms results in similar bond lengths (C51−O51 1.257 (5) Å/1.250(2) Å and C51−O52 1.270(5) Å and 1.263(2) Å for 1 and 5, respectively). The distortion parameter τ helps to distinguish between a square pyramidal coordination sphere (τ close to 0) and a trigonal bipyramidal coordination sphere (τ close to 1). It is defined as (α‐β)/60, with the largest angle of the coordination sphere being α and the second largest β.26 It has similar values for the both Zn(II) atoms in complex 1 (Zn1 0.15, and Zn2 0.21), this indicates a distorted square pyramidal coordination sphere. The values for complex 5 are different for the Zn(II) atoms of this complex; 0.6 for Zn1 and 0.02 for Zn2. This indicates a nearly ideal square pyramidal geometry for Zn2.
Figure 1.
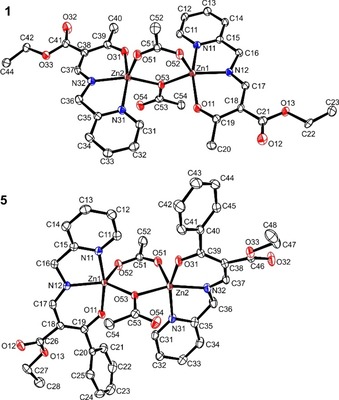
Molecular structures of complexes 1 (top) and 5 (bottom). Ellipsoids are drawn at 50 % probability level. Hydrogen atoms were omitted for clarity.
As the bond length Zn−Npy is still slightly longer compared to the remaining bond lengths in Zn1, the coordination geometry is likely to be square pyramidal as well. The significant differences in the τ values of complexes 5 can be explained with a C−H ⋅⋅⋅π interaction between an aromatic CH group of the pyridine ring of Zn2 (C32−H32) and the phenyl ring of Zn1 (see Figure S1, right); this interaction causes the tridentate ligand of Zn2 to be more bend than for Zn1. Details of all interactions are given in Table S3–S5. Pictures of the packing of the complexes in the crystal are given in Figure S1.
Powder X‐ray diffraction was performed to confirm the identical structure of the bulk and the single crystals. The diffraction patterns are given in the Supporting Information, Figure S2. It can be seen that the patterns for 1 and 5 are identical for the bulk complex and the calculated pattern for the crystal structure. Small differences can be explained with the different temperatures used for the measurements (single crystal at 133 K, powder at room temperature).
To determine the nuclearity of the complexes in solution, the conductivity of a 1.5 mM aqueous solution of compounds 2 and 4 was measured. Compared to the one of the used distilled water (1.6 μS/cm) it is enhanced (234.9 μS/cm for 2 and 217.9 μS/cm for 4). This is an indication for the formation of monomeric species in aqueous solution. The other compounds were not fully soluble in water.
2.3. Polymerization
All five complexes were tested regarding their activity in the ring‐opening polymerization of rac‐lactide (Tables 1 and 2). The corresponding polymerizations were carried out with non‐purified rac‐LA at a temperature of 150 °C. The [M]/[I] ratio was 500 : 1, assuming that both zinc atoms of one complex propagate a chain. An additional co‐initiator has been omitted. The kinetic measurements were accomplished by in situ Raman spectroscopy. In a steel reactor, the reaction progress was followed in melt at a stirring speed of 260 rpm. The kinetic evaluation was carried out by a semilogarithmic plot of the lactide concentration versus time (determination of k app). For the complexes 1, 2, 4 & 5 detailed results are given. Due to the intense fluorescence of complex 3, a kinetic study was not possible. All polymers have been characterized by gel permeation chromatography (GPC) to give information regarding their molar masses.
Table 1.
Polymerization data for rac‐LA with catalyst 2.
| [M]/[I] | k app (s−1)[b] | time (min) | conv. (%)[c] | M n,theo (g mol−1) | M n (g mol−1)[d] | PD |
|---|---|---|---|---|---|---|
| 500 | 1.14×10−3 | 25 | 62 | 45 000 | 65 000 | 1.5 |
| 625 | 8.60×10−4 | 30 | 78 | 70 000 | 54 000 | 1.8 |
| 1000 | 4.22×10−4 | 27 | 65 | 94 000 | 81 000 | 1.4 |
| 1500 | 2.23×10−4 | 61 | 57 | 123 000 | 43 000 | 1.8 |
| 2000 | 1.28×10−4 | 112 | 56 | 161 000 | 21 000 | 2.2 |
[a] Conditions: 150 °C, solvent free, non‐purified technical grade rac‐LA. [b] Determined from the slope of the plots of ln([LA]0/[LA]t) versus time. For spectra see SI. [c] As determined by 1H NMR spectroscopy. [d] Determined by GPC (in THF), M n,theo: 72 000 g mol−1 for 100 % conversion.
Table 2.
Polymerization data for rac‐LA with catalysts 1–5.[a]
| init. | k p (L mol−1 s−1)[b] | k app (s−1) [c] | time (min) | conv. (%)[d] | M n,theo (g mol−1)[e] | M n (g mol−1)[f] | PD |
|---|---|---|---|---|---|---|---|
| 1 | 1.22±0.15×10−3 | 41 | 79 | 57 000 | 62 000 | 1.6 | |
| 2 | 8.59±0.36×10−2 | 1.14±0.04×10−3 | 25 | 62 | 45 000 | 65 000 | 1.5 |
| 4 | 1.41±0.01×10−3 | 42 | 78 | 56 000 | 71 000 | 1.5 | |
| 5 | 6.08± 0.1×10−4 | 49 | 75 | 54 000 | 57 000 | 1.6 |
[a] Conditions: solvent free, non‐purified technical grade rac‐LA, 150 °C. [b] Determined by plotting k app versus [init.]. k p [I] [M]; k p=k app/[I]. [c] Determined from the slope of the plots of ln([LA]0/[LA]t) versus time for a ratio of [M]/[I]=500 : 1. [d] As determined by 1H NMR spectroscopy. [e] Calculated assuming that every zinc of each dinuclear complex propagates one chain M n,theo: 72 000 g mol−1 for 100 % conversion at a ratio of [M]/[I]=500 : 1. [f] Determined by GPC (in THF).
Regarding the different values for k app of the four different catalysts, it is clear that 5 is the slowest with a k app=6.08±0.1×10−4 s−1. On the other hand, the other complexes 1, 2 & 4 with values of k app=1.22±0.15×10−3 s−1 (1), k app=1.14±0.04×10−3 s−1 (2) & k app=1.41±0.01×10−3 s−1 (4) are of identical orders of magnitude. To understand the slower activity of 5, it helps to look at the structure of the complex. While the complexes 1, 2 & 4 bear short esters or an aldehyde plus a methyl group, complex 5 has an ester‐ and a phenyl group attached. This results in a higher steric demand and access of the lactide to the metal centre is made more difficult. To determine the polymerization rate constant kp detailed kinetic measurements with complex 2 were performed (Figure 2). By polymerization experiments at different catalyst concentrations (up to 2000 : 1 per zinc), it was possible to obtain the rate constant k p from the linear fit by plotting the different k app values against the catalyst concentration. Compared with the k p from the recently published zinc guanidine catalyst [ZnCl2(TMG5NMe2asme)] with a value of 6.10±0.34×10−2 L mol−1 s−1 23b complex 2 with k p=8.59±0.36×10−2 L mol−1 s−1 is slightly faster. In a comparison to the active zinc catalyst Zn(CH3COO)2 with a conversion of 69 % after 24 h ([M]/[I]=500 : 1) the herein presented systems with a conversion of 79 % after 41 min ([M]/[I]=500 : 1) are significantly faster.22d
Figure 2.
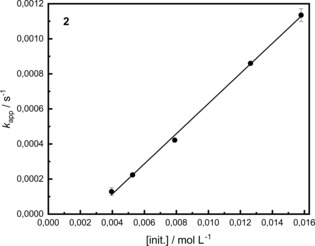
Plot of kapp versus [init.] for 2. Conditions: rac‐LA, 150 °C, 260 rpm, non‐purified; [M]/[I]=500:1, 625:1, 1000:1, 1500:1, 2000:1.
The analysis of the molar masses of the respective polylactides shows that all systems are able to synthesise high molar masses up to 71 000 g mol−1 (4). The theoretical molar masses propose that every available zinc atom propagates a chain. With polydispersities (PD) of 1.5–1.6, the values are very good for polymerization in melt. As mechanism, we propose the coordination‐insertion mechanism which will be detailed below. First, X‐ray data show that all complexes are dinuclear. However, if the kinetics of the polymerization catalyzed by complex 5 (Figure 3) are considered as example, an induction phase is conspicuous at the beginning of the polymerization. Typically, such induction phases are accounted to the formation of the active species. To investigate the reaction order, a plot of ln(k app) vs. ln([init.]) was used (see Figure S9). The slope of 1.57 was obtained indicating a fractional reaction rate. In this case a dissociation of the dinuclear complex is proposed.27 This is also supported by the obtained molar masses, which are closer to the theoretical value if based on the calculation per zinc atom. MALDI‐ToF measurements also confirm that a “half” complex is attached to the chain end (see Figure S11). While acetate primarily initiates the polymerization, the propagation of the chain takes place through half a complex. Due to a decomposition of the complex caused by impurities in the monomer, smaller amounts of ligand can be found at the end of the chains. Zinc acetate as the active species can be excluded due to its lower catalysis activity.22d All three observations lead to the result that by dissociation of the complex the active species is formed. Tacticity determinations by 1H NMR spectroscopy showed that the catalysts produce atactic polymer. To exclude potential epimerization during the polymerization, an experiment with L‐lactide using 2 has been performed. Homodecoupled 1H NMR revealed purely isotactic PLA.
Figure 3.
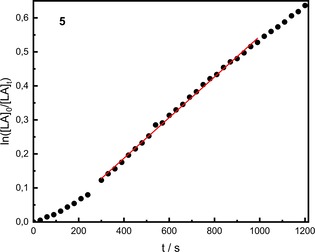
Semi‐logarithmic plot of the polymerization of non‐purified rac‐LA with 5 [M]/[I]=500:1, 150 °C, 260 rpm, conversion determined by in situ Raman spectroscopy.
TGA measurements of all five complexes show that the catalytic active systems remain stable at temperatures up to 225 °C. Therefore, they are suitable for industrial use at typical temperatures between 180 and 200 °C.
2.4. Cytotoxicity
In order to identify any potential toxicity of the complexes, the catalytically active complex 2 was tested against toxin‐sensitive 518A2 melanoma, HT‐29 and HCT‐116wt colon carcinoma, Hela cervix carcinoma cells and non‐malignant human fibroblasts using the MTT proliferation assay.30
Complex 2 showed virtually no cytotoxicity against any of these cells with 50 % growth inhibitory concentrations IC50>100 μM. It may therefore be considered non‐hazardous to health.
3. Conclusions
Dinuclear zinc acetate complexes with five different substituted Schiff base‐like ligands were prepared. The ligand and complex syntheses convince by their ease of preparation and their robustness towards higher temperature and lactide impurities. Four systems were found to be highly active in the catalytic ring‐opening polymerization of non‐purified lactide under industrial conditions. Their kinetic behaviour has been observed via in situ Raman spectroscopy. Despite an anionic ligand system, the complexes show a high degree of tolerance to the impurities in the monomer and produce industrially useful PLA with molar masses of up to 71 000 g mol−1 and a conversion of 78 %. With a k p=8.59±0.36×10−2 L mol−1 s−1, the systems are slightly faster than the recently published zinc guanidine complex23b and show that this class of ligands in combination with zinc also has a high potential to replace the currently industrially used catalyst Sn(Oct)2. Mechanistic investigations have shown that the dinuclear complex is present in melt of lactide as a mononuclear unit. As such, it forms the active species in the polymerization of lactide. Cytotoxic studies with sensitive non‐malignant fibroblasts and cancer cells also demonstrated the nontoxicity of the complexes, which thus represent an active, robust and green catalyst for the ROP of lactide. Together with the facile synthesis, a viable alternative for the cytotoxic Sn(Oct)2 opens up new avenues for lactide polymerization.
Experimental Section
HL1–HL5 were synthesized as previously reported.25a All other chemicals were commercially available and used without further purification. Elemental analysis were measured with Vario El III from Elementar AnalysenSysteme. Samples were prepared in a tin boat, and acetanilide was used as standard. Mass spectra were recorded with a Finnigan MAT 8500 with a data system MASPEC II. IR spectra were recorded with a Perkin Elmer Spectrum 100 FT‐IR spectrometer. TGA was measured with a Netzsch STA 449.
[ZnL1OAc] (1). Zn(AcO)2 ⋅ 2H2O (0.2 g, 0.91 mmol) and HL1 (0.377 g, 1.52 mmol) were dissolved in EtOH (5 mL) and the light orange solution was heated to reflux for 1 h. After cooling to RT and left to stand for 1 night the white precipitate was filtered, washed with a few mL of EtOH, and dried in air. Yield: 0.21 g (743.40 g mol−1, 31 %). Elemental analysis (C30H36Zn2N4O10, %) found C 48.52, H 4.91, N 7.51; calcd. C 48.47, H 4.88, N 7.54. MS (EI, pos.) m/z (%): 370 (C15H18ZnN2O5, 5), 310 (C13H15ZnN2O3, 93), 93 (C6H6N, 100). IR: ν=1680 (s, C=O), 1612 (s, C=O), 1572 (s, C=O) cm−1.
[ZnL2OAc] (2). Zn(AcO)2 ⋅ 2H2O (0.2 g, 0.91 mmol) and HL2 (0.331 g, 1.52 mmol) were dissolved in MeOH (5 mL) and the light yellow solution was heated to reflux for 1 h. After cooling to RT and left to stand for 1 night the white precipitate was filtered, washed with a few mL of MeOH, and dried in air. Yield: 0.25 g (683.34 g mol−1, 40 %). Elemental analysis (C28H32Zn2N4O10, %) found C 48.90, H 4.94, N 8.02; calcd. C 49.21, H 4.72, N 8.20. MS (EI, pos.) m/z (%): 340 (C14H16ZnN2O4, 5), 280 (C12H13ZnN2O2, 100), 93 (C6H6N, 65). IR: ν=1665 (s, C=O), 1567 (s, C=O) cm−1.
[ZnL3OAc] (3). Zn(AcO)2 ⋅ 2H2O (0.2 g, 0.91 mmol) and HL3 (0.176 g, 1.52 mmol) were dissolved in EtOH (5 mL) and the light yellow solution was heated to reflux for 1 h. After cooling to RT and left to stand for 1 night the white precipitate was filtered, washed with a few mL of EtOH, and dried in air. Yield: 0.22 g (709.34 g mol−1, 34 %). Elemental analysis (C28H30Zn2N6O8, %) found C 46.81, H 4.13, N 11.57; calcd. C 47.41, H 4.26, N 11.85. MS (EI, pos.) m/z (%): 353 (C14H15ZnN3O4, 6), 293 (C12H12ZnN3O2, 100). IR: ν=2193 (s, C≡N), 1650 (s, C=O), 1591 (s, C=O) cm−1.
[ZnL4OAc] (4). Zn(AcO)2 ⋅ 2H2O (0.2 g, 0.91 mmol) and HL4 (0.356 g, 1.52 mmol) were dissolved in MeOH (5 mL) and the light orange solution was heated to reflux for 1 h. After cooling to RT and left to stand for 1 night the white precipitate was filtered, washed with a few mL of MeOH, and dried in air. Yield: 0.23 g (715.34 g mol−1, 35 %). Elemental analysis (C28H32Zn2N4O10, %) found C 46.86, H 4.69, N 7.71; calcd. C 47.01, H 4.51, N 7.83. MS (EI, pos.) m/z (%): 356 (C14H16ZnN2O5, 7), 296 (C12H13ZnN2O3, 100), 93 (C6H6N, 45). IR: ν=1681 (s, C=O), 1611 (s, C=O), 1579 (s, C=O) cm−1.
[ZnL5OAc] (5). Zn(AcO)2 ⋅ 2H2O (0.2 g, 0.91 mmol) and HL5 (0.471 g, 1.52 mmol) were dissolved in EtOH (5 mL) and the light orange solution was heated to reflux for 1 h. After cooling to RT and left to stand for 1 night the white precipitate was filtered, washed with a few mL of EtOH, and dried in air. Yield: 0.32 g (867.54 g mol−1, 41 %). Elemental analysis (C40H40Zn2N4O10, %) found C 55.30, H 4.56, N 6.41; calcd. C 55.38, H 4.65, N 6.46. MS (EI, pos.) m/z (%): 432 (C20H20ZnN2O5, 6), 372 (C18H17ZnN2O3, 100), 93 (C6H6N, 38). IR: ν=1676 (s, C=O), 1608 (s, C=O), 1571 (s, C=O) cm−1.
X‐Ray Diffraction on Single Crystals
The X‐ray analysis was performed with a Stoe StadiVari diffractometer using graphite‐monochromated MoKα radiation. The data were corrected for Lorentz and polarization effects. The structures were solved by direct methods (SIR‐97)28 and refined by fullmatrix least‐square techniques against Fo2‐Fc2 (SHELXL‐97).29 All hydrogen atoms were calculated in idealized positions with fixed displacement parameters. ORTEP‐III30 was used for the structure representation, SCHAKAL‐9931 to illustrate molecule packing. CCDC 1901404 (1) and CCDC 1900405 (5) contain the supplementary crystallographic data for this paper.
Powder X‐Ray Diffraction
Powder diffractograms were measured with a STOE StadiP Powder Diffractometer (STOE, Darmstadt) using Cu[Kα1] radiation with a Ge Monochromator, and a Mythen 1 K Stripdetector in transmission geometry.
Reaction Monitoring
Raman spectra were obtained under process conditions using a RXN1 spectrometer from Kaiser Optical Systems. Ten accumulated measurements with 0.5 seconds measuring time were subsumed to one spectrum. The laser was used at a wavelength 785 nm and 459 mW through an immersion probe with a short‐focus sapphire lens (d=0.1 mm). The resulting time‐resolved data was processed with the PEAXACT 4.0 Software. The boundaries for the lactide integration were 627–713 cm−1.
Polymerization
All polymerizations at a ratio of [M]/[I]=500 : 1 and 2000 : 1 have been investigated twice.
Technical Grade Lactide: rac‐LA from Total Corbion PLA was used for the polymerizations. Therefore, d‐ and l‐lactide were mixed in a ratio of 1 : 1. Both d‐ and l‐lactide consisted of maximum free acids of 3 meq kg−1 and maximum water residues of 0.01 %.
Polymerization Followed by Raman Spectroscopy: In a nitrogen filled glovebox, the catalyst and rac‐LA (3,6‐dimethyl‐1,4‐dixane‐2,5‐dione, 12.0 g, 83.3 mmol) were weighed separately. The catalyst and the lactide were homogenized completely in an agate mortar and the mixture filled in a glass vial. The steel reactor was heated at 150 °C under vacuum and flashed three times with argon. For polymerization, the reaction mixture was filled in a steel reactor under argon conditions (99.998 % purity). The reactor was closed with a shaft drive stirrer with agitator speed control (“minisprint”, premix reactor AG, Switzerland) and the sample collection started after the reaction mixture insertion as soon as the reactor was closed. The Raman probe was installed close to the stirrer. The shaft drive stirrer with agitator speed control was used to stir the reaction at 260 rpm. The reaction mixture was removed from the reactor at 150 °C and 1H NMR was collected at room temperature on a Bruker Avance II (400 MHz) or a Bruker Avance III (400 MHz) to determine the conversion. The NMR signals were calibrated to the residual signals of the deuterated solvent [δ H(CDCl3)=7.26 ppm]. The reaction mixture was dissolved in an appropriate amount of DCM, the polymer was precipitated in ethanol (r.t.), dried in vacuo and characterized.
Gel Permeation Chromatography
The average molecular masses and the mass distributions of the obtained polylactide samples were determined by GPC in THF as the mobile phase at a flow rate of 1 mL min−1. The utilized GPCmax VE‐2001 from Viscotek was a combination of an HPLC pump, two Malvern Viscotek T columns (porous styrene divinylbenzene co‐polymer) with a maximum pore size of 500 and 5000 Å, a refractive index detector (VE‐3580), and a viscometer (Viscotek 270 Dual Detector). Universal calibration was applied to evaluate the chromatographic results.
MALDI‐ToF Mass Spectrometry: The end group analysis was performed by MALDI‐ToF on a Bruker ultrafleXtreme equipped with a 337 nm smartbeam laser in the reflective mode. THF solutions of trans‐2‐[3‐(4‐tert‐butylphenyl)‐2‐methyl‐2‐propenylidene]malononitrile (DCTB) (5 μL of a 20 mg/mL solution), sodium trifluoroacetate (0.1 μL of a 10 mg/mL solution), and analyte (5 μL of a 10 mg/mL) were mixed and a droplet thereof applied on the sample target. Protein 1 calibration standard is the name of the protein mixture used for calibration. For spectra 4000 laser shots with 24 % laser power were collected. The laser repetition rate was 1000 Hz. The homopolymer analysis was performed using Polymerix software (Sierra analytics).
Cell Culture
The human melanoma cell line 518 A2, the human colon carcinoma cell lines HT‐29 and HCT‐116, the cervix carcinoma cell line Hela, and the non‐malignant Hdfa fibroblasts were cultivated in Dulbecco's Modified Eagle Medium supplemented with 10 % FBS, and 1 % antibiotic‐antimycotic at 37 °C, 5 % CO2 and 95 % humidity. Only mycoplasma‐free cultures were used.
MTT Assay
The cytotoxicity of the compounds was studied via the MTT‐based proliferation assay32 on cells of 518 A2 melanoma (obtained from the department of Radiotherapy and Radiobiology, University Hospital Vienna, Austria), HT29 (DSMZ ACC‐299) and HCT116wt (DSMZ ACC‐581) colon carcinomas, Hela (DSMZ ACC‐57) cervix carcinoma, and Hdfa fibroblasts (Thermo Fisher). Briefly, cells (100 μL/well; 5×104 cells/mL for the four tumour cell lines, 1×105 for the Hdfa cells) were grown in 96‐well plates for 24 h and then treated with varying concentrations of the test compound or solvent control (DMSO) for 72 h. After centrifugation of the plates (300 g, 5 min, 4 °C), the supernatant was discarded and 50 μL/well of a 0.05 % MTT solution in PBS was added to the wells and incubated for 2 h. After another centrifugation step the supernatant was discarded and the formazan precipitate was dissolved in 25 μL DMSO containing 10 % SDS and 0.6 % acetic acid for at least 1 h at 37 °C and the absorbance of formazan (570 nm) and background (630 nm) was measured with a microplate reader (Tecan). The IC50 values were calculated as the mean ± standard deviation of four independent experiments.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
P.M.S. thanks the Hanns‐Seidel‐Foundation (fellowship) for funding (Bundesministerium für Bildung und Forschung, BMBF). The authors thank Total Corbion PLA for lactide donations. K.D. thanks the BayNAT graduate school for financial support. F. Puchtler and M. Schwarzmann (AC I, University of Bayreuth) are thanked for the PXRD measurements and the TGA.
P. M. Schäfer, K. Dankhoff, M. Rothemund, A. N. Ksiazkiewicz, A. Pich, R. Schobert, B. Weber, S. Herres-Pawlis, ChemistryOpen 2019, 8, 1020.
Contributor Information
Prof. Dr. Birgit Weber, Email: weber@uni-bayreuth.de
Prof. Dr. Sonja Herres‐Pawlis, Email: sonja.herres-pawlis@ac.rwth-aachen.de.
References
- 1.
- 1a. Wikström F., Verghese K., Auras R., Olsson A., Williams H., Wever R., Grönman K., Pettersen M. K., Møller H., Soukka R., J. Ind. Ecol., 0; [Google Scholar]
- 1b. Lavers J. L., Bond A. L., Proc. Natl. Acad. Sci. USA 2017, 114, 6052–6055; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Geyer R., Jambeck J. R., Law K. L., Sci. Adv. 2017, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gregory G. L., Lopez-Vidal E. M., Buchard A., Chem. Commun. 2017, 53, 2198–2217. [DOI] [PubMed] [Google Scholar]
- 3. Zhu Y., Romain C., Williams C. K., Nature 2016, 540, 354–362. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Castro-Aguirre E., Iñiguez-Franco F., Samsudin H., Fang X., Auras R., Adv. Drug Delivery Rev. 2016, 107, 333–366; [DOI] [PubMed] [Google Scholar]
- 4b. Auras R., Harte B., Selke S., Macromol. Biosci. 2004, 4, 835–864; [DOI] [PubMed] [Google Scholar]
- 4c. Drumright R. E., Gruber P. R., Henton D. E., Adv. Mater. 2000, 12, 1841–1846. [Google Scholar]
- 5.
- 5a. Dove A. P., Chem. Commun. 2008, 6446–6470; [DOI] [PubMed] [Google Scholar]
- 5b. Stanford M. J., Dove A. P., Chem. Soc. Rev. 2010, 39, 486–494; [DOI] [PubMed] [Google Scholar]
- 5c. Dechy-Cabaret O., Martin-Vaca B., Bourissou D., Chem. Rev. 2004, 104, 6147–6176; [DOI] [PubMed] [Google Scholar]
- 5d. Sarazin Y., Carpentier J.-F., Chem. Rev. 2015, 115, 3564–3614; [DOI] [PubMed] [Google Scholar]
- 5e. dos Santos Vieira I., Herres-Pawlis S., Eur. J. Inorg. Chem. 2012, 765–774. [Google Scholar]
- 6. Zhong Z., Dijkstra P. J., Feijen J., Angew. Chem. 2002, 114, 4692–4695; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2002, 41, 4510–4513. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Leenslag J. W., Pennings A. J., Makromol. Chem. 1987, 188, 1809–1814; [Google Scholar]
- 7b. Nijenhuis A. J., Grijpma D. W., Pennings A. J., Macromolecules 1992, 25, 6419–6424; [Google Scholar]
- 7c. Kricheldorf H. R., Dunsing R., Makromol. Chem. 1986, 187, 1611–1625; [Google Scholar]
- 7d. Kricheldorf H. R., Dunsing R., Serra A., Macromolecules 1987, 20, 2050–2057. [Google Scholar]
- 8.
- 8a. Kricheldorf H. R., Chemosphere 2001, 43, 49–54; [DOI] [PubMed] [Google Scholar]
- 8b. Stjerndahl A., Finne-Wistrand A., Albertsson A. C., Bäckesjö C. M., Lindgren U., J. Biomed. Mater. Res. Part A 2008, 87 A, 1086–1091. [DOI] [PubMed] [Google Scholar]
- 9. Chamberlain B. M., Cheng M., Moore D. R., Ovitt T. M., Lobkovsky E. B., Coates G. W., J. Am. Chem. Soc. 2001, 123, 3229–3238. [DOI] [PubMed] [Google Scholar]
- 10. Yang Y., Wang H., Ma H., Inorg. Chem. 2015, 54, 5839–5854. [DOI] [PubMed] [Google Scholar]
- 11. Ebrahimi T., Mamleeva E., Yu I., Hatzikiriakos S. G., Mehrkhodavandi P., Inorg. Chem. 2016, 55, 9445–9453. [DOI] [PubMed] [Google Scholar]
- 12. Williams C. K., Breyfogle L. E., Choi S. K., Nam W., Young V. G., Hillmyer M. A., Tolman W. B., J. Am. Chem. Soc. 2003, 125, 11350–11359. [DOI] [PubMed] [Google Scholar]
- 13. Wheaton C. A., Hayes P. G., Chem. Commun. 2010, 46, 8404–8406. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Scheiper C., Dittrich D., Wölper C., Bläser D., Roll J., Schulz S., Eur. J. Inorg. Chem. 2014, 2230–2240; [Google Scholar]
- 14b. Scheiper C., Schulz S., Wölper C., Bläser D., Roll J., Z. Anorg. Allg. Chem. 2013, 639, 1153–1159; [Google Scholar]
- 14c. Scheiper C., Wölper C., Bläser D., Roll J., Schulz S., Z. Naturforsch. 2014, 69b, 1365 -1374; [Google Scholar]
- 14d. Steiniger P., Schäfer P. M., Wölper C., Henkel J., Ksiazkiewicz A. N., Pich A., Herres-Pawlis S., Schulz S., Eur. J. Inorg. Chem. 2018, 2018, 4014–4021. [Google Scholar]
- 15. Chisholm M. H., Eilerts N. W., Huffman J. C., Iyer S. S., Pacold M., Phomphrai K., J. Am. Chem. Soc. 2000, 122, 11845–11854. [Google Scholar]
- 16. Huang Y., Wang W., Lin C.-C., Blake M. P., Clark L., Schwarz A. D., Mountford P., Dalton Trans. 2013, 42, 9313–9324. [DOI] [PubMed] [Google Scholar]
- 17. Thevenon A., Romain C., Bennington M. S., White A. J. P., Davidson H. J., Brooker S., Williams C. K., Angew. Chem. Int. Ed. 2016, 55, 8680–8685; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8822–8827. [Google Scholar]
- 18. Chmura A. J., Davidson M. G., Frankis C. J., Jones M. D., Lunn M. D., Chem. Commun. 2008, 1293–1295. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Forder T. R., Mahon M. F., Davidson M. G., Woodman T., Jones M. D., Dalton Trans. 2014, 43, 12095–12099; [DOI] [PubMed] [Google Scholar]
- 19b. Hancock S. L., Mahon M. F., Jones M. D., Dalton Trans. 2013, 42, 9279–9285; [DOI] [PubMed] [Google Scholar]
- 19c. Jones M. D., Brady L., McKeown P., Buchard A., Schäfer P. M., Thomas L. H., Mahon M. F., Woodman T. J., Lowe J. P., Chem. Sci. 2015, 6, 5034–5039; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19d. Jones M. D., Davidson M. G., Keir C. G., Hughes L. M., Mahon M. F., Apperley D. C., Eur. J. Inorg. Chem. 2009, 635–642; [Google Scholar]
- 19e. Jones M. D., Hancock S. L., McKeown P., Schäfer P. M., Buchard A., Thomas L. H., Mahon M. F., Lowe J. P., Chem. Commun. 2014, 50, 15967–15970; [DOI] [PubMed] [Google Scholar]
- 19f. McKeown P., Davidson M. G., Kociok-Kohn G., Jones M. D., Chem. Commun. 2016, 52, 10431–10434. [DOI] [PubMed] [Google Scholar]
- 20. McKeown P., McCormick S. N., Mahon M. F., Jones M. D., Polym. Chem. 2018, 9, 5339–5347. [Google Scholar]
- 21.
- 21a. Bienemann O., Froin A.-K., dos Santos Vieira I., Wortmann R., Hoffmann A., Herres-Pawlis S., Z. Anorg. Allg. Chem. 2012, 638, 1683–1690; [Google Scholar]
- 21b. Herres-Pawlis S., Neuba A., Seewald O., Seshadri T., Egold H., Flörke U., Henkel G., Eur. J. Org. Chem. 2005, 4879–4890; [Google Scholar]
- 21c. Rösener T., Bienemann O., Sigl K., Schopp N., Schnitter F., Flörke U., Hoffmann A., Döring A., Kuckling D., Herres-Pawlis S., Chem. Eur. J. 2016, 22, 13550–13562; [DOI] [PubMed] [Google Scholar]
- 21d. Rösener T., Hoffmann A., Herres-Pawlis S., Eur. J. Inorg. Chem. 2018, 3164–3175; [Google Scholar]
- 21e. Stanek J., Rösener T., Metz A., Mannsperger J., Hoffmann A., Herres-Pawlis S., in Topics in Heterocyclic Chemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 1–70. [Google Scholar]
- 22.
- 22a. Metz A., Heck J., Gohlke C., Kröckert K., Louven Y., McKeown P., Hoffmann A., Jones M., Herres-Pawlis S., Inorganics 2017, 5, 85; [Google Scholar]
- 22b. Metz A., McKeown P., Esser B., Gohlke C., Kröckert K., Laurini L., Scheckenbach M., McCormick S. N., Oswald M., Hoffmann A., Jones M. D., Herres-Pawlis S., Eur. J. Inorg. Chem. 2017, 5557–5570; [Google Scholar]
- 22c. Metz A., Plothe R., Glowacki B., Koszalkowski A., Scheckenbach M., Beringer A., Rösener T., de Vasconcellos J. Michaelis, Haase R., Flörke U., Hoffmann A., Herres-Pawlis S., Eur. J. Inorg. Chem. 2016, 4974–4987; [Google Scholar]
- 22d. Börner J., Flörke U., Huber K., Döring A., Kuckling D., Herres-Pawlis S., Chem. Eur. J. 2009, 15, 2362–2376. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Schäfer P. M., Fuchs M., Ohligschläger A., Rittinghaus R., McKeown P., Akin E., Schmidt M., Hoffmann A., Liauw M. A., Jones M. D., Herres-Pawlis S., ChemSusChem 2017, 10, 3547–3556; [DOI] [PubMed] [Google Scholar]
- 23b. Schäfer P. M., McKeown P., Fuchs M., Rittinghaus R. D., Hermann A., Henkel J., Seidel S., Roitzheim C., Ksiazkiewicz A. N., Hoffmann A., Pich A., Jones M. D., Herres-Pawlis S., Dalton Trans. 2019, 48, 6071–6082. [DOI] [PubMed] [Google Scholar]
- 24. Rittinghaus R. D., Schäfer P. M., Albrecht P., Conrads C., Hoffmann A., Ksiazkiewicz A., Bienemann O., Pich A., Herres-Pawlis S., ChemSusChem 2019, 12, 2161–2165. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Dankhoff K., Weber B., CrystEngComm 2018, 20, 818–828; [Google Scholar]
- 25b. Dankhoff K., Schneider S., Nowak R., Weber B., Z. Anorg. Allg. Chem. 2018, 644, 1839–1848. [Google Scholar]
- 26. Yang L., Powell D. R., Houser R. P., Dalton Trans. 2007, 955–964. [DOI] [PubMed] [Google Scholar]
- 27. Munzeiwa W. A., Nyamori V. O., Omondi B., Appl. Organomet. Chem. 2018, 32, e4247. [Google Scholar]
- 28. Altomare A., Burla M. C., Camalli M., Cascarano G. L., Giacovazzo C., Guagliardi A., Moliterni A. G. G., Polidori G., Spagna R., J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar]
- 29. Sheldrick G., Acta Crystallogr. Sect. A 2008, 64, 112–122. [DOI] [PubMed] [Google Scholar]
- 30.M. N. Burnett, C. K. Johnson, Oak Ridge National Lab., TN (United States), 1996, p. 176.
- 31.E. Keller, Schakal-99, University of Freiburg, Germany, Freiburg, 1999.
- 32. Mosmann T., J. Immunol. Methods 1983, 65, 55–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary