

Abstract
The ability of Mycobacterium tuberculosis to respond and adapt to various stresses such as oxygen/nitrogen radicals and low pH inside macrophages is critical for the persistence of this human pathogen inside its host. We have previously shown that an AraC/XylS-type transcriptional regulator, VirS, which is induced in low pH, is involved in remodeling the architecture of the bacterial cell envelope. However, how VirS influences gene expression to coordinate these pH responses remains unclear. Here, using a genetic biosensor of cytoplasmic pH, we demonstrate that VirS is required for the intracellular pH maintenance in response to acidic stress and inside acidified macrophages. Furthermore, we observed that VirS plays an important role in blocking phagosomal-lysosomal fusions. Transcriptomics experiments revealed that VirS affects the expression of genes encoding metabolic enzymes, cell-wall envelope proteins, efflux pumps, ion transporters, detoxification enzymes, and transcriptional regulators expressed under low-pH stress. Employing electrophoretic mobility-shift assays, DNA footprinting, and in silico analysis, we identified a DNA sequence to which VirS binds and key residues in VirS required for its interaction with DNA. A significant role of VirS in M. tuberculosis survival in adverse conditions suggested it as a potential anti-mycobacterial drug target. To that end, we identified VirS inhibitors in a virtual screen; the top hit compounds inhibited its DNA-binding activity and also M. tuberculosis growth in vitro and inside macrophages. Our findings establish that VirS mediates M. tuberculosis responses to acidic stress and identify VirS-inhibiting compounds that may form the basis for developing more effective anti-mycobacterial agents.
Keywords: Mycobacterium tuberculosis, gene microarray, transcription factor, tuberculosis, inhibitor, AraC/XylS type transcription factor, immune evasion, site-directed mutagenesis, VirS, virtual screening
Introduction
Mycobacterium tuberculosis is known to resist the acidic stress encountered in macrophages and multiply in these hostile conditions; however, the mechanisms for its survival in acidic conditions are poorly understood. There are a few genes that have been implicated in acid resistance in M. tuberculosis, which include serine protease Rv3671c, the OmpATb operon, and a putative magnesium transporter, MgtC (1–3). A transposon mutant of Rv3671c was shown to be impaired in the maintenance of intrabacterial pH under acidic conditions, suggesting its involvement in the acid resistance (1). OmpATb, an acid-responsive porin, was shown to be involved in the adaptation to acidic environment by mediating secretion of ammonia; however, its role was considered to be redundant, as knockout of OmpATb did not influence the virulence of the pathogen in mice (2, 4). M. tuberculosis lacking MgtC, a putative magnesium transporter, was found to be attenuated for growth under mild acidic conditions at low Mg2+ (3). Apart from M. tuberculosis, acid resistance has also been shown to be important for other bacteria, such as Helicobacter pylori, which colonizes in the human stomach, having an acidic environment; Streptococcus pneumoniae; and pathogenic strains of Escherichia coli and Salmonella enterica. Studies have shown the involvement of a few proteins in the acid resistance of these bacteria that include urease and ExbD in H. pylori, Gad proteins, and F0F1-ATPase in E. coli and Mg2+ transporter in Salmonella (5–9).
VirS (Rv3082c) of M. tuberculosis belongs to the AraC family of transcriptional regulators (10, 11). VirS is present divergently upstream of an acid-inducible operon termed the mymA operon, which comprises of seven genes (Rv3083–Rv3089) (12). The transcription of the mymA operon under acidic stress has been earlier shown to be regulated by VirS, which itself is regulated by acidic pH (12). Studies demonstrated that the virS mutant of M. tuberculosis exhibited altered cell-wall structure, altered mycolic acid content, defective intramacrophage survival, and reduced hematogenous dissemination in vivo (13). Importantly, virS expression was induced during chronic and reactivation phases of murine tuberculosis, implicating VirS in persistence and reactivation of tuberculosis (14). Despite these findings, mechanisms of how VirS exerts its influence on gene expression to elicit the response of M. tuberculosis under acid stress remain uncharacterized.
Here, our study has delineated the contribution of VirS in acid stress and how it mediates its influence on gene expression to coordinate pH responses in M. tuberculosis. Our study shows the requirement of VirS in controlling cytoplasmic pH balance and phagosomal maturation during acidic stress. Further, structure-guided mutational studies revealed key residues of VirS required for its interaction with DNA, and structure-based virtual screening identified some molecules inhibiting the DNA binding activity of VirS that could be a subject of lead optimization for future drug design against M. tuberculosis.
Results
VirS is required to survive under acidic stress and to block phagosomal maturation
VirS has been previously reported to be up-regulated under acidic conditions (12). To understand the involvement of VirS in acidic responses of M. tuberculosis, an in vitro growth study was carried out by growing M. tuberculosis Erdman (WT), virS mutant, and virS complemented strain under varying pH conditions (pH 4.5, 5.0, 5.5, and 6.6) in MB7H9 medium, and the survival of the cells under these conditions was evaluated. The growth of all three strains was comparable at pH 6.6 and 5.5. However, the growth of the virS mutant was significantly reduced at pH 5.0 and 4.5, with a pronounced defect at pH 4.5 as compared with parental M. tuberculosis and virS complemented strain (Fig. 1). We also performed survival studies of WT, virS mutant, and virS complemented strain at acidic conditions of pH 4.5 in 7H9 medium containing nonhydrolyzable tyloxapol (7H9–4.5-Ty) as the dispersing agent instead of Tween 80 to negate the possibility of a growth defect due to hydrolysis of Tween 80 under acidic conditions to free fatty acids, which can be toxic to the cells (1). We monitored survival of these strains after 6 and 9 days of incubation in 7H9–4.5-Ty medium by cfu enumeration. It was observed that virS mutant displayed a growth defect in acidified medium after 6 and 9 days of incubation, whereas parental and complemented strain did not exhibit any marked effect on their growth under acidic conditions, suggesting the involvement of VirS in the survival of the bacteria under acidic conditions (Fig. S1). Moreover, it was reported earlier that virS mutant showed survival defects specifically in immune-activated macrophages (13). M. tuberculosis is known to survive under acidic conditions in macrophages by arresting the phagosome-lysosome fusion. Hence, we evaluated the role of VirS in arresting phagosomal maturation in M. tuberculosis. For this, we employed FITC-labeled M. tuberculosis, virS mutant, and virS complemented strain and studied the localization of the pathogen in the lysosomal acidic compartments by using LysoTracker Red dye in THP-1 macrophages. In resting macrophages, all three of the strains exhibited similar colocalization of phagosomes with the LysoTracker-rich compartments (data not shown). However, in the case of activated macrophages, parental M. tuberculosis cells exhibited ∼9% colocalization with the LysoTracker-stained acidified compartments and largely remained in the nonacidified phagosomes, whereas in contrast to the WT strain, virS mutant displayed significantly greater colocalization (∼32%) with the acidified compartments (Fig. 2, A and B). The complemented strain also exhibited ∼6% colocalization, which is similar to that observed in the case of WT. These observations suggest the involvement of VirS in the arrest of phagosomal maturation in the activated macrophages, providing a survival advantage to the pathogen under harsher conditions.
Figure 1.

Involvement of VirS in M. tuberculosis survival at acidic pH. In vitro growth curves of M. tuberculosis Erdman (red), virS mutant (green), and virS complemented (blue) strain under varying pH conditions are shown. The x axis represents number of days, whereas the y axis represents A600 nm. The values are represented as the mean ± S.D. (error bars) of at least two independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001, two-way ANOVA.
Figure 2.

Role of VirS in phagosomal maturation. The influence of disruption of virS on the ability of M. tuberculosis to arrest phagosomal acidification was evaluated in THP-1 macrophages activated with IFN-γ. Macrophages were infected with FITC-labeled M. tuberculosis strains (green) separately. Blue, DAPI staining of the cells. The cells were then subjected to LysoTracker Red staining (red) followed by observation under a confocal microscope. B, bar diagram represents percentage colocalization of phagosomes containing FITC-labeled bacteria with LysoTracker Red. Data represent the mean ± S.D. (error bars) of two independent experiments carried out in triplicate (**, p < 0.01; ***, p < 0.001, one-way ANOVA). DIC, differential interference contrast.
Involvement of VirS in maintaining the intrabacterial pH of M. tuberculosis in acidic conditions
Activated macrophages are known to have an acidic environment, and M. tuberculosis robustly persists in this acidic milieu, indicating the role of mycobacterial factors in intramycobacterial pH homeostasis. Hence, we next sought to determine the role of VirS in maintaining the pH homeostasis by measuring the intrabacterial pH (pHIB)4 of various strains by using a pH-sensitive ratiometric GFP at various intervals under normal and acidic conditions. The pHIB was calculated by measuring the ratio of the fluorescence intensities at excitation wavelengths of 395 and 475 nm with an emission wavelength of 510 nm (1). A standard curve of the ratio of the intensities (excitation wavelengths, 395 and 475 nm; emission wavelength, 510 nm) at varying pH conditions was plotted to determine the unknown pHIB values (Fig. S2A). It was observed that under normal buffer conditions (pH 7.4), the pHIB was maintained nearly neutral in the parental (pHIB ∼7.0) as well as in the complemented strain even after 8 and 24 h of incubation (Fig. 3A). However, the virS mutant consistently maintained marginally lower intrabacterial pH (pHIB 6.75) even in normal buffer conditions as compared with the parental and complemented strains (Fig. 3A). At pH 4.5, the initial pHIB values of all three strains were similar to that observed in normal conditions (Fig. 3B). However, after 8 and 24 h of incubation in pH 4.5 buffer, the intrabacterial pH of the mutant dropped to ∼6.4, whereas the pHIB values of parental and complemented strains were still maintained at nearly neutral conditions of ∼6.9 (Fig. 3B). This suggested that both the parental and complemented strains maintain pH homeostasis upon acidic stress, whereas the virS mutant was relatively defective. Taken together, these observations suggest the importance of VirS in maintaining the intrabacterial pH under acidic conditions.
Figure 3.

virS disruption causes change in intrabacterial pH of the pathogen. The intrabacterial pH of the WT, virS mutant, and virS complemented strain was measured in nearly neutral conditions (i.e. pH 7.4) (A) and in acidified medium (i.e. pH 4.5) (B). Intrabacterial pH was measured at different time intervals (i.e. 0, 8, and 24 h after exposure to acidic pH). Disruption of VirS caused a reduction in intrabacterial pH at pH 4.5, which was maintained in the WT and complemented strain. Data represent the mean ± S.D. (error bars) of at least two independent experiments carried out in duplicates (***, p < 0.001, two-way ANOVA). The intrabacterial pH of the WT, virS mutant, and virS complemented strain was measured inside IFN-γ activated macrophages 24 h postinfection (C) and 48 h postinfection (D). virS mutant failed to maintain its intrabacterial pH inside activated macrophages, whereas WT and virS complemented strain were able to maintain their pHIB levels. There were ∼115 bacterial groups examined, and data represent two independent experiments.
We next sought to determine the pHIB of mycobacteria residing in phagosomes of IFN-γ–activated macrophages. The unknown pHIB values were determined by using a standard curve of the ratio of intensities at excitation wavelengths 405 and 476 nm, emission wavelength 535 nm at varying pH conditions (Fig. S2B). It was observed that WT and virS complemented strains mostly maintained their pHIB at pH 6.5–7.25 after 24 h postinfection. However, the majority of the virS mutant mostly maintained its pHIB at pH 6.26–7.0 after 24 h postinfection (Fig. 3C) with a small fraction of mutant cells having a pHIB in the range of <5.5–6.2, suggesting that at 24 h postinfection, a few mutant cells were unable to maintain their intrabacterial pH near neutral. This effect was more pronounced after 48 h postinfection, when we found that the pHIB of parental and virS complemented strain mostly had a pHIB of 6.51–7.25, whereas virS mutant mostly had a pHIB of 5.5 or lower in activated macrophages, suggesting that after 48 h of postinfection, the virS mutant strain was unable to maintain its intrabacterial pH (Fig. 3D). This inability of virS mutant to maintain its intrabacterial pH in activated macrophages indicates its role in the maintenance of pH homeostasis upon acidic stress and could be a probable reason for its poor survival under acidic conditions.
Identification of genes regulated by VirS (VirS-regulon) under acidic conditions by microarray studies
To understand the role of VirS in regulating acidic pH–mediated gene expression of M. tuberculosis, we carried out microarray studies by isolating RNA from M. tuberculosis Erdman, virS mutant, and virS complemented strain grown in MB7H9 medium at pH 6.6 as well as at pH 4.5. We shortlisted the VirS-dependent genes induced or repressed at acidic pH relative to neutral pH with >2.0-fold difference in the expression level when compared with M. tuberculosis Erdman and virS mutant. We then further shortlisted these genes on the basis of their similar expression profile at acidic pH in M. tuberculosis Erdman and virS complemented strain compared with virS mutant, suggesting that indeed the disruption of virS was involved in the change of expression of such genes at acidic pH. We also carried out the classification of all of these induced and repressed genes into various classes based on their function and homology; however, the hypothetical proteins, unannotated genes, and genes of unknown function were not considered for further analysis. All of these analyses revealed that VirS induces expression of 54 genes and represses 39 genes (>2.0-fold difference; p < 0.05) at acidic pH (Fig. 4, A and B). The major classes of genes (after removing genes belonging to hypothetical proteins, genes of unknown function, and unannotated genes) found to be induced and repressed were related to cellular metabolism followed by cell wall–associated genes involved in fatty acid oxidation and biosynthesis, which is in agreement with the role of VirS in the remodeling of cell-envelope architecture as reported earlier (13). The other classes of genes induced by VirS at acidic pH were related to the PE/PPE family, efflux pumps and ion transporters, detoxification, and transcriptional regulation. Additionally, genes related to energy pathways and respiration and genes related to information pathways were also found to be regulated by VirS at acidic pH. Fig. 4C represents the heat map showing the expression of various genes that are differentially regulated by VirS in WT M. tuberculosis, M.tbΔvirS, and M.tbΔvirSComp in response to acidic pH relative to neutral pH. Tables 1 and 2 describe a few of the genes from the most represented categories induced or repressed by VirS at acidic pH. A detailed description of the genes and their speculated role in counteracting acidic responses is provided under “Discussion.” The complete list of the genes that showed up-regulation and down-regulation with >2.0-fold difference are listed as a supporting Excel Sheet X1.
Figure 4.

VirS-dependent gene regulation at acidic pH. Microarray studies revealed VirS-mediated differential gene regulation at acidic pH. Up-regulation of VirS at acidic pH causes change in the expression of many genes that were found to be induced and repressed at acidic pH. These genes were categorized into various classes based on their description given by Tuberculist. A, pie chart represents the categorization of various genes that are induced by VirS at acidic pH. B, pie chart represents the categorization of various genes that are repressed by VirS at acidic pH. The percentage distribution of genes is represented in both of the pie charts. C, heat map showing the expression of VirS regulated genes in M. tuberculosis Erdman, M.tbΔvirS, and M.tbΔvirSComp in response to acidic pH relative to neutral pH (>2-fold difference, p < 0.05). The figure here depicts complete linkage and was generated by employing Heatmapper software (57).
Table 1.
List of VirS-dependent genes identified by microarray studies that were found to be up-regulated >2.0-fold at acidic pH
Only a few genes from the most represented categories are listed here.
| Gene | Rv no. | -Fold regulation | Description |
|---|---|---|---|
| Cell wall–associated | |||
| fadB2 | Rv0468 | 8.65 | 3-Hydroxyacyl-CoA dehydrogenase |
| fadE35 | Rv3797 | 3.88 | Acyl-CoA dehydrogenase |
| Rv2672 | Rv2672 | 2.85 | Protease and lipase |
| Efflux pumps and transporters | |||
| ctpG | Rv1992c | 2.87 | Cation transport ATPase |
| kdpC | Rv1031 | 2.69 | Potassium-transporting ATPase C chain |
| Metabolic enzymes | |||
| Rv1405c | Rv1405c | 54.07 | Similar to phosphatidylethanolamine N-methyltransferase |
| icl | Rv0467 | 11.28 | Isocitrate lyase |
| pdc | Rv0853c | 3.32 | Pyruvate decarboxylase |
| adhE1 | Rv0162c | 2.81 | Alcohol dehydrogenase |
| Detoxification | |||
| ahpD | Rv2429 | 6.04 | Member of AhpC/TSA family |
| ahpC | Rv2428 | 5.38 | Alkyl hydroperoxide reductase |
| Rv3177 | Rv3177 | 4.69 | Probable non-heme haloperoxidase |
| Transcriptional regulator | |||
| Rv1994c | Rv1994c | 3.22 | Transcriptional regulator (MerR family) |
Table 2.
List of VirS-dependent genes identified by microarray studies that were found to be down-regulated >2.0-fold at acidic pH
Only a few genes from the most represented categories are listed here.
| Gene | Rv no. | -Fold regulation | Description |
|---|---|---|---|
| Cell wall–associated | |||
| papA1 | Rv3824c | 5.43 | PKS-associated protein |
| fadD9 | Rv2590 | 4.79 | Acyl-CoA synthase |
| pks2 | Rv3825c | 3.58 | Polyketide synthase |
| mmpS5 | Rv0677c | 3.59 | Membrane protein |
| mmpL5 | Rv0676c | 3.23 | Membrane protein |
| Metabolic enzymes | |||
| lipF | Rv3487c | 7.42 | Esterase |
| sirA | Rv2391 | 2.81 | Sulfite reductase |
| cysH | Rv2392 | 2.63 | 3-Phosphoadenylsulfate (PAPS) reductase |
| Information pathways | |||
| rpsK | Rv3459c | 2.74 | 30S ribosomal protein S11 |
| infC | Rv1641 | 2.61 | Initiation factor IF-3 |
| Energy pathways and respiration | |||
| nuoK | Rv3155 | 2.81 | NADH dehydrogenase chain K |
| cydD | Rv1621c | 2.76 | ABC transporter |
| narX | Rv1736c | 2.32 | Nitrate reductase |
Identification of VirS-binding region of DNA
Based on the above observations, VirS appears to be a promising drug target regulating a number of important downstream targets. Hence, we further attempted to characterize VirS by evaluating its DNA-binding activity, its DNA-binding site, and crucial residues of VirS required for its activity and to identify its inhibitors. Toward this, we cloned the virS gene in the pET43.1a vector as described under “Experimental procedures” for the expression of fusion protein NusA-VirS, because our earlier studies showed VirS to be an insoluble protein when expressed in E. coli,5 and the NusA tag is considered to be an effective solubility tag. NusA-VirS was indeed found to be localized in the soluble fraction; however, the NusA-VirS fusion protein failed to bind to the resin, probably due to the nonexposure of the His tag. Hence, cell lysates were used for further studies (data not shown). The DNA-binding activity of NusA-VirS fusion was determined by employing electrophoretic mobility-shift assay (EMSA) by using a 325 bp DNA fragment, including the intergenic region between the virS and mymA genes, as VirS regulates its own expression and activates mymA operon (12). The fusion protein displayed a shift in the mobility of DNA suggesting the binding of VirS to the DNA fragment employed in the assay (Fig. 5A). To rule out the possibility of involvement of NusA tag in the observed shift in the DNA, the lysate with overexpressed NusA was employed as a control that did not display any shift in the mobility of DNA, indicating that the observed shift in the mobility was due to the binding of VirS to the DNA (Fig. 5A). Further, a DNase I protection assay was performed to determine the VirS-binding region of DNA. The lysate of overexpressed fusion protein (i.e. NusA-VirS) was used for determining the binding region of VirS that displayed a protected region (Fig. 5B). The lysate of only overexpressed NusA protein was used as a negative control, which did not display any protection. It was found that VirS protected a 46-bp sequence that was determined to be 5′-CTGTCGGAAAATGATAAAAGCGTGTCGCAAAGTGTCAATACGTGGC-3′ (Fig. 5B). Based on this VirS-binding DNA sequence, we determined a VirS-binding consensus logo by using the software Weblogo (15) taking the upstream regions of a few identified downstream targets of VirS for alignment (16) (Fig. 5C). For this, we performed multiple-sequence alignment of this DNA sequence with the DNA sequence of micF and mar promoters used in the crystal structure determination of Rob and MarA, respectively, which provided us with a 28-bp sequence exhibiting alignment and conservation in DNA-binding regions of these three proteins. This 28-bp region was employed to search for homology in the upstream regions of a few of the VirS-regulated genes, namely icl, ctpG, ahpC, infC, sirA, pdc, Rv1405c, adh, fadD9, and fadE35, identified by microarray. These genes were randomly selected to represent a variety of different functional categories. We observed that all of these genes carried the 28-bp sequence required for binding of VirS although with a few variations in the sequences (Fig. 5C).
Figure 5.

Determination of DNA-binding activity of VirS and its binding region. A, determination of DNA-binding activity of VirS by employing EMSA. The DNA-binding activity of lysate overexpressing fusion protein NusA-VirS was analyzed with a 325-bp amplified 32P-radiolabeled DNA fragment by EMSA. The lysate overexpressing only the NusA tag and samples containing DNA (no protein) were utilized as negative controls. Lanes 1, 2, and 3, 13, 26, and 52 μg of E. coli lysate overexpressing NusA-VirS protein. Lanes 4, 5, and 6, 13, 26, and 52 μg of E. coli lysate overexpressing NusA protein. Lane 7, free DNA. B, VirS-binding region of DNA was determined by DNase I footprinting assay. Lanes 1 and 9, G+A DNA ladder; lane 2, free DNA; lanes 3–5, DNA bound to NusA-VirS; lanes 6–8, DNA in the presence of E. coli lysate overexpressing NusA protein. Lanes 3–5, protected region bound by VirS. C, a consensus DNA sequence for binding of VirS was identified. The protected region identified by footprint analysis was used to define the VirS-binding sites in its downstream targets. The consensus sequence logo was generated by the software WebLogo, in which the height of the stack indicates the degree of conservation at that position in the consensus and the height of an individual letter within a stack represents the relative frequency of that nucleotide at that position. D, the upstream regions of a few of the downstream targets of VirS (ahpD, ppe23, cmtR, ctpG, ppe38, and fadD9) were selected and evaluated for their binding to 1 μg of purified VirS by employing EMSA. All of the genes showed a positive binding with VirS, suggesting their direct interaction with VirS. E, as a negative control, an intergenic region between MbtA and MbtB containing the IdeR-binding region was employed, which did not display any shift in the mobility of the DNA in the presence of VirS. As a positive control, VirS and its binding sequence were used, which showed shift in the mobility of its DNA.
We further explored the regulation of a few of the genes identified by microarray studies by VirS. The genes selected were Rv2428 (ahpD), Rv1706 (ppe23), Rv1994c (cmtR), Rv1992c (ctpG), Rv2352c (ppe38), and Rv2590 (fadD9). For determining whether these genes are directly or indirectly regulated by VirS, we selected a ∼400–600-bp upstream region of these genes that comprised their respective promoters as predicted by the online software NNPP (http://www.fruitfly.org/seq_tools/promoter.html)6 (29). These upstream regions were separately amplified by employing a Cy5 PCR Labeling Kit (Jena Biosciences). The labeled product was then evaluated for its ability to bind VirS by employing EMSA as described under “Experimental procedures.” It was observed that the upstream regions of all of the selected genes displayed a shift in mobility, suggesting their direct interaction with VirS (Fig. 5D). As a negative control to negate a general binding of VirS to DNA, we used a Cy5-labeled 32-bp intergenic region between MbtA and MbtB that contains the IdeR (iron-dependent transcription regulator)-binding region, which did not display any shift in its mobility in the presence of VirS, indicating the specificity of VirS to its own downstream targets (Fig. 5E).
Homology modeling of VirS
We generated a three-dimensional comparative homology model of VirS for performing virtual screening studies and also for carrying out identification of the crucial residues involved in its DNA-binding activity. Because the crystal structure of M. tuberculosis VirS is not available, we generated the three-dimensional structure of VirS by employing comparative homology modeling, as described under “Experimental procedures.” It was found by bioinformatic analysis that the C terminus of VirS (amino acids 250–340) contained the DNA-binding region and that it was homologous to the C terminus of the transcriptional regulators of the AraC/XylS family. Hence, this C-terminal region was employed for multiple-sequence alignment by ClustalW (16) and for model generation by Modeler 9.0 (17) (Fig. 6A). The generated 3D homology model of VirS contained two helix-turn-helix motifs (Fig. 6B). The coordinates of DNA bound to the MarA structure were used to predict the DNA contact sites of VirS (Fig. 6C). The predicted structure of VirS was also validated by using Rampage (18), Errat (19), Verify 3D (20, 21), Pro-Q (22), and Prosa (23, 24) software (Table S1). The results obtained from these programs implied that the generated structure of VirS was in a good configuration.
Figure 6.

Prediction of a 3D model of VirS. A, multiple-sequence alignment of amino acid sequences of VirS and its homologous proteins was generated by employing ClustalW. This alignment file was then used to generate the 3D structure of VirS by using the software Modeler 9.0. B, the 3D structure of VirS was generated by comparative homology modeling by using the software Modeler 9.0. The predicted structure of VirS contains two HTH motifs. C, structure of VirS with the predicted DNA-binding sites. B and C were generated by PyMOL (58).
Identification of the critical residues of VirS involved in its DNA-binding activity
On the basis of multiple-sequence alignment and the predicted structure of VirS, 15 residues were selected that might affect the DNA-binding activity of VirS, and structure-guided mutational studies were performed (Fig. 7A and Table 3). These residues were substituted with alanine for mutational studies because alanine is a nonpolar and small amino acid that takes care of charge as well as the size of the side chain.
Figure 7.

Determination of crucial residues of VirS. A, 15 amino acid residues of VirS selected for mutational studies are highlighted in the homology model of VirS. B, the effect of mutations on the DNA-binding activity of VirS was determined by employing EMSA. The WT 3a-VirS protein was used as a positive control, and only DNA was used as the negative control. The dashed line shows that lanes from two different polyacrylamide gels are brought together. DNA-binding activity was calculated by comparing the activity in WT 3a-VirS loaded in the same gel. 0.5 μg of WT 3a-VirS and mutants was subjected to the evaluation of the effect on DNA-binding activity. 20 μg of cell lysates from WT 3a-VirS and mutants Y61A, R73A, and R80A were used to evaluate the effect of mutation on the DNA-binding activity of VirS. C, bar graph displays percentage activity exhibited by various mutants when compared with WT VirS. Data represent the mean ± S.D. (error bars) of at least two independent experiments (*, p < 0.05; ***, p < 0.001, two-way ANOVA).
Table 3.
VirS residues selected for site-directed mutational studies
| S. no. | Amino acid position | Amino acid | Amino acid position in C terminus of VirS | Mutation |
|---|---|---|---|---|
| 1 | 254 | Glu | 5 | E5A |
| 2 | 265 | Arg | 16 | R16A |
| 3 | 268 | Gln | 19 | Q19A |
| 4 | 269 | Arg | 20 | R20A |
| 5 | 277 | Arg | 28 | R28A |
| 6 | 279 | His | 30 | H30A |
| 7 | 283 | Glu | 34 | E34A |
| 8 | 287 | Arg | 38 | R38A |
| 9 | 310 | Tyr | 61 | Y61A |
| 10 | 318 | Arg | 69 | R69A |
| 11 | 319 | Ser | 70 | S70A |
| 12 | 321 | Arg | 72 | R72A |
| 13 | 322 | Arg | 73 | R73A |
| 14 | 329 | Arg | 80 | R80A |
| 15 | 330 | Gln | 81 | Q81A |
For carrying out the site-directed mutagenesis study, we subcloned the virS gene from the pET43.1a-virS construct to another vector, pBEn-SET3a, as described under “Experimental procedures,” because the size of the pBEn-3a-virS construct (6.8 kb) is smaller than the pET43.1a-virS (8.3 kb) and thus more appropriate for site-directed mutagenesis studies. The 3a-VirS fusion protein was purified and assessed for its DNA-binding activity with the VirS-binding region of DNA identified by a DNA footprint assay containing 46 bp flanked by 10 bp at each side (66 bp). For ease of work, the ends of this 66-bp region were end-labeled with fluorophore Cy5. The Cy5-labeled DNA was then employed for the determination of DNA-binding activity of VirS by EMSA studies. The purified protein was found to be functionally active and displayed the gel shift that also verified the identified binding region of DNA (data not shown). 0.5 μg of protein was selected for further work.
Fifteen VirS mutants were generated by using site-directed mutagenesis as described under “Experimental procedures.” The sequences of the primers used for mutagenesis are given in Table S2. All of the mutations were confirmed by DNA sequencing, and the mutant proteins were separately expressed and purified successfully by using nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography (data not shown). Mutants Y61A, R73A, and R80A showed poor binding to Ni-NTA resin due to probable changes in the protein folding as a result of mutation. Thus, their lysates were prepared and employed for EMSA studies, and lysate of WT 3a-VirS was used as a control for comparison. The mutants were evaluated for their DNA-binding activity by using EMSA (Fig. 7B).
Mutants R16A, Y61A, and R69A displayed more than 90% reduction in the DNA-binding activity of VirS, suggesting that the corresponding residues are critical for the DNA binding (Fig. 7B). Mutant R20A displayed 84% reduction in the activity, indicating the importance of Arg-20 in DNA binding. Mutants R28A and R73A showed moderate effect on the activity, as they displayed reductions of 34 and 30%, respectively (Fig. 7B). Fig. 7C depicts the bar graph representing the percentage of DNA-binding activity exhibited by the mutants when compared with the WT VirS protein. Mutants H30A, E34A, R38A, and S70A did not display any influence on the DNA-binding activity, suggesting that these residues are not important for the DNA-binding activity of VirS. In conclusion, four residues (Arg-16, Arg-20, Tyr-61, and Arg-69), when substituted with alanine, resulted in the loss of VirS DNA binding activity by more than 84% compared with the WT VirS. These residues of VirS might directly make contacts with the cognate DNA and can be used as specific target sites for rational design of inhibitors against VirS for anti-tubercular drug development.
Identification of the inhibitory molecules against VirS by structure-based virtual screening and their evaluation against DNA-binding activity of VirS and M. tuberculosis growth
Among the homologous proteins of VirS, only three structures (i.e. MarA (PDB entry 1BL0), Rob (PDB entry 1D5Y), and AdpA (PDB entry 3W6V)) were available in ligand-bound form (25–27). The DNA contact sites of VirS were predicted by superimposition with DNA-bound MarA structure (Fig. 8A). On superimposing the 3D structure of VirS with the structure of MarA, two DNA contact sites in VirS were observed (Fig. 8B). Hence, virtual screening was carried out at both of the DNA contact sites of VirS by using Autodock4.2 software (http://autodock.scripps.edu/)6 (28). The grid covering DNA contact site 1 of VirS is represented in Fig. 8, C and D. The residues (i.e. Glu-5, Gln-19, Arg-28, and His-30), were surrounding the grid and found to be covered in the generated grid (Fig. 8E). Similarly, another grid was generated against DNA contact site 2 of VirS (Fig. 8F), and its enlarged representation is shown in Fig. 8G. The residues found to surround and be covered in the grid were Ala-56, Ser-53, Tyr-61, Ser-62, Glu-63, and Gln-64 (Fig. 8H). A filtered library of the NCI (National Institutes of Health) Open Database was employed for virtual screening against the DNA contact sites of VirS (30). A total of 114 compounds, 57 compounds each from docking at site 1 and site 2, were procured from NCI-DTP, numbered as V1A–V57A (site 1) and V1B–V57B (site 2), and evaluated for their ability to inhibit the DNA-binding activity of VirS at a concentration of 100 μg/ml (Fig. 9). 26 compounds (i.e. 10 from site 1 and 16 from site 2) displayed more than 60% inhibition with 15 compounds (4 from site 1 and 11 from site 2) exhibiting greater than 80% inhibition at 100 μg/ml (Fig. 9, A and B and Table S3). These 26 compounds were further subjected to dose-response studies for the determination of their IC50 values (Table S3). Compound V7B with an IC50 value of 1.8 μg/ml was found to be the most potent one, whereas compound V29B displayed an IC50 value of 16.38 μg/ml. The rest of the compounds showed IC50 values in the range of 17–75 μg/ml (Table S3).
Figure 8.

Target-based virtual screening against the DNA-binding region of VirS. A, the figure represents the predicted 3D structure of VirS interacting with DNA. B, the structure of VirS depicts the presence of two DNA contact sites. C, virtual screening was carried out against these two DNA contact sites by using Autodock4.2. A surface representation of the VirS structure is shown here with a grid covering DNA contact site 1 of VirS. D, enlarged representation showing the grid covering the DNA contact site 1 and nearby residues of VirS. E, the figure represents the residues that are near the DNA and are covered in the grid. F, the grid generation against the second DNA contact site of VirS is shown here. G, enlarged representation of the grid around the DNA contact site 2 of VirS. H, residues surrounding the second DNA contact site, covered in the grid, are shown here. The figures were prepared by using the Autodock4.2.
Figure 9.
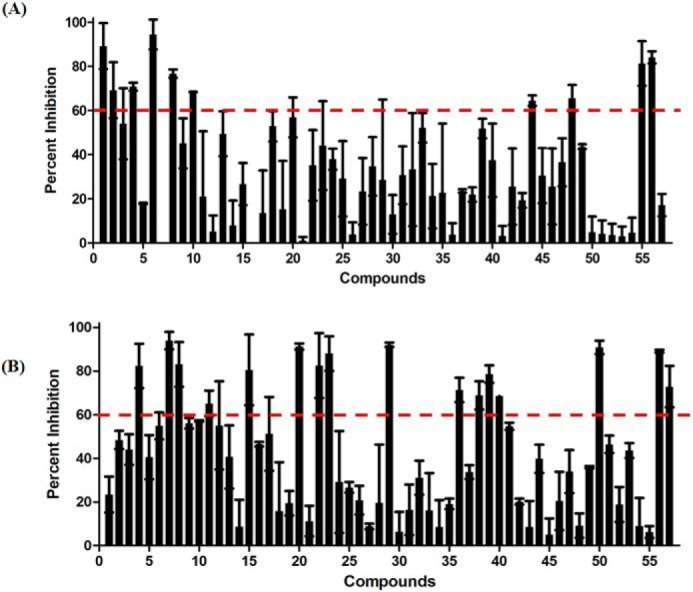
Evaluation of the compounds for their inhibitory potential against the DNA binding activity of VirS. A and B, bar graph displaying percentage inhibition exhibited by compounds at 100 μg/ml, obtained on the basis of docking at site 1 (A) and obtained on the basis of docking at site 2 (B). DMSO was used as a negative control in EMSA, which displayed no inhibition of DNA-binding activity of VirS. Error bars, S.D.
These 26 compounds were also evaluated for their ability to inhibit the growth of M. tuberculosis in broth culture by subjecting them to a resazurin microtiter assay (31–33). Rifampicin was used as a positive control in the assay, which exhibited an MIC90 of 0.25 μg/ml, and DMSO was used as a vehicle control. Fourteen compounds displayed growth inhibition of M. tuberculosis with varying MIC90 values (Table 4 and Fig. S3). The most potent inhibition against M. tuberculosis was exhibited by compound V15B with an MIC90 value of 5 μg/ml. Compound V7B, which was the most potent compound in the in vitro assay, displayed an MIC90 of 40 μg/ml. To summarize, we identified 14 compounds (listed in Table 4) that exhibited growth inhibition against M. tuberculosis as well as inhibited DNA-binding activity of VirS in biochemical assays.
Table 4.
List of the compounds that displayed M. tuberculosis growth inhibition
| S. no. | Compound | MIC90 exhibited by compounds against M. tuberculosis growth in in vitro broth culture | IC50 exhibited by compounds in EMSA |
|---|---|---|---|
| μg/ml | μg/ml | ||
| 1 | V1A | 160 | 72.01 |
| 2 | V4A | 40 | 58.33 |
| 3 | V6A | 25 | 34.59 |
| 4 | V8A | 20 | 33.63 |
| 5 | V44A | 12.5 | 24.91 |
| 6 | V48A | 50 | 27.86 |
| 7 | V55A | 20 | 74.86 |
| 8 | V7B | 40 | 1.8 |
| 9 | V8B | 55% inhibition at 200 μg/ml | 17.44 |
| 10 | V11B | 61% inhibition at 160 μg/ml | 50.45 |
| 11 | V15B | 5 | 31.21 |
| 12 | V20B | 80 | 38.44 |
| 13 | V22B | 61% inhibition at 80 μg/ml | 72.44 |
| 14 | V56B | 25 | 37.19 |
We then further evaluated the cytotoxicity of eight compounds (V4A, V6A, V8A, V44A, V55A, V7B, V15B, and V56B) that exhibited an MIC90 ≤ 40 μg/ml and were also active against the DNA-binding activity of VirS. Various mammalian cell lines were employed, and IC50 values of the compounds were determined as described under “Experimental procedures.” The most potent inhibitor of VirS i.e. V7B (IC50 = 1.8 μg/ml, MIC90 = 40 μg/ml) was found to be noncytotoxic until a concentration of 100 μg/ml in all of the cell lines (Table S4). Another potent inhibitor of M. tuberculosis growth (i.e. V15B (MIC90 = 5 μg/ml and IC50 = 31.21 μg/ml)) was also found to be noncytotoxic until a concentration of 150 μg/ml in all of the cell lines (Table S4). Compounds V4A and V8A also displayed negligible cytotoxicity in all of the employed cell lines until 200 and 150 μg/ml, respectively. The rest of the compounds displayed moderate cytotoxicity in at least one or two of the three employed cell lines (Table S4).
Four compounds (V4A, V8A, V7B, and V15B) that displayed inhibition of VirS DNA-binding activity as well as growth of the pathogen and did not display any significant cytotoxicity were further evaluated for their potency to inhibit the growth of the pathogen inside macrophages as described under “Experimental procedures.” It was found that compounds V7B and V15B inhibited the intracellular growth of the pathogen at ≥80 and ≥40 μg/ml, respectively (Fig. 10). To rule out the possibility of this inhibition due to cytotoxic effects of the compounds, uninfected THP-1 cells were also subjected to the same concentrations of the compounds. V7B and V15B were found to be noncytotoxic to THP-1 cells until 320 and 100 μg/ml, respectively. Compounds V4A and V8A failed to inhibit the intracellular growth of the pathogen even at a concentration of 100 μg/ml (data not shown). Thus, these results suggested that the compounds V7B and V15B also possess the ability to inhibit the intracellular growth of the pathogen; however, further optimization of these compounds is required to improve their potency.
Figure 10.

Evaluation of the inhibitory potential of compounds against intracellular survival of the pathogen. Compounds V7B and V15B were evaluated for their inhibitory potential against intracellular survival of M. tuberculosis inside macrophages. A, compound V7B displayed inhibition of growth of the pathogen at 80 μg/ml and above. Cytotoxicity of compound V7B was determined by resazurin dye. The bottom panel depicts the cytotoxicity exhibited by compound V7B to the macrophages. Except at the highest concentration employed, compound V7B did not display any cytotoxicity to the macrophages. B, compound V15B displayed inhibition of growth of the pathogen at 40 μg/ml and above. The bottom panel depicts the cytotoxicity exhibited by compound V15B to the macrophages. Compound V15B did not display any cytotoxicity to the macrophages.
Identification of the binding mode of the hit compounds
Based on the IC50, MIC90, cytotoxicity, and macrophage experiment results, compounds V7B and V15B emerged as potential hits; hence, we predicted their probable mode of binding at the active site of VirS. The compound V7B was docked at the DNA contact site 2 of VirS and surrounded by residues Gln-1, Ser-53, Ala-56, Val-57, Tyr-61, Ser-62, Glu-63, Gln-64, and Ser-65 (Fig. 11A). Most of these residues were also covered in the grid generated against DNA contact site 2 of VirS. Compound V7B showed van der Waals interactions with Ser-53, Glu-63, and Tyr-61 (Fig. 11A). It was found that compound V7B interacted with residues Gln-64, Ser-65, and Val-57 by forming hydrogen bonds (Fig. 11B). Compound V15B was found to be surrounded by Gln-1, Ser-53, Ala-56, Val-57, Gly-60, Tyr-61, Ser-62, Glu-63, and Gln-64 (Fig. 11C). Compound V15B exhibited van der Waals interactions with residues Gln-1, Gly-60, and Glu-63. It also showed hydrogen bonding with Tyr-61 at DNA-binding site 2 (Fig. 11D). V15B also displayed π–σ bond formation with Val-57. It also showed hydrophobic interactions like π–alkyl bond formation with Ala-56 and Val-57 (Fig. 11D).
Figure 11.

Binding mode of the hit molecules. The predicted binding mode of the compounds V7B (A) and V15B (C) to the VirS is shown here. B and D, ligand interaction diagrams of compounds V7B and V15B, respectively, showing distance measurement with the neighboring residues of VirS. The ligand interaction diagrams were generated using Accelrys Discovery Studio (59).
Structure–activity relationship studies for compounds V7B and V15B
On the basis of IC50, MIC90, cytotoxicity, and macrophage studies, two hit molecules (i.e. compounds V7B and V15B) were shortlisted for further SAR studies. For this, they were subjected to a similarity search for identifying their analogs by using the NCI database employing the software Open Babel. The molecules were selected on the basis of a Tanimoto coefficient of ≥0.7. Seventeen molecules similar to compound V7B and 10 molecules similar to compound V15B were procured from NCI-DTP and evaluated for their ability to inhibit VirS DNA-binding activity to identify the structural features responsible for inhibiting VirS activity. These molecules were named V7B-1 to V7B-17 and V15B-1 to V15B-10.
The procured molecules were evaluated for their ability to inhibit DNA-binding activity of VirS at a concentration of 100 μg/ml, and it was found that four analogs of V7B (V7B-4, V7B-5, V7B-8, and V7B-9) and one analog of V15B (V15B-2) displayed >60% inhibition of VirS DNA-binding activity at 100 μg/ml, whereas the rest of them exhibited lesser inhibition (Tables S5 and S6). These five analogs were further evaluated for the determination of their IC50 values. They were found to display a reduced inhibitory potential against VirS DNA-binding activity compared with their parent molecules, which is tabulated in Table S5 and S6. It was found that none of these molecules displayed an enhanced potential to inhibit VirS; however, their structures were carefully analyzed to derive the structure–activity relationship.
A detailed comparison of the structures of V7B and its analogs revealed the presence of a benzene sulfonamide moiety in all of the structures except in V7B-5 and V7B-17. The structure comparison of a few of the analogs of V7B with its parent compound is shown in Fig. 12. The absence of acetamide moiety in compound V7B-9 (IC50 = 2.9 μg/ml) led to reduction in the potency of V7B-9 compared with its parent compound V7B (IC50 = 1.8 μg/ml). Compound V7B-4 contains 2-amino-3-chloro-1,4-napthoquinone moiety attached to benzene sulfonamide, which resulted in a poor IC50. Substitution of methane diamine by benzene sulfonamide attached to 2-amino-3-chloro-1,4-napthoquinone moiety in compound V7B-8 also resulted in poor IC50 (12.4 μg/ml) compared with its parent molecule. Replacement of benzene sulfonamide with sulfonyl benzene and modification of 1,4-dihydronapthalene-1-one to 1,4-dihydronapthalene-1,4-dione in parent compound resulted in reduced potency to inhibit VirS DNA-binding activity, as observed in compound V7B-5. Thus, the structural comparisons of all of these molecules with their activity emphasize the importance of the acetamide ring attached to benzene sulfonamide, which might be essential for the potent inhibition of DNA-binding activity of VirS. Second, it was also found that bulkier modifications to the acetamide group attached to benzene sulfonamide may lead to poor inhibition of VirS DNA-binding activity, as observed in the cases of V7B-10, V7B-13, V7B-15, and V7B-17.
Figure 12.

Structure comparison of compound V7B and its analogs.
Among the procured analogs of compound V15B, only one compound was found to be functionally active against the VirS DNA-binding activity whose structure comparison with its parent compound is shown in Fig. 13. Replacement of the isoquinoline ring from the parent molecule (V15B) with a benzene ring, pyridine ring, and methyl pyridine displayed poor inhibition of VirS DNA-binding activity compared with its parent molecule, as observed in compounds V15B-1, V15B-3, V15B-5, and V15B-6. Further modification of the isoquinoline ring to 1-methyl-3,4-dihydroisoquinoline-4-one and its attachment to the 2-(cyclohexa-1,3-dien-1-yl) pyridine moiety in compound V15B-2 showed a potential to inhibit the DNA-binding activity of VirS; however, it displayed a slightly lower IC50 than the parent molecule. It was also observed that modifications to the 2-(4,5-dihydro-1H-pyrrol-2-y) pyridine moiety failed to show any inhibition against DNA-binding activity of VirS, as observed in the cases of V15B-8 and V15B-10. Thus, the structural comparisons of all of these molecules with their activities emphasize the importance of the isoquinoline ring attached to 3-nitroso-2-phenyl-pyrrole as present in the parent molecule, which is essential for inhibition of the DNA-binding activity of VirS.
Figure 13.

Comparison of structure of compound V15B and its analogs. ND, not determined.
Discussion
In this study, we attempted to dissect the role of VirS in the survival of M. tuberculosis under acidic stress and in the maintenance of pH homeostasis of the pathogen. We found that the loss of virS reduced the ability of M. tuberculosis to block phagosomal-lysosomal fusion in the activated macrophages as well as its ability to survive in acidic conditions, and its absence also led to a disturbance in the maintenance of the intrabacterial pH under acidic stress and inside activated macrophages, thereby suggesting the role of VirS in evading stressful acidic conditions.
To understand how VirS mediates the regulation of gene expression in acid stress, microarray studies were carried out. Transcriptome profiling has provided insights into the role of VirS in the regulation of various pathways, such as those involved in cellular metabolism; cell-wall and lipid metabolism; PE/PPE family proteins; energy pathways and respiration; efflux pumps and ion transporters; detoxification; information pathways; and transcription regulators in acidic stress.
First, we found a striking influence of VirS on the regulation of a number of genes (induced or repressed) involved in fatty acid oxidation and biosynthesis that can contribute to remodeling of the M. tuberculosis cell wall. It has been a general belief that the cell wall of mycobacteria remodels itself when exposed to hostile environments to become impervious to outside harsher conditions (13, 34). We observed VirS-dependent up-regulation of genes involved in fatty acid β-oxidation (fadB2 and fadE35), lipase and hydrolyzing enzymes (Rv2672 and Rv0867c), and membrane-associated proteins (Rv2911, Rv0314c, Rv0531, and Rv2180c) and down-regulation of genes encoding enzymes involved in polyketide synthase (pks2, pks3, pks6, and papA1), membrane proteins (mmpS3, Rv2869c, Rv0412c, Rv2091c, and Rv0677c), phenolic glycolipids (fadD22), biosynthesis and export of siderophores (mmpS5 and mmpL5), and lipoprotein (Rv2290). VirS-regulated expression of genes involved in lipid metabolism suggests its pivotal role in remodeling of the cell envelope that can facilitate intracellular survival under acidic stress.
Our expression profile also revealed the regulation of metabolic enzymes by VirS at acidic pH. It has been reported that there is an induction of genes involved in metabolism via an anaplerotic node at acidic pH in M. tuberculosis. We observed VirS-dependent induction of a glyoxylate shunt comprising isocitrate lyase (icl) at acidic pH, which replenishes intermediates of the TCA cycle and gluconeogenesis and decreases carbon flux through the TCA cycle (35). The gene most up-regulated by VirS in acid stress is Rv1405c, which has also been reported previously to be induced under acid stress and is implicated in adaptation of M. tuberculosis to the intramacrophage environment; however, the substrate for Rv1405c is still not known (36–38). It has been reported that fermentative pathways become up-regulated under acidic stress in B. subtilis and Bacillus cereus (39, 40). The genes for alcohol dehydrogenase were found to be induced that catalyze the conversion of pyruvate to ethanol, generating CO2 and dissipating H+ to deal with low intracellular pH or restoration of NAD+/NADH balance (41). Consistent with this fact, we found VirS-dependent induction of pyruvate decarboxylase (pdc) and alcohol dehydrogenases (adhE1 and Rv1530) at acidic pH, which may reduce acidity through NAD(P)H that transfers electrons to electron transport chain and pumps protons out of the cell (40). We also found a strong influence of VirS on the expression of genes related to cellular metabolism. Genes encoding enzymes for the synthesis of amino acids feeding into the TCA cycle (argJ), nitrate reductase (narX), for the synthesis of heme and porphyrin (hemB) were found to be down-regulated by VirS at acidic pH. Genes for sulfur reduction are essential for the synthesis of cysteine, methionine, mycothiol, and sulfolipids, and these reduced sulfur products contribute to M. tuberculosis pathogenesis. We found that genes for sulfur reduction (sirA and cysH) were also down-regulated in a VirS-dependent manner. These data suggest that changes in the central metabolism by VirS enable M. tuberculosis to adapt to the acidic intraphagosomal environment.
In the context of maintenance of intrabacterial pH, we observed VirS-dependent induction of efflux pumps and ion transporters (kdpC and ctpG). It is reported that a decrease in pH leads to reduction in membrane potential and K+ uptake, which induces the genes encoding K+ transporters (42). Induction of potassium transporters is necessary for the maintenance of internal pH homeostasis and promotes survival in the acidic environment (42). Kdp is a K+ uptake system known to be induced in an acidic environment, which may be required for bacterial adaptation at low pH levels (42). As Kdp is a K+/H+ antiporter, a maintenance of cytoplasmic pH is expected by pumping protons out of the cell. The metal transporter CtpG that transports Cd2+ across the mycobacterial cell membrane (43), which is also regulated by VirS. Such transporters have been previously reported to be critical for ion homeostasis and bacterial survival (43, 44). Thus, regulation of these cell-wall transporters suggests the role of VirS in response to phagosomal acidification and in the maintenance of intrabacterial pH.
Transcriptome profiling also revealed that VirS further activates other transcriptional regulators (Rv1994c and Rv3765c); thus, it might be acting as a master regulator under these conditions inducing the acidic responses of the cell. This is in agreement with the earlier study by Talaat et al. (14), which implicated that VirS could be the master regulator of reactivation of tuberculosis. CmtR (Rv1994c) is an AsrR-SmtB repressor that senses cadmium and modulates the expression of genes related to the efflux and detoxification of this metal ion (45). The regulation of CmtR and CtpG by VirS indicates its importance in the efflux of the cadmium ions at acidic pH to regulate ion homeostasis.
A down-regulation in the VirS-dependent gene expression was observed in ribosome biogenesis pathway (rpsk) and protein translation (infC). Because the genes involved in ribosome biogenesis and protein translation are the major consumer of cellular energy, their down-regulation is also expected (46, 47). Repression of these genes indicates a slowing down of the bacterial growth at acidic stress, which is mediated by VirS. It is well-understood that there is a strong connection between acid stress and oxidative stress (40, 48), and our transcriptome profiling revealed VirS-dependent up-regulation of genes involved in protection from oxidative stress (i.e. ahpC, ahpD, and Rv3177), which may aid bacterial survival at acidic pH. We also observed a decreased expression of aerobic respiration (nuok), and such repression at acidic pH has also been documented earlier (49). We also found VirS-dependent down-regulation of cytochrome assembly ATP-binding protein (cydD) at acidic pH, which promotes proton export for the maintenance of intrabacterial pH (40).
Based on the analysis of our transcriptional profile, we propose a model for the survival of M. tuberculosis under acidic conditions mediated by VirS, although further investigations are needed to confirm these proposed mechanisms and the involvement of the downstream genes of VirS in pH homeostasis (Fig. 14).
Figure 14.

Proposed model for VirS-mediated downstream actions at acidic pH. Exposure to acidic pH causes up-regulation of VirS, which in turn regulates a number of genes probably involved in combating acidic stress faced by the pathogen. First, VirS regulates genes related to cellular metabolism that may help bacteria to adapt to acidic stress. Second, it regulates the genes related to various cell-wall components that might help in remodeling of the cell envelope to make it impervious to acidic stress. It also up-regulates the expression of efflux pumps and ion transporters that may help in the maintenance of intrabacterial pH by exporting an excess of protons outside the cell. VirS also regulates genes related to detoxification, the information pathway, and other transcriptional regulators that may help bacteria to resist and adapt to acidic stress.
The importance of VirS for the survival of bacterial cells under acidic stress and its role in regulating multiple downstream targets signify that it is an attractive drug target for the development of new anti-tuberculosis drugs. Toward this, we characterized it further by evaluating the DNA-binding activity of VirS and by determining the DNA sequence to which VirS binds by employing DNA footprinting. Further, we generated a consensus logo by WebLogo to map the conservation of nucleotides in the sequences employed (Fig. 5C) (15). Additionally, we performed EMSA with the upstream regions of a few downstream targets of VirS, which were found to exhibit direct interaction with VirS, as evident by a shift in the EMSA studies (Fig. 5D).
Site-directed mutational studies revealed four residues (i.e. Arg-16, Arg-20, Tyr-61, and Arg-69) as the critical residues for binding of VirS to its cognate DNA. Residue Tyr-61 was found to be a conserved residue in other homologous proteins. Arg-20 of VirS corresponds to Arg-46 in MarA, which penetrates into the major groove of DNA and makes hydrogen bonds with bases. Similarly, Arg-69 of VirS is homologous to Arg-96 of MarA, which hydrogen-bonds with the base of DNA. Similarly, Arg-16 of VirS corresponds to Trp-42 in MarA; side chains of Trp-42 are involved in the van der Waals contacts with the DNA. The essentiality of the identified critical residues for DNA-binding activity suggests their importance in the interaction of VirS with the DNA. Hence, these residues could be utilized for rational design of inhibitors to target the DNA-binding activity of VirS.
Because VirS appears as an attractive target for the discovery of new inhibitors against M. tuberculosis, we performed structure-based virtual screening for the identification of small molecules against the DNA binding activity of VirS, which may serve as hit molecules for novel anti-tuberculosis drug development. We identified two hit molecules (i.e. V7B and V15B) that exhibited potent inhibition of VirS activity and inhibited the growth of M. tuberculosis in vitro as well as inside macrophages. However, IC50 and MIC values of both of these compounds did not correlate well, and the most probable reason for such disparity could be attributed to the chemical nature of the potent enzyme inhibitors leading to their poor penetration into M. tuberculosis cells, resulting in weak MIC values. Also, the target specificity of these compounds inside M. tuberculosis still needs to be validated. Hence, these two compounds can be further explored to increase their potency and could be a subject of lead optimization.
Thus, we have explored the promising drug target VirS by identifying a few crucial residues for its activity that could help in designing rational inhibitors as well as by identifying few moieties that inhibit VirS activity as well as M. tuberculosis growth. This is the first report of exploring VirS as a target for drug development against M. tuberculosis.
Experimental procedures
Oligonucleotides for PCR and for site-directed mutagenesis, FITC, IFN-γ, poly(dI-dC), tyloxapol, and complete mini EDTA-free protease inhibitor mixture tablets were purchased from Sigma-Aldrich. DpnI and Pfu polymerase were from New England Biolabs (Ipswich, MA). Cy5-labeled dsDNA was customized from IDT (Coralville, IA). Ni-NTA was obtained from IBA Life Sciences (Goettingen, Germany). RPMI 1640, Dulbecco's modified Eagle's medium, LysoTracker Red DND-99, Prolong Gold antifade reagent with DAPI, antibiotic-antimycotic, and fetal bovine serum (FBS) were obtained from Thermo Fisher Scientific (Waltham, MA). Difco Middlebrook 7H9 (MB7H9) medium, 7H11 agar, and ADC were obtained from BD Biosciences (Franklin Lakes, NJ). The compounds were obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, NCI, National Institutes of Health (Bethesda, MD).
Bacterial strains and growth conditions
M. tuberculosis strains were grown in MB7H9 medium supplemented with 1× ADC, 0.2% glycerol and 0.2% Tween 80 with constant shaking at 200 rpm or on 7H11 agar plates containing 10% oleic acid/albumin/dextrose/catalase (OADC) and 0.2% glycerol at 37 °C. The pH of MB7H9 medium was adjusted by using HCl, and instead of Tween 80, 0.02% tyloxapol was used, wherever required. Hygromycin B and kanamycin were used at a concentration of 50 and 25 μg/ml, respectively, for mycobacteria. Phosphate-citrate buffers were prepared from 200 mm sodium phosphate and 100 mm citric acid. E. coli strains XL-1 Blue and BL21(λDE3) were grown in Luria–Bertani (LB) broth or on LB agar. Ampicillin was used at a concentration of 50 μg/ml for E. coli.
Growth kinetics and colocalization studies by confocal microscopy
The virS mutant of M. tuberculosis and virS complemented strain were earlier generated in our laboratory (12, 13). The M. tuberculosis Erdman, virS mutant, and virS complemented strain were grown in MB7H9 medium adjusted to a pH of 6.6, 5.5, 5.0, and 4.5 at 37 °C with constant shaking at 200 rpm. The growth was monitored daily by measuring the A600 nm. In case of cfu studies, the cells were appropriately diluted and plated at various time points on MB7H11 agar plates. The plates were kept at 37 °C for 3–4 weeks for the appearance of colonies. The phagosomal maturation arrest of M. tuberculosis was studied as described previously (50). For colocalization studies in activated macrophages, PMA-activated macrophages were washed once with plain RPMI 1640 medium, and cells were further incubated for 24 h in RPMI 1640 medium containing 10% FBS and 10 ng/ml IFN-γ before infection. After infection and LysoTracker Red treatment, coverslips were mounted by using Prolong Gold antifade reagent and visualized by a Leica TCS Sp5 confocal laser-scanning microscope (Leica Microsystems, Mannheim, Germany). The percentage colocalization of FITC-labeled mycobacteria with LysoTracker Red was calculated by analyzing ∼100 phagosomes.
Intrabacterial pH measurement assays
The plasmid encoding ratiometric pH-GFP (a kind gift from Dr. Sabine Ehrt, Department of Microbiology and Immunology, Weill Cornell Medical College, New York; received from Dr. Amit Singh, Department of Microbiology and Cell Biology, Indian Institute of Science, Bengaluru, India) was electroporated into M. tuberculosis Erdman, virS mutant, and complemented strain, and the recombinants were selected on the basis of fluorescence of GFP on 7H11-OADC-hygromycin B plates. The selected recombinant clones were grown in MB7H9 medium in the presence of hygromycin until A600 nm of 0.3–0.5. The cultures were harvested, and pellets were resuspended in phosphate-citrate buffer, pH 7.4 and 4.5, supplemented with ADC, 0.2% Tween 80, and hygromycin B. The intrabacterial pH measurements were carried out after incubation of the bacteria in the respective buffers for different time intervals. The bacterial cultures at different time points were harvested and concentrated to 20 times by centrifugation to increase GFP signal. Fluorescence was analyzed in a Cary Varian spectrofluorometer with excitation wavelengths of 395 and 475 nm at an emission wavelength of 510 nm. The intrabacterial pH was derived by interpolating the ratio of 395/475-nm fluorescence intensity on a standard curve. The standard curve for intrabacterial pH measurement was generated by incubating the lysate containing GFP with the phosphate-citrate buffers of pH ranging from 5.5 to 8.5 in a 96-well plate (1). The plate was read at excitation wavelengths of 395 and 475 nm and an emission wavelength of 510 nm.
For pHIB measurements of bacteria residing inside macrophages, 5 × 105 PMA-differentiated THP-1 cells were seeded in poly-l-lysine–coated glass bottom culture dishes of 35-mm thickness. PMA-activated macrophages were washed once with plain RPMI 1640 medium, and cells were further incubated for 24 h in RPMI 1640 medium containing 10% FBS and 10 ng/ml IFN-γ before infection. Macrophages were infected with M. tuberculosis at an MOI of 1:5 (macrophage/bacteria) for 4 h followed by amikacin treatment of 1 h. After different time points postinfection, macrophages were analyzed under confocal microscopy to determine the intensities of GFP (exciters D405 and D476, emitter 535). All of the images were acquired within an experiment under identical conditions. A bacterial group was defined as at least one bacterium but may consist of 2–5 bacilli. The bacterial groups were analyzed for their emission intensities when excited at 405 and 476 nm, and the ratios of intensities at 405:476 nm were calculated. pHIB were derived by interpolating the 405:476 nm intensity ratios on a standard curve.
Microarray studies
M. tuberculosis Erdman, virS mutant, and virS complemented strain were grown until an A600 nm of 0.3–0.5, and cells were harvested by centrifugation at 4500 rpm for 10 min. Harvested cells were exposed to MB7H9 medium adjusted to pH 6.6 and 4.5 and grown at 37 °C with constant shaking at 200 rpm for 1 h. Total RNA was isolated from all of the mycobacterial strains (grown at pH 6.6 and 4.5) and was processed and hybridized to a M. tuberculosis whole-genome Gene Expression Profiling microarray G2509F (AMADID: G2509F_43893, Agilent Technologies). DNA microarrays and data analysis were performed at the University of Delhi South Campus Microarray Centre (UDSCMAC). Slides were scanned on a microarray scanner (Agilent Technologies) and analyzed using the GeneSpring software.
Cloning and expression of virS
The virS gene was PCR-amplified by employing M. tuberculosis H37Rv genomic DNA as the template and by using the primers F-virS containing SpeI, thrombin site, and EcoRI followed by a His6 tag and R-virS–containing HindIII site. The sequence of the primers is given in Table S2. The amplified PCR product after digestion with SpeI and HindIII was cloned into pET43.1a vector digested with the same enzymes to create pET43.1a-virS (NusA-VirS) for the synthesis of N-terminal His-tagged VirS. To subclone virS from pET43.1a-virS construct into vector pBEn-SET3a, the positive clone of pET43.1a-virS was digested with EcoRI and HindIII to obtain a fallout of ∼1.0 kb, corresponding to the size of virS. The pBEn-SET3a vector was also digested with the same enzymes. The digested vector pBEn-SET3a and digested virS were ligated to obtain the recombinant clone pBEn-virS (3a-VirS). Positive clones were confirmed by restriction digestion. For expression, E. coli BL21(λDE3) cells transformed with pET43.1a-virS and pBEn-virS, were grown at 37 °C in Luria–Bertani medium containing 50 μg/ml ampicillin until A600 nm of 0.8. The cultures (i.e. NusA-VirS and 3a-VirS) were then induced with 1 mm isopropyl-1-thio-β-d-galactopyranoside and were allowed to grow for 16 h at 16 and 18 °C, respectively. The cells were harvested by centrifugation at 4 °C and 6000 × g for 10 min.
Purification of 3a-VirS
E. coli BL21(λDE3) cells were transformed with pBEn-virS, and the cells were grown and induced as described above. For purification, the cells from the induced culture were harvested and resuspended in lysis buffer containing 1× PBS, pH 7.4, 10 mm imidazole, 5 mm β-mercaptoethanol, 1 mm phenylmethylsulfonyl fluoride, and complete mini EDTA-free protease inhibitor mixture tablet and lysed by sonication followed by centrifugation to remove cell debris (15,000 × g, 45 min, 4 °C). Clarified lysate was loaded onto a Ni-NTA resin pre-equilibrated with lysis buffer. Washing of the column was performed initially with buffer containing 1× PBS, 10 mm imidazole followed by one wash each with the same buffer containing 20 mm and 50 mm imidazole. The protein was eluted with buffer containing 250 mm imidazole, and the purity of the protein was analyzed by SDS-PAGE by using a 12.5% polyacrylamide gel. The fractions containing VirS were pooled, and 10% glycerol was added. The pooled protein was dialyzed overnight against 20 mm Tris, pH 8.0, and stored at −80 °C.
Gel-shift assay for NusA-VirS fusion protein
DNA probes for gel mobility-shift assays were amplified by PCR from vector pSG10 comprising the intergenic region between virS and mymA, which is hypothesized to be bound by VirS (12). Two primers (UP6 and DW2) were utilized for the amplification of a 325-bp intergenic region. (Table S2). Primer DW2 was end-labeled with [γ-32P]ATP by using T4 polynucleotide kinase. The PCR product was run on 1.5% agarose gel, and the amplified band was cut and gel-eluted by using GFX columns and quantitated by a β-counter (TRI-CARB 2900TR Liquid Scintillation Counter, PerkinElmer Life Sciences). Lysates of E. coli cells overexpressing either the NusA-VirS fusion protein or only NusA protein were prepared. The reaction mixture consisted of 120,000 cpm of labeled DNA, binding mix, and different amounts (i.e. 13, 26, and 52 μg) of the lysate. The binding mix consisted of 1× binding buffer, 1 mg/ml BSA, 1 mm DTT, 4% PEG-8000, and 1 μg of poly(dI-dC). The samples were incubated for 30 min at 37 °C. Samples were then electrophoresed on a 3.5% polyacrylamide gel, dried, and autoradiographed.
DNA footprinting assay
Binding reaction mixture was prepared as described above in a total volume of 30 μl that consisted of 126,000 cpm of labeled DNA probe and 52 μg of lysate and incubated at 37 °C for 30 min. After incubation, the binding reaction mix was subjected to DNase I digestion (0.2 units) for 1 min, and digestion was terminated by the addition of 180 μl of stop solution (300 mm sodium acetate, pH 7.0, 10 mm EDTA, pH 8.0, 0.3 mg/ml yeast t-RNA, 0.01% SDS). Samples were extracted with phenol-chloroform, and precipitation of nucleic acid was performed with ethanol. DNA was resuspended in water, and formamide dye was added. A G+A ladder was also prepared by using the same probe, and formamide dye was mixed. The samples and G+A ladder were preheated at 90 °C for 10 min before loading. These samples were separated on 6% polyacrylamide sequencing gel and electrophoresed (in 0.5× TBE buffer and 0.5 m sodium acetate in the lower chamber and 0.5× TBE in the upper chamber) at 1050 V. Gel was dried and exposed for 14 h, followed by autoradiography.
Homology modeling of VirS
The amino acid sequence similarity search was performed by using the NCBI-BLAST server in the PDB structure database. Based on the BLAST results, the first few homologs of VirS were identified, namely ToxT of Vibrio cholera (PDB entry 3GBG) (51), gadX of E. coli (PDB entry 3MKL),7 AdpA of Streptomyces griseus (PDB entry 3W6V) (27), unknown protein of Listeria innocula (PDB entry 3OOU),8 AraC of E. coli (PDB entry 2K9S) (52), MarA of E. coli (PDB entry 1BL0) (25), yesn of Fusobacterium nucleatum (PDB entry 3LSG),9 ternary complex of MarA, DNA, and C terminus of RNA polymerase of E. coli (PDB entry 1XS9) (53), Rob of E. coli (PDB entry 1D5Y) (26), transcription regulator of Chromobacterium violaceum (PDB entry 3OIO),10 and XylR of E. coli (PDB entry 4FE4) (54). The sequences of all of these homologs were aligned with the C-terminal region (amino acids 250–340) of the M. tuberculosis VirS sequence by using the software ClustalW. The result of sequence alignments was then submitted to the software Modeler 9.0 to generate a 3D structure of VirS, and the generated homology model was validated by employing Rampage, Errat, Verify 3D, Pro-Q, and Prosa software.
Site-directed mutagenesis
Amino acid substitutions were generated by site-directed mutagenesis kit provided by Stratagene (La Jolla, CA) according to the manufacturer's instructions. The E. coli XL1 Blue cells were transformed to get the colonies on ampicillin plates. These colonies were grown in LB medium, and DNA was isolated. The mutations in the DNA were confirmed by DNA sequencing. The primers used in mutagenesis are listed in Table S2.
EMSA for purified 3a-VirS
0.5 μg of purified 3a-VirS protein was taken in a reaction mixture that contained 1× binding buffer (10 mm Tris acetate, pH 8.0, 50 mm KCl, 5% glycerol), 4% PEG, 1 μg of poly(dI-dC), 100 μg/ml BSA, 1 mm DTT, and 0.8 pmol of 5′ Cy5 fluorophore–labeled DNA and incubated for 30 min at 37 °C. This labeled dsDNA represents the sequence to which VirS binds as identified by a DNA footprinting study with 10-bp flanking regions on both sides (66-bp dsDNA), which were labeled with Cy5 fluorophore at the 5′ ends. After incubation, samples were loaded onto a 5% polyacrylamide gel and electrophoresed in 1× TAE buffer overnight at 32 V. The next day, fluorescence was detected by a phosphor imager (model FLA-9000, FUJIFILM Corp., Minato-ku, Tokyo, Japan).
Regulation of downstream genes by VirS using EMSA
Upstream regions of the genes were PCR-amplified by using the Cy5 PCR Labeling Kit (Jena Biosciences). The amplified products were gel-eluted and quantified. Forty fmol of labeled product was incubated with 1 μg of purified VirS in the binding buffer (composition same as mentioned above) for 30 min at 37 °C. Samples were electrophoresed onto a 5% polyacrylamide gel in 1× TAE buffer overnight. The bands were visualized using a phosphor imager.
Virtual screening
The 3D structure generated by Modeler 9.0 was superimposed with the crystal structure of MarA (PDB entry 1BL0), and the coordinates of DNA bound to MarA structure were used to predict the DNA contact sites of VirS. Virtual screening was carried out against both of the DNA contact sites by using a small-molecule library of the NCI Open Database consisting of 260,071 compounds. These compounds were shortlisted by using the FAF server, which resulted in 95,748 compounds that were used for screening against the DNA contact sites of VirS by using Autodock4.2 (30). The top compounds were procured from NCI-DTP, depending upon the availability for conducting inhibition studies. Fifty-seven compounds each on the basis of docking at site 1 and site 2 were procured from NCI-DTP. Compounds obtained from docking at site 1 were denoted as V1A–V57A. Compound V7A was not processed further due to its toxicity. Compounds obtained from docking at site 2 were denoted as V1B–V57B.
Assay for inhibition of DNA-binding activity of VirS
All the compounds were dissolved in DMSO to prepare a stock solution of 2 mg/ml and were screened for their ability to inhibit the DNA-binding activity of VirS at a concentration of 100 μg/ml. The reaction mixture was same as described above. Binding buffer, purified protein (3a-VirS), and the individual test compounds were incubated at 37 °C for 30 min followed by the addition of the rest of the components and further incubated for 30 min at 37 °C. The rest of the steps were the same as mentioned above. Sample containing DMSO was used as a negative control for the assay. IC50 is defined as the concentration of the compound that exhibits 50% inhibition of the activity.
Assay for inhibition of growth of M. tuberculosis
The inhibition of M. tuberculosis growth was evaluated by employing a resazurin microtiter assay as described previously (30, 55). Blue color depicts no growth, whereas conversion to pink color depicts viable cells. Fluorescence was measured at an emission wavelength of 590 nm with an excitation wavelength of 530 nm, and percentage inhibition was calculated. Rifampicin was employed as a positive control, whereas wells with DMSO were used as a negative control. MIC90 is defined as the concentration of compound at which growth of the pathogen was inhibited by 90%.
Evaluation of cytotoxicity of the compounds
Compounds were evaluated for their cytotoxicity against three mammalian cell lines, namely THP-1 (human monocytic cell line), HEK (human embryonic kidney cell line), and HeLa (cervical cell line), by employing the protocol described previously (30, 55). IC50 is defined as the concentration of the compound at which 50% growth of the cell line is inhibited.
Evaluation of inhibitory potential of the compounds against intracellular survival of M. tuberculosis inside macrophages
The ability of the compounds to inhibit the intracellular growth of the pathogen was evaluated as described previously (56).
Author contributions
S. S., G. K., and A. K. T. designed the experiments. S. S. and N. G. performed the experiments. S. S. wrote the manuscript; G. K. and A. K. T. proofread, corrected, and edited the manuscript. G. K. and A. K. T. provided overall supervision of the work.
Supplementary Material
Acknowledgments
We thank the University of Delhi South Campus Microarray Center (UDSCMAC) (New Delhi, India) and Bionivid Technologies Pvt Ltd. (Bengaluru, India) for conducting microarray experiments and data analysis. We thank Dr. Sabine Ehrt (Department of Microbiology and Immunology, Weill Cornell Medical College, New York) and Dr. Amit Singh (Department of Microbiology and Cell Biology, Indian Institute of Science, Bengaluru, India) for providing the plasmid encoding ratiometric pH-GFP. We are thankful to Dr. Amit Singh for critical reading of the manuscript. The Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis (NCI, National Institutes of Health, Bethesda, MD) is gratefully acknowledged for providing the compounds. We thank the Central Instrumentation Facility (CIF), University of Delhi South Campus (UDSC), for providing the grid facility for docking studies. We also thank the UDSC-BSL3 facility for providing necessary facilities for conducting experiments related to the use of M. tuberculosis. CIF is also acknowledged for confocal microscopy studies and the FLA-9000 phosphor imager. Priti Singh and Tannu Priya Gosain are acknowledged for excellent technical help. The DNA sequencing facility, UDSC, is acknowledged for DNA sequencing. The distributed information subcenter, Department of Biotechnology, Ministry of Science and Technology (DBT), UDSC, is acknowledged for providing computers and internet services.
This work was supported by Department of Biotechnology, Government of India, Grant BT/01/COE/05/06-II (to A. K. T.); Department of Science and Technology, Government of India, J. C. Bose Fellowship SR/S2/JCB-39/2009 (to A. K. T.), and Department of Biotechnology, Government of India, Innovative Young Biotechnologist Award BT/07/IYBA/2013-5 (to G. K.). The authors declare that they have no conflicts of interest with the contents of this article.
The microarray data have been deposited to the GEO database and are available under accession ID GSE118508.
This article contains Tables S1–S6, Figs. S1–S3, and Excel Sheet XI.
G. Khare, and A. K. Tyagi, unpublished observations.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
C. Chang, J. Mack, S. Clancy, and A. Joachimiak, unpublished data.
Y. Fan, J. Mack, B. Feldman, and A. Joachimiak, unpublished data.
K. Tan, E. Rakowski, S. Clancy, and A. Joachimiak, unpublished data.
C. Chang, J. Mack, B. Feldman, and A. Joachimiak, unpublished data.
- pHIB
- intrabacterial pH
- 3D
- three-dimensional
- Ni-NTA
- nickel-nitrilotriacetic acid
- TCA
- tricarboxylic acid
- IFN
- interferon
- FBS
- fetal bovine serum
- OADC
- oleic acid/albumin/dextrose/catalase
- ADC
- albumin/dextrose/catalase
- LB
- Luria–Bertani
- PMA
- phorbol 12-myristate 13-acetate
- ANOVA
- analysis of variance.
References
- 1. Vandal O. H., Pierini L. M., Schnappinger D., Nathan C. F., and Ehrt S. (2008) A membrane protein preserves intrabacterial pH in intraphagosomal Mycobacterium tuberculosis. Nat. Med. 14, 849–854 10.1038/nm.1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Song H., Huff J., Janik K., Walter K., Keller C., Ehlers S., Bossmann S. H., and Niederweis M. (2011) Expression of the ompATb operon accelerates ammonia secretion and adaptation of Mycobacterium tuberculosis to acidic environments. Mol. Microbiol. 80, 900–918 10.1111/j.1365-2958.2011.07619.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buchmeier N., Blanc-Potard A., Ehrt S., Piddington D., Riley L., and Groisman E. A. (2000) A parallel intraphagosomal survival strategy shared by Mycobacterium tuberculosis and Salmonella enterica. Mol. Microbiol. 35, 1375–1382 [DOI] [PubMed] [Google Scholar]
- 4. Raynaud C., Papavinasasundaram K. G., Speight R. A., Springer B., Sander P., Böttger E. C., Colston M. J., and Draper P. (2002) The functions of OmpATb, a pore-forming protein of Mycobacterium tuberculosis. Mol. Microbiol. 46, 191–201 10.1046/j.1365-2958.2002.03152.x [DOI] [PubMed] [Google Scholar]
- 5. Athmann C., Zeng N., Kang T., Marcus E. A., Scott D. R., Rektorschek M., Buhmann A., Melchers K., and Sachs G. (2000) Local pH elevation mediated by the intrabacterial urease of Helicobacter pylori cocultured with gastric cells. J. Clin. Invest. 106, 339–347 10.1172/JCI9351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marcus E. A., Sachs G., and Scott D. R. (2013) The role of ExbD in periplasmic pH homeostasis in Helicobacter pylori. Helicobacter 18, 363–372 10.1111/hel.12055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stincone A., Daudi N., Rahman A. S., Antczak P., Henderson I., Cole J., Johnson M. D., Lund P., and Falciani F. (2011) A systems biology approach sheds new light on Escherichia coli acid resistance. Nucleic Acids Res. 39, 7512–7528 10.1093/nar/gkr338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martín-Galiano A. J., Ferrándiz M. J., and de la Campa A. G. (2001) The promoter of the operon encoding the F0F1 ATPase of Streptococcus pneumoniae is inducible by pH. Mol. Microbiol. 41, 1327–1338 10.1046/j.1365-2958.2001.02597.x [DOI] [PubMed] [Google Scholar]
- 9. Krulwich T. A., Sachs G., and Padan E. (2011) Molecular aspects of bacterial pH sensing and homeostasis. Nat. Rev. Microbiol. 9, 330–343 10.1038/nrmicro2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gupta S., Jain S., and Tyagi A. K. (1999) Analysis, expression and prevalence of the Mycobacterium tuberculosis homolog of bacterial virulence regulating proteins. FEMS Microbiol. Lett. 172, 137–143 10.1111/j.1574-6968.1999.tb13461.x [DOI] [PubMed] [Google Scholar]
- 11. Gupta S., and Tyagi A. K. (1993) Sequence of a newly identified Mycobacterium tuberculosis gene encoding a protein with sequence homology to virulence-regulating proteins. Gene 126, 157–158 10.1016/0378-1119(93)90607-5 [DOI] [PubMed] [Google Scholar]
- 12. Singh A., Jain S., Gupta S., Das T., and Tyagi A. K. (2003) mymA operon of Mycobacterium tuberculosis: its regulation and importance in the cell envelope. FEMS Microbiol. Lett. 227, 53–63 10.1016/S0378-1097(03)00648-7 [DOI] [PubMed] [Google Scholar]
- 13. Singh A., Gupta R., Vishwakarma R. A., Narayanan P. R., Paramasivan C. N., Ramanathan V. D., and Tyagi A. K. (2005) Requirement of the mymA operon for appropriate cell wall ultrastructure and persistence of Mycobacterium tuberculosis in the spleens of guinea pigs. J. Bacteriol. 187, 4173–4186 10.1128/JB.187.12.4173-4186.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Talaat A. M., Ward S. K., Wu C. W., Rondon E., Tavano C., Bannantine J. P., Lyons R., and Johnston S. A. (2007) Mycobacterial bacilli are metabolically active during chronic tuberculosis in murine lungs: insights from genome-wide transcriptional profiling. J. Bacteriol. 189, 4265–4274 10.1128/JB.00011-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crooks G. E., Hon G., Chandonia J. M., and Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thompson J. D., Higgins D. G., and Gibson T. J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., and Sali A. (2007) Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. Chapter 2, Unit 2.9 10.1002/0471140864.ps0209s50 [DOI] [PubMed] [Google Scholar]
- 18. Lovell S. C., Davis I. W., Arendall W. B. 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., and Richardson D. C. (2003) Structure validation by Cα geometry: φ,ψ and Cβ deviation. Proteins 50, 437–450 10.1002/prot.10286 [DOI] [PubMed] [Google Scholar]
- 19. Wallner B., and Elofsson A. (2003) Can correct protein models be identified? Protein Sci. 12, 1073–1086 10.1110/ps.0236803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bowie J. U., Lüthy R., and Eisenberg D. (1991) A method to identify protein sequences that fold into a known three-dimensional structure. Science 253, 164–170 10.1126/science.1853201 [DOI] [PubMed] [Google Scholar]
- 21. Lüthy R., Bowie J. U., and Eisenberg D. (1992) Assessment of protein models with three-dimensional profiles. Nature 356, 83–85 10.1038/356083a0 [DOI] [PubMed] [Google Scholar]
- 22. Colovos C., and Yeates T. O. (1993) Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 2, 1511–1519 10.1002/pro.5560020916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sippl M. J. (1993) Recognition of errors in three-dimensional structures of proteins. Proteins 17, 355–362 10.1002/prot.340170404 [DOI] [PubMed] [Google Scholar]
- 24. Wiederstein M., and Sippl M. J. (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410 10.1093/nar/gkm290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rhee S., Martin R. G., Rosner J. L., and Davies D. R. (1998) A novel DNA-binding motif in MarA: the first structure for an AraC family transcriptional activator. Proc. Natl. Acad. Sci. U.S.A. 95, 10413–10418 10.1073/pnas.95.18.10413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kwon H. J., Bennik M. H., Demple B., and Ellenberger T. (2000) Crystal structure of the Escherichia coli Rob transcription factor in complex with DNA. Nat. Struct. Biol. 7, 424–430 10.1038/75213 [DOI] [PubMed] [Google Scholar]
- 27. Yao M. D., Ohtsuka J., Nagata K., Miyazono K., Zhi Y., Ohnishi Y., and Tanokura M. (2013) Complex structure of the DNA-binding domain of AdpA, the global transcription factor in Streptomyces griseus, and a target duplex DNA reveals the structural basis of its tolerant DNA sequence specificity. J. Biol. Chem. 288, 31019–31029 10.1074/jbc.M113.473611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., and Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reese M. G. (2001) Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem. 26, 51–56 [DOI] [PubMed] [Google Scholar]
- 30. Rohilla A., Khare G., and Tyagi A. K. (2017) Virtual screening, pharmacophore development and structure based similarity search to identify inhibitors against IdeR, a transcription factor of Mycobacterium tuberculosis. Sci. Rep. 7, 4653 10.1038/s41598-017-04748-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Palomino J. C., Martin A., Camacho M., Guerra H., Swings J., and Portaels F. (2002) Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 46, 2720–2722 10.1128/AAC.46.8.2720-2722.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martin A., Camacho M., Portaels F., and Palomino J. C. (2003) Resazurin microtiter assay plate testing of Mycobacterium tuberculosis susceptibilities to second-line drugs: rapid, simple, and inexpensive method. Antimicrob. Agents Chemother. 47, 3616–3619 10.1128/AAC.47.11.3616-3619.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Collins L., and Franzblau S. G. (1997) Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 41, 1004–1009 10.1128/AAC.41.5.1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schnappinger D., Ehrt S., Voskuil M. I., Liu Y., Mangan J. A., Monahan I. M., Dolganov G., Efron B., Butcher P. D., Nathan C., and Schoolnik G. K. (2003) Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198, 693–704 10.1084/jem.20030846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baker J. J., Johnson B. K., and Abramovitch R. B. (2014) Slow growth of Mycobacterium tuberculosis at acidic pH is regulated by phoPR and host-associated carbon sources. Mol. Microbiol. 94, 56–69 10.1111/mmi.12688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Healy C., Golby P., MacHugh D. E., and Gordon S. V. (2016) The MarR family transcription factor Rv1404 coordinates adaptation of Mycobacterium tuberculosis to acid stress via controlled expression of Rv1405c, a virulence-associated methyltransferase. Tuberculosis (Edinb.) 97, 154–162 10.1016/j.tube.2015.10.003 [DOI] [PubMed] [Google Scholar]
- 37. Rohde K. H., Abramovitch R. B., and Russell D. G. (2007) Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues. Cell Host Microbe 2, 352–364 10.1016/j.chom.2007.09.006 [DOI] [PubMed] [Google Scholar]
- 38. Golby P., Hatch K. A., Bacon J., Cooney R., Riley P., Allnutt J., Hinds J., Nunez J., Marsh P. D., Hewinson R. G., and Gordon S. V. (2007) Comparative transcriptomics reveals key gene expression differences between the human and bovine pathogens of the Mycobacterium tuberculosis complex. Microbiology 153, 3323–3336 10.1099/mic.0.2007/009894-0 [DOI] [PubMed] [Google Scholar]
- 39. Desriac N., Broussolle V., Postollec F., Mathot A. G., Sohier D., Coroller L., and Leguerinel I. (2013) Bacillus cereus cell response upon exposure to acid environment: toward the identification of potential biomarkers. Front. Microbiol. 4, 284 10.3389/fmicb.2013.00284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wilks J. C., Kitko R. D., Cleeton S. H., Lee G. E., Ugwu C. S., Jones B. D., BonDurant S. S., and Slonczewski J. L. (2009) Acid and base stress and transcriptomic responses in Bacillus subtilis. Appl. Environ. Microbiol. 75, 981–990 10.1128/AEM.01652-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mols M., van Kranenburg R., van Melis C. C., Moezelaar R., and Abee T. (2010) Analysis of acid-stressed Bacillus cereus reveals a major oxidative response and inactivation-associated radical formation. Environ. Microbiol. 12, 873–885 10.1111/j.1462-2920.2009.02132.x [DOI] [PubMed] [Google Scholar]
- 42. Cholo M. C., van Rensburg E. J., Osman A. G., and Anderson R. (2015) Expression of the genes encoding the Trk and Kdp potassium transport systems of Mycobacterium tuberculosis during growth in vitro. Biomed. Res. Int. 2015, 608682 10.1155/2015/608682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. López M., Quitian L. V., Calderón M. N., and Soto C. Y. (2018) The P-type ATPase CtpG preferentially transports Cd2+ across the Mycobacterium tuberculosis plasma membrane. Arch. Microbiol. 200, 483–492 10.1007/s00203-017-1465-z [DOI] [PubMed] [Google Scholar]
- 44. Raimunda D., Long J. E., Sassetti C. M., and Argüello J. M. (2012) Role in metal homeostasis of CtpD, a Co2+ transporting P(1B4)-ATPase of Mycobacterium smegmatis. Mol. Microbiol. 84, 1139–1149 10.1111/j.1365-2958.2012.08082.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chauhan S., Kumar A., Singhal A., Tyagi J. S., and Krishna Prasad H. (2009) CmtR, a cadmium-sensing ArsR-SmtB repressor, cooperatively interacts with multiple operator sites to autorepress its transcription in Mycobacterium tuberculosis. FEBS J. 276, 3428–3439 10.1111/j.1742-4658.2009.07066.x [DOI] [PubMed] [Google Scholar]
- 46. Lemke J. J., Sanchez-Vazquez P., Burgos H. L., Hedberg G., Ross W., and Gourse R. L. (2011) Direct regulation of Escherichia coli ribosomal protein promoters by the transcription factors ppGpp and DksA. Proc. Natl. Acad. Sci. U.S.A. 108, 5712–5717 10.1073/pnas.1019383108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nomura M., Gourse R., and Baughman G. (1984) Regulation of the synthesis of ribosomes and ribosomal components. Annu. Rev. Biochem. 53, 75–117 10.1146/annurev.bi.53.070184.000451 [DOI] [PubMed] [Google Scholar]
- 48. Maurer L. M., Yohannes E., Bondurant S. S., Radmacher M., and Slonczewski J. L. (2005) pH regulates genes for flagellar motility, catabolism, and oxidative stress in Escherichia coli K-12. J. Bacteriol. 187, 304–319 10.1128/JB.187.1.304-319.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fisher M. A., Plikaytis B. B., and Shinnick T. M. (2002) Microarray analysis of the Mycobacterium tuberculosis transcriptional response to the acidic conditions found in phagosomes. J. Bacteriol. 184, 4025–4032 10.1128/JB.184.14.4025-4032.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Khare G., Reddy P. V., Sidhwani P., and Tyagi A. K. (2013) KefB inhibits phagosomal acidification but its role is unrelated to M. tuberculosis survival in host. Sci. Rep. 3, 3527 10.1038/srep03527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lowden M. J., Skorupski K., Pellegrini M., Chiorazzo M. G., Taylor R. K., and Kull F. J. (2010) Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proc. Natl. Acad. Sci. U.S.A. 107, 2860–2865 10.1073/pnas.0915021107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rodgers M. E., and Schleif R. (2009) Solution structure of the DNA binding domain of AraC protein. Proteins 77, 202–208 10.1002/prot.22431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dangi B., Gronenborn A. M., Rosner J. L., and Martin R. G. (2004) Versatility of the carboxy-terminal domain of the alpha subunit of RNA polymerase in transcriptional activation: use of the DNA contact site as a protein contact site for MarA. Mol. Microbiol. 54, 45–59 10.1111/j.1365-2958.2004.04250.x [DOI] [PubMed] [Google Scholar]
- 54. Ni L., Tonthat N. K., Chinnam N., and Schumacher M. A. (2013) Structures of the Escherichia coli transcription activator and regulator of diauxie, XylR: an AraC DNA-binding family member with a LacI/GalR ligand-binding domain. Nucleic Acids Res. 41, 1998–2008 10.1093/nar/gks1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Singh S., Khare G., Bahal R. K., Ghosh P. C., and Tyagi A. K. (2018) Identification of Mycobacterium tuberculosis BioA inhibitors by using structure-based virtual screening. Drug Des. Devel. Ther. 12, 1065–1079 10.2147/DDDT.S144240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Khare G., Kumar P., and Tyagi A. K. (2013) Whole-cell screening-based identification of inhibitors against the intraphagosomal survival of Mycobacterium tuberculosis. Antimicrob. Agents Chemother 57, 6372–6377 10.1128/AAC.01444-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Babicki S., Arndt D., Marcu A., Liang Y., Grant J. R., Maciejewski A., and Wishart D. S. (2016) Heatmapper: web enabled heatmapping for all. Nucleic Acid Res. 44, W147–W153 10.1093/nar/gkw419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.1, Schroedinger, LLC, New York [Google Scholar]
- 59. Dassault Systèmes (2016) BIOVIA, Discovery Studio Modeling Environment, release 2017, Dassault Systèmes, San Diego [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.