

Abstract
Parkinson's disease (PD) is a major human disease associated with degeneration of the central nervous system. Evidence suggests that several endogenously formed 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)–mimicking chemicals that are metabolic conversion products, especially β-carbolines and isoquinolines, act as neurotoxins that induce PD or enhance progression of the disease. We have demonstrated previously that mitochondrially targeted human cytochrome P450 2D6 (CYP2D6), supported by mitochondrial adrenodoxin and adrenodoxin reductase, can efficiently catalyze the conversion of MPTP to the toxic 1-methyl-4-phenylpyridinium ion. In this study, we show that the mitochondrially targeted CYP2D6 can efficiently catalyze MPTP-mimicking compounds, i.e. 2-methyl-1,2,3,4-tetrahydroisoquinoline, 2-methyl-1,2,3,4-tetrahydro-β-carboline, and 9-methyl-norharmon, suspected to induce PD in humans. Our results reveal that activity and respiration in mouse brain mitochondrial complex I are significantly affected by these toxins in WT mice but remain unchanged in Cyp2d6 locus knockout mice, indicating a possible role of CYP2D6 in the metabolism of these compounds both in vivo and in vitro. These metabolic effects were minimized in the presence of two CYP2D6 inhibitors, quinidine and ajmalicine. Neuro-2a cells stably expressing predominantly mitochondrially targeted CYP2D6 were more sensitive to toxin-mediated respiratory dysfunction and complex I inhibition than cells expressing predominantly endoplasmic reticulum–targeted CYP2D6. Exposure to these toxins also induced the autophagic marker Parkin and the mitochondrial fission marker Dynamin-related protein 1 (Drp1) in differentiated neurons expressing mitochondrial CYP2D6. Our results show that monomethylamines are converted to their toxic cationic form by mitochondrially directed CYP2D6 and result in neuronal degradation in mice.
Keywords: drug metabolism, neurotoxin, mitochondria, complex I, cytochrome P450, Parkinson's disease, monomethylamines, neurodegeneration, substantia nigra, tyrosine hydroxylase
Introduction
Human PD3 is associated with degeneration of the central nervous system, causing movement disorders, resting tremors, and neuropsychiatric disorders, including mood, cognition, and behavioral abnormalities and dementia (1, 2). Two major factors, including genetic and environmental causes, have been implicated in PD (3–6). Idiopathic PD is caused by environmental and endogenous factors, although the precise mechanisms are not clear. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a neurotoxic intermediary product formed during chemical synthesis of the opioid analgesic desmethylprodine, has been helpful in elucidating mechanisms of chemically induced PD in rodents and primate models (7–9). Recent studies show that several MPTP-mimicking compounds produced endogenously in the brain may play a role in idiopathic PD. There is compelling evidence suggesting that MPTP-mimicking chemicals, such as N-methylated β-carbolines, isoquinolines, and norharmans, either induce PD or enhance the progression of the disease in animal models and in the human population (10–13).
9-Methyl norharmans and methylated β-carbolines and isoquinolines are derived endogenously by transmethylation of various alkaloids and other aliphatic or aromatic compounds present in food material and cigarette smoke and from indole and tryptophan present in dietary sources. Another source of methylation is acetaldehyde, present in the blood and tissues of chronic alcoholics (14–20). Although the precise metabolic profiles of these monomethylamine neurotoxins remain unclear, they are converted to their respective cationic forms 2-methyl β-carbolinium and N-methyl isoquinolinium ions, whose chemical and toxicological properties mimic MPP+. β-Carbolines and isoquinolines and their cationic metabolites have been detected previously in human, rat, and monkey brains (10–12, 18, 19). Studies have shown that the levels of β-carbolines and isoquinolines are elevated in the brains of alcoholics and long-term smokers and under certain dietary conditions (14–16, 20). Several reports suggest that β-carbolines and isoquinolines induce PD in rodents (10, 21); however, the mechanism of action still remains unclear.
Human cytochrome P450 (CYP) 2D6 metabolizes nearly 25% of drugs used in human medicine (22–24), including tricyclic antidepressants, β-blockers, antihypertensive drugs, opioids, dopamine antagonists, selective estrogen receptor modulators, and antiarrhythmic and antidiabetic drugs. Compelling evidence suggests that CYP2D6 is targeted to mitochondria in addition to its well-established ER destination (25–27). Analysis of human liver samples established that the mitochondrial CYP2D6 content ranged from 10%–60% of the total tissue pool, and in 25% of human liver samples from a human tissue bank, the mtCYP2D6 content was nearly equal or higher than that of microsomal CYP2D6 (25). Studies from our laboratory also showed that mtCYP2D6, in the presence of the mitochondrial electron transfer proteins adrenodoxin (Adx) and adrenodoxin reductase (AdxR), efficiently metabolizes many conventional CYP2D6 substrates (28).
In a previous study, we showed that mitochondrially targeted CYP2D6 (Mt2D6) plays a pivotal role in the metabolism of MPTP to its toxic cationic form, MPP+ (28). Although in agreement with studies from other groups (29–33), the microsomal CYP2D6 supported by cytochrome P450 reductase (CYPOR) catalyzed the demethylation of MPTP to nontoxic metabolites. Our results showing the ability of Mt2D6 to metabolize MPTP to the cationic form MPP+ are a paradigm shift because it is widely believed that monoamine oxidase B, present on the mitochondrial outer membrane of glial cells, carries out oxidation of MPTP, and the metabolites are then transported to dopaminergic neurons through dopamine transporters (28, 34–37). Both monoamine oxidase B and dopamine transporters have been targets of drug development for PD over the past two to three decades. In this study, we used cultured Neuro-2a cells, which mimic human dopamine neurons, and a transgenic Cyp2d6 locus knockout (Cyp2d6KO) mouse model to demonstrate that Mt2D6 plays a direct role in the metabolic conversion of MPTP-mimicking toxins, β-carbolines, isoquinolines, and 9-methyl-norharmon to toxic intermediates, which, in turn, induce oxidative stress and neuronal degeneration in a mouse model.
Results
Effects of monomethylamine neurotoxins on mouse brain mitochondrial function
We investigated the effect of three neurotoxins, 2-methyl-1,2,3,4-tetrahydroisoquinoline HCl (TISQ), 2-methyl-1,2,3,4-tetrahydro-β-carboline (MTHBC), and 9-methyl-9H-pyrido(3,4-β)indole (β-Carb) (Table 1), all of which are produced endogenously in rodent and primate brains (10–12) from dietary or environmental components. To evaluate the effects on mitochondrial function, we incubated brain mitochondria from WT mice and Cyp2d6 locus KO (Cyp2d6KO) mice with these three neurotoxins. In WT brain mitochondria, complex I activity was significantly inhibited (∼55%) by all three toxins compared with control untreated mitochondria. However, the activity was not inhibited by these toxins in brain mitochondria from Cyp2d6KO mice (Fig. 1A), suggesting that mitochondrial CYP2D6 is associated with monomethylamine-induced complex I inhibition. Complex IV and II+III activities, on the other hand, remained unaffected by these toxins in both WT and Cyp2d6KO brain mitochondria (Fig. 1, B and C). We further ascertained the role of CYP2D6 on MTHBC-mediated inhibition of complex I in brain mitochondria from WT mice using quinidine as a selective inhibitor of CYP2D6. We observed a steady decrease in complex I activity with increasing concentrations of MTHBC, and quinidine provided significant protection (Fig. 1D).
Table 1.
The list of monomethylamine toxins used
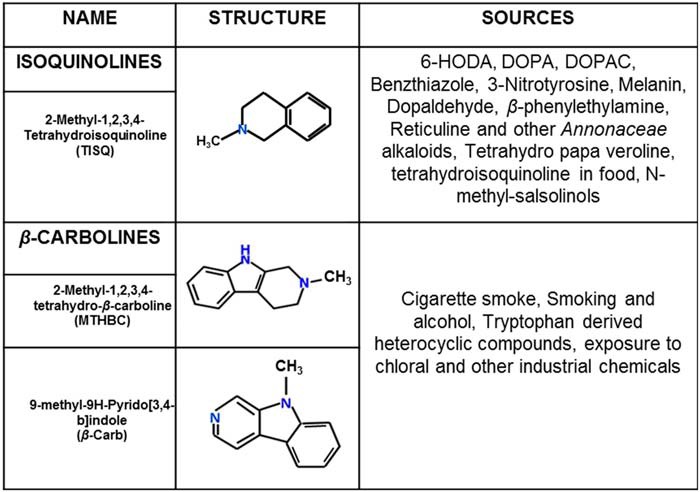
Figure 1.

Inhibition of brain mitochondrial complex I activity in vitro by monomethylamine toxins. A–C, mitochondria were isolated from WT and Cyp2d6KO mice by differential centrifugation as described under “Experimental procedures” and incubated without (Control) or with 5 mm TISQ, MTHBC, or β-Carb individually for 20 min at 37 °C, and complex I (A), complex IV (B), and complex II+III (C) activities were measured. n = 3; ***, p < 0.001 versus control. D, isolated mitochondria were incubated with varying concentrations of (0–7.5 mm) MTHBC without or with (50 μm) quinidine for 20 min at 37 °C, and complex I activity was measured. **, p < 0.01; ***, p < 0.001 versus control. ##, p < 0.01 versus MTHBC. All values are represented as mean ± S.D. of three independent experiments.
The in vivo effects of these toxins on mitochondrial function were tested in WT and Cyp2d6KO mice by intraperitoneal injection of TISQ (64 mg/kg/day) for 21 days, and the mitochondrial respiration profiles were measured in a Seahorse XF-24 flux analyzer. We measured baseline respiration, ADP-coupled respiration (state III respiration), and maximal uncoupled respiration (FCCP-uncoupled) (Fig. 2, A–E). The results show that TISQ treatment significantly reduced baseline respiration (∼28%), state III respiration (∼44%), and maximal respiration (∼35%) in the WT mouse brain but had no effect on the Cyp2d6KO mouse brain (Fig. 2, A–D). State III/IV respiration was marginally but not significantly affected by TISQ treatment in WT mice (Fig. 2E). These results clearly indicate that the TISQ-induced respiratory defect in WT mice is associated with mitochondrial CYP2D6.
Figure 2.

In vivo effects of TISQ on brain mitochondrial respiratory controls. WT (n = 6) and Cyp2d6KO (n = 6) mice were injected intraperitoneally with TISQ (64 mg/kg b.w., n = 3) or vehicle (n = 3) for 21 days. Mitochondria freshly isolated from the brains were then used to measure OCRs in the Seahorse flux analyzer as described under “Experimental procedures.” A, respiratory profiles of brain mitochondria from vehicle-treated WT mice (WT Vehicle), TISQ-treated WT mice (WT + TISQ), vehicle-treated Cyp2d6KO mice (Cy2d6KO Vehicle), and TISQ-treated Cyp2d6KO mice (Cyp2d6KO + TISQ) are shown. B–E, basal respiration (B), state III (ADP-linked coupled) respiration (C), maximal respiration (D), and state III/IV respiration (E) using isolated brain mitochondria. *, p < 0.05; **, p < 0.01 versus vehicle. All values are represented as mean ± S.D. of three independent experiments. n represents the number of mice used in each group.
Fig. 3, A–C, shows that in vivo treatment with TISQ inhibited complex I activity in WT mice but not in Cyp2d6KO mice. Complex I activity was inhibited by about 45% by TISQ in WT mice but not in Cyp2d6KO mice (Fig. 3A). These results fully support our in vitro results shown in Fig. 1. Furthermore, TISQ had no significant inhibitory effect on complex IV and complex II+III activities in both WT and Cyp2d6KO mouse brains (Fig. 3, B and C). These results suggest that the inhibitory effects of monomethylamine neurotoxins are specific for complex I activity and that they are likely mediated through mitochondrial CYP2D6. The immunoblots in Fig. 3, D and E, show that in vivo treatment with TISQ for 21 days increased the levels of Parkin and Drp1, markers for autophagy and mitochondrial fission, respectively, in the brains of WT but not in the same fractions of Cyp2d6KO mouse brains. Furthermore, TISQ induced mitochondrial fission in a CYP2D6-dependent manner.
Figure 3.

Effects of monomethylamine treatment on brain mitochondrial electron transfer complexes and mitochondrial fusion. A–C, WT (n = 6) and Cyp2d6KO (n = 6) mice were injected intraperitoneally with TISQ (64 mg/kg b.w., n = 3) or vehicle (n = 3) for 21 days (once a day) and complex I activity (A), complex IV activity (B), and complex II and III activity (C) were estimated in brain mitochondria isolated from these mice. The groups consisted of vehicle-treated WT mice (WT vehicle), TISQ-treated WT mice (WT + TISQ), vehicle-treated Cyp2d6KO mice (Cyp2d6KO Vehicle), and TISQ-treated Cyp2d6KO mice (Cyp2d6KO + TISQ). ***, p < 0.001 versus vehicle. All values are represented as mean ± S.D. of three independent experiments. n represents the number of mice used in each group. (D, E) mitochondrial proteins (30 μg each) from the abovementioned groups were analyzed by immunoblotting with (D) anti-Parkin (1:1000 dilution, v/v) and (E) anti-Drp1 (1:1000 dilution, v/v).
Fig. 4 shows the histopathology of brain sections from midbrain sections of WT and Cyp2d6KO mice treated with vehicle alone or TISQ for 21 days (64 mg/kg/day). Midbrain sections from WT mice stained prominently, indicating a large cluster of tyrosine hydroxylase (TH)–positive neurons that are markedly diminished by treatment with TISQ (Fig. 4, A and B). Quantitation of staining intensities, presented in Fig. 4E, shows that the brain of a TISQ-treated WT animal has markedly diminished numbers of TH-positive neurons. Surprisingly, a Cyp2d6KO brain showed reduced TH staining even without any treatment. The reason for this reduction remains unclear. However, TISQ treatment had no effect on the extent of staining or the number of TH-positive neurons in Cyp2d6KO brains (Fig. 4, C and D). These results further confirm our biochemical data showing that the WT brain is most sensitive to monomethylamine toxicity, whereas the Cyp2d6KO brain is insensitive to the action of TISQ.
Figure 4.

The role of CYP2D6 in TISQ-induced brain damage and neuronal loss. WT (n = 6) and Cyp2d6KO (n = 6) mice were injected i.p. with TISQ (64 mg/kg b.w., n = 3) or vehicle (n = 3) once a day for 21 days. Brains were extracted following euthanasia, and formalin-fixed brains were sliced using the coronal brain matrix system as described under “Experimental procedures.” The brain slices were stained with TH antibody as described under “Experimental procedures.” IHC evaluation was performed on two slides per sample, two serial sections per slide, with an ∼20-μm step between slides. A and C, WT and Cyp2d6KO mouse brains stained with TH antibody. B and D, TISQ-treated mouse brains. E, % TH positive by area, mean ± S.D. of TH-positive neurons. **, p < 0.01 versus vehicle. n represents the number of mice used in each group.
Differential effects of mitochondrially and ER-targeted CYP2D6 on monomethylamine toxicity in Neuro-2a cells
Our results from in vitro and in vivo treatment experiments suggest that monomethylamine neurotoxins inhibit mitochondrial respiratory and electron transport functions in a CYP2D6-dependent way. To test the hypothesis about the role of mitochondrial CYP2D6, we used Neuro-2a cells stably expressing WT, Mc (ER-targeted), or Mt (mitochondrially targeted) human CYP2D6, which mimic human dopaminergic neurons when induced with dibromo-cAMP. These stably transduced Neuro-2a cells were used in our previous study (25, 28). As shown in Fig. S2A, the WT CYP2D6 construct contained unmodified human CYP2D6 cDNA. The Mc2D6 construct contained substitutions E4L and P8L, which makes the signal more hydrophobic, with a higher ER targeting potential. The MtCYP2D6 construct contained V11R, F16K, and L19R substitutions with a higher mitochondrion-targeting potential (28). The schematic representation of the various CYP2D6 constructs in Fig. S2A is from a previous publication by our group (28). The level of mitochondrial CYP2D6 protein was lowest in Mc2D6 cells and highest in Mt2D6-expressing cells (Fig. 5A). In contrast, the level of microsomal CYP2D6 was highest in cells transduced with the Mc2D6 construct and lowest in Mt2D6-expressing cells. To determine mitochondrial purity, Tom20 (a mitochondrion-specific protein) and Cypor (a microsome-specific protein) levels were estimated for all samples (Fig. 5A). As shown, Tom20 and Cypor were used as markers for mitochondrial and microsomal fractions, respectively. The immunoblot shows the relative purities of the two fractions used.
Figure 5.

Mitochondrial CYP2D6 content and P450 activity correlate with monomethylamine-induced complex I inhibition. A, MtCYP2D6 and McCYP2D6 levels in mock-transfected and human CYP2D6 (WT2D6, Mc2D6, and Mt2D6)–expressing Neuro-2a cells were estimated by immunoblot analysis (30 μg of mitochondrial and microsomal proteins each) as described under “Experimental procedures.” The blots were probed with a mAb to human CYP2D6 (1:1000 dilution, v/v) and co-developed with Cypor antibody (1:1000 dilution, v/v) to assess the levels of microsomal contamination in mitochondrial preparations and with Tom20 antibody (1:1000 dilution, v/v) to estimate the level of mitochondrial contamination in microsomal preparations. B, mitochondrial protein (300 μg) from the indicated Neuro-2a cell lines was reconstituted with 0.2 nmol of purified Adx and 0.02 nmol of AdxR in a final volume of 200 μl of 50 mm phosphate buffer containing 5 mm MgCl2, and mitochondrial CYP2D6 activity was estimated using MAMC (100 μm), a specific substrate for CYP2D6. In the presence of added NADPH (4 mm), MAMC is converted to HAMC, which is measured fluorometrically. *, p < 0.05 versus WT2D6; ***, p < 0.001 versus Mt2D6. C, Neuro-2a cells expressing different forms of CYP2D6 were treated with 200 μm TISQ, MTHBC, and β-Carb for 16 h, and live cells were counted. *, p < 0.05 versus control. D, undifferentiated WT2D6-, Mc2D6- and Mt2D6-expressing Neuro-2a cells were treated without (Control) or with 200 μm TISQ in the absence or presence of the CYP2D6 inhibitor quinidine (10 μm) for 16 h. Mitochondria were isolated, and complex I activity was measured. **, p < 0.01 versus control; *, p < 0.05 versus TISQ. E, WT2D6-, Mc2D6-, and Mt2D6-expressing Neuro-2a cells were induced to differentiate for 72 h with 1 mm dibromo-cAMP. In some plates, 200 μm TISQ was added after 56 h of differentiation, without or with 10 μm quinidine. Mitochondria were then isolated from these cells, and complex I activity was measured. ***, p < 0.001 versus control; ***, p < 0.001; **, p < 0.01 versus TISQ. F and G, Mt2D6-expressing Neuro-2a cells were differentiated for 72 h with 1 mm dibromo-CAMP, and TISQ (200 μm, F) or 50–200 μm β-Carb (G) was added after 56 h of differentiation for 16 h without or with 10 μm quinidine, 10 μm proadifen, or 10 μm deprenyl. Mitochondria were then isolated, and complex I activity was measured. F, ***, p < 0.001 versus control; ***, p < 0.001; **, p < 0.01; *, p < 0.05 versus TISQ. G, *, p < 0.05; **, p < 0.01 versus β-Carb. H, WT2D6 and Mc2D6- and Mt2D6-expressing Neuro-2a cells were induced to differentiate for 72 h with 1 mm dibromo-cAMP, and 200 μm TISQ was added after 56 h of differentiation without or with 10 μm quinidine or 20 μm ajmalicine. Mitochondria were then isolated from these cells, and complex I activity was measured. ***, p < 0.001 versus control; *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus TISQ. All values are represented as mean ± S.D. of three independent experiments.
The mitochondrial CYP2D6 activity of Neuro-2a cells expressing different CYP2D6 constructs was estimated using 7-methoxy-4-(aminomethyl) coumarin (MAMC), a specific substrate for CYP2D6 (Fig. 5B). Demethylase activity was found to be highest in Mt2D6-expressing cells, intermediate in WT2D6-expressing cells, and lowest in Mc2D6-expressing cells, and mock cells did not show any activity, validating our Western blot data. Neuro-2a cells stably expressing cDNA constructs were treated with different toxins for 16 h to monitor cell death (Fig. 5C). A maximum of about 20% cell death was observed in cells treated with the three toxins, and maximum cell death was seen in Mt2D6-expressing cells. The monomethylamines inhibited complex I activity in both undifferentiated and differentiated neurons (Fig. 5, D–G). Undifferentiated as well as differentiated Neuro-2a cells expressing stable constructs were treated with TISQ for 16 h (Fig. 5, D–F). Complex I activity was inhibited by TISQ; maximum inhibition was observed in Mt2D6-expressing cells, followed by WT2D6-expressing cells, but inhibition was minimal in Mc2D6-expressing cells. Furthermore, the CYP2D6 inhibitor quinidine reversed the inhibitory effects in all cell types. Proadifen (a CYP inhibitor) and deprenyl (an MAO inhibitor) also showed similar patterns but were less effective than quinidine (Fig. 5F). Similar patterns of inhibition by MTHBC (Fig. S1, A and B) and β-Carb (Fig. 5F) and reversal by quinidine were observed in undifferentiated and differentiated Neuro-2a neurons. β-Carb showed concentration-dependent inhibition of complex I activity in differentiated Mt2D6-expressing Neuro-2a cells (Fig. 5G). Ajmalicine, a more potent inhibitor of CYP2D6 (38, 39), showed similar protective effects on monomethylamine-induced mitochondrial functional parameters (Fig. 5H and Fig. S2). These results suggest that complex I inhibition and inhibition of respiratory parameters by monomethylamine toxins in Neuro-2a cells were more effective when mitochondrial CYP2D6 content was higher.
Analysis of respiratory pattern using a Seahorse flux analyzer showed substantial differences in basal OCR, ADP-coupled OCR, and maximal OCR among the three cell lines. In general, cells expressing Mt2D6 with higher levels of mitochondrial CYP2D6 protein showed significantly lower respiration compared with Mc2D6-expressing cells with the lowest level of mitochondrial CYP2D6 (Fig. S3, A–I). Although the precise reason for this difference is not clear, it is likely that Mt2D6, with its metabolic conversion of monomethylamine toxins, affects complex I more severely. All three neurotoxins affected basal respiration, ATP-linked respiration, and maximal respiration in all three cell types, although Mt2D6-expressing cells were most affected. Furthermore, of the three toxins, β-Carb and MTHBC were more potent inhibitors than TISQ. In all cases, quinidine, a known inhibitor of CYP2D6, reversed the inhibitory effects in a significant manner. A notable observation was that monomethylamine-induced changes were relatively minimal in Mc2D6-expressing Neuro-2a cells, although there were marked changes in Mt2D6-expressing Neuro-2a cells (Fig. S3, A–I).
Effects of monomethylamine neurotoxins on differentiation of Neuro-2a cells
Differentiated neurons with different metabolic activity have different responses to different inhibitors. We therefore compared the effects of monomethylamines on differentiation of Neuro-2a cells expressing Mt-2D6 and Mc2D6. Cells were incubated with 1 mm dibromo-cAMP for 72 h to induce differentiation. Both Mc2D6- and Mt2D6-expressing cells underwent efficient differentiation in the presence of dibromo-cAMP, as observed by immunoblot analysis of TH, a biomarker for differentiation of dopaminergic neurons (Fig. 6, A and B). Immunoblots showed that TH is induced by serum deprivation and treatment with dibromo-cAMP for 3 days in both Mt- and Mc2D6-expressing cells. Treatment with TISQ reduced the level of TH protein drastically in Mt2D6-expressing cells but only marginally in Mc2D6-expressing cells (Fig. 6A, quantitation in Fig. 6B). Immunofluorescence analysis (Fig. 6, C and D) showed essentially similar results with MTHBC. MTHBC marginally reduced TH-positive neurons in Mc2D6-expressing Neuro-2a cells but had a marked effect on Mt2D6-expressing Neuro-2a cells, which showed nearly 70% inhibition. Quinidine prevented this loss significantly in Mt2D6-expressing cells (Fig. 6E). These results further support our previous observation that mitochondrial CYP2D6-induced monomethylamine toxicity disrupts mitochondrial activity and neuronal function.
Figure 6.

Effects of TISQ and MTHBC on TH expression and TH-positive neurons in Neuro-2a cells. Mc2D6- and Mt2D6-expressing Neuro-2a cells were differentiated (Diff) for 72 h with 1 mm dibromo-CAMP. A, TISQ (200 μm) was added after 56 h of differentiation, without or with 10 μm quinidine. Whole-cell extracts (30 μg) were subjected to immunoblot analysis against TH (1:1000 dilution, v/v) and co-developed with anti-actin (1:1000 dilution, v/v) as a loading control. UD, undifferentiated; B, quantification of an immunoblot probed with TH antibody. **, p < 0.01; ***, p < 0.001 versus control; *, p < 0.05; **, p < 0.01 versus TISQ. C and D, 200 μm MTHBC was added after 56 h of differentiation without or with 10 μm quinidine. Immunofluorescence microscopy was carried out to count TH-positive neurons. The differentiated cells were probed with anti-TH antibody (1:1000 dilution, v/v) and co-stained with 4′,6-diamidino-2-phenylindole (1 μg/ml). Shown are TH-stained Mc2D6 (C) and Mt2D6 (D) neurons without (Control) or with 200 μm MTHBC treatment in the absence or presence (MTHBC+quinidine) of 10 μm quinidine. E, the presence of TH-positive neurons with treatments was determined as a percentage of the control. ***, p < 0.001 versus control; **, p < 0.01 versus MTHBC. All values are represented as mean ± S.D. of three independent experiments.
Induction of autophagy markers in Neuro-2a cells expressing Mt2D6 by monomethylamine toxins
Recruitment of Parkin to the mitochondrial compartment is a marker for cells undergoing autophagy. Immunofluorescence patterns of Mc2D6- and Mt2D6-expressing Neuro-2a cells stained with antibody for parkin and co-stained with a mitochondrial marker, ATP-B antibody, are shown in Fig. 7A. TISQ had only a minimal effect on Parkin induction in Mc2D6-expressing cells, whereas there was extensive recruitment of Parkin in Mt2D6-expressing cells, suggesting induced autophagy in the latter in response to monomethylamine toxin. Quinidine had a significant protective effect. Immunoblot analysis of cells treated with MTHBC and β-Carb (Fig. 7, B and C) showed similar effects and protection by quinidine. These results clearly indicate that cells expressing Mt2D6 are much more susceptible to monomethylamine-induced toxicity and susceptibility to undergo autophagy than Mc2D6-expressing Neuro-2a cells.
Figure 7.

Induced expression of Parkin in response to monomethylamine toxins in Mt2D6-expressing Neuro-2a cells. Differentiated Mc2D6 and Mt2D6 expressing Neuro-2a cells were incubated for 16 h without (Control) or with 200 μm TISQ/MTHBC/β-Carb in the absence or presence of 10 μm quinidine as described under “Experimental procedures.” A, immunofluorescence microscopy was carried out in treated cells. The cells were incubated with anti-Parkin (1:200 dilution, v/v) and co-stained with anti-ATP-B (1:200 dilution, v/v) antibody. B and C, mitochondria isolated from similarly treated cells were subjected to immunoblot analysis using 30 μg of protein each, and the respective densitometry analyses were done. **, p < 0.01; ***, p < 0.001 versus control; *, p < 0.05 versus MTHBC; ***, p < 0.001 versus β-Carb. All values are represented as mean ± S.D. of three independent experiments.
Mitochondria maintain their morphology via fission and fusion (40). Drp1 is a known fission marker and also an indicator of the mitochondrial quality control system (41). Both immunocytochemistry and Western blot analysis showed that mitochondrial Drp-1 levels were markedly increased by all three toxins in Mt2D6-expressing cells, but there was only a slight increase in Mc2D6-expressing cells (Fig. 8, A–C). However, treatment with quinidine markedly reduced mitochondrial Drp-1 levels (Fig. 8, A–C). All of these results confirm that mitochondrial CYP2D6 plays a pivotal role in monomethylamine neurotoxin-induced mitochondrial dysfunction.
Figure 8.

Induction of mitochondrial fission markers in Mt2D6-expressing Neuro-2a cells in response to monomethylamine toxins. Mc2D6- and Mt2D6-expressing Neuro-2a cells were differentiated for 72 h without (Control) or with 200 μm TISQ/MTHBC/β-Carb in the absence or presence of 10 μm quinidine for 16 h as described under “Experimental procedures.” A, immunofluorescence microscopy was carried out in the cells. The cells were incubated with anti-Drp1antiody (1:200 dilution, v/v) and co-stained with anti-ATP-B (1:200 dilution, v/v) antibody. B and C, mitochondria isolated from the treated cells were subjected to immunoblot analysis (30 μg of protein each) and subjected to densitometry analysis. **, p < 0.01; ***, p < 0.001 versus control; *, p < 0.05; **, p < 0.01 versus TISQ; *, p < 0.05; **, p < 0.01 versus MTHBC. All values are represented as mean ± S.D. of three independent experiments.
Discussion
The mechanism of MPTP-mediated neuronal toxicity and idiopathic Parkinson's disease has been extensively studied in rodent models and in primates (7–9). A large majority of studies of this topic suggest that MPTP is metabolized to MPDP + (1-methyl-4-phenyl-2,3-dihydropyridium) by monoamine oxidase B, present on the outer mitochondrial membranes of glial cells and the oxidized product released outside, which is then oxidized to MPP+ in a nonenzymatic reaction. The toxic metabolite, taken up by dopaminergic neurons through dopamine transporters, is then translocated to neuronal mitochondria because of its cationic form, where it causes inhibition of complex I; this is regarded as a critical factor in the initiation of PD (34–37). In significant divergence from this model, we showed that MPTP taken up directly by dopaminergic neurons is metabolized to MPP+ by mitochondrially targeted CYP2D6, which then inhibits complex I activity (28). Our results therefore suggest that neuronal mitochondria themselves are the sites of metabolism of MPTP to toxic MPP+. In this study, we tested the metabolism and associated toxicity of three different N-methylated β-carbolines, isoquinoline, and norharmans, which are endogenously produced in rodent and primate brains and are also suspected to cause idiopathic PD (10–12, 18, 19).
All three monomethylamine toxins inhibited respiratory parameters in isolated brain mitochondria from WT mice but were relatively ineffective in Cyp2d6KO mice. Similarly, all three toxins inhibited complex I activity in mitochondria from WT mice but not from Cyp2d6KO mice. The effects of neurotoxins administered i.p. in mice were similar. Interestingly, the inhibitory effects in vitro in isolated mitochondria from WT mice or the in vivo effects in injected mouse brains were significantly protected by the selective CYP2D6 inhibitor quinidine, suggesting involvement of mitochondrial CYP2D6, which was not seen in Cyp2d6KO mice. Consistent with this conclusion, TISQ injected into mice markedly reduced TH-positive neurons in the substantia nigra region of WT mice (which express CYP2D6), but the effect was not observed in Cyp2d6KO mice. Notably, the level of TH staining per se was lower in Cyp2d6KO mice, and toxin treatment did not reduce this level any further. The reasons for the lower level of TH antibody staining in Cyp2d6KO mouse brains remain unclear, although it is consistent with lower levels of basal, state III, and maximal respiration in mitochondria from Cyp2d6KO mice. It is likely that CYP2D6-linked metabolism of some unknown substrate(s) is responsible for this difference. However, the results do show that monomethylamine neurotoxins damage dopaminergic neurons in a CYP2D6-dependent manner.
Neuro-2a cells, when induced to differentiate in the presence of added dibromo-cAMP, exhibit many properties of dopaminergic neurons, including induced expression of TH and axonal outgrowths. We used these cells for the expression of CYP2D6 forms predominantly targeted to the ER or mitochondria for assessing the role of Mt2D6 in mediating monomethylamine toxicity. The mitochondrial and microsomal distribution of CYP2D6 showed marked differences between the cell lines, with WT2D6-expressing cells showing nearly equal distribution, Mc2D6-expressing cells showing markedly higher microsomal CYP2D6, and Mt2D6-expressing cells showing higher levels of mitochondrial CYP2D6 (Fig. 5A). The MAMC activity of isolated mitochondria showed Adx plus AdxR-supported activities that were consistent with the CYP2D6 levels observed in the immunoblots. The monomethylamine neurotoxin–induced inhibition of complex I activity in Neuro-2a cells was proportional to their mitochondrial CYP2D6 levels. Furthermore, TISQ inhibited TH expression maximally in Mt2D6-expressing cells, and the inhibition was reversed by quinidine. These results, along with the patterns of expression of Parkin and Drp1, strongly suggest a role of MtCYP2D6 in monomethylamine-induced damage of dopaminergic neurons. A generally lower level of toxin-induced depletion of dopaminergic neurons suggests that McCYP2D6 plays a relatively minor role in modulating the toxicity. It is likely that a more unfolded nature of mtCYP (27, 42) or the mode of electron transfer by the soluble Adx+AdxR system may be responsible for the difference in toxicity modulation.
In summary, our results show that mitochondrial CYP2D6 plays an important role in monomethylamine-induced neuronal toxicity and Parkinson's syndrome. Our results also suggest that mitochondrial CYP2D6 may be a direct molecular target for drug development for treating idiopathic PD; pharmacological agents targeting monoamine oxidase B and dopamine transporters have only had mixed success.
Experimental procedures
Materials
All three monomethylamine neurotoxins were custom-synthesized, purified to near homogeneity by HPLC, and further characterized by MS analysis to confirm structure. TISQ was synthesized by AstaTech Inc. (Bristol, PA). The two indole derivatives, β-Carb and MTHBC, were synthesized by Accel Pharmtech (East Brunswick, NJ).
Mouse models
In this study, we used WT (C57BL/6J) mice procured from The Jackson Laboratory and CYP2D6 locus KO (Cyp2d6KO; C57BL/6-Del(15Cyp2d22-Cyp2d26)1Arte) (43) mice from Taconic Biosciences to evaluate the role of CYP2D6 in monomethylamine-induced mitochondrial function. Cyp2d6KO mice lack the entire Cyp2d6 locus, which codes for nine members of expressed and pseudogenes (43). To our knowledge, the deletion strategy does not inactivate any other non-Cyp2d6 genes. Six- to eight-week-old male WT and Cyp2d6KO mice weighing ∼20–25 g were conditioned at 25 °C ± 2 °C with a 12-h light/12-h dark cycle and fed a standard diet ad libitum. A subset of WT (n = 3) and Cyp2d6KO (n = 3) mice were injected intraperitoneally with TISQ (64 mg/kg b.w.) for 21 consecutive days. Another subset of WT (n = 3) and Cyp2d6KO (n = 3) mice was injected with saline (50 mm NaCl, 10 mm K2HPO4 (pH 7.0)) (vehicle control). Mice were euthanized by CO2 asphyxiation using a Crainey Tech asphyxiation chamber in accordance with the American Veterinary Medical Association and National Institutes of Health–approved guidelines and a protocol approved by the institutional animal care and use committee at the University of Pennsylvania. Brain tissues from control and treated mice were collected and used to prepare subcellular fractions. Identically treated mouse brains were fixed in paraformaldehyde for histopathological studies as described later.
Cell lines and culture conditions
Mouse Neuro-2a cells (ATCC, CCL-131) were grown in DMEM (Invitrogen) containing 10% fetal bovine serum (v/v) and 0.1% gentamycin (w/v). Neuro-2a cells were transduced with CYP2D6 cDNAs cloned in a retroviral vector (pBABE-puro), and stable clones were selected based on resistance to puromycin as described earlier (28). To ensure integrity of the viral vectors, puromycin (Sigma) was added after every two passages. To rule out adverse effects of puromycin on mitochondrial function, all experiments were conducted in cells that were cultured without puromycin for at least three passages. The cells were differentiated by changing the medium to DMEM with 0.5% FBS (differentiation medium, v/v) after overnight growth and grown for an additional 3 days with 1 mm dibromo-cAMP (Sigma). For in vitro experiments, differentiated as well as undifferentiated cells were incubated with 200 μm TISQ, MTHBC, or β-Carb for 16 h with or without 10 μm quinidine (Sigma), 10 μm proadifen (Sigma), 20 μm ajmalicine (Sigma), or 10 μm deprenyl (Sigma).
Cell fractionation and isolation of respiration-competent mitochondria
The procedure for mitochondrial isolation from brain tissue was modified from those reported earlier (28, 44). Brain tissue was rapidly harvested and washed in ice-cold PBS buffer containing 0.25 mg/ml soybean trypsin inhibitor. The tissue was then finely minced in 3–4 ml of ice-cold H medium (70 mm sucrose, 210 mm mannitol,1 mm EGTA, and 10 mm HEPES-KOH (pH 7.4)) containing 0.5% (w/v) BSA; 1 μg/ml each of pepstatin, aprotinin, antipain, and leupeptin; 1 mm PMSF; 10 mm sodium fluoride; and 1 mm sodium orthovanadate. The minced tissue was homogenized with 8–10 strokes in a Dounce glass homogenizer using a glass pestle. The homogenate was centrifuged at 1000 × g for 10 min to remove cell debris and nuclei. This step was repeated to remove any remaining debris, and the supernatant was then centrifuged at 8000 × g for 20 min to pellet crude mitochondria. The supernatant was kept aside for microsome isolation. The mitochondrial pellet was washed at 8000 × g for 20 min with excess H medium without BSA. The final pellet was resuspended in a small volume (100–400 μl) of H medium and used immediately for respirometry. To obtain highly pure mitochondria, the mitochondrial pellet was treated with 75 μg of digitonin per milligram of mitochondrial protein for 20 min on ice, and mitochondria were pelleted through 0.8 m sucrose by centrifugation at 14,000 × g for 20 min at 4 °C to remove any cytosolic contamination. The resulting pellet was washed once with H medium. For metabolic assays, the final mitochondrial pellet was resuspended in 50 mm potassium phosphate buffer (pH 7.5) containing 20% glycerol (v/v), 0.1 mm EDTA, 0.1 mm DTT, and 0.1 mm phenylmethylsulfonyl fluoride and stored at −80 °C. In some of the respiratory experiments, mitochondria were incubated with 0–7.5 mm TISQ, MTHBC, or β-Carb at 37 °C for 20 min with or without 50 μm quinidine, and mitochondria were reisolated as above before loading on the plates. The 8000 × g supernatant fraction was centrifuged at 120,000 × g for 90 min to obtain microsomes.
Spectrofluorometric assay of MAMC demethylation
Mitochondria were assayed for O-demethylation activity using MAMC as a substrate, following a method published previously (25, 45) with slight modifications. The mitochondria (300 μg of protein) were disrupted by repeated freezing and thawing in 10 mm potassium phosphate (pH 7.4). The O-demethylation reactions were performed in a final volume of 200 μl of 50 mm potassium phosphate buffer (pH 7.4) containing 5 mm MgCl2, 300 μg of mitochondria (protein basis), 0.2 nmol of purified Adx, 0.02 nmol of AdxR, and 100 μm MAMC. Reactions were initiated by addition of 1 mm NADPH and then incubated for 20 min at 37 °C. The reaction was stopped by addition of 50 mm glycine. Insoluble material was sedimented by centrifugation at 10,000 × g for 10 min, and the supernatant containing 7-hydroxy-4-aminomethylcoumarin (HAMC) was measured fluorometrically (25, 45) with the excitation wavelength set at 405 nm and emission set at 480 nm. A standard curve was established using different concentrations of HAMC to quantify the reaction.
Measurement of mitochondrial complex I activity
Complex I activity was measured following a method described by Birch-Machin and Turnbull (46), with modifications, using a Cary-1E spectrophotometer. The reaction was initiated by addition of a mitochondrial suspension (10–30 μg of protein) to the assay mixture containing 25 mm phosphate buffer (pH 7.4), 5 mm MgCl2, 2 mm NaCN, 375.93 μm (25 mg/ml) BSA, 0.13 mm NADH, 9.11 μm (5 μg/ml) antimycin A, 65 μm ubiquinone, and 0.45 mm dodecyl maltoside in a 1-ml cuvette. The decrease in absorbance at 340 nm was recorded for 3 min, indicating the extent of NADH oxidation (slope 1), and then 12.67 μm rotenone (5 μg/ml) was added to each reaction, and the absorbance was measured at 340 nm for another 2 min (slope 2). The rotenone-sensitive activity (slope 1 − slope 2) was used to calculate the final complex I activity for each sample, and then absorbance units were converted to molar amounts of NADH oxidized using the molar extinction co-efficient (ϵ) of NADH, 6.22 mm−1 cm−1, as described previously (28).
Measurement of mitochondrial complex II+III activity
Complex II+III activity (succinate–cytochrome c reductase) was estimated by incubating 10–30 μg (protein basis) of freeze-thawed mitochondrial extract in 1 ml of assay medium consisting of 25 mm potassium phosphate (pH 7.4), 2 mm NaCN, 20 mm succinate, 5 μm (2 μg/ml) rotenone, and 37.5 μm oxidized cytochrome c and measuring the increase in absorbance at 550 nm. The enzyme activity was calculated by taking into consideration the mitochondrial protein in the cuvette and the molar extinction coefficient of reduced cytochrome c at 550 nm (21.1 mm−1 cm−1).
Measurement of cytochrome c oxidase activity
Cytochrome c oxidase activity (complex IV activity) was measured following a method published earlier (47, 48). The rate of oxidation of ferrocytochrome c was measured by following the decrease in absorbance at 550 nm. Freeze-thawed mitochondrial extract (2–10 μg of protein) was incubated in 1 ml of assay medium containing 25 mm potassium phosphate (pH 7.4) and 0.45 mm dodecyl maltoside. Ferrocytochrome c (15 μm) was added, and reaction rates were measured using a Cary-1E spectrophotometer. Cary-Win kinetics software was used to calculate first-order rate constants. The enzyme activity was calculated by taking into consideration the mitochondrial protein in the cuvette and the molar extinction coefficient of reduced cytochrome c at 550 nm (21.1 mm−1 cm−1).
Measurement of mitochondrial respiration
A Seahorse XF24 extracellular flux analyzer (Seahorse Biosciences, North Billerica, MA) was used to determine mitochondrial respiratory function. Freshly isolated mitochondria (5 μg of protein/well) from control and treated mouse brains were plated onto poly-l-lysine–coated Seahorse plates. Mitochondria were energized by adding 8 mm succinate, and respiration was measured in energized mitochondria (basal respiration). Then 4 μm ADP was added, and state 3 (phosphorylating respiration) was measured, followed by state 4 (resting respiration) after addition of 3.16 μm (2.5 μg/ml) oligomycin, an inhibitor of mitochondrial ATPase. Uncoupled maximal respiration was determined by adding 4 μm FCCP. Finally, 4 μm complex III inhibitor antimycin A was added to measure nonmitochondrial respiration.
Using intact cells, OCRs were measured as described by Wu et al. (49). Neuro-2a cells were cultured in DMEM in the absence or presence of TISQ, MTHBC, or β-Carb (200 μm) without or with 10 μm quinidine. In each case, 25,000 cells were plated onto Seahorse plates in DMEM overnight. One hour before measurement, DMEM was replaced with XF assay medium (low buffered bicarbonate-free DMEM (pH) 7.4). The final concentrations of inhibitors used were 6.32 μm (5 μg/ml) oligomycin, 0.33 μm FCCP (used as an uncoupler), and 1 μm complex I inhibitor rotenone. Each plate (along with the cartridge) was loaded into the XF analyzer, and the OCR was measured under basal conditions and after sequential addition of oligomycin, 2,4-dinitrophenol, and rotenone.
Immunofluorescence microscopy
Cells were rinsed twice with PBS, fixed with 2% paraformaldehyde, permeabilized with 0.2% Triton X-100 (v/v), blocked with 5% goat serum (v/v), and stained with the appropriate primary (1:200) and Alexa-conjugated secondary antibodies (1:500, v/v). Confocal fluorescence microscopy was done using a Leica Sp5 fluorescence lifetime imaging microscope inverted microscope. TH-positive cells were counted under an Olympus BX61 microscope.
SDS-PAGE and immunoblot analysis
For SDS-PAGE analysis, protein from mitochondria, microsomes, or cell extracts were solubilized in Laemmli sample buffer (50), resolved on 10–12% SDS-polyacrylamide gels, transferred to nitrocellulose membranes (Bio-Rad), and probed with the appropriate antibodies. Antibodies specific for CYP2D6 (ab185625), Parkin (ab15954), Drp1 (ab184247), ATP-B (ab14730), VDAC/Porin (ab15895), and TH (ab112) were obtained from Abcam (Cambridge, MA). Antibodies for Cypor (sc-25270), Tom20 (sc-11415), and Actin (sc-8432) were procured from Santa Cruz Biotechnology, Inc. (Dallas, TX). The blots were then probed with the appropriate primary antibody (1:1000, v/v), followed by corresponding LI-COR infrared-conjugated secondary antibodies (1:10,000, v/v). The blots were imaged on an Odyssey Licor instrument (LI-COR Biotechnology, Lincoln, NE).
Histopathology
Histopathology was carried out by the Penn Vet Comparative Pathology Core. Formalin-fixed brains were sliced using the coronal brain matrix system (Zivic Instruments, BSMYS001-1). Two anatomical landmarks were used as references to identify the coronal slice, including the entire midbrain with the substantia nigra: the cranial border of the pons varolii and the caudal limit of the optic chiasm. Sliced brains were routinely processed for paraffin embedding and sectioned for immunohistochemistry (IHC) examination. Microanatomically comparable sections were then selected from the designated slice of brain. TH staining was performed using the EMD Millipore AB152 rabbit polyclonal antibody on a fully automated Leica BOND RXm. The IHC evaluation was finally performed on two slides per sample, two serial sections per slide, and an ∼20-μm step between slides.
Sample evaluation was performed using a digital image analysis module integrated in the Aperio VERSA 200 platform. For each brain section, the total TH-immunoreactive area was first calculated and then normalized to the total area of midbrain included in the section.
Statistical analysis
The mean values ± S.D. were calculated from three to five independent experiments. Group differences were assessed by one-way analysis of variance or unpaired two-tailed Student's t test and, where necessary, with Tukey tests for multiple comparisons. Differences were considered significant at p ≤ 0.05.
Author contributions
M. C. and A. R. C. data curation; M. C., A. R. C., T. F., E. R., and F. P. G. formal analysis; M. C., A. R. C., T. F., C.-A. A., and E. R. investigation; M. C., A. R. C., T. F., and C.-A. A. methodology; M. C., A. R. C., T. F., and N. G. A. writing-original draft; F. P. G. and N. G. A. writing-review and editing; N. G. A. conceptualization; N. G. A. supervision; N. G. A. funding acquisition; N. G. A. validation.
Supplementary Material
Acknowledgments
We thank members of the Avadhani laboratory for useful suggestions and discussions. We also acknowledge help from the Comparative Pathology Core at the School of Veterinary Medicine, University of Pennsylvania.
This work was supported in part by National Institutes of Health Grant RO1 GM034883 and an endowment from the Harriet Ellison Woodward trust (to N. G. A.) and National Institutes of Health Grant R01 GM118122 (to F. P. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- PD
- Parkinson's disease
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MPP+
- 1-methyl-4-phenylpyridinium
- CYP
- cytochrome P450
- ER
- endoplasmic reticulum
- Adx
- adrenodoxin
- AdxR
- adrenodoxin reductase
- CYPOR
- cytochrome P450 reductase
- TISQ
- 2-methyl-1,2,3,4-tetrahydroisoquinoline
- MTHBC
- 2-methyl-1,2,3,4-tetrahydro-β-carboline
- β-Carb
- 9-methyl-9H-pyrido(3,4-β)indole
- TH
- tyrosine hydroxylase
- cDNA
- complementary DNA
- MAMC
- 7-methoxy-4-(aminomethyl) coumarin
- OCR
- oxygen consumption rate.
References
- 1. Lew M. (2007) Overview of Parkinson's disease. Pharmacotherapy 27, 155S–160S 10.1592/phco.27.12part2.155S [DOI] [PubMed] [Google Scholar]
- 2. Nagatsu T., and Sawada M. J. (2007) Biochemistry of postmortem brains in Parkinson's disease: historical overview and future prospects. J. Neural Transm. Suppl. 72, 113–120 [DOI] [PubMed] [Google Scholar]
- 3. Cannon J. R., and Greenamyre J. T. (2013) Gene-environment interactions in Parkinson's disease: specific evidence in humans and mammalian models. Neurobiol. Dis. 57, 38–46 10.1016/j.nbd.2012.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kieburtz K., and Wunderle K. B. (2013) Parkinson's disease: evidence for environmental risk factors. Mov. Disord. 28, 8–13 10.1002/mds.25150 [DOI] [PubMed] [Google Scholar]
- 5. Wirdefeldt K., Adami H. O., Cole P., Trichopoulos D., and Mandel J. (2011) Epidemiology and etiology of Parkinson's disease: a review of the evidence. Eur. J. Epidemiol. 26, S1–S58 [DOI] [PubMed] [Google Scholar]
- 6. Kalia L. V., and Lang A. E. (2015) Parkinson's disease. Lancet 386, 896–912 10.1016/S0140-6736(14)61393-3 [DOI] [PubMed] [Google Scholar]
- 7. Fox S. H., and Brotchie J. M. (2010) The MPTP-lesioned non-human primate models of Parkinson's disease. Past, present, and future. Prog. Brain. Res. 184, 133–157 10.1016/S0079-6123(10)84007-5 [DOI] [PubMed] [Google Scholar]
- 8. Porras G., Li Q., and Bezard E. (2012) Modeling Parkinson's disease in primates: the MPTP model. Cold Spring Harb. Perspect. Med. 2, a009308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meredith G. E., and Rademacher D. J. (2011) MPTP mouse models of Parkinson's disease: an update. J. Parkinsons Dis. 1, 19–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ostergren A., Fredriksson A., and Brittebo E. B. (2006) Norharman-induced motoric impairment in mice: neurodegeneration and glial activation in substantia nigra. J. Neural Transm. 113, 313–329 10.1007/s00702-005-0334-0 [DOI] [PubMed] [Google Scholar]
- 11. Nagatsu T. (1997) Isoquinoline neurotoxins in the brain and Parkinson's disease. Neurosci. Res. 29, 99–111 10.1016/S0168-0102(97)00083-7 [DOI] [PubMed] [Google Scholar]
- 12. Matsubara K., Gonda T., Sawada H., Uezono T., Kobayashi Y., Kawamura T., Ohtaki K., Kimura K., and Akaike A. (1998) Endogenously occurring β-carboline induces parkinsonism in nonprimate animals: a possible causative protoxin in idiopathic Parkinson's disease. J. Neurochem. 70, 727–735 [DOI] [PubMed] [Google Scholar]
- 13. Louis E. D., Michalec M., Jiang W., Factor-Litvak P., and Zheng W. (2014) Elevated blood harmane (1-methyl-9H-pyrido[3,4-b] indole) concentrations in Parkinson's disease. Neurotoxicology 40, 52–56 10.1016/j.neuro.2013.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herraiz T. (2004) Relative exposure to β-carbolines norharman and harman from foods and tobacco smoke. Food Addit. Contam. 21, 1041–1050 10.1080/02652030400019844 [DOI] [PubMed] [Google Scholar]
- 15. Pfau W., and Skog K. (2004) Exposure to β-carbolines norharman and harman. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 802, 115–126 10.1016/j.jchromb.2003.10.044 [DOI] [PubMed] [Google Scholar]
- 16. Rommelspacher H., Schmidt L. G., and May T. (1991) Plasma norharman (β-carboline) levels are elevated in chronic alcoholics. Alcohol Clin. Exp. Res. 15, 553–559 10.1111/j.1530-0277.1991.tb00559.x [DOI] [PubMed] [Google Scholar]
- 17. Matsubara K., Aoyama K., Suno M., and Awaya T. (2002) N-methylation underlying Parkinson's disease. Neurotoxicol. Teratol. 24, 593–598 10.1016/S0892-0362(02)00212-X [DOI] [PubMed] [Google Scholar]
- 18. Matsubara K., Collins M. A., Akane A., Ikebuchi J., Neafsey E. J., Kagawa M., and Shiono H. (1993) Potential bioactivated neurotoxicants, N-methylated β-carbolinium ions, are present in human brain. Brain Res. 610, 90–96 10.1016/0006-8993(93)91221-D [DOI] [PubMed] [Google Scholar]
- 19. Matsubara K., Neafsey E. J., and Collins M. A. (1992) Novel S-adenosylmethionine-dependent indole-N-methylation of β-carbolines in brain particulate fractions. J. Neurochem. 59, 511–518 10.1111/j.1471-4159.1992.tb09400.x [DOI] [PubMed] [Google Scholar]
- 20. Myers R. D. (1989) Isoquinolines, β-carbolines and alcohol drinking: involvement of opioid and dopaminergic mechanisms. Experientia 45, 436–443 10.1007/BF01952025 [DOI] [PubMed] [Google Scholar]
- 21. Lorenc-Koci E., Rommelspacher H., Schulze G., Wernicke C., Kuter K., Smiałowska M., Wierońska J., Zieba B., and Ossowska K. (2006) Parkinson's disease-like syndrome in rats induced by 2,9-dimethyl-β-carbolinium ion, a β-carboline occurring in the human brain. Behav. Pharmacol. 17, 463–473 10.1097/00008877-200609000-00012 [DOI] [PubMed] [Google Scholar]
- 22. Guengerich F. P. (2015) Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th Ed., Springer, New York [Google Scholar]
- 23. Zanger U. M., Raimundo S., and Eichelbaum M. (2004) Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch. Pharmacol. 369, 23–37 10.1007/s00210-003-0832-2 [DOI] [PubMed] [Google Scholar]
- 24. Yu A. M., Idle J. R., and Gonzalez F. J. (2004) Polymorphic cytochrome P450 2D6: humanized mouse model and endogenous substrates. Drug Metab. Rev. 36, 243–277 10.1081/DMR-120034000 [DOI] [PubMed] [Google Scholar]
- 25. Sangar M. C., Anandatheerthavarada H. K., Tang W., Prabu S. K., Martin M. V., Dostalek M., Guengerich F. P., and Avadhani N. G. (2009) Human liver mitochondrial cytochrome P450 2D6: individual variations and implications in drug metabolism. FEBS J. 276, 3440–3453 10.1111/j.1742-4658.2009.07067.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sangar M. C., Bansal S., and Avadhani N. G. (2010) Bimodal targeting of microsomal cytochrome P450s to mitochondria: implications in drug metabolism and toxicity. Expert Opin. Drug Metab. Toxicol. 6, 1231–1251 10.1517/17425255.2010.503955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Avadhani N. G., Sangar M. C., Bansal S., and Bajpai P. (2011) Bimodal targeting of cytochrome P450s to endoplasmic reticulum and mitochondria: the concept of chimeric signals. FEBS J. 278, 4218–4229 10.1111/j.1742-4658.2011.08356.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bajpai P., Sangar M. C., Singh S., Tang W., Bansal S., Chowdhury G., Cheng Q., Fang J. K., Martin M. V., Guengerich F. P., and Avadhani N. G. (2013) Metabolism of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine by mitochondrion-targeted cytochrome P450 2D6: implications in Parkinson disease. J. Biol. Chem. 288, 4436–4451 10.1074/jbc.M112.402123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Uehara S., Uno Y., Inoue T., Murayama N., Shimizu M., Sasaki E., and Yamazaki H. (2015) Activation and deactivation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine by cytochrome P450 enzymes and flavin-containing monooxygenases in common marmosets (Callithrix jacchus). Drug Metab. Dispos. 43, 735–742 10.1124/dmd.115.063594 [DOI] [PubMed] [Google Scholar]
- 30. Gilham D. E., Cairns W., Paine M. J., Modi S., Poulsom R., Roberts G. C., and Wolf C. R. (1997) Metabolism of MPTP by cytochrome P4502D6 and the demonstration of 2D6 mRNA in human foetal and adult brain by in situ hybridization. Xenobiotica 27, 111–125 10.1080/004982597240802 [DOI] [PubMed] [Google Scholar]
- 31. Coleman T., Ellis S. W., Martin I. J., Lennard M. S., and Tucker G. T. (1996) 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is N-demethylated by cytochromes P450 2D6, 1A2, and 3A4: implications for susceptibility to Parkinson disease. J. Pharmacol. Exp. Ther. 277, 685–690 [PubMed] [Google Scholar]
- 32. Herraiz T., Guillén H., Arán V. J., Idle J. R., and Gonzalez F. J. (2006) Comparative aromatic hydroxylation and N-demethylation of MPTP neurotoxin and its analogs, N-methylated β-carboline and isoquinoline alkaloids, by human cytochrome P450 2D6. Toxicol. Appl. Pharmacol. 216, 387–398 10.1016/j.taap.2006.06.003 [DOI] [PubMed] [Google Scholar]
- 33. Modi S., Gilham D. E., Sutcliffe M. J., Lian L. Y., Primrose W. U., Wolf C. R., and Roberts G. C. (1997) N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine as a substrate of cytochrome P450 2D6: allosteric effects of NADPH-cytochrome P450 reductase. Biochemistry 36, 4461–4470 10.1021/bi962633p [DOI] [PubMed] [Google Scholar]
- 34. Chiba K., Trevor A., and Castagnoli N. Jr. (1984) Metabolism of the neurotoxic tertiary amine, MPTP, by brain monoamine oxidase. Biochem. Biophys. Res. Commun. 120, 574–578 10.1016/0006-291X(84)91293-2 [DOI] [PubMed] [Google Scholar]
- 35. Bezard E., Gross C. E., Fournier M. C., Dovero S., Bloch B., and Jaber M. (1999) Absence of MPTP-induced neuronal death in mice lacking the dopamine transporter. Exp. Neurol. 155, 268–273 10.1006/exnr.1998.6995 [DOI] [PubMed] [Google Scholar]
- 36. Ramsay R. R., Dadgar J., Trevor A., and Singer T. P. (1986) Energy-driven uptake of N-methyl-4-phenylpyridine by brain mitochondria mediates the neurotoxicity of MPTP. Life Sci. 39, 581–588 10.1016/0024-3205(86)90037-8 [DOI] [PubMed] [Google Scholar]
- 37. Ramsay R. R., Kowal A. T., Johnson M. K., Salach J. I., and Singer T. P. (1987) The inhibition site of MPP+, the neurotoxic bioactivation product of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine is near the Q-binding site of NADH dehydrogenase. Arch. Biochem. Biophys. 259, 645–649 10.1016/0003-9861(87)90531-5 [DOI] [PubMed] [Google Scholar]
- 38. Usia T., Watabe T., Kadota S., and Tezuka Y. (2005) Cytochrome P450 2D6 (CYP2D6) inhibitory constituents of Catharanthus roseus. Biol. Pharm. Bull. 28, 1021–1024 10.1248/bpb.28.1021 [DOI] [PubMed] [Google Scholar]
- 39. Strobl G. R., von Kruedener S., Stöckigt J., Guengerich F. P., and Wolff T. (1993) Development of a pharmacophore for inhibition of human liver cytochrome P-450 2D6: molecular modeling and inhibition studies. J. Med. Chem. 36, 1136–1145 10.1021/jm00061a004 [DOI] [PubMed] [Google Scholar]
- 40. Westermann B. (2010) Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872–884 10.1038/nrm3013 [DOI] [PubMed] [Google Scholar]
- 41. Kohutnicka M., Lewandowska E., Kurkowska-Jastrzebska I., Członkowski A., and Członkowska A. (1998) Microglial and astrocytic involvement in a murine model of Parkinson disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Immunopharmacology 39, 167–180 10.1016/S0162-3109(98)00022-8 [DOI] [PubMed] [Google Scholar]
- 42. Eilers M., and Schatz G. (1986) Binding of a specific ligand inhibits import of a purified precursor protein into mitochondria. Nature 322, 228–232 10.1038/322228a0 [DOI] [PubMed] [Google Scholar]
- 43. Scheer N., Kapelyukh Y., McEwan J., Beuger V., Stanley L. A., Rode A., and Wolf C. R. (2012) Modeling human cytochrome P450 2D6 metabolism and drug-drug interaction by a novel panel of knockout and humanized mouse lines. Mol. Pharmacol. 81, 63–72 10.1124/mol.111.075192 [DOI] [PubMed] [Google Scholar]
- 44. Niranjan B. G., Wilson N. M., Jefcoate C. R., and Avadhani N. G. (1984) Hepatic mitochondrial cytochrome P-450 system: distinctive features of cytochrome P-450 involved in the activation of aflatoxin B1 and benzo (a) pyrene. J. Biol. Chem. 259, 12495–12501 [PubMed] [Google Scholar]
- 45. Onderwater R. C., Venhorst J., Commandeur J. N., and Vermeulen N. (1999) Design, synthesis, and characterization of 7-methoxy-4-(aminomethyl)coumarin as a novel and selective cytochrome P450 2D6 substrate suitable for high-throughput screening. Chem. Res. Toxicol. 12, 555–559 10.1021/tx980248q [DOI] [PubMed] [Google Scholar]
- 46. Birch-Machin M. A., and Turnbull D. M. (2001) Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 65, 97–117 10.1016/S0091-679X(01)65006-4 [DOI] [PubMed] [Google Scholar]
- 47. Smith L. (1955) Spectrophotometric assay of cytochrome c oxidase. Methods Biochem. Anal. 2, 427–434 [DOI] [PubMed] [Google Scholar]
- 48. Ghosh J., Chowdhury A. R., Srinivasan S., Chattopadhyay M., Bose M., Bhattacharya S., Raza H., Fuchs S. Y., Rustgi A. K., Gonzalez F. J., and Avadhani N. G. (2018) Cigarette smoke toxins-induced mitochondrial dysfunction and pancreatitis involves aryl hydrocarbon receptor mediated Cyp1 gene expression: protective effects of resveratrol. Toxicol. Sci. 166, 428–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu M., Neilson A., Swift A. L., Moran R., Tamagnine J., Parslow D., Armistead S., Lemire K., Orrell J., Teich J., Chomicz S., and Ferrick D. A. (2007) Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 292, C125–C136 10.1152/ajpcell.00247.2006 [DOI] [PubMed] [Google Scholar]
- 50. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.