

Abstract
CRISPR-Cas systems are RNA-based immune systems that protect many prokaryotes from invasion by viruses and plasmids. Type III CRISPR systems are unique, as their targeting mechanism requires target transcription. Upon transcript binding, DNA cleavage by type III effector complexes is activated. Type III systems must differentiate between invader and native transcripts to prevent autoimmunity. Transcript origin is dictated by the sequence that flanks the 3′ end of the RNA target site (called the PFS). However, how the PFS is recognized may vary among different type III systems. Here, using purified proteins and in vitro assays, we define how the type III-B effector from the hyperthermophilic bacterium Thermotoga maritima discriminates between native and invader transcripts. We show that native transcripts are recognized by base pairing at positions −2 to −5 of the PFS and by a guanine at position −1, which is not recognized by base pairing. We also show that mismatches with the RNA target are highly tolerated in this system, except for those nucleotides adjacent to the PFS. These findings define the target requirement for the type III-B system from T. maritima and provide a framework for understanding the target requirements of type III systems as a whole.
Keywords: CRISPR-Cas, CRISPR-RNA, crRNA, deoxyribonuclease (DNase), ribonuclease, RNA, DNA, bacterial immunity, Cmr effector complex, nucleotide mismatch, sequence similarity
Introduction
CRISPR arrays and CRISPR-associated (Cas)2 genes provide prokaryotes with adaptive immunity to invading genetic elements such as bacteriophage (1). CRISPR arrays consist of short DNA sequences of foreign origin, called spacers, separated by host repeat sequences (2–4). Cas proteins assemble with CRISPR RNAs (crRNAs) to form effector complexes (5–7). These complexes identify and destroy invading nucleic acids that are complementary to their crRNAs (5, 8). CRISPR-Cas systems are extremely diverse, with six distinct types currently recognized (types I–VI) (9). Types I, II, and V (and possibly IV) degrade DNA (5, 10, 11) whereas type VI degrades RNA (12). Type III systems, which are identified by their signature cas10 gene, are unique in that they degrade both DNA and RNA (13–15). Type III systems are further divided into four subtypes, III-A and III-D, which employ the Csm effector complex, and III-B and III-C, which employ the Cmr effector complex (9).
CRISPR arrays are transcribed to produce a single transcript (the pre–CRISPR RNA) containing multiple spacer sequences. In type III systems, mature crRNAs are generated from pre–CRISPR RNA in two steps. The Cas6 endoribonuclease cleaves the transcript within the repeat sequences, producing individual crRNAs consisting of a spacer sequence with repeat sequence at each end (6). These Cas6 cleavage products are then trimmed to remove the 3′ repeat sequence by host nucleases (6, 16, 17). Following these processing events, a mature crRNA contains eight nucleotides of repeat sequence on the 5′ end, called the crRNA tag, followed by the spacer region (Fig. 1A).
Figure 1.
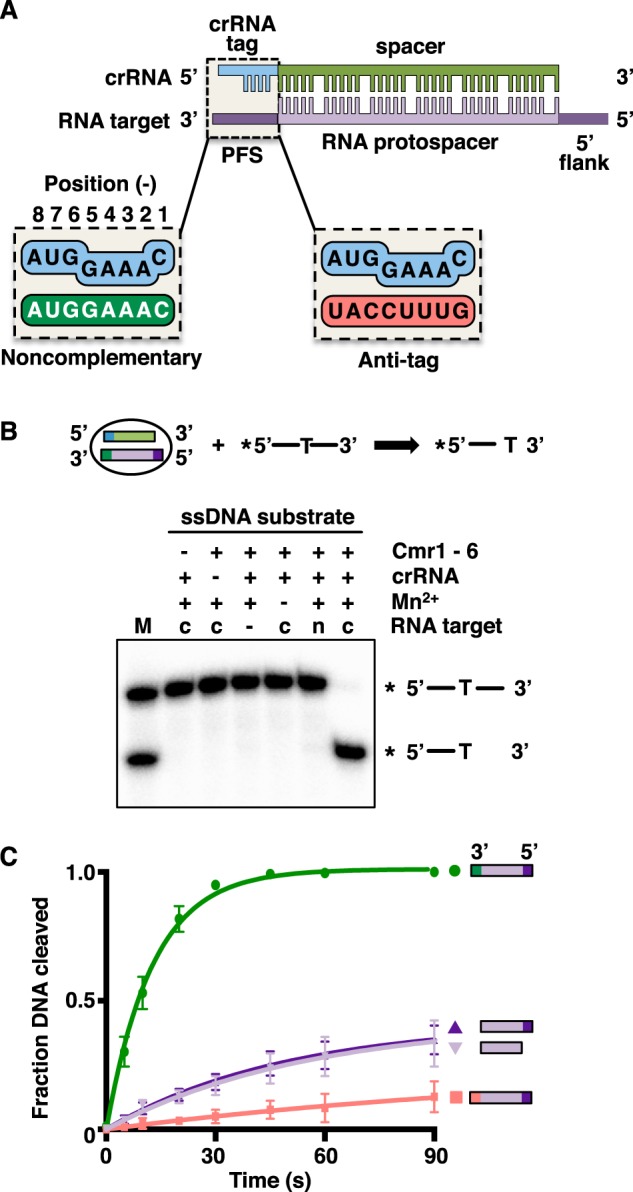
DNA cleavage by the TmaCmr complex is transcript-dependent. A, schematic depicting the interaction between the crRNA and the RNA protospacer. B, 5′-labeled ssDNA substrate was incubated with the TmaCmr complex, Mn2+ and either a complementary (c) or noncomplementary (n) RNA target with 10 nucleotide noncomplementary 5′ and 3′ flanks. The reaction products were analyzed by denaturing PAGE. M denotes markers for the substrate and expected product. C, quantification of DNA cleavage products by the TmaCmr complex over time upon activation with RNA targets containing various 3′ and 5′ flanks. RNA targets represented by green circles have a noncomplementary PFS, purple triangles represent targets without a PFS (dark purple includes a 5′ flank, light purple lacks a 5′ flank), and pink squares represent targets with an anti-tag PFS.
Immunity provided by type III CRISPR systems depends on transcription (18–21). Transcription produces an RNA target containing a crRNA-binding site (the RNA protospacer). Upon binding to an RNA protospacer, several enzymatic activities within the Csm/Cmr complexes are activated. The RNA target is cleaved at six nucleotide intervals by multiple copies of the Csm3/Cmr4 subunit (8, 21–25). The Cas10 subunit nonspecifically degrades invading single-stranded DNA (ssDNA) (20, 21, 25, 26) and generates a signaling molecule, cyclic oligoadenylate (cOA), from ATP. cOA then stimulates the trans-acting endoribonuclease Csm6/Csx1 for nonspecific RNA degradation (27–30). Together, these activities provide a robust transcription-coupled immune response.
To prevent autoimmune cleavage of host CRISPR arrays, types I, II, and V effector complexes identify short (2–4 nucleotides) DNA sequences called protospacer adjacent motifs (PAMs), which flank invading target sequences but are absent from host CRISPR arrays (31). Type III systems do not use PAMs to avoid autoimmunity because they are activated by RNA binding rather than DNA binding. CRISPR transcripts do not trigger autoimmunity because they have identical sequences to their respective crRNAs and therefore are not bound by the complex. However, autoimmunity could arise if a CRISPR array is transcribed in the antisense direction, as these anti-CRISPR transcripts are complementary to crRNA (26). Although their function is unknown, anti-CRISPR transcripts have been observed in a number of species (32–34). All characterized Csm/Cmr complexes fail to activate DNA cleavage (20, 25, 26, 35, 36) or cOA production (27–29) when bound to anti-CRISPR transcripts, although the transcripts themselves are still cleaved.
Anti-CRISPR transcripts are identified by the sequence that flanks the 3′ end of the RNA protospacer, which is called the protospacer flanking site (PFS). Two mechanisms have been described by which the PFS in anti-CRISPR transcripts inhibits autoimmunity in type III systems. In type III-A systems, base pairing between the crRNA tag and a complementary PFS, called an anti-tag, deactivates the Csm complex (37–40). In the type III-B systems the mechanism is less clear. In Pyrococcus furiosus (Pfu) the sequence of positions −1 to −3 of the PFS (that is, the three nucleotides 3′ to the start of the RNA protospacer) dictate whether DNA cleavage is activated, with anti-CRISPR transcripts failing to activate (20). Here, recognition of the anti-tag appears not to be driven by base pairing (at least not cognate Watson-Crick base pairing) but is presumably mediated by subunits of the PfuCmr complex (likely Cas10 and/or Cmr3) (15, 20). It is currently unclear if the mechanism of discrimination is subtype-specific (i.e. that type III-A systems use base pairing, whereas type III-B systems do not) or if either mechanism can be utilized by any given type III system.
The tolerance for mismatches between the crRNA and its target sequence varies among the different CRISPR-Cas systems. Most type III systems are highly tolerant of mismatches; targets containing multiple mismatches can trigger RNA cleavage, DNA cleavage, and cOA production (20, 24–26, 39, 41) and do not compromise immunity in vivo (19, 38, 42–44). Consequently, viral escape from type III immunity is observed to be more difficult than escape from other CRISPR systems (38, 58). Much of these data come from studies of type III-A systems; how mismatches modulate the activation of DNA cleavage by type III-B systems is not understood as well.
In this study, we define how the Cmr complex from Thermotoga maritima (Tma) discriminates between host (anti-CRISPR) and invader transcripts. We find that base pairing is necessary at positions −2 to −5 of the PFS but not at position −1. We also examine how mismatches between the crRNA spacer and RNA protospacer are tolerated in this system and show that mismatches are highly tolerated across the RNA protospacer, except for those nucleotides adjacent to the PFS.
Results
Activation of DNA cleavage requires a noncomplementary 3′ flanking sequence
Activation of DNA cleavage by type III systems requires pairing of the crRNA with an RNA protospacer and is regulated by the PFS. DNA cleavage is deactivated in all type III systems if the RNA target contains an anti-tag sequence in the PFS (20, 25, 26, 35, 37, 45, 46) (Fig. 1A). However, the role of the PFS in the activation of DNA cleavage is unclear. We have reported previously that RNA targets lacking flanking sequences (and therefore a PFS) can activate the DNase function of the TmaCmr complex (25) but in other type III systems, a PFS that lacks anti-tag sequence is required for activation (26, 35, 46). To investigate this further, we monitored cleavage of ssDNA by the TmaCmr complex in the presence of a series of RNA targets. The TmaCmr complex was assembled from recombinant Cmr proteins (Fig. S1A) and crRNA generated by in vitro transcription and subsequent processing with recombinant TmaCas6 (Fig. S1, B and C). We next 5′ end–labeled an ssDNA oligonucleotide containing a single thymine at its center. We have shown previously that TmaCmr cleaves ssDNA specifically after thymine bases (25). We then incubated this labeled ssDNA with the TmaCmr complex and RNA target consisting of a complementary protospacer with 10 nucleotides of noncomplementary sequence at each flank (Fig. 1B). DNA cleavage was monitored by denaturing PAGE followed by autoradiography (Fig. 1B). After 1 min at 80°C, the expected cleavage product was observed when all required components (crRNA, TmaCmr complex, complementary RNA target, and Mn2+) were present (Fig. 1B).
The fraction of ssDNA cleaved was then monitored over time in the presence of a series of RNA targets containing different flanking sequences (Fig. 1C and Fig. S1D). An RNA target containing a noncomplementary PFS and a 5′ flank (the same RNA target used in the previous experiment, Fig. 1B) triggered rapid cleavage of the DNA; all DNA was cleaved within 60 s. However, RNA targets lacking a PFS (either with or without a 5′ flank) triggered slower DNA cleavage, with less than half the DNA being cleaved after 90 s. Almost no DNA cleavage was observed in the presence of an RNA target containing an anti-tag PFS (Fig. 1C). These results indicate that DNA cleavage by the TmaCmr complex is partially activated by a complementary RNA protospacer, and a PFS that lacks anti-tag sequence is required for full activation whereas an anti-tag PFS deactivates DNA cleavage.
Recent structural and single-molecule fluorescence studies indicate that the PFS of the RNA target allosterically regulates the DNase activity of Csm complexes through conformational changes in the Cas10 subunit (39, 40, 42). The DNase activity of Csm/Cmr complexes is also regulated by cleavage of the bound RNA target (20, 25, 26); once the target RNA is cleaved and/or dissociates, DNase activity is deactivated. To determine whether RNA cleavage altered the rate of DNA cleavage in our experiments, we 5′ end–labeled the RNA targets, rather than the ssDNA, and monitored RNA cleavage. Under the same conditions as the DNA cleavage reaction (where the RNA target is in ∼8-fold excess of the TmaCmr complex) we observed that less than 5% of each RNA target is cleaved after 60 s (Fig. S2). We also compared the rates of DNA cleavage by WT TmaCmr complex and by a TmaCmr complex formed with the Cmr4 D26A mutant, which can bind to but cannot cleave RNA (25, 47–49), and found no significant differences (Fig. S3). Thus, we concluded that the difference in the rate of DNA cleavage in the presence of the different RNA targets was not because of differing rates of RNA cleavage.
Finally, we determined the affinity of the TmaCmr complex for each of the RNA targets using EMSAs (Fig. S2). All targets are bound with similar affinities (apparent KDs ∼0.3 nm). Therefore, RNA binding does not explain the observed differences in DNA cleavage (Fig. S2). Together these data indicate that the PFS directly regulates the DNase activity of the TmaCmr complex, likely via conformational changes in the Cas10 subunit as observed in type III-A systems (39, 40, 42).
Positions −1 to −3 of the PFS are important for regulating DNA cleavage
Plasmid transformation experiments in Pfu have suggested a specific role for the sequence of positions −1 to −3 of the PFS in activating type III-B immunity (20). To determine which positions in the anti-tag inhibit the DNase activity of the TmaCmr complex, we measured DNA cleavage in the presence of RNA targets containing varying complementarity to the crRNA tag (Fig. 2A). Beginning with a target containing an anti-tag PFS, which triggered very little DNA cleavage (Fig. 2A, target 1), we removed complementarity one base at a time, making cumulative changes from position −8 to position −1 (Fig. 2A, targets 2–8). With these targets, we observed no significant increase in DNA cleavage until only positions −1 and −2 contained anti-tag sequence (that is positions −3 to −8 were noncomplementary), whereupon DNA cleavage was triggered to nearly the same extent as with a target containing a noncomplementary PFS (Fig. 2A, targets 7 and 9). Moreover, a target with anti-tag sequence at all positions but −1 to −3 triggered DNA cleavage to the same extent as a target with a fully noncomplementary PFS (Fig. 2A, target 10). These observations parallel data obtained from transformation assays in Pfu (20) and suggest that the three nucleotides at the 3′ end of the PFS (positions −1 to −3) are important for regulating the DNase activity of the TmaCmr complex.
Figure 2.

Importance of positions −1 to −3 of the PFS in invader host transcript discrimination. A, scatter plots showing the fraction of DNA cleaved by the TmaCmr complex when activated by RNA targets with varying amounts of anti-tag sequence in the PFS. Black letters indicate positions of anti-tag sequence. Individual values are plotted as solid black circles. Bar chart represents the mean of these values. Independent experiments were repeated at least three times. Error bars are S.D. B, heat map of normalized TmaCmr DNA cleavage upon activation with RNA targets with all sequences in positions −1 to −3 of the PFS. All targets contain the noncomplementary sequence, AGGUA, in positions −4 to −8 of the PFS. Dark purple indicates a high level of activation, whereas white indicates low activation. Individual plots of these data are shown in Fig. S4. Values shown are the average of at least three replicates.
To understand how the sequence in positions −1 to −3 of the RNA target affects DNA cleavage, we generated a series of RNA targets containing all possible sequences in positions −1 to −3. In each of these 64 targets, the sequence of positions −4 to −8 is noncomplementary to the crRNA tag. The extent of DNA cleavage triggered by each target was measured as before. Nearly every triplet sequence triggered DNA cleavage (Fig. 2B and Fig. S4). Only anti-tag sequence in positions −1 to −3 (GUU) failed to trigger significant DNA cleavage. DNA cleavage was also somewhat reduced in the closely related AUU and GCU sequences (Fig. 2B and Fig. S4).
A guanine base at position −1 of the PFS helps to prevent DNA cleavage
In the previous experiment (Fig. 2B), a GUU sequence in positions −1 to −3 of the RNA target failed to activate DNA cleavage whereas UUU, CUU, and AUU sequences at the same positions all activated DNA cleavage. Structural data shows that the base at position −1 of a bound RNA target is flipped out and not able to base-pair with the crRNA tag (39, 40). Together, these observations suggest that a guanine at position −1, and not base pairing, is important for deactivation of DNA cleavage on binding an anti-CRISPR transcript. To confirm this, we generated crRNAs with each nucleotide at position −1. WT crRNA contains a cytosine at this position so we generated C-1U, C-1A, and C-1G variants. We formed TmaCmr complexes containing WT or each crRNA variant and measured their DNase activation when supplied with RNA targets containing each base at position −1 (all of these targets contained anti-tag sequence at position −2 and −3). Regardless of crRNA sequence, and therefore base pair potential, a guanine at position −1 of the RNA target triggered poor DNA cleavage (Fig. 3A). Conversely, when position −1 of the target contained a cytosine, uracil, or to a lesser extent adenine, robust DNA cleavage was triggered (Fig. 3A). TmaCmr complexes formed with each crRNA stimulated robust DNA cleavage in the presence of the RNA target containing a uracil at position −1 (Fig. 3A), indicating that all crRNA variants were functional. These results demonstrate that in the TmaCmr complex, position −1 of the RNA target cannot base pair with the crRNA tag and that a guanine at this position helps to deactivate DNA cleavage.
Figure 3.

A guanine base at position −1 of the PFS helps to prevent DNA cleavage. A, scatter plots showing the fraction of DNA cleaved by the TmaCmr complex loaded with WT and crRNAs with mutations at position −1 (C-1U, C-1A, and C-1G). RNA targets contained U, C, A, or G at position −1. All targets have the sequence UUAGGUA in positions −2 to −8. B, scatter plots of the fraction DNA cleaved by TmaCmr complex when activated by transcripts containing PFSs with varying amounts of anti-tag sequence. A and B, black letters indicate positions of anti-tag sequence. Individual values are plotted as solid black circles. Bar chart represents the mean of these values. Independent experiments were repeated at least three times. Error bars are S.D.
Base pairing between the PFS and the crRNA in positions −2 to −5 deactivates DNA cleavage
Our data indicate the importance of positions −1 to −3, but does not rule out that other positions in the PFS also play a role. Thus, we tested the role of anti-tag sequence in other positions of the PFS. We found that substituting with a cytosine at position −1 of an otherwise anti-tag sequence did not fully restore DNA cleavage (Fig. 3B, target 2). Thus, a guanine at position −1 is not necessary to prevent DNA cleavage if the rest of the PFS contains anti-tag sequence. Removal of any more anti-tag sequence starting at position −2 fully restored DNase activity (Fig. 3B, targets 3–8).
Structural studies in type III-A systems have shown that positions −2 to −5 of the crRNA tag are exposed and form base pairs with anti-tag sequence. However, positions −1 and −6 to −8 are buried and thus cannot form base pairs (39, 40). In vivo data also implicate base pairing in positions −2 to −5 in inhibiting immunity (37, 38). To test if positions −2 to −5 are important for regulation of DNase activity of the TmaCmr complex, we measured the extent of DNA cleavage triggered by RNA targets with varying amounts of anti-tag sequence in these positions. We found that an RNA target with anti-tag sequence in positions −1 and −6 to −8 permitted full DNA cleavage (Fig. 4A, target 2). However, RNA targets with anti-tag sequence at positions −2 to −5 and either guanine or cytosine at position −1, inhibited DNA cleavage (Fig. 4A, targets 3 and 4). Consistent with our earlier observations (Fig. 3B), the target with a guanine at position −1 inhibited DNA cleavage more than the target with a cytosine at the same position. We conclude that, as in type III-A systems (37, 39, 40), positions −2 to −5 of the RNA target regulate the DNase activity of the TmaCmr complex.
Figure 4.

Role of PFS positions −2 to −5 in distinguishing invader and host transcripts. A and B, scatter plots of the fraction of DNA cleaved by the TmaCmr complex when activated by RNA targets with varying amounts of anti-tag sequence in positions −1 to −5. C, scatter plots of normalized fraction of DNA cleaved by the TmaCmr complex. RNA targets with anti-tag sequence in three of four positions from −2 to −5 were paired with WT crRNA or a crRNA mutated such that the target gained full complementarity in this region. Black letters indicate positions of anti-tag sequence. Individual values are plotted as solid black circles. Bar chart represents the mean of these values. Independent experiments were repeated at least three times. Error bars are S.D. D, scatter plots of fraction DNA cleaved by the TmaCmr complex programmed with mutant crRNA.
To understand how much anti-tag sequence is needed between positions −2 and −5 to inhibit DNA cleavage, we designed RNA targets that only contain anti-tag sequence at three of these four positions. We tested DNase activation of the TmaCmr complex by each of these targets and found that they all triggered as much DNA cleavage as an RNA target containing no anti-tag sequence (Fig. 4A; compare targets 5–8 to target 9). Thus, if position −1 is a cytosine, all four bases in positions −2 to −5 must have anti-tag sequence to inhibit DNA cleavage. Given that a guanine in position −1 helps to prevent activation of DNA cleavage, we also generated a series of RNA targets containing a guanine at position −1 and one or two bases of anti-tag sequence in positions −2 to −5. Analysis of DNA cleavage in the presence of these targets revealed that RNA targets with anti-tag sequence in positions −4 and −5 permitted more DNase activity than those with anti-tag sequence in positions −2 and −3 (Fig. 4B, targets 2–11). Thus, positions −2 and −3 appear more important for inhibiting the DNase activity of TmaCmr than positions −4 and −5.
The above results implicate anti-tag sequence in positions −2 to −5 of the RNA target in inhibiting the DNase activity of the TmaCmr complex. However, the data do not directly address whether these nucleotides base-pair with the crRNA tag. To explore this, we generated four variant crRNAs, each with a single nucleotide substitution in one position between −2 and −5. We then monitored DNA cleavage by the TmaCmr complexes containing WT and variant crRNAs in the presence of RNA targets that possess compensatory substitutions. These targets trigger robust DNA cleavage with the TmaCmr complex loaded with WT crRNA (Fig. 4C). In agreement with the base-pairing model and data from type III-A systems, DNA cleavage was inhibited only when base pairing was possible (Fig. 4C). All crRNA variants were functional as each TmaCmr complex robustly cleaved DNA in the presence of an RNA target with a noncomplementary PFS (Fig. 4D).
The DNase activity of the TmaCmr complex tolerates mismatches between the RNA protospacer and crRNA guide
DNase activity and immunity in type III systems are highly tolerant of mismatches between the crRNA and RNA protospacer. The structure of the crRNA:RNA target duplex in type III systems (and all Class I systems) is arranged into segments of 5 bp separated by disrupted 1-nucleotide gaps (39, 40, 50, 51). To determine where mismatches might be tolerated for DNase activation of the TmaCmr complex, we introduced 5-nucleotide blocks of mismatches at each of these segments. We monitored DNA cleavage by the TmaCmr complex in the presence of these RNA targets and found that mismatches within segment 1 (the segment adjacent to the PFS) inhibited DNA cleavage (Fig. 5A, target 2), whereas an RNA target containing any other mismatched segment fully activated DNA cleavage (Fig. 5A, targets 3–7). We performed EMSAs to confirm that the target containing a mismatched segment 1 was bound by the complex and found that the affinities of the TmaCmr complex for this target and a target with mismatched segment 6 were within experimental error of a fully complementary target (compare Figs. S5A and S2B). We also observed that this RNA target was cleaved to the same extent (<5% after 60 s) (Fig. S5B) as a fully complementary target (Fig. S2A). We conclude that base pairing in segment 1 of a bound RNA target is important for the activation of DNA cleavage. To see how many base pairs are needed, we also monitored DNA cleavage in the presence of RNA targets containing single point or accumulating mismatches within segment 1 and found that as few as 1 to 2 bp in this segment is sufficient to activate DNA cleavage (Fig. 5B, targets 19 and 20).
Figure 5.

Tolerance for mismatches between the RNA protospacer and crRNA. A, scatter plots of the fraction of DNA cleaved by TmaCmr complex activated by targets containing protospacers that are mismatched to the crRNA in one or multiple segments. Segments are defined by the 5-nucleotide stretches of base pairs within the structure of the crRNA-target duplex. Individual values are plotted as solid black circles. Bar chart represents the mean of these values. Independent experiments were repeated at least three times. B, scatter plots of DNA cleavage by the TmaCmr complex upon activation with targets containing one or more mismatches within segment 1. C, scatter plots of TmaCmr complex binding to mismatched targets 8, 9, 10, and 11 from panel A. Independent experiments were repeated at least three times. Error bars are S.D. All targets contain a noncomplementary PFS.
Our observations show that base pairing in segment 1 is necessary to activate DNA cleavage. To determine how many other segments must be complementary to activate DNA cleavage, we tested RNA targets containing an increasing number of mismatched segments, starting at segment 6 (the 5′ end of the RNA protospacer). We found that as the number of mismatched segments increases DNA cleavage decreases, and when only segments 1 and 2 remain complementary we detect no significant DNA cleavage (Fig. 5A, targets 7–11). The TmaCmr complex is likely to have a lower affinity for targets that contain extensive mismatches to the crRNA, so we measured target binding by EMSA. We found that targets which failed to activate DNA cleavage were also poorly bound by the TmaCmr complex (Fig. 5C and Fig. S5, F–H). Binding was weak for the RNA target containing mismatches in segments 4 through 6 (Fig. 5, target 9), yet this RNA stimulated DNase activity, albeit to a lesser extent than a fully complementary target (Fig. 5A). We presume that this target likely binds transiently to TmaCmr and that this transient interaction is not well-detected by EMSA. We conclude that DNA cleavage by TmaCmr is highly tolerant of mismatches with the RNA protospacer and it is likely that DNA cleavage can be activated by any invading transcript with complementarity in segment 1 and enough additional base pairs to promote complex binding.
Discussion
For type III CRISPR-Cas systems, accurately distinguishing invader from host anti-CRISPR transcripts is essential to avoid cleavage of the host genome. Systems that only target DNA (types I, II, and V) use PAM sequences to distinguish invading DNA; host CRISPR arrays lack PAM sequences. Type III systems are activated by RNA binding, not DNA binding, and therefore do not use PAMs. Instead the PFS dictates whether the RNA target is of invader or host origin. Genetic studies with the type III-A system from Staphylococcus epidermidis (Sep) initially demonstrated that base pairing between an anti-tag PFS and the crRNA tag at positions −2 to −5 inhibits immunity (37, 38). Subsequent structure-function studies confirmed this base pairing in a number of type III-A systems (39, 40). A study in the type III-B system in Pfu, however, suggests that the PFS does not base-pair with anti-CRISPR transcripts in this system (20). Although it was previously unclear if these mechanisms were specific to the subtypes in which they were discovered, we present data that the type III-B system from T. maritima uses a PFS that spans positions −1 to −5 of the RNA target and that its anti-tag PFS is recognized by both base pairing and non–base pairing interactions.
The role of the PFS in activating DNA cleavage varies among type III systems. We reported previously that RNA targets lacking any flanking sequence (and therefore a PFS) activated DNA cleavage by the TmaCmr complex (25). However, in several systems, a PFS lacking anti-tag sequence is required for activation (26, 35, 46). Here we show that RNA targets lacking a PFS partially activate DNA cleavage by the TmaCmr complex and that full activation requires a noncomplementary PFS (Fig. 1C). Thus, all type III systems characterized required a noncomplementary PFS for full activation. With RNA targets lacking a PFS, the rate of DNA cleavage is slower than the rate of RNA target cleavage (25). However, with RNA targets containing a PFS, DNA cleavage is faster than RNA cleavage (Fig. 1C and Fig. S2). DNA cleavage being more rapid than RNA cleavage is consistent with the model that RNA cleavage and/or dissociation deactivates DNase activity (and cOA production) in type III systems and acts as a temporal control of the system (20, 25, 26, 29).
In the Tma type III-B system, different positions within the anti-tag sequence are identified by distinct mechanisms. We found that, like Pfu, anti-tag sequence in the first three nucleotides of the PFS is sufficient to deactivate DNA cleavage by the TmaCmr complex. However, unlike Pfu, other sequences in these positions do not deactivate DNA cleavage. In Pfu 28 of the 64 possible sequence combinations in these positions inhibit immunity (20). In contrast, the DNase activity of the TmaCmr complex was only fully inhibited by the anti-tag sequence (with two other closely related sequences allowing for reduced DNA cleavage) (Fig. 2B and Fig. S4). We show that a guanine in position −1 inhibits DNA cleavage regardless of which base is in this position of the crRNA (Fig. 3A), indicating that this position is not identified through base pairing and presumably identified by contacts with Cmr subunits. Structures of the Csm complex bound to an RNA target show that the base in position −1 is flipped out and sits in a pocket formed by Cas10, Csm4, and Csm3 (39, 40). Although sequence divergence makes identification of specific residues difficult, in the Cmr complex the base in position −1 likely sits in a pocket formed by Cas10, Cmr5, and Cmr4. Our observations contrast with data from the Sep type III-A system and the Pfu type III-B system, which suggest no sequence requirement in position −1 (20, 38).
Upon further analysis, we found that targets containing anti-tag sequence in positions −2 to −5 of the PFS also inhibit DNA cleavage by the TmaCmr complex (Figs. 3B and 4A). By making compensatory mutations in the crRNA tag, we show that anti-tag sequence in these positions is identified through base pairing (Fig. 4C). Therefore, the TmaCmr complex employs base pairing and non–base pairing interactions with an anti-tag sequence. Thus, some type III effector complexes (e.g. SepCsm) may recognize the anti-tag through base pairing alone and others may recognize the anti-tag through interactions with protein subunits (e.g. PfuCmr), yet others may combine these mechanisms to different extents (e.g. TmaCmr).
We noted that although nucleotides in the PFS control discrimination between invader and host anti-CRISPR transcripts, not all positions contribute equally. We show that if position −1 is a guanine, less base pairing is necessary at positions −2 to −5 to inhibit cleavage. Furthermore, we observed that RNA targets with anti-tag sequence at either position −2 or −3 permit less DNA cleavage than targets with anti-tag sequence at position −4 or −5 (Fig. 4B). Similar results were obtained with the Csm complexes from Streptococcus thermophilus (Sth) and Sep (37–39). This suggests that positions −2 and −3 of the PFS play a more important role in regulating the DNase activity (and cOA production) in those type III systems that employ base pairing to recognize the anti-tag.
CRISPR systems that only target DNA (types, I, II, and V) follow stringent rules in activating DNA cleavage. Targets must contain a PAM and cannot contain mismatches in the seed region (52–55). In contrast, the TmaCmr complex uses stringent rules to deactivate DNA cleavage and has relaxed rules for activation of DNA cleavage. Complementary targets that contain an anti-tag sequence in the PFS fail to activate DNA cleavage, whereas the vast majority of possible PFS sequences appear to activate DNA cleavage. Similar observations have been made through genetic studies of type III-A immunity in Sep (38). Therefore, type III systems assess if the target is of host origin (i.e. an anti-CRISPR transcript), whereas type I, II, and V systems verify that the target is from an invader. An exception is PfuCmr, which was suggested to be activated by particular sequences (rather than being deactivated by specific host sequences) (20); however, regardless of the mechanism of transcript identification, type III systems universally appear to be active given a wide variety of target flanking sequences, in contrast to DNA targeting subtypes. Additionally, recent data from a type VI system suggests that extensive complementarity between the crRNA tag and an anti-tag PFS inhibits activation of Cas13 (56), suggesting that broad requirements for activation could be a defining characteristic among RNA-targeting CRISPR-Cas systems.
Complementarity between the crRNA and target sequence is a key component of all CRISPR-Cas systems, yet the tolerance for mismatches among the types varies. Most types (including type I, II, and V) use a seed region to initiate binding to the target. Mismatches in the seed region are therefore not tolerated as they inhibit initial binding to the target (52–55). In contrast to the other types, many type III systems have been shown to be highly tolerant of mismatches. Here we find that activation of DNA cleavage by the TmaCmr complex also tolerates extensive mismatching. Base pairing is most important in the segment adjacent to the PFS (segment 1), but DNA cleavage is still activated with targets that contain up to four mismatches in this segment (Fig. 5). Although an RNA target with mismatched segment 1 does not activate DNA cleavage, this target is still bound, thus this segment does not constitute a seed region in which complementarity is required for binding. This result is consistent with studies in type III-A systems and suggests that the absence of a seed region may be a hallmark of all type III systems. We also find that targets containing extensive mismatches in segments 2 through 6 activate DNA cleavage as long as the RNA protospacer maintains enough complementarity to bind to the complex.
Together, our analysis of the sequence requirements of the PFS and RNA protospacer suggest that type III RNA targets can be functionally divided into two regions (Fig. 6). In the first, nucleotides encompassing segments 2 to 6 drive binding of the effector complex to the RNA target, without a seed region. In the second, the PFS and segment 1 (positions −5 to 5 of the RNA target) modulate DNA cleavage (and likely cOA production) by the Cas10 subunit. Base pairing in segment 1 activates DNA cleavage whereas base pairing in the PFS deactivates DNA cleavage, with the nucleotide at position −1 modulating the extent of this deactivation in some systems (e.g. TmaCmr) (Fig. 6).
Figure 6.
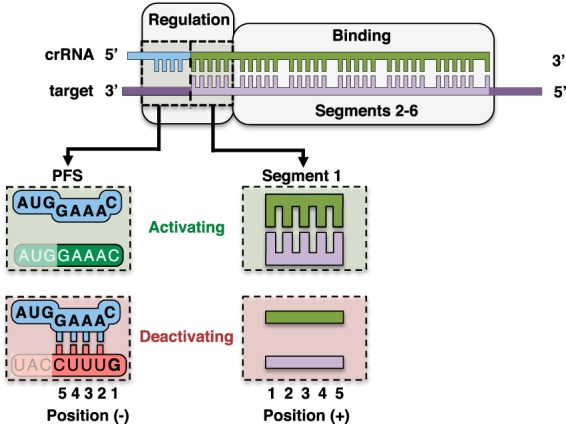
Functional regions of a type III RNA target. Segments 2–6 drive binding of the TmaCmr complex to the RNA target, whereas the PFS (positions −1 to −5) and segment 1 (positions 1 to 5) of the RNA target regulate activation of Cas10. Noncomplementary PFSs are activating, whereas an anti-tag PFS is deactivating; this sequence is recognized by base pairing in positions −2 to −5 and a guanine base in position −1. Base pairing in segment 1 allows activation, and mismatches across this segment prevent activation.
Experimental procedures
Expression and purification of recombinant Cmr proteins
The WT and mutant TmaCmr2–6 complexes were made by expressing each subunit individually in T7Express cells (New England Biolabs) and then pooling all cultures before purification. Cmr2 to Cmr5 were cloned separately into pRSFDuet-1 (Novagen), with Cmr2 and Cmr3 codon optimized for expression in Escherichia coli (GeneArt). Cmr6 was cloned into pHAT2 (25). 1 liter of each culture was grown with appropriate antibiotics at 37 °C to A600 = 0.4 in Luria-Bertani media followed by induction with 200 μm isopropyl β-d-1-thiogalactopyranoside (IPTG) and overnight growth at 20 °C. Cells of each were pelleted and resuspended together in lysis buffer (1 m KCl, 20 mm tris, pH 8.0, 10 mm imidazole, 1 mm tris(2-carboxyethyl)phosphine (TCEP)) with added protease inhibitors E-64, phenylmethylsulfonyl fluoride (PMSF), bestatin, and pepstatin A. The cells were then lysed using a microfluidizer. The lysate was heat treated at 80 °C in a water bath for 10 mins, then clarified by centrifugation at 20 °C. This sample was passed over a 5-ml immobilized metal affinity chromatography (IMAC) column (Bio-Rad) charged with nickel sulfate and equilibrated with lysis buffer. After washing with 25 column volumes lysis buffer, the sample was eluted (500 mm KCl, 20 mm tris, pH 8.0, 250 mm imidazole, 1 mm TCEP) and injected onto a HiLoad 26/60 S200 size exclusion column (GE Healthcare) equilibrated with gel filtration buffer (350 mm KCl, 20 mm tris, pH 8.0, 1 mm TCEP). Cmr1 and Cas6 were cloned into pHAT2 and purified separately as described above without heat treatment (57).
Expression and purification of T7 RNA polymerase
His6-tagged T7 RNA polymerase (P266L) was expressed in T7Express (New England Biolabs). 1 liter was grown to A600 = 0.3 at 37 °C, induced with 200 μm IPTG, and grown overnight at 20 °C. The cells were pelleted and resuspended in lysis buffer (250 mm NaCl, 10 mm imidazole, 20 mm HEPES, pH 7.0, 10 mm DTT, 5% glycerol) with protease inhibitors listed above. Cells were lysed using a microfluidizer and clarified by centrifugation at 4 °C. The sample was added to 3 ml of nickel IMAC resin (Bio-Rad) and gently rocked at 4 °C for 30 min. The supernatant was removed and the resin was washed with 25 column volumes lysis buffer. The protein was then eluted (250 mm NaCl, 250 mm imidazole, 20 mm HEPES, pH 7.0, 10 mm DTT, 5% glycerol).
Synthesis of RNA
All RNAs less than 45 nucleotides were purchased (Sigma), whereas longer RNAs were synthesized by in vitro transcription. 5 μm complementary DNA templates (Sigma) were annealed (50 mm NaCl, 10 mm tris, pH 8.0, 1 mm EDTA) by slow cooling from 95 °C to room temperature. 100 nm dsDNA was then incubated with 5 mm each ribonucleotide triphosphate (rNTP), 15 mm MgCl2, 0.1 mg/ml P266L T7 RNA polymerase (59), and transcription mix (25 mm tris pH 8.0, 2 mm spermidine, 40 mm DTT) for 2 to 3 h at 37 °C.
crRNAs were transcribed as described above. The 5′ end contained repeat sequence and was cleaved with 200 nm Cas6 by incubating in cleavage buffer (65 mm KCl, 20 mm HEPES, pH 7.0, 20 mm EDTA, 10% glycerol) at 80 °C for 30 min. Transcripts and crRNAs were then gel extracted, ethanol precipitated, and resuspended in RNA storage solution (Thermo Fisher) with 1 unit RiboLock RNase Inhibitor (Thermo Fisher). Sequences of all oligonucleotides used in this study are provided in the supporting information.
Radiolabeling of oligonucleotides
DNA was purchased from Sigma, gel purified, and radiolabeled with 1–2 pmol [γ-32P]ATP by incubating 200 nm DNA in 1× T4 PNK buffer with 10 units T4 polynucleotide kinase (PNK) (New England Biolabs) at 37 °C for 30 min, followed by a 20-min heat inactivation at 65 °C. 5′ ends of transcripts were first dephosphorylated by incubating 200 nm RNA in 1× CutSmart buffer with 1 unit shrimp alkaline phosphatase (rSAP) (New England Biolabs) for 30 min at 37 °C, followed by a 20-min heat inactivation at 75 °C. 10 units T4 PNK, 1× PNK buffer, and 2 pmol [γ-32P]ATP (Perkin-Elmer) were added to this reaction and incubated at 37 °C for 30 min, followed by heat inactivation for 20 min at 65 °C. The transcripts were then gel extracted, ethanol precipitated, and resuspended in RNA storage solution.
DNA and RNA cleavage assays
TmaCmr complex was first formed by incubating crRNA, Cmr 1, and Cmr 2–6 at 80 °C for 20 min. Following complex formation, 95 nm unlabeled DNA substrate, and 5 nm 5′ radiolabeled DNA were added. Reactions were initiated upon the addition of 200 nm RNA (or RNA storage solution in no transcript controls) and quenched (90% formamide, 2.5% glycerol, 0.01% SDS, 0.01% bromphenol blue, 0.01% xylene cyanol, 1 mm EDTA) after 1 min (unless otherwise indicated) at 80 °C. Reactions were done in reaction buffer (100 mm KCl, 50 mm HEPES, pH 7.0, 1 mm TCEP, 1 mm MnCl2) with 25 nm Cmr1–6:crRNA complex. These samples were then run on 15% polyacrylamide denaturing gels and visualized by phosphorimaging (Fujifilm FLA-7000). Images were quantified using Image Gauge (Fujifilm) and data analysis was done in Prism (GraphPad Software). All data points are the average of at least three replicates and error bars represent standard error of the mean. RNA cleavage experiments were carried out as described for DNA cleavage experiments utilizing 5 nm radiolabeled RNA, 195 nm unlabeled RNA, and 100 nm unlabeled DNA. 5′ end–labeled synthetic RNAs of expected products were used as markers to confirm appropriate product size.
EMSAs
D26A TmaCmr complex was formed as above and then gel filtered. The complex was placed on ice and 2-fold serial dilutions were made in binding buffer (100 mm KCl, 20 mm HEPES, pH 7.0, 1 mm DTT, 1 mm MnCl2, 50 μg/ml BSA, 5% glycerol). 10 pm 5′ radiolabeled RNA target was added, and in the case of low affinity RNA targets, 5 μm poly-dT40 nonspecific competitor was also added. Reactions equilibrated for an hour on ice prior to being run on 5% polyacrylamide native gels. Reactions were visualized, quantified, and analyzed as above.
Author contributions
K. J. and B. A. L. data curation; K. J. and S. B. formal analysis; K. J. and B. A. L. investigation; K. J. writing-original draft; K. J. and S. B. writing-review and editing; B. A. L. and M. A. E. methodology; S. B. conceptualization; S. B. supervision; S. B. funding acquisition.
Supplementary Material
Acknowledgment
We thank members of the Bailey lab for helpful discussion.
This work was supported by National Institutes of Health Grants GM097330 (to S. B.) and Ruth L. Kirschstein National Institutes of Health F31 fellowship GM105364 (to M. A. E.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5.
- Cas
- CRISPR-associated
- crRNA
- CRISPR RNA
- ssDNA
- single-stranded DNA
- cOA
- cyclic oligoadenylate
- PAM
- protospacer adjacent motif
- PFS
- protospacer flanking sequence
- Pfu
- Pyrococcus furiosus
- Tma
- Thermotoga maritima
- Sep
- Staphylococcus epidermidis
- Cmr
- CRISPR RAMP module
- Csm
- CRISPR-Cas subtype Mycobacterium tuberculosis
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- TCEP
- tris(2-carboxyethyl)phosphine
- IMAC
- immobilized metal affinity chromatography
- PNK
- polynucleotide kinase.
References
- 1. Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D. A., and Horvath P. (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 10.1126/science.1138140 [DOI] [PubMed] [Google Scholar]
- 2. Bolotin A. (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2551–2561 10.1099/mic.0.28048-0 [DOI] [PubMed] [Google Scholar]
- 3. Mojica F. J. M., Díez-Villaseñor C. S., García-Martínez J., and Soria E. (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 60, 174–182 10.1007/s00239-004-0046-3 [DOI] [PubMed] [Google Scholar]
- 4. Pourcel C., Salvignol G., and Vergnaud G. (2005) CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151, 653–663 10.1099/mic.0.27437-0 [DOI] [PubMed] [Google Scholar]
- 5. Brouns S. J. J., Jore M. M., Lundgren M., Westra E. R., Slijkhuis R. J. H., Snijders A. P. L., Dickman M. J., Makarova K. S., Koonin E. V., and van der Oost J. (2008) Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964 10.1126/science.1159689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carte J., Wang R., Li H., Terns R. M., and Terns M. P. (2008) Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 22, 3489–3496 10.1101/gad.1742908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deltcheva E., Chylinski K., Sharma C. M., Gonzales K., Chao Y., Pirzada Z. A., Eckert M. R., Vogel J., and Charpentier E. (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607 10.1038/nature09886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hale C. R., Zhao P., Olson S., Duff M. O., Graveley B. R., Wells L., Terns R. M., and Terns M. P. (2009) RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139, 945–956 10.1016/j.cell.2009.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Makarova K. S., Wolf Y. I., Alkhnbashi O. S., Costa F., Shah S. A., Saunders S. J., Barrangou R., Brouns S. J. J., Charpentier E., Haft D. H., Horvath P., Moineau S., Mojica F. J. M., Terns R. M., Terns M. P., et al. (2015) An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 13, 722–736 10.1038/nrmicro3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garneau J. E., Dupuis M-È., Villion M., Romero D. A., Barrangou R., Boyaval P., Fremaux C., Horvath P., Magadán A. H., and Moineau S. (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71 10.1038/nature09523 [DOI] [PubMed] [Google Scholar]
- 11. Zetsche B., Gootenberg J. S., Abudayyeh O. O., Slaymaker I. M., Makarova K. S., Essletzbichler P., Volz S. E., Joung J., van der Oost J., Regev A., Koonin E. V., and Zhang F. (2015) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 10.1016/j.cell.2015.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abudayyeh O. O., Gootenberg J. S., Konermann S., Joung J., Slaymaker I. M., Cox D. B. T., Shmakov S., Makarova K. S., Semenova E., Minakhin L., Severinov K., Regev A., Lander E. S., Koonin E. V., and Zhang F. (2016) C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573 10.1126/science.aaf5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Terns M. P. (2018) CRISPR-based technologies: Impact of RNA-targeting systems. Mol. Cell 72, 404–412 10.1016/j.molcel.2018.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pyenson N. C., and Marraffini L. A. (2017) Type III CRISPR-Cas systems: When DNA cleavage just isn't enough. Curr. Opin. Microbiol. 37, 150–154 10.1016/j.mib.2017.08.003 [DOI] [PubMed] [Google Scholar]
- 15. Tamulaitis G., Venclovas Č., Siksnys V. (2017) Type III CRISPR-Cas immunity: Major differences brushed aside. Trends Microbiol. 25, 49–61 10.1016/j.tim.2016.09.012 [DOI] [PubMed] [Google Scholar]
- 16. Hatoum-Aslan A., Maniv I., and Marraffini L. A. (2011) Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc. Natl. Acad. Sci. U.S.A. 108, 21218–21222 10.1073/pnas.1112832108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walker F. C., Chou-Zheng L., Dunkle J. A., and Hatoum-Aslan A. (2017) Molecular determinants for CRISPR RNA maturation in the Cas10–Csm complex and roles for non-Cas nucleases. Nucleic Acids Res. 45, 2112–2123 10.1093/nar/gkw891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deng L., Garrett R. A., Shah S. A., Peng X., and She Q. (2013) A novel interference mechanism by a type IIIB CRISPR-Cmr module in Sulfolobus. Mol. Microbiol. 87, 1088–1099 10.1111/mmi.12152 [DOI] [PubMed] [Google Scholar]
- 19. Goldberg G. W., Jiang W., Bikard D., and Marraffini L. A. (2014) Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature 514, 633–637 10.1038/nature13637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elmore J. R., Sheppard N. F., Ramia N., Deighan T., Li H., Terns R. M., and Terns M. P. (2016) Bipartite recognition of target RNAs activates DNA cleavage by the Type III-B CRISPR-Cas system. Genes Dev. 30, 447–459 10.1101/gad.272153.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Samai P., Pyenson N., Jiang W., Goldberg G. W., Hatoum-Aslan A., and Marraffini L. A. (2015) Co-transcriptional DNA and RNA cleavage during type III CRISPR-Cas immunity. Cell 161, 1164–1174 10.1016/j.cell.2015.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang J., Rouillon C., Kerou M., Reeks J., Brugger K., Graham S., Reimann J., Cannone G., Liu H., Albers S.-V., Naismith J. H., Spagnolo L., and White M. F. (2012) Structure and mechanism of the CMR complex for CRISPR-mediated antiviral immunity. Mol. Cell 45, 303–313 10.1016/j.molcel.2011.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Staals R. H. J., Agari Y., Maki-Yonekura S., Zhu Y., Taylor D. W., van Duijn E., Barendregt A., Vlot M., Koehorst J. J., Sakamoto K., Masuda A., Dohmae N., Schaap P. J., Doudna J. A., Heck A. J. R., Yonekura K., van der Oost J., and Shinkai A. (2013) Structure and activity of the RNA-targeting type III CRISPR-Cas complex of Thermus thermophilus. Mol. Cell 52, 135–145 10.1016/j.molcel.2013.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tamulaitis G., Kazlauskiene M., Manakova E., Venclovas Č., Nwokeoji A. O., Dickman M. J., Horvath P., and Siksnys V. (2014) Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol. Cell 56, 506–517 10.1016/j.molcel.2014.09.027 [DOI] [PubMed] [Google Scholar]
- 25. Estrella M. A., Kuo F.-T., and Bailey S. (2016) RNA-activated DNA cleavage by the Type III-B CRISPR-Cas effector complex. Genes Dev. 30, 460–470 10.1101/gad.273722.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kazlauskiene M., Tamulaitis G., Kostiuk G., Venclovas Č., and Siksnys V. (2016) Spatiotemporal control of type III-A CRISPR-Cas immunity: Coupling DNA degradation with the target RNA recognition. Mol. Cell 62, 295–306 10.1016/j.molcel.2016.03.024 [DOI] [PubMed] [Google Scholar]
- 27. Kazlauskiene M., Kostiuk G., Venclovas Č., Tamulaitis G., and Siksnys V. (2017) A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science 357, 605–609 10.1126/science.aao0100 [DOI] [PubMed] [Google Scholar]
- 28. Niewoehner O., Garcia-Doval C., Rostøl J. T., Berk C., Schwede F., Bigler L., Hall J., Marraffini L. A., and Jinek M. (2017) Type III CRISPR–Cas systems produce cyclic oligoadenylate second messengers. Nature 548, 543–548 10.1038/nature23467 [DOI] [PubMed] [Google Scholar]
- 29. Rouillon C., Athukoralage J. S., Graham S., Grüschow S., and White M. F. (2018) Control of cyclic oligoadenylate synthesis in a type III CRISPR system. eLife 7, e36734 10.7554/eLife.36734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Han W., Stella S., Zhang Y., Guo T., Sulek K., Peng-Lundgren L., Montoya G., and She Q. (2018) A type III-B Cmr effector complex catalyzes the synthesis of cyclic oligoadenylate second messengers by cooperative substrate binding. Nucleic Acids Res. 46, 10319–10330 10.1093/nar/gky844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mojica F. J. M., Díez-Villaseñor C., García-Martínez J., and Almendros C. (2009) Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155, 733–740 10.1099/mic.0.023960-0 [DOI] [PubMed] [Google Scholar]
- 32. Lillestøl R. K., Shah S. A., Brügger K., Redder P., Phan H., Christiansen J., and Garrett R. A. (2009) CRISPR families of the crenarchaeal genus Sulfolobus: Bidirectional transcription and dynamic properties. Mol. Microbiol. 72, 259–272 10.1111/j.1365-2958.2009.06641.x [DOI] [PubMed] [Google Scholar]
- 33. Hale C. R., Majumdar S., Elmore J., Pfister N., Compton M., Olson S., Resch A. M., Glover C. V. C. 3rd, Graveley B. R., Terns R. M., and Terns M. P. (2012) Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol. Cell 45, 292–302 10.1016/j.molcel.2011.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wei W., Zhang S., Fleming J., Chen Y., Li Z., Fan S., Liu Y., Wang W., Wang T., Liu Y., Ren B., Wang M., Jiao J., Chen Y., Zhou Y., et al. (2019) Mycobacterium tuberculosis type III-A CRISPR/Cas system crRNA and its maturation have atypical features. FASEB J. 33, 1496–1509 10.1096/fj.201800557RR [DOI] [PubMed] [Google Scholar]
- 35. Liu T. Y., Iavarone A. T., and Doudna J. A. (2017) RNA and DNA targeting by a reconstituted Thermus thermophilus type III-A CRISPR-Cas system. PLoS One 12, e0170552 10.1371/journal.pone.0170552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Park K.-H., An Y., Jung T.-Y., Baek I. Y., Noh H., Ahn W. C., Hebert H., Song J. J., Kim J. H., Oh B.-H., and Woo E. J. (2017) RNA activation-independent DNA targeting of the type III CRISPR-Cas system by a Csm complex. EMBO Rep. 18, 826–840 10.15252/embr.201643700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marraffini L. A., and Sontheimer E. J. (2010) Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463, 568–571 10.1038/nature08703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pyenson N. C., Gayvert K., Varble A., Elemento O., and Marraffini L. A. (2017) Broad targeting specificity during bacterial type III CRISPR-Cas immunity constrains viral escape. Cell Host Microbe 22, 343–353.e3 10.1016/j.chom.2017.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. You L., Ma J., Wang J., Artamonova D., Wang M., Liu L., Xiang H., Severinov K., Zhang X., and Wang Y. (2019) Structure studies of the CRISPR-Csm complex reveal mechanism of co-transcriptional interference. Cell 176, 239–253.e16 10.1016/j.cell.2018.10.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jia N., Mo C. Y., Wang C., Eng E. T., Marraffini L. A., and Patel D. J. (2019) Type III-A CRISPR-Cas Csm complexes: Assembly, periodic RNA cleavage, DNase activity regulation, and autoimmunity. Mol. Cell 73, 264–277.e5 10.1016/j.molcel.2018.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Staals R. H. J., Zhu Y., Taylor D. W., Kornfeld J. E., Sharma K., Barendregt A., Koehorst J. J., Vlot M., Neupane N., Varossieau K., Sakamoto K., Suzuki T., Dohmae N., Yokoyama S., Schaap P. J., et al. (2014) RNA targeting by the type III-A CRISPR-Cas Csm complex of Thermus thermophilus. Mol. Cell 56, 518–530 10.1016/j.molcel.2014.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang L., Mo C. Y., Wasserman M. R., Rostøl J. T., Marraffini L. A., and Liu S. (2018) Dynamics of Cas10 govern discrimination between self and non-self in type III CRISPR-Cas immunity. Mol. Cell 73, 278–290.e4 10.1016/j.molcel.2018.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maniv I., Jiang W., Bikard D., and Marraffini L. A. (2016) Impact of different target sequences on type III CRISPR-Cas immunity. J. Bacteriol. 198, 941–950 10.1128/JB.00897-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peng W., Feng M., Feng X., Liang Y. X., and She Q. (2015) An archaeal CRISPR type III-B system exhibiting distinctive RNA targeting features and mediating dual RNA and DNA interference. Nucleic Acids Res. 43, 406–417 10.1093/nar/gku1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang J., Graham S., Tello A., Liu H., and White M. F. (2016) Multiple nucleic acid cleavage modes in divergent type III CRISPR systems. Nucleic Acids Res. 44, 1789–1799 10.1093/nar/gkw020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Han W., Li Y., Deng L., Feng M., Peng W., Hallstrøm S., Zhang J., Peng N., Liang Y. X., White M. F., and She Q. (2017) A type III-B CRISPR-Cas effector complex mediating massive target DNA destruction. Nucleic Acids Res. 45, 1983–1993 10.1093/nar/gkw1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Benda C., Ebert J., Scheltema R. A., Schiller H. B., Baumgärtner M., Bonneau F., Mann M., and Conti E. (2014) Structural model of a CRISPR RNA-silencing complex reveals the RNA-target cleavage activity in Cmr4. Mol. Cell 56, 43–54 10.1016/j.molcel.2014.09.002 [DOI] [PubMed] [Google Scholar]
- 48. Ramia N. F., Spilman M., Tang L., Shao Y., Elmore J., Hale C., Cocozaki A., Bhattacharya N., Terns R. M., Terns M. P., Li H., and Stagg S. M. (2014) Essential structural and functional roles of the Cmr4 subunit in RNA cleavage by the Cmr CRISPR-Cas complex. Cell Rep. 9, 1610–1617 10.1016/j.celrep.2014.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhu X., and Ye K. (2015) Cmr4 is the slicer in the RNA-targeting Cmr CRISPR complex. Nucleic Acids Res. 43, 1257–1267 10.1093/nar/gku1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Taylor D. W., Zhu Y., Staals R. H. J., Kornfeld J. E., Shinkai A., van der Oost J., Nogales E., and Doudna J. A. (2015) Structures of the CRISPR-Cmr complex reveal mode of RNA target positioning. Science 348, 581–585 10.1126/science.aaa4535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mulepati S., Héroux A., and Bailey S. (2014) Crystal structure of a CRISPR RNA-guided surveillance complex bound to a ssDNA target. Science 345, 1479–1484 10.1126/science.1256996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Semenova E., Jore M. M., Datsenko K. A., Semenova A., Westra E. R., Wanner B., van der Oost J., Brouns S. J. J., and Severinov K. (2011) Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U.S.A. 108, 10098–10103 10.1073/pnas.1104144108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wiedenheft B., van Duijn E., Bultema J. B., Bultema J., Waghmare S. P., Waghmare S., Zhou K., Barendregt A., Westphal W., Heck A. J. R., Heck A., Boekema E. J., Boekema E., Dickman M. J., Dickman M., and Doudna J. A. (2011) RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc. Natl. Acad. Sci. U.S.A. 108, 10092–10097 10.1073/pnas.1102716108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., and Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Swarts D. C., van der Oost J., and Jinek M. (2017) Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol. Cell 66, 221–233.e4 10.1016/j.molcel.2017.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Meeske A. J., and Marraffini L. A. (2018) RNA guide complementarity prevents self-targeting in type VI CRISPR systems. Mol. Cell 71, 791–801.e3 10.1016/j.molcel.2018.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Peränen J., Rikkonen M., Hyvönen M., and Kääriäinen L. (1996) T7 vectors with a modified T7lacPromoter for expression of proteins in Escherichia coli. Anal. Biochem. 236, 371–373 10.1006/abio.1996.0187 [DOI] [PubMed] [Google Scholar]
- 58. Silas S., Lucas-Elio P., Jackson S. A., Aroca-Crevillén A., Hansen L. L., Fineran P. C., Fire A. Z., and Sánchez-Amat A. (2017) Type III CRISPR-Cas systems can provide redundancy to counteract viral escape from type I systems. eLife 6, e27601 10.7554/eLife.27601 ; Correction (2018) eLife7, e36853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guillerez J., Lopez P. J., Proux F., Launay H., and Dreyfus M. (2005) A mutation in T7 RNA polymerase that facilitates promoter clearance. Proc. Natl. Acad. Sci. 102, 5958–5963 10.1073/pnas.0407141102 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.