

Abstract
Cytochrome P450 (P450, CYP) enzymes are the major catalysts involved in the oxidation of steroids as well as many other compounds. Their versatility has been explained in part by flexibility of the proteins and complexity of the binding mechanisms. However, whether these proteins bind their substrates via induced fit or conformational selection is not understood. P450 17A1 has a major role in steroidogenesis, catalyzing the two-step oxidations of progesterone and pregnenolone to androstenedione and dehydroepiandrosterone, respectively, via 17α-hydroxy (OH) intermediates. We examined the interaction of P450 17A1 with its steroid substrates by analyzing progress curves (UV-visible spectroscopy), revealing that the rates of binding of any of these substrates decreased with increasing substrate concentration, a hallmark of conformational selection. Further, when the concentration of 17α-OH pregnenolone was held constant and the P450 concentration increased, the binding rate increased, and such opposite patterns are also diagnostic of conformational selection. Kinetic simulation modeling was also more consistent with conformational selection than with an induced-fit mechanism. Cytochrome b5 partially enhances P450 17A1 lyase activity by altering the P450 17A1 conformation but did not measurably alter the binding of 17α-OH pregnenolone or 17α-OH progesterone, as judged by the apparent Kd and binding kinetics. The P450 17A1 inhibitor abiraterone also bound to P450 17A1 in a multistep manner, and modeling indicated that the selective inhibition of the two P450 17A1 steps by the drug orteronel can be rationalized only by a multiple-conformation model. In conclusion, P450 17A1 binds its steroid substrates via conformational selection.
Keywords: cytochrome P450, steroidogenesis, enzyme kinetics, pre-steady-state kinetics, conformational change, androgen, conformational selection, CYP17A1, induced-fit binding, P450 17A1, pregnenolone, steroid
Introduction
Cytochrome P450 (P450)2 enzymes are the main catalysts involved in the oxidation of steroids, drugs, fat-soluble vitamins, chemical carcinogens, and many other chemicals (1, 2). Collectively, they account for >95% of the oxidations and reductions of all chemicals reported to date (3). The oxidations are accomplished by a complex catalytic cycle (4), which usually begins with binding of the substrate to the enzyme in its ferric oxidation state. Because of the catalytic promiscuity, there has been considerable interest in the features of these enzymes that allow for such diversity of substrates and reactions, in terms of fundamental as well as practical issues.
Considerable evidence exists for multiple conformations of P450 enzymes. The well-studied bacterial model P450cam (P450 101A1) has been shown to exist in both open and closed states, and the relevance of these in different steps in the catalytic cycle is a subject of current interest (5–9). Another bacterial P450, OleP, has been recently reported to show multiple conformations in the presence of a substrate analog (10). Mammalian P450s have also been found to exist in open, closed, and “mixed” conformational states, at least in crystal structures (11–17). Further, P450 3A4 has been found to exist in different conformational states, depending upon the particular drug or other ligand bound to it (16–20).
The kinetics of binding of substrates and other ligands to P450s has also been studied, beginning with bacterial P450cam (21). Although substrate binding to some mammalian P450s can apparently be described by a simple two-state model (E + S ⇄ ES) (22, 23), in other cases, the binding process is quite complex and clearly involves multiple steps (e.g. P450s 1A2 (24), 3A4 (25–27), and 17A1 (28)). Part of the ambiguity may be related to multiple occupancy with some P450s (e.g. 1A2 and 3A4 (24–26)). However, multiple occupancy is probably not causal for the complex kinetics in the case of binding of bromocriptine to P450 3A4 (25, 27).
Complexity in the binding of substrates to enzymes is not unique to P450s and is widespread among enzymes (29, 30). The matter is at the heart of considerations about catalytic selectivity of enzymes. The classic “lock-and-key” concept (31) is considered far too simple and has been largely replaced by the concepts of induced fit and conformational selection, which have their roots in concepts developed >50 years ago (32–34) (Fig. 1). As discussed elsewhere, these two concepts are the extremes of a continuum of possible events (35, 36), and induced fit has been considered a special case of conformational selection (37). The case has been made that conformational selection is far more common than the induced-fit mechanism (38), but strong evidence for induced fit in enzymes has been reported (39, 40).
Figure 1.
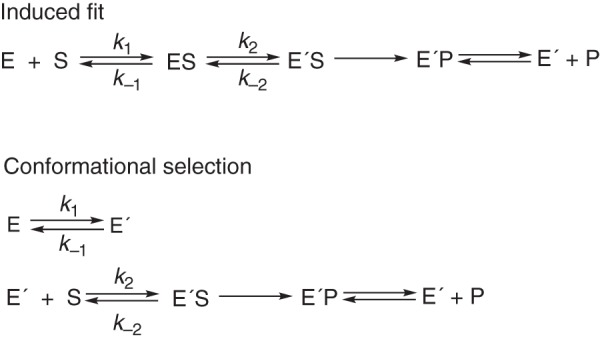
Basic induced-fit and conformational selection hypotheses.
Static structural approaches cannot discern between induced-fit and conformational selection models, in that these are the thermodynamic methods, but kinetic properties are the issue. The E′S complex (Fig. 2) can be generated from E by either of two routes, which have the same overall free energy change. In some kinetic studies with P450s (and many other enzymes), the complexity of substrate binding has probably been oversimplified, including our own (41, 42). However, there is an inherent danger in overdeveloping kinetic schemes in that the addition of indefensible steps, often without knowledge of extinction coefficients, is not justified (43). In this regard, we developed kinetic schemes for P450s 3A4 (25, 26) and 17A1 (28) that involved both conformational selection and induced-fit components for each. Although these could fit the spectral data reasonably well, we could not be certain which spectral events were being observed or what the relevant extinction coefficients are. Accordingly, we have now tried to discern whether induced fit or conformational selection (Figs. 1 and 2 and Scheme S1) is dominant in the binding of substrates to these enzymes, and this report is focused on several aspects of human P450 17A1.
Figure 2.
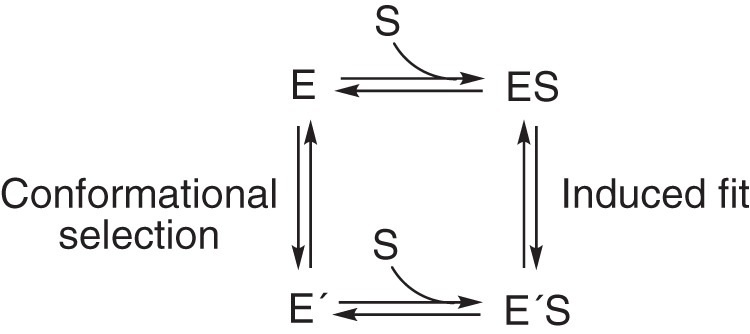
Thermodynamic box diagram for basic induced-fit and conformational selection models.
Although the literature on the nature of substrate binding may seem complex, there are three primary and relatively straightforward approaches to discerning between dominant roles of induced-fit versus conformational selection models, beginning with kinetic traces of the progress of substrate binding at multiple reagent concentrations. Complex behavior is generally characterized by the need to use at least two exponentials to fit the traces (25), although even single-exponential fits may be complex. (i) If plots show that the rates of binding decrease as a function of ligand concentration, then conformational selection is clearly indicated (38). However, increases in binding rates (versus ligand concentration) are not necessarily indicative of an induced-fit mechanism (44). (ii) When plots of rates of enzyme-ligand binding are compared by (a) using increasing concentrations of substrate and (b) using increasing concentrations of enzymes, an induced-fit mechanism will give the same plot, but these will differ for a conformational selection model (44). (iii) Simulation approaches, employing realistic rates of diffusion-limited binding (45), will yield credible fits for an appropriate mechanism (46). Again, a minimal kinetic mechanism is the goal.
We utilized these approaches to study aspects of binding of substrates and other ligands (Fig. 3) to human P450 17A1. P450 17A1 hydroxylates both progesterone and pregnenolone at the 17α position and also cleaves two carbons from the D ring of the steroids in a so-called “lyase” reaction. We conclude that the dominant phenomenon in binding of all steroid substrates to this P450 is conformational selection.
Figure 3.
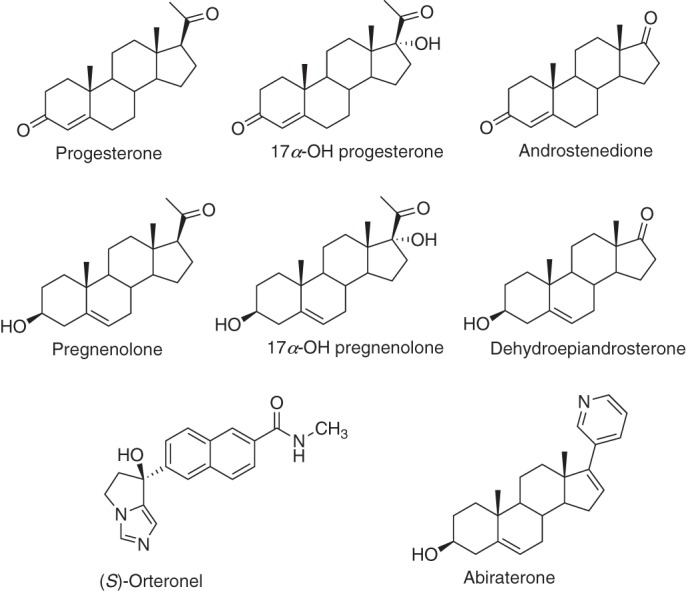
Structure of steroids and other molecules used with P450 17A1. Note the stereochemistry of S-orteronel. The stereochemistry of the structures shown in Fig. 8 of Ref. (47) was inadvertently reversed.
Results
P450 17A1 and progesterone binding
Previous work from this laboratory dealt with human P450 17A1 and the binding of several steroids (28). The data for binding pregnenolone, 17α-OH pregnenolone, and dehydroepiandrosterone (DHEA) were best fit with a kinetic model that had both conformational selection and induced-fit mechanisms and two spectrally equivalent complexes. What was not addressed was whether a conformational selection or an induced-fit model was dominant in explaining the results. Also, whereas the work treated the Δ5 steroids (i.e. pregnenolone etc.) in detail, analysis of binding of the Δ4 steroids (progesterone and its oxidation products) was not considered.
We re-evaluated data obtained in that study, in the context of the general modeling plan. The raw traces for binding of progesterone could be fit to double-exponential plots (Fig. 4A), indicative of a multistep binding process. The amplitudes of both steps could be fit to hyperbolae (results not shown). Plots of either single-exponential rates (Fig. 4B) or the two-exponential rates versus progesterone concentration showed decreases in (both the faster and slower) rates with increasing ligand concentration (Fig. 4, C and D), a result only attributable to a conformational selection model (38, 44).
Figure 4.

Binding of progesterone to P450 17A1. Data are from Ref. 28. The final P450 concentration was 1 μm; the final progesterone concentrations were 0.2, 0.5, 1.0, 1.5, 2, 4, 10, and 20 μm; and the time period was 7 s. A, double-exponential fits of the data curves. B, plot of the single-exponential rates from the data in A versus progesterone concentration. C, plot of the faster phase biexponential rates from A versus progesterone concentration. D, plot of the slower phase biexponential rates from A versus progesterone concentration. E–H, modeling of binding of progesterone to P450 17A1 (28). The colors of the fitted lines match the data sets. E, induced-fit model 1, developed to balance initial rates at higher substrate concentrations: k1 = 3.0 × 106 m−1 s−1, k−1 = 100 s−1, k2 = 10 s−1, k−2 = 0.85 s−1 (ϵ390–418 55 mm−1 cm−1). F, induced-fit model 2, developed to balance amplitudes at lower progesterone concentrations: k1 = 100 × 106 m−1 s−1, k−1 = 50 s−1, k2 = 1.4 s−1, k−2 = 0.38 s−1 (ϵ390–418 55 mm−1 cm−1). G, conformational selection model: k1 = 0.91 s−1, k−1 = 1.2 s−1, k2 = 1.3 × 106 m−1 s−1, k−2 = 1.6 s−1 (ϵ390–418 = 45 mm−1 cm−1). H, combined conformational selection/induced-fit model: k1 = 0.80 s−1, k−1 = 1.1 s−1, k2 = 12 × 106 m−1 s−1, k−2 = 34 s−1, k3 = 24 s−1, k−3 = 2.7 s−1 (ϵ390–418 = 50 mm−1 cm−1).
Attempts to use pure induced-fit models for progesterone binding to P450 17A1 are shown in Fig. 4, E and F, utilizing two different sets of rate constants. In Fig. 4E, the rate constants are too slow to match the initial binding traces at any progesterone concentration. With the rate constants used in Fig. 4F, fits were best at low substrate concentrations but poorer at higher ones.
A pure conformational selection model was used in Fig. 4G, which had a credible kon rate of 1.3 × 106 m−1 s−1 (45) and reasonable fits at all progesterone concentrations. Adding an induced-fit step to the conformational selection model (and adjustment of kon to 12 × 106 m−1 s−1) led to an improved fit at some progesterone concentrations but not others. Overall, the fit with this “combined” model (Fig. 4H) led to a slightly improved global fit over the pure conformation selection model (Fig. 4G).
The derivations of Vogt and Di Cera (38) provide an approach to estimating k1 and k−1 in the conformational selection model. The hyperbolic relationship for the single-exponential rates (Fig. 4C) yields a y-intercept of ∼2.5 s−1 (discarding the experimental data point for the 0.2 μm concentration of progesterone) and an asymptote of ∼0.75 s−1. Using the published approach, k1 (kr in Ref. 38) = ∼0.75, and k1 + k−1 (k−r) = ∼2.5, so that k−1 = ∼1.75 and k1/k−1 = ∼0.75/1.75 = ∼0.4, which compares favorably with the value of 0.9/1.2 = 0.75 used independently in the modeling (Fig. 4G). As pointed out below, these rates could be used for the binding of other steroids (Figs. S1–S5), in that the rates of equilibration of the conformations should be independent of the ligand. Further, the apparent Kd for ligand binding can be predicted from a plot such as Fig. 4B (38) and gives a value of ∼1.5 μm, consistent with that estimated previously (0.47 ± 0.04 μm, corrected using a quadratic equation (28)) and the values of 1.3 ± 0.3 and 3.0 ± 1.0 μm estimated from the fast- and slow-phase amplitudes in Fig. 4, C and D (uncorrected for bound ligand). Similar estimates of ratios of k1/k−1 (i.e. somewhat less than unity) could be estimated from the binding plots of the steroids in Figs. S1–S5, showing consistency in the model.
Binding of other steroids to P450 17A1
If the conformational selection model (Figs. 1, 2, and 4) is more relevant than an induced-fit model for P450 17A1 binding of progesterone, then the rates of conformational change should be independent of the substrate bound, with only kon and koff rate constants for ligand binding changing. This test was applied to the Δ4-steroid substrate 17α-OH progesterone, the product androstenedione, and the Δ5 steroids pregnenolone, 17α-OH pregnenolone, and DHEA, the binding of which had been considered previously in the combined kinetic model (28). Only the plots of single-exponential rates versus steroid concentrations are shown. The results are shown in Figs. S1–S5 for the best fits that utilize the induced-fit and conformational selection models, retaining the same rate constants for the steps of the interconversion of the two forms of unbound P450 17A1 (Fig. 4G). (Some of the fits at the lower concentrations were considered less reliable due to sensitivity and the fact that the steroid was not in excess (e.g. with the 17α-OH steroids).)
As in our previous report with 17α-OH progesterone and androstenedione (28), the fitting of all steroid binding traces required biexponential fits (Figs. S1–S5). With 17α-OH progesterone, the rate increased and then decreased when the substrate was in excess (Fig. S1B). The fit to an induced-fit plot (Fig. S1C) was inferior to that obtained with a conformational selection model (Fig. S1D). The plots for pregnenolone binding to P450 17A1 (Fig. S2) were similar to those for 17α-OH progesterone, again decreasing when the substrate was in excess (Fig. S2B). A better overall fit was also obtained with a conformational selection model (Fig. S2D), although observed rates at the three highest concentrations were not as fast as predicted. Similar patterns were seen with 17α-OH pregnenolone (Fig. S3) as with 17α-OH progesterone (Fig. S1).
The binding rate plots of the products androstenedione and DHEA were similar (Figs. S4 and S5), with decreasing rates with increasing steroid concentration in both cases (Figs. S4B and S5B). Better fits were obtained with conformational selection models (panel D of Figs. S4 and S5) than induced-fit models (panel C of Figs. S4 and S5), although the fit was poorer at the higher substrate concentrations.
Comparison of plots of steroid binding as a function of varying concentrations of 17α-OH pregnenolone versus P450 17A1
One of the hallmarks of a conformational selection model is decreasing rates of binding as a formation of ligand concentration (38), which we established for P450 17A1 and several steroid substrates (and products) (Fig. 4 and Figs. S1–S5). Another hallmark is differences in the dependence of rates of binding on varying both ligand and enzyme, each varied in the presence of a fixed concentration of the other component (44). We applied this approach in the case of P450 17A1 and 17α-OH pregnenolone (Fig. 5, A and B). The negative and positive slopes for the two cases clearly show differing patterns (Fig. 5, C and D), consistent only with the dominance of a conformational selection model (44).
Figure 5.

Comparisons of changes in rates of P450 17A1–17α-OH pregnenolone binding by varying the individual components. A, ΔA390 − A418 traces observed by mixing 2 μm P450 17A1 with varying concentrations of 17α-OH pregnenolone (0.2, 0.5, 1.0, 1.5, 2, 4, 10, and 20 μm; data the same as presented in Fig. 6A). B, ΔA390 − A418 traces obtained after mixing 2 μm 17α-OH pregnenolone with varying concentrations of P450 17A1 (2, 4, 6, 8, 11, and 15 μm), with amplitude increasing with concentration. The data are fit with single-exponential rates. C, plots of the fast (■) and slow (○) rates from a biexponential fit of the data in A. D, plot of the rates of the single-exponential fits of the data of B. The calculated errors (S.D.) are within the sizes of the points in C and D.
Influence of b5 on steroid binding to P450 17A1
Multiple explanations for the selective stimulatory effect of b5 on P450 17A1 lyase activity (relative to the 17α-hydroxylation step) have been proposed (48), the main two being direct electron transfer (49, 50) and facilitation of a conformational change to favor the lyase reaction (51, 52). We repeated the experiments done with apo-b5 (51) in our system and found a partial stimulation (∼75%) of 17α-OH progesterone lyase activity, consistent in part with both hypotheses (Fig. 6A). We had previously shown that the presence of b5 did not change the apparent koff rates of any of the substrates or products of P450 17A1 (28). However, if a conformational selection model is in order for P450 17A1 (Figs. 4 and 5), then the possibility can be raised that b5 might influence the steps involved in substrate binding (e.g. b5 could alter the conformation of P450 17A1 to one amenable to productive steroid binding).
Figure 6.

Interactions of P450 and b5. A, stimulation of P450 17A1–catalyzed 17α-OH progesterone conversion to androstenedione by b5 and apo-b5. The rate of androstenedione formation in the absence of b5 was ≤0.08 min−1 (limit of detection under these conditions). Results are represented as means ± S.D. (error bars) for experiments done in triplicate. B, equilibrium binding of 17α-OH progesterone to P450 17A1 in the absence (○) and presence (●) of an equimolar concentration of b5. Calculated Kd values for 17α-OH progesterone binding were 0.52 ± 0.06 μm in the absence and 0.67 ± 0.04 μm in the presence of b5 (S.D. values estimated from fitting to hyperbolic plots). C and D, traces of absorbance changes for binding of 17α-OH pregnenolone in the absence (C, same data set as in Fig. 5C, obtained on the same day) and presence (D) of an equimolar concentration of b5 (2 μm). Concentrations of 17α-OH pregnenolone are indicated, with the colors matching the concentrations to the individual traces. E and F, plots of rates of 17α-OH pregnenolone binding to P450 17A1 in the absence (E) and presence (F) of an equimolar concentration of b5 (data from C and D). The fast (■) and slow (○) rates as plotted versus the final concentration of 17α-OH pregnenolone.
The binding kinetics for 17α-OH pregnenolone and 17α-OH progesterone to P450 17A1 were compared in the absence and presence of an equimolar concentration of b5 (with P450 17A1) (Fig. 6A). No difference was seen in the Kd values for 17α-OH progesterone or 17α-OH pregnenolone binding due to the presence of b5 (Fig. 6B, 17α-OH pregnenolone data not shown). The binding kinetics were also examined in the absence and presence of b5 (Fig. 6, C and D). (Note that data from Fig. 5A were also used in Fig. 6C, an experiment done during the same day as the one in Fig. 6D.) Although there was a slight change in the amplitudes, the patterns were very similar in these plots and in further KinTek Explorer analyses (not shown). A conformational effect of b5 on P450 17A1 is still postulated but was not observable in any of these kinetic (Fig. 6, C and D) or equilibrium (Fig. 6B) experiments.
Binding of the Nile Red to P450 17A1
Nile Red is a dye, used extensively in histochemical staining, which is also a substrate for P450 3A4 (Fig. 7) and has been employed in biophysical studies with P450 3A4 by the Atkins laboratory (53, 55). In agreement with the report of Faletrov et al. (54), we found that P450 17A1 had high affinity for Nile Red (Fig. 8, Kd(app) = 1.0 ± 0.2 μm) and was also a substrate for slow N-deethylation by the enzyme, with a kcat of 0.14 ± 0.01 min−1 and Km of 12 ± 3 μm (Fig. S6). The fluorescence of Nile Red increased upon binding to P450 17A1 (Fig. S7A), presumably due to its movement from a hydrophilic environment to a more hydrophobic one. Nile Red fluorescence was more intense in the presence of P450 17A1 if the active site was occupied by abiraterone, a strong inhibitor of P450 17A1 (Fig. S7, B and C). This result implies that Nile Red can interact with P450 17A1 inside or outside its active site, with varying fluorescence intensities.
Figure 7.

N-Deethylation of Nile Red by P450s (17A1 and 3A4) (53, 54).
Figure 8.

Binding of the dye and substrate Nile Red to P450 17A1. A, equilibrium titration of P450 17A1 (1 μm) with varying concentrations of Nile Red, with spectra recorded at each. B, quadratic fit to the changes in absorbance in the titration of A. The colors match the concentrations to the individual traces.
The kinetics of binding of Nile Red to P450 17A1 were investigated using both fluorescence and absorbance changes (Fig. 9 and Fig. S8). A rapid increase in Nile Red fluorescence (excitation wavelength 550 nm, emission >590 nm) occurred upon mixing (Fig. 9A). The rate of the increase was >40 s−1 at a concentration of 4 μm (Fig. S8), suggesting a binding rate constant of >107 m−1 s−1, and was diffusion-limited. The fluorescence decreased with a rate of ∼10 s−1 irrespective of Nile Red concentration (Fig. 9A and Fig. S8) and remained at a higher level than the starting value.
Figure 9.
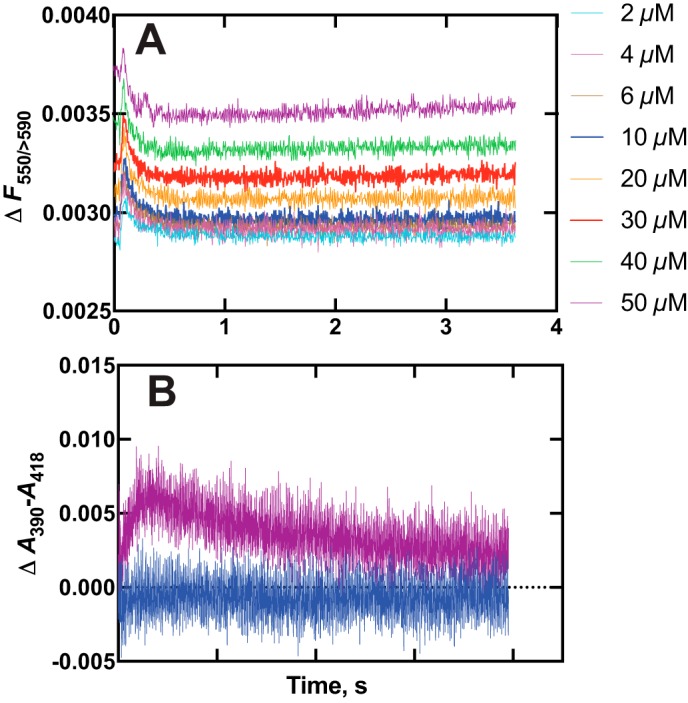
Kinetics of binding of the dye and substrate Nile Red to P450 17A1. A, varying concentrations of Nile Red were mixed with 2 μm P450 17A1, and traces of fluorescence were recorded (Fexcitation wavelength 550 nm, emission >590 nm measured). The colors match the concentrations to the individual traces. B, absorbance changes were recorded following mixing under the conditions used in A, and ΔA390 − A418 was plotted. Only the traces obtained upon mixing with 2 and 50 μm Nile Red are shown for clarity.
The absorbance spectra revealed a transient increase in ΔA390 − A418 (type I difference spectrum) that was more pronounced with increasing concentrations of Nile Red (substrate concentrations of 2 and 50 μm Nile Red shown; Fig. 9B). The kinetic course of the increase in ΔA390 − A418 was such that it began during the decrease in Nile Red fluorescence shown in Fig. 8C, with rates of ∼10 s−1 (Fig. S8). The ΔA390 − A418 then decreased at a rate of ∼5 s−1. The final value of ΔA390 − A418 increased with Nile Red concentration (Fig. 8B). Overall, the binding of Nile Red to P450 17A1 could be described with the scheme shown in Fig. 10.
Figure 10.

Course of events in binding of the dye and substrate Nile Red to P450 17A1.
Kinetic modeling of selective inhibition of the two reaction steps of P450 17A1
P450 17A1 is a target for discovery and development of drugs to treat prostate cancer, and several candidates have been advanced (56–58). One goal has been to achieve selective inhibition of the second step, the steroid 17,20-lyase reaction, to lower androgen levels but to spare 17α-OH progesterone for use in the biosynthesis of mineralocorticoids and glucocorticoids (59). Selective inhibition of the second step has been reported for some drug candidates, including both enantiomers (R- and S-) of orteronel (TAK-700) (28, 56, 59, 60). However, it should be emphasized that the degree of enantioselective inhibition of the activities appears to vary considerably, depending upon the particular assay system used and the experimental conditions (28, 56, 59, 60).
Selective inhibition of two distinct catalytic activities of a single enzyme raises mechanistic questions, in that structural studies with P450 17A1 to date have shown a single binding site shared by all substrates and ligands (47, 61–63), with only limited evidence for an allosteric mechanism involving a second binding site for a substrate or other small molecule (56). This question led to modeling of the kinetics to examine the question of whether an enzyme with a single active site could show multiple IC50 values for two reactions in the absence of conformational changes.
The system is shown in the scheme in Fig. 11, with the possibility of two forms of P450 17A1 (E and E′) at various steps. Most of the rate constants for individual reaction steps are derived from kinetic studies reported previously (28). As noted above, the kon rates for the steroids are complex, and we utilized the apparent koff/Kd values (28) for calculating a simplified kon rate for use in the modeling (Table 1). The kon rates of R- and S-orteronel binding to P450 17A1 were estimated from fitting of the data for the binding of P450 17A1 and orteronel (28). Values used for kon were 6.4 and 3.0 × 106 m−1 s−1 for the R- and S-enantiomers, respectively. The S-enantiomer has been found to be somewhat more inhibitory than the R-enantiomer (28, 56), and we restricted the analysis to the S-enantiomer. The reported estimate of Kd of 40 nm for the S-enantiomer is probably too high, in that our kinetic fitting indicated that the koff rate constant should be <0.01 s−1 (i.e. Kd = koff/kon ≅ 3 nm).
Figure 11.

Kinetic scheme for inhibition of the two P450 17A1 reactions (17α-hydroxylation and 17α,20-lyase (cleavage) by orteronel).
Table 1.
Models used to fit selective inhibition of the two reactions of P450 17A1 by S-orteronel
S, pregnenolone; P, 17α-OH pregnenolone; Q, DHEA; E, P450 17A1; E′, conformational variant of P450 17A1; I, S-orteronel. See Ref. 28 and Figs. S2 and S3 for input of many of the rate constants.
| Basic model |
Model with multiple conformations |
||||||
|---|---|---|---|---|---|---|---|
| Step | Reaction | k+ | k− | Step | Reaction | k+ | k− |
| m−1 s−1 | s−1 | m−1 s−1 | s−1 | ||||
| 1 | E + S ⇄ ES | 0.62 × 106 | 0.29 | 1 | E + S ⇄ ES | 0.62 × 106 | 0.29 |
| 2 | ES → EP | 0.43a | 2 | ES → EP | 0.43a | ||
| 3 | E + P ⇄ EP | 0.79 × 106 | 0.41 | 3 | E + P ⇄ EP | 0.79 × 106 | 0.41 |
| 6 | EP → EQ | 0.077a | 4 | EP ⇄ E′P | 1.0 | 1.0 | |
| 8 | E + Q ⇄ EQ | 0.45 × 106 | 0.77 | 5 | E′ + P ⇄ E′P | 0.79 | 0.42 |
| 10 | E + I⇄ EI | 3.0 × 106 | 0.01 | 6 | EP → EQ | 0.077a | |
| 7 | E′P → E′Q | 0.077a | |||||
| 8 | E + Q ⇄ EQ | 0.45 × 106 | 0.77 | ||||
| 9 | E′ + Q ⇄ E′Q | 0.45 × 106 | 0.77 | ||||
| 10 | E + I ⇄ EI | 3.0 × 106 | 0.01 | ||||
| 11 | E′ + I ⇄ E′I | 3.0 × 106 | 0.001 | ||||
a First-order rate constant for irreversible reaction, in s−1.
Reaction modeling of the steps in Fig. 11 with the basic model of Table 1 yielded identical IC50 values (43 nm) for both P450 17A1 reactions (Fig. 12A). Many of the IC50 assays have involved the lyase reaction being measured separately, so we also initiated the reaction model with 1 μm 17α-OH pregnenolone instead of (1 μm) pregnenolone. Interestingly, the same IC50 value was obtained under these conditions.
Figure 12.

Kinetic modeling of orteronel inhibition of individual reactions of P450 17A1. The models described in Fig. 11 and Table 1 were used to predict the rates of conversion of pregnenolone to 17α-OH pregnenolone (♦) and 17α-OH pregnenolone to DHEA (○). A, basic model without conformational change: IC50 = 44 nm for both reactions. B, model with conformational change occurring after the formation of 17α-OH pregnenolone. The fit shown here was obtained with the values for steps 4, 5, 7, 9, and 11 shown in Table 1. The IC50 values for 17α-OH pregnenolone and DHEA formation were 48 and 22 nm, respectively.
The model was expanded to include a second conformation of P450 17A1, which only formed upon the production of 17α-OH pregnenolone (Fig. 11), with the addition of several steps in the model (Table 1). Some of the steps with the conformationally distinct P450 (i.e. E′ in Fig. 11 and Table 1) were used with the same rate constants (as with E) (e.g. steps 5 (equal to step 3) and 9 (equal to step 8). To achieve separation of the IC50 values, step 4 was altered in favor of the E′S complex, the rate of formation of DHEA by E′ was increased (step 7), the binding of DHEA (Q) by E′ (step 9) was attenuated ∼20-fold, and the affinity of E′ for the inhibitor (I) (step 11) was increased 10-fold. Collectively, the introduction of these extra steps yielded modeled IC50 values of 48 nm for the 17α-hydroxylation step and 22 nm for the lyase reaction (forming DHEA, Fig. 12B). This ≥2-fold difference is similar to that found experimentally (28, 56). As before, separating the model to initiate the reaction with 1 μm 17-OH pregnenolone instead of (1 μm) pregnenolone did not lower the IC50 (34 nm).
Some other points are worthy of note at this point. Reaction linearity is not perfect in the system beginning with pregnenolone, in that 17α-OH pregnenolone is not an end product (Fig. 3). However, analysis of IC50 values at different reaction time points led to different IC50 values (perhaps surprisingly) but did not have a major effect on the ratio of the IC50 values for the two steps. Finally, achieving a further discrimination of the IC50 values for the two steps may require major changes to the model, which is already complex and relies on major assumptions.
Binding of the inhibitor abiraterone to P450 17A1
Abiraterone is the only P450 17A1 inhibitor presently in clinical use and is prescribed for the treatment of castration-insensitive prostate cancer (64). This is a steroidal inhibitor (Fig. 3); its mechanism of clinical activity is complicated by its inhibition of the androgen receptor and by the activities of metabolites (65, 66).
The C17-pyridinium moiety is responsible for the observed “type II” binding spectrum seen upon binding of abiraterone to P450 17A1, due to the interaction of the pyridine nitrogen with the heme iron atom. This change was monitored at varying concentrations (Fig. 13). The plots could be fit with biexponential values, and the fast-phase rates decreased with increasing abiraterone concentration (Fig. 13B). The slow-phase rates also decreased with increasing abiraterone concentration (Fig. 13C) (the rates estimated at the two lowest abiraterone concentrations were not considered very reliable). The data could not be fit to an induced-fit model (Fig. 13D).
Although the KinTek Explorer fitting of the data to the mid-range concentrations of ligand is less than ideal (Fig. 13E), overall, these results indicate that the binding of abiraterone, like other steroids (Fig. 4 and Figs. S1–S5), is more consistent with a conformational selection model than an induced-fit model.
Figure 13.

Binding of the inhibitor abiraterone to P450 17A1. P450 17A1 (2 μm), in 100 mm potassium phosphate buffer (pH 7.4), was mixed with varying concentrations (A) of abiraterone dissolved in the same buffer (2, 4, 8, 12, 16, and 40 μm). A, traces of ΔA430 − A410 versus time. B, rates of the fast phase of binding (A) plotted versus final substrate concentration. C, rates of the slow phase of binding (A) plotted versus final substrate concentration. D, fitting of data (A) to an induced-fit model (Model 1) with k1 = 1.2 × 106 m−1 s−1, k−1 = 5.5 s−1, k2 = 0.16 s−1, and k−2 = 0.60 s−1 (ϵ430–410 = 55 mm−1 cm−1). E, fitting of data (A) to a conformational selection model (Model 2) with k1 = 0.61 s−1, k−1 = 0.80 s−1, k2 = 0.61 × 106 m−1 s−1, and k−2 = 5.6 s−1 (ϵ430–410 = 29 mm−1 cm−1).
Discussion
The issue of the kinetic mode of steroid binding to the important enzyme P450 17A1 was addressed, particularly with regard to nature of the multistep binding indicated by multiexponential kinetics (28). We conclude that the binding of all steroids examined can be rationalized mainly in the context of a conformational selection model (Figs. 1 and 2). Our conclusion is based on three lines of experimental evidence. (i) plots of rates of binding decrease with increasing steroid concentration (Figs. 4–6 and 13 and Figs. S1–S5), in line with the principles outlined by Vogt and Di Cera (38). (ii) Rate profiles obtained by fixing the steroid concentration and varying the P450 concentration are opposite those obtained with a fixed P450 concentration and varying the steroid (Fig. 5). This is also behavior indicative of a conformational change model, as described by Gianni et al. (44). (iii) Comparisons of fits to conformational selection and induced-fit models in KinTek Explorer software provided consistently better fits than for induced-fit models (Figs. 4 and 13 and Figs. S1–S5).
The complexity of binding of Δ5 steroids to P450 17A1 was reported in a previous study from this laboratory (28). The model used to achieve fits utilized a mixture of conformational selection and induced fit, with multiple species assigned to be responsible for the observed chromophoric change (with undetermined extinction coefficients). However, the model, although fitting the Δ5 steroids reasonably well (28), is probably too complex to prove. In the present work, simpler mechanisms were desired (i.e. “minimal” mechanisms), even if the fits were less than ideal. In the binding of progesterone (Fig. 4), the expansion of the basic conformational model (Fig. 4, from G to H) only yielded a slight improvement. Thus, we conclude that a conformational selection model is the most appropriate for binding of steroids to P450 17A1.
NMR evidence for the existence of multiple forms of P450 17A1 in the presence of substrates has been presented by the Scott laboratory (67), and different conformational states were seen in the X-ray crystal structures of P450 17A1 bound to the R- and S-enantiomers of orteronel (56). The multiple structures found in the presence of a ligand indicate that multiple conformations can exist, but these provide no guidance on the path to them (Fig. 2). The existence of multiple conformations in the presence of ligand (56, 67) is consistent with a conformational selection model, although it does not in itself prove that multiple conformations interconvert on a reasonable time scale in the absence of ligand; nor does it demonstrate that both bind substrate (Fig. 2). No ligand-free structures of P450 17A1 have been published, and to our knowledge, no reports of conformational heterogeneity in ligand-free P450 17A1 have been reported.
We cannot rule out a role for some induced-fit contribution with our models, which we conclude are driven largely by conformational selection. The fits are less than ideal in some cases, in that a major goal of our work was to minimalize the complexity of the binding mechanism, and we have not subjected the fitting to FitSpace analysis in KinTek Explorer (68).
The role of b5 in the P450 17A1 reactions has been a source of debate. An electron transfer role (the “second electron” source) was first postulated based on studies with other P450s (69–71). The second P450 17A1 reaction (i.e. the lyase activity) is stimulated more by b5 than the 17α-hydroxylation (72), a result we are in agreement with (28). Auchus et al. (51) reported that apo-b5 could stimulate the lyase reaction, and Lee-Robichaud et al. (52) reported a similar enhancement with manganese-substituted b5. The extent to which this stimulation occurs is, however, not clear in either of these studies. The Sligar laboratory (49) reported that manganese-substituted b5 was incapable of stimulating the lyase reaction and that b5 was capable of being oxidized by a ferrous O2 complex of P450 17A1 (50). The former study has a major caveat in the very low activity reported with intact b5 (49). The latter result (50) may possibly be relevant to the situation in the steady-state system. We carried out studies with apo-b5 and found partial, although not complete, stimulation of P450 17A1 (Fig. 6A), providing some support for both previous results (i.e. partial roles for both electron transfer and conformational effects). Although we are of the opinion that b5-induced conformational changes are involved in b5 stimulation, we did not find evidence for stimulation of binding of 17α-OH pregnenolone or 17α-OH progesterone in either thermodynamic (Fig. 6B) or kinetic (Fig. 6, C–F) experiments.
Estrada et al. (67) reported that b5 altered the conformation of a P450 17A1–abiraterone complex (as judged by 15N NMR changes) and that b5 was less easily displaced from a P450 17A1–b5 complex by the FMN domain of NADPH-P450 reductase in the presence of pregnenolone or 17α-OH pregnenolone (73). Recently, Ershov et al. (74) reported that the Kd of the P450 17A1–b5 complex was lowered 5–10-fold by the presence of the substrates progesterone, 17α-OH progesterone, and pregnenolone, although the surface plasmon resonance measurements are subject to their usual caveats about surface artifacts. The changes were attributed to much faster kon rates in the presence of the steroids. Although we have not attempted such measurements in solution, in principle, tighter binding of b5 and P450 17A1 in the presence of a steroid should be reflected in tighter binding of the steroid to P450 17A1 in the presence of b5 in the usual thermodynamic box analysis (Fig. 2). However, this result was not seen (Fig. 6B), and further analysis requires determination of a P450 17A1–b5 Kd by an independent method.
A microbial P450 involved in erythromycin A biosynthesis, Saccharopolyspora erythrea CYP113A1, is monomeric and has been concluded to bind its substrate erythromycin D, utilizing a conformational selection mode (75). One question that can be raised about our own work with human P450 17A1 is whether shifts in oligomeric composition could be a part of the conformational equilibrium, a point raised in the review of this manuscript. Most mammalian P450s form oligomers in solution, with or without NADPH-P450 reductase (e.g. P450s 2B1 (rat) and 2B4 (rabbit) have been reported to have “average” molecular weights of 500,000 and 300,000, respectively (76, 77)). When we examined our P450 17A1 by size-exclusion chromatography (GE Superdex 200 column), about one-half of the enzyme eluted near standard ovalbumin (45 kDa, near the size of the P450 monomer), and the remainder eluted in two earlier regions. We conclude that P450 17A1 is not all monomeric under the conditions in which we are using it, and we cannot rule out a role of oligomerization in the conformational selection process.
Others had reported that the dye Nile Red is a substrate for P450 17A1 (N-deethylation) (Fig. 7) (54). We also observed this reaction (Fig. S6A), which showed only marginal stimulation by b5 (results not presented). The binding of Nile Red could be monitored by both absorbance and fluorescence measurements (Figs. 8 and 9 and Figs. S7 and S8). The kinetic scheme is interpreted in the context of Fig. 10. The first event is the diffusion-limited binding of Nile Red somewhere on P450 17A1, where it shows increased fluorescence due to being in a more lipophilic environment. The molecule then moves to a position within the active site in which a Type I spectral shift (i.e. partial conversion of low- to high-spin iron) is observed (Figs. 8A and 9 and Fig. S6). The process is then completed by the movement of the dye to a different position, as judged by the decrease in the high-spin iron state (Fig. 9B), leading to a final position in which the high-spin content is still present, as judged by the steady-state titration of Nile Red binding to P450 17A1 (Fig. 8A). This pathway is depicted in Fig. 10.
The Nile Red pathway (Fig. 10) can be considered in the context of steroid binding to P450 17A1. We did not see clear evidence for a conformational selection process with Nile Red, although this might have been precluded by the very rapid (and transient) fluorescence increases (Fig. 9A and Fig. S8) or the limited magnitude of the transient ΔA390 − A418 increases (Fig. 9B). However, it is very possible for a steroid to follow the same pathway. The initial rapid binding could be invisible in that the time frame of the ΔA390 − A418 increase seen with Nile Red (Fig. 8B) is similar to that observed with the steroids (Figs. 4A, 5A, and 6D and Figs. S1–S5). We did not observe the proposed repositioning, which we attribute the ΔA390 − A418 decrease to, with any steroids. However, these may be appropriately positioned for catalysis in the initial docking into the P450 17A1 active site. Presumably, the last step in Nile Red binding (Fig. 10) positions it for N-deethylation (although we do not have specific proof).
Abiraterone is a clinically effective inhibitor of P450 17A1, and its interaction with P450 17A1 appeared to resemble that of other steroids (Fig. 13). The structure of P450 17A1 is similar with either abiraterone (61) or the steroid substrates (62) bound to it. Both abiraterone and orteronel have been reported to selectively inhibit the lyase reaction of P450 17A1 relative to the 17α-hydroxylation step (56, 60), which is a desirable goal in reducing endocrinological side effects in patients being treated for prostate cancer. However, the concept of a drug molecule selectively inhibiting one reaction of an enzyme known to have an active site with only single occupancy is challenging, in that these drugs are known to occupy the same space that the substrates do in the enzyme (56, 61). We considered kinetic models and did not find a difference in the predicted IC50 values when only a single conformational form of P450 was included (Fig. 12A). Selective inhibition of the lyase reaction was possible, however, if a conformational change of the enzyme was introduced after the first reaction (17α-hydroxylation) was completed (Table 1 and Figs. 11 and 12B). The difference in the IC50 values for the two steps (2.5-fold) is similar to that reported for orteronel in some experimental studies (28, 56). We obtained similar results regardless of whether IC50 values for the lyase reaction were calculated beginning with pregnenolone or 17α-OH pregnenolone as the initial substrate. It should be pointed out that the model presented in Table 1 and Fig. 11 is hypothetical and that more possibilities exist. Although we have not included models with multiple ligand occupancy, the possibility cannot be discounted in that Petrunak et al. (56) reported some electron density in some of their P450 17A1-orteronel structures in the region between the F/G loop and the N-terminal residues. However, the point is made that it does not seem possible to generate different IC50 values for the two reaction steps in the absence of conformationally distinct forms of P450 17A1.
We conclude that P450 17A1 binds its steroidal substrates and analogs using a conformational selection model (Figs. 1 and 2), based upon multiple lines of evidence. This information is relevant to the field of mammalian P450s. However, although P450 17A1 does bind and oxidize unnatural molecules such as Nile Red (Figs. 8 and 9 and Figs. S6–S8), the enzyme has high selectivity for a few steroids. Our conclusions about the importance of a conformational selection model over an induced fit one cannot be extended to other P450s at this time, particularly those that have broad catalytic selectivity and also show evidence of multiphasic substrate binding kinetics (e.g. P450 3A4) (25–27).
One argument against the invocation of a conformational selection model for P450 3A4 is the diversity of rates of binding of different substrates (25) and inhibitors (26) that have been seen and would not be explained by a single E ⇄ E′ equilibrium, as was done here for P450 17A1 (Figs. 4 and Figs. S1–S5). However, a conformational selection model could be considered with more than two conformational possibilities, although the complexity would probably outstrip any possibilities of proof. Some evidence for induced fit has been presented with P450 3A4 (26), but further studies with some of the new approaches (38, 44) are in order.
Multiple conformations of P450 17A1 are also consistent with the multiplicity of products derived from both the Δ4 and Δ5 steroids, including the unexpected 6β-hydroxylation of 16α,17α-(OH)2 progesterone (78). The 16α-hydroxylation could also be the result of a conformational change, and a role of Ala-105 has been proposed on the basis of site-directed mutagenesis work (62). These minor products, however, could be explained on the basis of rearrangements of the substrates within the active site, as opposed to protein conformational changes. Ideally, documenting the existence of multiple conformation of ligand-free P450 17A1 by physical methods would be in order, preferably demonstrating that one or more of these was altered by the addition of substrate. Two other points should be reiterated. One is that the conformational selection hypothesis is not restricted to only two conformations, and the possibility exists that others can be captured by other molecules (e.g. nonsteroids). Also, elements of induced fit may also exist, along with the conformational selection process. However, on the basis of the kinetic evidence we currently have available, we conclude that the binding of steroids to P450 17A1 is dominated by a conformational selection process.
Experimental procedures
Chemicals
Racemic orteronel (TAK-700) was purchased from ApexBio (Houston, TX) (catalogue no. A4326). The material was resolved into the R- and S-enantiomers using chiral HPLC (28, 56). Nile Red was purchased from Sigma-Aldrich, and its purity was confirmed by analytical UV-visible UPLC (octadecylsilane column). Abiraterone was purchased from Selleckchem (Houston, TX) and checked by high-resolution MS (m/z observed 350.2454 (MH+), calc. 350.2498, Δ 6.5 ppm)). All other ligands were purchased from Sigma-Aldrich.
Enzymes
P450 17A1 (78) was expressed with a C-terminal His6 tag and purified to near electrophoretic homogeneity as described previously. Recombinant rat NADPH-P450 reductase (79) and human b5 (80) were expressed in Escherichia coli and purified as described. Heme was removed from b5 to form apo-b5 as described elsewhere (81).
Measurements of equilibrium binding
Absorbance measurements were made with an Aminco DW2/OLIS spectrophotometer (On-Line Instrument Systems, Bogart, GA). In a 1-cm cell, either Nile Red or 17α-OH progesterone or 17α-OH pregnenolone was incrementally added (from a concentrated stock in C2H5OH) to 1.0 μm human P450 17A1 in 100 mm potassium phosphate buffer (pH 7.4). Titrations (utilizing b5) contained 1 μm b5. Samples did not contain lipid or detergent.
Fluorescence measurements were made with an OLIS DM45 fluorimeter. The excitation wavelength was 550 nm, and the emission range of 575–700 nm was scanned. Both excitation and emission slits were set at 6.32 mm. The integration time was set at 0.25 s, and the emission was measured 90° to the excitation source. A 1 × 1-cm quartz cell (Starna Cells (Atascadero, CA), catalogue no. 23-Q-10-MC) was used that contained mirrored surfaces opposite the excitation source and detector to maximize detection. Fluorescence titrations were performed using the same conditions as in the absorbance-based titrations. Titrations utilizing abiraterone contained 25 μm abiraterone previously mixed with P450 17A1 prior to the addition of any Nile Red.
Catalytic assays
Steady-state catalytic assays with 17α-OH progesterone were performed as described previously (28), with the following specifications: 0.5 μm human P450 17A1, 2 μm E. coli recombinant rat NADPH-P450 reductase, 16 μm l-α-1,2,-dilauroyl-sn-glycero-3-phosphocholine (added as lipid vesicles after sonication of a 1.6 mm stock in H2O), 5 μm 17α-OH progesterone, and 0.5 μm b5, apo-b5, or no b5 were included in 0.5 ml of 50 mm potassium phosphate buffer (pH 7.4). Reactions were initiated with an NADPH-generating system (10 mm glucose 6-phosphate, 0.5 mm NADP+, and 2 μg/ml yeast glucose-6-phosphate dehydrogenase) (82) and quenched with 2 ml of CH2Cl2 after 2 min. Extracts were dissolved in 120 μl of a CH3CN-H2O mixture (2:3, v/v). A 20-μl aliquot of each sample was injected on a Waters Acquity BEH C18 UPLC octadecylsilane column (2.1 × 50 mm, 1.7 μm) with mobile phases A (5% CH3CN, 95% H2O, v/v) and B (95% CH3CN, 5% H2O, v/v). The mobile phase linear gradient was as follows: 0–0.5 min, 80% A (v/v), 0.2 ml/min; 10 min, 40% A (v/v), 0.2 ml/min; 10.5–12.5 min, 0% A, 0.4 ml/min; 13–15 min, 80% A (v/v), 0.2 ml/min. Androstenedione formation was identified by co-elution with a commercial standard and quantified on the basis of A243 peak areas.
Steady-state catalytic assays with Nile Red were performed in a final volume of 150 μl containing 0.2 μm human P450 17A1, 2 μm E. coli recombinant rat NADPH-P450 reductase, and 48 μm l-α-1,2,-dilauroyl-sn-glycerol-3-phosphocholine (added as lipid vesicles after sonication of a 1.6 mm stock in H2O) in 50 mm potassium phosphate buffer (pH 7.4). Substrate was added from a stock (in 95% C2H5OH) to yield final concentrations ranging from 0.2 to 40 μm. Samples were equilibrated at 37 °C for 5 min in a shaking water bath. Reactions were initiated with the addition of an NADPH-generating system (82). After the addition of the NADPH-generating system, samples were incubated for 5 min at 37 °C in a shaking water bath. Reactions were quenched by the addition of 300 μl of CH2Cl2, mixed using a vortex device, and placed on ice. Samples were then centrifuged at 2500 × g for 5 min. The organic (bottom) layer (250 μl) was transferred to a new container and dried under a nitrogen stream. Samples were resuspended in 150 μl of a 1:1 (v/v) mixture of LC mobile phases. 10-μl aliquots of each sample were injected on a Waters Acquity BEH C18 UPLC octadecylsilane (C18) column (1.0 × 100 mm, 1.7 μm) (Waters, Milford, MA). The mobile phase solvents used were A (95% H2O, 5% CH3CN, 0.1% HCO2H (v/v)) and B (99% CH3CN, 1% H2O, 0.1% HCO2H (v/v)) at a flow rate of 0.15 ml/min. The mobile phase linear gradient was as follows: 0 min, 100% A; 12.5 min, 0% B; 12.65 min, 100% A; 14 min, 100% A (all v/v). The retention times of Nile Red and the N-deethylation product (Fig. 7, identified by its mass spectrum, m/z 291.2 (MH+, positive electrospray)) were 9.32 and 7.82 min, respectively.
Measurement of kinetics of binding
All absorbance measurements were made in 100 mm potassium phosphate buffer (pH 7.4) using an OLIS RSM-1000 stopped-flow spectrophotometer (On-Line Instrument Systems, Bogart, GA) in the rapid scanning mode with a path length of 20 mm (20 × 4-mm cell), 1.24-mm slits, and 600-line/500-nm gratings at 23 °C. In the cases in which the P450 Soret absorbance was high (>1), a 4-mm path length cell (4 × 4-mm cell) was used to decrease the absorbance. For collection time periods of ≤4 s, data were collected at 1000 scans/s. For time periods of ≥4 s, 62 scans/s were collected in the signal averaging mode. The wavelength range was 330–570 nm.
For fluorescence kinetic measurements with Nile Red, the same stopped-flow instrument was used with a 4 × 4-mm cell, an excitation wavelength of 550 nm, and a >590-nm emission end-on filter (Oriel, Stratford, CT) attached to the photomultiplier tube, set at a 90° angle to the incident light beam. The slits were also 1.24 mm, and the recording time was 4 s. The general measurement mode involved mixing one syringe containing 2–4 μm P450 (in 100 mm potassium phosphate buffer, pH 7.4) with an equal volume of the same buffer containing varying concentrations of substrate or other ligand.
The data were converted to ΔAmax − ΔAmin files (e.g. ΔA390 − ΔA418, depending upon the P450-ligand system) or ΔF files and were saved as Excel files. Data from at least four individual traces were averaged using OLIS software before export. The files were corrected to ΔAt = 0 = 0 and saved as txt files for direct import into KinTek Explorer.
Kinetic modeling
All work was done with KinTek Explorer® software (Kintek, Snowshoe PA) using an Apple iMac OSX version 10.13.6 system and Explorer version 8.0 (46).
The general procedure involved an initial overall analysis of a family of traces of ΔA versus time (varying substrate concentration) with a series of single exponential fits for each. The individual rates were plotted versus the substrate concentration. This analysis was followed by a series of double-exponential fits of all traces and then plotting both rates (fast and slow phases) versus the substrate concentration.
Attempts were made to globally fit the data to either an induced-fit mode (Model 3),
where E is P450, S is substrate, ES is the initial substrate complex, E′S is the final substrate complex, and only E′S is observed (indicated with an asterisk), or to a conformation selection model (Model 4),
where E and E′ are alternate conformational forms of P450 and only E′ binds the substrate, S is the substrate, and E′S is the only observed P450–substrate complex (indicated with an asterisk).
More complex models included both induced-fit and conformational selection modes (Model 5) and have been used in the past with P450s 3A4 and 17A1 (25, 28) (Model 5).
In this configuration, only E′S is observed (*), but it can be generated by either of two routes. Individual rate constants for both forward and reverse steps were adjusted manually to obtain the most general fits with the various models. In the case of the fitting of orteronel inhibition of P450 17A1 reactions, a more complex model was used, as discussed (Fig. 10 and Table 1).
Author contributions
F. P. G. conceptualization; F. P. G., C. J. W., S. M. G., and M. J. R. data curation; F. P. G. formal analysis; F. P. G. supervision; F. P. G. funding acquisition; F. P. G. validation; F. P. G., C. J. W., S. M. G., and M. J. R. investigation; F. P. G. visualization; F. P. G. and S. M. G. methodology; F. P. G. writing-original draft; F. P. G. project administration; F. P. G., S. M. G., and M. J. R. writing-review and editing; C. J. W. resources.
Supplementary Material
Acknowledgments
We thank Dr. E. Gonzalez for acquiring the previously unpublished data used in Figs. S1 and S4 and K. Trisler for assistance in preparation of the manuscript.
This work was supported by National Institutes of Health Grants R01 GM118122 (to F. P. G.) and T32 ES007028 (to F. P. G., support of S. M. G. and M. J. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Scheme S1 and Figs. S1–S8.
- P450 (or CYP)
- cytochrome P450
- DHEA
- dehydroepiandrosterone
- b5
- cytochrome b5
- OH
- hydroxy.
References
- 1. Ortiz de Montellano P. R. (ed) (2015) Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th Ed., Springer, New York [Google Scholar]
- 2. Guengerich F. P. (2015) Human cytochrome P450 enzymes. in Cytochrome P450: Structure, Mechanism, and Biochemistry (Ortiz de Montellano P. R., ed) pp. 523–785, 4th Ed., Springer, New York [Google Scholar]
- 3. Rendic S., and Guengerich F. P. (2015) Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem. Res. Toxicol. 28, 38–42 10.1021/tx500444e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guengerich F. P. (2018) Mechanisms of cytochrome P450-catalyzed oxidations. ACS Catalysis 8, 10964–10976 10.1021/acscatal.8b03401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tripathi S., Li H., and Poulos T. L. (2013) Structural basis for effector control and redox partner recognition in cytochrome P450. Science 340, 1227–1230 10.1126/science.1235797 [DOI] [PubMed] [Google Scholar]
- 6. Lee Y. T., Wilson R. F., Rupniewski I., and Goodin D. B. (2010) P450cam visits an open conformation in the absence of substrate. Biochemistry 49, 3412–3419 10.1021/bi100183g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liou S. H., Mahomed M., Lee Y. T., and Goodin D. B. (2016) Effector roles of putidaredoxin on cytochrome P450cam conformational states. J. Am. Chem. Soc. 138, 10163–10172 10.1021/jacs.6b04110 [DOI] [PubMed] [Google Scholar]
- 8. Follmer A. H., Mahomed M., Goodin D. B., and Poulos T. L. (2018) Substrate-dependent allosteric regulation in cytochrome P450cam (CYP101A1). J. Am. Chem. Soc. 140, 16222–16228 10.1021/jacs.8b09441 [DOI] [PubMed] [Google Scholar]
- 9. Follmer A. H., Tripathi S., and Poulos T. L. (2019) Ligand and redox partner binding generates a new conformational state in cytochrome P450cam (CYP101A1). J. Am. Chem. Soc. 141, 2678–2683 10.1021/jacs.8b13079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Montemiglio L. C., Giuffrè A., Macone A., Scaglione A., Parisi G., Giuffrè A., Cerutti G., Exertier C., Savino C., and Vallone B. (2019) Substrate-induced conformational change in cytochrome P450 OleP. FASEB J. 33, 1787–1800 10.1096/fj.201800450RR [DOI] [PubMed] [Google Scholar]
- 11. Scott E. E., He Y. A., Wester M. R., White M. A., Chin C. C., Halpert J. R., Johnson E. F., and Stout C. D. (2003) An open conformation of mammalian cytochrome P450 2B4 at 1.6-Å resolution. Proc. Natl. Acad. Sci. U.S.A. 100, 13196–13201 10.1073/pnas.2133986100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gay S. C., Sun L., Maekawa K., Halpert J. R., and Stout C. D. (2009) Crystal structures of cytochrome P450 2B4 in complex with the inhibitor 1-biphenyl-4-methyl-1H-imidazole: ligand-induced structural response through α-helical repositioning. Biochemistry 48, 4762–4771 10.1021/bi9003765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Muralidhara B. K., Negi S., Chin C. C., Braun W., and Halpert J. R. (2006) Conformational flexibility of mammalian cytochrome P450 2B4 in binding imidazole inhibitors with different ring chemistry and side chains: solution thermodynamics and molecular modeling. J. Biol. Chem. 281, 8051–8061 10.1074/jbc.M509696200 [DOI] [PubMed] [Google Scholar]
- 14. Wang A., Savas U., Hsu M. H., Stout C. D., and Johnson E. F. (2012) Crystal structure of human cytochrome P450 2D6 with prinomastat bound. J. Biol. Chem. 287, 10834–10843 10.1074/jbc.M111.307918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mast N., White M. A., Bjorkhem I., Johnson E. F., Stout C. D., and Pikuleva I. A. (2008) Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc. Natl. Acad. Sci. U.S.A. 105, 9546–9551 10.1073/pnas.0803717105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Poulos T. L., and Johnson E. F. (2015) Structures of cytochrome P450 enzymes. in Cytochrome P450: Structure, Function, and Biochemistry (Ortiz de Montellano P. R., ed) pp. 3–32, Springer, New York [Google Scholar]
- 17. Wang A., Stout C. D., Zhang Q., and Johnson E. F. (2015) Contributions of ionic interactions and protein dynamics to cytochrome P450 2D6 (CYP2D6) substrate and inhibitor binding. J. Biol. Chem. 290, 5092–5104 10.1074/jbc.M114.627661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ekroos M., and Sjögren T. (2006) Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U.S.A. 103, 13682–13687 10.1073/pnas.0603236103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schoch G. A., Yano J. K., Sansen S., Dansette P. M., Stout C. D., and Johnson E. F. (2008) Determinants of cytochrome P450 2C8 substrate binding: structures of complexes with montelukast, troglitazone, felodipine, and 9-cis-retinoic acid. J. Biol. Chem. 283, 17227–17237 10.1074/jbc.M802180200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sevrioukova I. F., and Poulos T. L. (2013) Dissecting cytochrome P450 3A4-ligand interactions using ritonavir analogues. Biochemistry 52, 4474–4481 10.1021/bi4005396 [DOI] [PubMed] [Google Scholar]
- 21. Griffin B. W., and Peterson J. A. (1972) Camphor binding by Pseudomonas putida cytochrome P-450: kinetics and thermodynamics of the reaction. Biochemistry 11, 4740–4746 10.1021/bi00775a017 [DOI] [PubMed] [Google Scholar]
- 22. Yun C. H., Kim K. H., Calcutt M. W., and Guengerich F. P. (2005) Kinetic analysis of oxidation of coumarins by human cytochrome P450 2A6. J. Biol. Chem. 280, 12279–12291 10.1074/jbc.M411019200 [DOI] [PubMed] [Google Scholar]
- 23. Johnson K. M., Phan T. T. N., Albertolle M. E., and Guengerich F. P. (2017) Human mitochondrial cytochrome P450 27C1 is localized in skin and preferentially desaturates trans-retinol to 3,4-dehydroretinol. J. Biol. Chem. 292, 13672–13687 10.1074/jbc.M116.773937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sohl C. D., Isin E. M., Eoff R. L., Marsch G. A., Stec D. F., and Guengerich F. P. (2008) Cooperativity in oxidation reactions catalyzed by cytochrome P450 1A2: highly cooperative pyrene hydroxylation and multiphasic kinetics of ligand binding. J. Biol. Chem. 283, 7293–7308 10.1074/jbc.M709783200 [DOI] [PubMed] [Google Scholar]
- 25. Isin E. M., and Guengerich F. P. (2006) Kinetics and thermodynamics of ligand binding by cytochrome P450 3A4. J. Biol. Chem. 281, 9127–9136 10.1074/jbc.M511375200 [DOI] [PubMed] [Google Scholar]
- 26. Isin E. M., and Guengerich F. P. (2007) Multiple sequential steps involved in the binding of inhibitors to cytochrome P450 3A4. J. Biol. Chem. 282, 6863–6874 10.1074/jbc.M610346200 [DOI] [PubMed] [Google Scholar]
- 27. Sevrioukova I. F., and Poulos T. L. (2012) Structural and mechanistic insights into the interaction of cytochrome P4503A4 with bromoergocryptine, a Type I ligand. J. Biol. Chem. 287, 3510–3517 10.1074/jbc.M111.317081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gonzalez E., and Guengerich F. P. (2017) Kinetic processivity of the two-step oxidations of progesterone and pregnenolone to androgens by human cytochrome P450 17A1. J. Biol. Chem. 292, 13168–13185 10.1074/jbc.M117.794917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Benkovic S. J., and Hammes-Schiffer S. (2006) Biochemistry: enzyme motions inside and out. Science 312, 208–209 10.1126/science.1127654 [DOI] [PubMed] [Google Scholar]
- 30. Hammes G. G., Benkovic S. J., and Hammes-Schiffer S. (2011) Flexibility, diversity, and cooperativity: pillars of enzyme catalysis. Biochemistry 50, 10422–10430 10.1021/bi201486f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fischer E. (1894) Einfluss der configuration auf die wirkung der enzyme. Berichte der deutschen chemischen Gesellschaft 27, 2985–2993 [Google Scholar]
- 32. Koshland D. E. Jr., Némethy G., and Filmer D. (1966) Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5, 365–385 10.1021/bi00865a047 [DOI] [PubMed] [Google Scholar]
- 33. Monod J., Wyman J., and Changeaux J. P. (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118 10.1016/S0022-2836(65)80285-6 [DOI] [PubMed] [Google Scholar]
- 34. Changeux J. P., and Edelstein S. (2011) Conformational selection or induced fit? 50 years of debate resolved. F1000 Biol. Rep. 3, 19 10.3410/B3-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hammes G. G., Chang Y. C., and Oas T. G. (2009) Conformational selection or induced fit: a flux description of reaction mechanism. Proc. Natl. Acad. Sci. U.S.A. 106, 13737–13741 10.1073/pnas.0907195106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou H. X. (2010) From induced fit to conformational selection: a continuum of binding mechanism controlled by the timescale of conformational transitions. Biophys. J. 98, L15–L17 10.1016/j.bpj.2009.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chakraborty P., and Di Cera E. (2017) Induced fit is a special case of conformational selection. Biochemistry 56, 2853–2859 10.1021/acs.biochem.7b00340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vogt A. D., and Di Cera E. (2012) Conformational selection or induced fit? A critical appraisal of the kinetic mechanism. Biochemistry 51, 5894–5902 10.1021/bi3006913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johnson K. A. (2008) Role of induced fit in enzyme specificity: A molecular forward/reverse switch. J. Biol. Chem. 283, 26297–26301 10.1074/jbc.R800034200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Agafonov R. V., Wilson C., Otten R., Buosi V., and Kern D. (2014) Energetic dissection of Gleevec's selectivity toward human tyrosine kinases. Nat. Struct. Mol. Biol. 21, 848–853 10.1038/nsmb.2891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pallan P. S., Wang C., Lei L., Yoshimoto F. K., Auchus R. J., Waterman M. R., Guengerich F. P., and Egli M. (2015) Human cytochrome P450 21A2, the major steroid 21-hydroxylase: structure of the enzyme·progesterone substrate complex and rate-limiting C-H bond cleavage. J. Biol. Chem. 290, 13128–13143 10.1074/jbc.M115.646307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim D., Cha G. S., Nagy L. D., Yun C. H., and Guengerich F. P. (2014) Kinetic analysis of lauric acid hydroxylation by human cytochrome P450 4A11. Biochemistry 53, 6161–6172 10.1021/bi500710e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Johnson K. A. (2003) Introduction to kinetic analysis of enzyme systems. in Kinetic Analysis of Macromolecules: A Practical Approach (Johnson K. A., ed) pp. 1–18, Oxford University Press, Oxford, UK [Google Scholar]
- 44. Gianni S., Dogan J., and Jemth P. (2014) Distinguishing induced fit from conformational selection. Biophys. Chem. 189, 33–39 10.1016/j.bpc.2014.03.003 [DOI] [PubMed] [Google Scholar]
- 45. Schreiber G., Haran G., and Zhou H. X. (2009) Fundamental aspects of protein-protein association kinetics. Chem. Rev. 109, 839–860 10.1021/cr800373w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Johnson K. A., Simpson Z. B., and Blom T. (2009) Global Kinetic Explorer: A new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 10.1016/j.ab.2008.12.024 [DOI] [PubMed] [Google Scholar]
- 47. Gonzalez E., Johnson K. M., Pallan P. S., Phan T. T. N., Zhang W., Lei L., Wawrzak Z., Yoshimoto F. K., Egli M., and Guengerich F. P. (2018) Inherent steroid 17α,20-lyase activity in defunct cytochrome P450 17A enzymes. J. Biol. Chem. 293, 541–556 10.1074/jbc.RA117.000504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Peng H. M., Im S. C., Pearl N. M., Turcu A. F., Rege J., Waskell L., and Auchus R. J. (2016) Cytochrome b5 activates the 17,20-lyase activity of human cytochrome P450 17A1 by increasing the coupling of NADPH consumption to androgen production. Biochemistry 55, 4356–4365 10.1021/acs.biochem.6b00532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Duggal R., Liu Y., Gregory M. C., Denisov I. G., Kincaid J. R., and Sligar S. G. (2016) Evidence that cytochrome b5 acts as a redox donor in CYP17A1 mediated androgen synthesis. Biochem. Biophys. Res. Commun. 477, 202–208 10.1016/j.bbrc.2016.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Duggal R., Denisov I. G., and Sligar S. G. (2018) Cytochrome b5 enhances androgen synthesis by rapidly reducing the CYP17A1 oxy-complex in the lyase step. FEBS Lett. 592, 2282–2288 10.1002/1873-3468.13153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Auchus R. J., Lee T. C., and Miller W. L. (1998) Cytochrome b5 augments the 17,20-lyase activity of human P450c17 without direct electron transfer. J. Biol. Chem. 273, 3158–3165 10.1074/jbc.273.6.3158 [DOI] [PubMed] [Google Scholar]
- 52. Lee-Robichaud P., Akhtar M. E., and Akhtar M. (1998) Control of androgen biosynthesis in the human through the interaction of Arg347 and Arg358 of CYP17 with cytochrome b5. Biochem. J. 332, 293–296 10.1042/bj3320293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lampe J. N., Fernandez C., Nath A., and Atkins W. M. (2008) Nile Red is a fluorescent allosteric substrate of cytochrome P450 3A4. Biochemistry 47, 509–516 10.1021/bi7013807 [DOI] [PubMed] [Google Scholar]
- 54. Faletrov Y. V., Frolova N. S., Hlushko H. V., Rudaya E. V., Edimecheva I. P., Mauersberger S., and Shkumatov V. M. (2013) Evaluation of the fluorescent probes Nile Red and 25-NBD-cholesterol as substrates for steroid-converting oxidoreductases using pure enzymes and microorganisms. FEBS J. 280, 3109–3119 10.1111/febs.12265 [DOI] [PubMed] [Google Scholar]
- 55. Nath A., Fernández C., Lampe J. N., and Atkins W. M. (2008) Spectral resolution of a second binding site for Nile Red on cytochrome P4503A4. Arch. Biochem. Biophys. 474, 198–204 10.1016/j.abb.2008.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Petrunak E. M., Rogers S. A., Aubé J., and Scott E. E. (2017) Structural and functional evaluation of clinically relevant inhibitors of steroidogenic cytochrome P450 17A1. Drug. Metab. Dispos. 45, 635–645 10.1124/dmd.117.075317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fehl C., Vogt C. D., Yadav R., Li K., Scott E. E., and Aubé J. (2018) structure-based design of inhibitors with improved selectivity for steroidogenic cytochrome P450 17A1 over cytochrome P450 21A2. J. Med. Chem. 61, 4946–4960 10.1021/acs.jmedchem.8b00419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bonomo S., Hansen C. H., Petrunak E. M., Scott E. E., Styrishave B., Jørgensen F. S., and Olsen L. (2016) promising tools in prostate cancer research: Selective non-steroidal cytochrome P450 17A1 inhibitors. Sci. Rep. 6, 29468 10.1038/srep29468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yamaoka M., Hara T., Hitaka T., Kaku T., Takeuchi T., Takahashi J., Asahi S., Miki H., Tasaka A., and Kusaka M. (2012) Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. J. Steroid Biochem. Mol. Biol. 129, 115–128 10.1016/j.jsbmb.2012.01.001 [DOI] [PubMed] [Google Scholar]
- 60. Kaku T., Hitaka T., Ojida A., Matsunaga N., Adachi M., Tanaka T., Hara T., Yamaoka M., Kusaka M., Okuda T., Asahi S., Furuya S., and Tasaka A. (2011) Discovery of orteronel (TAK-700), a naphthylmethylimidazole derivative, as a highly selective 17,20-lyase inhibitor with potential utility in the treatment of prostate cancer. Bioorg. Med. Chem. 19, 6383–6399 10.1016/j.bmc.2011.08.066 [DOI] [PubMed] [Google Scholar]
- 61. DeVore N. M., and Scott E. E. (2012) Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 482, 116–119 10.1038/nature10743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Petrunak E. M., DeVore N. M., Porubsky P. R., and Scott E. E. (2014) Structures of human steroidogenic cytochrome P450 17A1 with substrates. J. Biol. Chem. 289, 32952–32964 10.1074/jbc.M114.610998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pallan P. S., Nagy L. D., Lei L., Gonzalez E., Kramlinger V. M., Azumaya C. M., Wawrzak Z., Waterman M. R., Guengerich F. P., and Egli M. (2015) Structural and kinetic basis of steroid 17α,20-lyase activity in teleost fish cytochrome P450 17A1 and its absence in cytochrome P450 17A2. J. Biol. Chem. 290, 3248–3268 10.1074/jbc.M114.627265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Scott L. J. (2017) Abiraterone acetate: a review in metastatic castration-resistant prostate cancer. Drugs 77, 1565–1576 10.1007/s40265-017-0799-9 [DOI] [PubMed] [Google Scholar]
- 65. Norris J. D., Ellison S. J., Baker J. G., Stagg D. B., Wardell S. E., Park S., Alley H. M., Baldi R. M., Yllanes A., Andreano K. J., Stice J. P., Lawrence S. A., Eisner J. R., Price D. K., Moore W. R., et al. (2017) Androgen receptor antagonism drives cytochrome P450 17A1 inhibitor efficacy in prostate cancer. J. Clin. Invest. 127, 2326–2338 10.1172/JCI87328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Caron P., Turcotte V., Lévesque E., and Guillemette C. (2019) An LC-MS/MS method for quantification of abiraterone, its active metabolites Δ4-abiraterone (Δ4A) and 5α-abiraterone, and their inactive glucuronide derivatives. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 1104, 249–255 10.1016/j.jchromb.2018.12.003 [DOI] [PubMed] [Google Scholar]
- 67. Estrada D. F., Skinner A. L., Laurence J. S., and Scott E. E. (2014) Human cytochrome P450 17A1 conformational selection: modulation by ligand and cytochrome b5. J. Biol. Chem. 289, 14310–14320 10.1074/jbc.M114.560144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Johnson K. A., Simpson Z. B., and Blom T. (2009) FitSpace Explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 387, 30–41 10.1016/j.ab.2008.12.025 [DOI] [PubMed] [Google Scholar]
- 69. Hildebrandt A., and Estabrook R. W. (1971) Evidence for the participation of cytochrome b5 in hepatic microsomal mixed-function oxidation reactions. Arch. Biochem. Biophys. 143, 66–79 10.1016/0003-9861(71)90186-X [DOI] [PubMed] [Google Scholar]
- 70. Correia M. A., and Mannering G. J. (1973) Reduced diphosphopyridine nucleotide synergism of the reduced triphosphopyridine nucleotide-dependent mixed-function oxidase system of hepatic microsomes. II. Role of the Type I drug-binding site of cytochrome P-450. Mol. Pharmacol. 9, 470–485 [PubMed] [Google Scholar]
- 71. Hall P. F. (1985) Role of cytochromes P-450 in the biosynthesis of steroid hormones. Vitam. Horm. 42, 315–368 10.1016/S0083-6729(08)60065-5 [DOI] [PubMed] [Google Scholar]
- 72. Katagiri M., Kagawa N., and Waterman M. R. (1995) The role of cytochrome b5 in the biosynthesis of androgens by human P450c17. Arch. Biochem. Biophys. 317, 343–347 10.1006/abbi.1995.1173 [DOI] [PubMed] [Google Scholar]
- 73. Estrada D. F., Laurence J. S., and Scott E. E. (2013) Substrate-modulated cytochrome P450 17A1 and cytochrome b5 interactions revealed by NMR. J. Biol. Chem. 288, 17008–17018 10.1074/jbc.M113.468926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ershov P. V., Yablokov E. O., Florinskaya A. V., Mezentsev Y. V., Kaluzhskiy L. A., Tumilovich A. M., Gilep A. A., Usanov S. A., and Ivanov A. S. (2019) SPR-based study of affinity of cytochrome P450s/redox partners interactions modulated by steroidal substrates. J. Steroid Biochem. Mol. Biol. 187, 124–129 10.1016/j.jsbmb.2018.11.009 [DOI] [PubMed] [Google Scholar]
- 75. Savino C., Montemiglio L. C., Sciara G., Miele A. E., Kendrew S. G., Jemth P., Gianni S., and Vallone B. (2009) Investigating the structural plasticity of a cytochrome P450: three-dimensional structures of P450 EryK and binding to its physiological substrate. J. Biol. Chem. 284, 29170–29179 10.1074/jbc.M109.003590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Guengerich F. P., and Holladay L. A. (1979) Hydrodynamic characterization of highly purified and functionally active liver microsomal cytochrome P-450. Biochemistry 18, 5442–5449 10.1021/bi00591a029 [DOI] [PubMed] [Google Scholar]
- 77. French J. S., Guengerich F. P., and Coon M. J. (1980) Interactions of cytochrome P-450, NADPH-cytochrome P-450 reductase, phospholipid, and substrate in the reconstituted liver microsomal enzyme system. J. Biol. Chem. 255, 4112–4119 [PubMed] [Google Scholar]
- 78. Yoshimoto F. K., Gonzalez E., Auchus R. J., and Guengerich F. P. (2016) Mechanism of 17α,20-lyase and new hydroxylation reactions of human cytochrome P450 17A1: 18O labeling and oxygen surrogate evidence for a role of a perferryl oxygen. J. Biol. Chem. 291, 17143–17164 10.1074/jbc.M116.732966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hanna I. H., Teiber J. F., Kokones K. L., and Hollenberg P. F. (1998) Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch. Biochem. Biophys. 350, 324–332 10.1006/abbi.1997.0534 [DOI] [PubMed] [Google Scholar]
- 80. Guengerich F. P. (2005) Reduction of cytochrome b5 by NADPH-cytochrome P450 reductase. Arch. Biochem. Biophys. 440, 204–211 10.1016/j.abb.2005.06.019 [DOI] [PubMed] [Google Scholar]
- 81. Yamazaki H., Johnson W. W., Ueng Y. F., Shimada T., and Guengerich F. P. (1996) Lack of electron transfer from cytochrome b5 in stimulation of catalytic activities of cytochrome P450 3A4. Characterization of a reconstituted cytochrome P450 3A4/NADPH-cytochrome P450 reductase system and studies with apo-cytochrome b5. J. Biol. Chem. 271, 27438–27444 10.1074/jbc.271.44.27438 [DOI] [PubMed] [Google Scholar]
- 82. Guengerich F. P. (2014) Analysis and characterization of enzymes and nucleic acids relevant to toxicology, in Hayes' Principles and Methods of Toxicology, 6th Ed. (Hayes A. W., and Kruger C. L., eds) pp. 1905–1964, CRC Press-Taylor & Francis; Boca Raton, FL [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.