

Abstract
Recently, oxidized phospholipid species have emerged as important signaling lipids in activated immune cells and platelets. The canonical pathway for the synthesis of oxidized phospholipids is through the release of arachidonic acid by cytosolic phospholipase A2α (cPLA2α) followed by its enzymatic oxidation, activation of the carboxylate anion by acyl-CoA synthetase(s), and re-esterification to the sn-2 position by sn-2 acyltransferase activity (i.e. the Lands cycle). However, recent studies have demonstrated the unanticipated significance of sn-1 hydrolysis of arachidonoyl-containing choline and ethanolamine glycerophospholipids by other phospholipases to generate the corresponding 2-arachidonoyl-lysolipids. Herein, we identified a pathway for oxidized phospholipid synthesis comprising sequential sn-1 hydrolysis by a phospholipase A1 (e.g. by patatin-like phospholipase domain–containing 8 (PNPLA8)), direct enzymatic oxidation of the resultant 2-arachidonoyl-lysophospholipids, and the esterification of oxidized 2-arachidonoyl-lysophospholipids by acyl-CoA–dependent sn-1 acyltransferase(s). To circumvent ambiguities associated with acyl migration or hydrolysis, we developed a synthesis for optically active (d- and l-enantiomers) nonhydrolyzable analogs of 2-arachidonoyl-lysophosphatidylcholine (2-AA-LPC). sn-1 acyltransferase activity in murine liver microsomes stereospecifically and preferentially utilized the naturally occurring l-enantiomer of the ether analog of lysophosphatidylcholine. Next, we demonstrated the high selectivity of the sn-1 acyltransferase activity for saturated acyl-CoA species. Importantly, we established that 2–15-hydroxyeicosatetraenoic acid (HETE) ether-LPC sn-1 esterification is markedly activated by thrombin treatment of murine platelets to generate oxidized PC. Collectively, these findings demonstrate the enantiomeric specificity and saturated acyl-CoA selectivity of microsomal sn-1 acyltransferase(s) and reveal its participation in a previously uncharacterized pathway for the synthesis of oxidized phospholipids with cell-signaling properties.
Keywords: lipid synthesis, lipid peroxidation, acyltransferase, phospholipase A, phospholipid, lysophospholipid, stereoselectivity, mass spectrometry (MS), lipid metabolism, lipidomics, oxidized lysophospholipid, oxidized phospholipid, sn-1 acyltransferase
Introduction
Phospholipids are of critical importance in eukaryotic cell biology through their propensity to form membrane bilayers that serve as a permeability barrier separating intracellular compartments, solvate transmembrane proteins, and function as cryptic reservoirs for the release of lipid second messengers after cellular stimulation. Moreover, oxidized phospholipids, predominantly oxidized phosphatidylcholines and oxidized phosphatidylethanolamines, are produced in immune cells and platelets following activation by agonists (1–4). Many studies have shown that the presence of oxidized phospholipids in membrane bilayers results in changes in membrane molecular dynamics that modulate the activity of transmembrane proteins (5, 6). Furthermore, oxidized phospholipids have important signaling functions in diverse biological processes such as inflammation, adaptive immunity, proliferation, ferroptosis, necroptosis, and pyroptosis (7–12).
Multiple studies have examined the biosynthetic pathway of oxidized phospholipid production (1, 2, 13, 14). The canonical pathway comprises four sequential steps. The first step is the hydrolysis of a nonoxidized phospholipid by cytosolic phospholipase A2α (cPLA2α),2 resulting in the release of polyunsaturated fatty acids (e.g. arachidonic acid) from the sn-2 position of the phospholipid. The second step is the oxidation of the released arachidonic acid (or other polyunsaturated fatty acids) by cyclooxygenases, lipoxygenases, or cytochromes P450 to form nonesterified eicosanoids. The third step is the thioesterification of the carboxylate anion by acyl-CoA synthetases. The final step in the canonical pathway is acylation of the eicosanoid-CoAs to the sn-2 position of 1-acyl-lysophospholipids (Fig. 1, top pathway).
Figure 1.

A novel enzyme-catalyzed pathway for oxidized phospholipid synthesis. The canonical pathway (top) of oxidized phospholipid synthesis is a four-step process composed of the sn-2 hydrolysis of phospholipid by a PLA2, the oxidization of the released nonesterified arachidonic acid by LOX or COX, activation of the oxidized arachidonic acid (oxAA) by acyl-CoA synthetase (ACS), and finally acylation of eicosanoid-CoA to the sn-2 position of 1-palmitoyl (or 1-stearoyl)-LPC. In this study, we have described a novel pathway (bottom) initiated by an sn-1 phospholipase activity that generates 2-AA-LPC, which can be directly oxidized to 2–15-HETE-LPC followed by the acylation of 2–15-HETE-LPC by palmitoyl-CoA or stearoyl-CoA catalyzed by sn-1 LPC acyltransferase activity. FA, fatty acid.
Previously, we identified calcium-independent phospholipase A2γ (iPLA2γ; also known as patatin-like phospholipase domain–containing 8 (PNPLA8)) that contained dual mitochondrial and peroxisomal localization sites (15, 16). Intriguingly, iPLA2γ possessed a heretofore unprecedented regiospecificity of hydrolysis (17). Specifically, when the sn-2 position of phosphatidylcholine (PC) or phosphatidylethanolamine (PE) is polyunsaturated, then iPLA2γ acts nearly exclusively as an sn-1 phospholipase (17) that is activated by divalent cations (18), resulting in the production of 2-arachidonoyl-lysophosphatidylcholine (2-AA-LPC) or 2-arachidonoyl-lysophosphatidylethanolamine (2-AA-LPE). Because iPLA2γ is the predominant phospholipase in cardiac mitochondria and presumably peroxisomes, the generation of 2-AA-LPC was recognized as a metabolic node integrating the actions of multiple enzymes. Thus potentially generate a plethora of signaling molecules comprising different oxidized aliphatic chains covalently bound to lysolipids. For example, we demonstrated that 2-arachidonoyl-lysophospholipids are excellent substrates for direct oxidation by cyclooxygenase-2 (COX-2) and lipoxygenases (e.g. 15-lipoxygenase (15-LOX)) (19). Thus 2-eicosanoid lysophospholipids could serve as lipid second messengers directly, be further hydrolyzed to release nonesterified eicosanoids for downstream signaling, or reacylated by sn-1 acyltransferase(s) to form the corresponding oxidized phospholipids. Thus, we postulated that a previously undescribed pathway of oxidized phospholipid synthesis was present in mammalian cells.
Initial studies with isotope-labeled 1-hydroxy-2–15(S)-hydroxyeicosatetraenoic acid-sn-glycero-3-phosphocholine (2–15-HETE-LPC) and 2–15(S)-hydroxyeicosatetraenoic acid-sn-glycero-3-phosphoethanolamine (2–15-HETE-LPE) demonstrated that this novel pathway was active in hepatic microsomes. However, the robust amounts of lysophospholipase activity in mammalian cells preclude definitive mechanistic identification of the pathway involved. In fact, the lysophospholipase activity of cPLA2α for 2-arachidonoyl-lysolipids is over 50-fold higher than that of its phospholipase A2 activity for arachidonate-containing phospholipids when measured in vitro (20). To remove mechanistic ambiguities resulting from the potential hydrolysis of 2-acyl-LPCs, α-hydroxy migration, and/or transacylation reactions, we endeavored to synthesize optically active nonhydrolyzable ether analogs of 2-arachidonoyl-lysophosphatidylcholine (2-AA-ether-LPC) to delineate the different possible metabolic pathways involved in oxidized phospholipid synthesis. Unfortunately, traditional chemical approaches to synthesize 2-AA-ether-LPC were precluded by the instability of the polyunsaturated arachidonoyl acyl chain in most protection/deprotection reactions such as H2/Pd and 2,3-dichloro-5,6-dicyano-p-benzoquinone. Moreover, we found that the arachidonoyl acyl chain readily undergoes isomerization reactions when treated with Lewis acids such as BF3 and p-toluenesulfinic acid as reported previously (21). Therefore, these challenges needed to be traversed to stereospecifically synthesize 2-AA-ether-LPC and determine its metabolic fate in mammalian cells.
In this work, we demonstrate 1) the robust activity of sn-1 acyltransferase activity for oxidized lysolipids, 2) the first reported enantiomerically specific synthesis of nonhydrolyzable l- and d-forms of 2-arachidonyl-ether-LPC, and 3) the utilization of sn-2 oxidized lysolipids to generate oxidized phospholipids both in subcellular fractions and in activated platelets. Importantly, comparisons of the l- versus d-isomers of 2-arachidonyl-ether-LPC identified the stereospecific production of phospholipids from l-lysophosphatidylcholine but only modestly when d-lysophosphatidylcholine was used as substrate. Furthermore, detailed characterization of the acyl-chain specificity of the microsomal sn-1 acyltransferase activity demonstrated that it was highly selective for saturated acyl-CoAs. Finally, we demonstrated that this pathway is activated in platelets by thrombin to produce the stereospecific synthesis of enzymatically generated stereospecific oxidized phospholipids.
Results
Synthesis of oxidized phosphatidylcholine and oxidized phosphatidylethanolamine by sn-1 acyltransferase using 2-eicosanoid lysophosphatidylcholine and 2-eicosanoid lysophosphatidylethanolamine as substrates
To determine the potential presence of this pathway in mammalian cells, we used a stable isotope labeling approach to determine the metabolic fate of 2-AA-LPC-d9 or 2–15-HETE-LPC-d9 incubated with murine hepatic microsomes in the presence of stearoyl-CoA. The resulting lipids were extracted and analyzed by LC-MS. The results showed that 1-stearoyl-2-arachidonoyl-sn-PC-d9 (18:0/20:4-PC-d9) was synthesized from 2-AA-LPC-d9 by hepatic microsomal sn-1 acyltransferase activity, whereas 18:0/15-HETE-PC-d9 was synthesized from 2–15-HETE-LPC-d9 (Fig. 2A). The specific activities of the initial rates for these two reactions were 0.5 and 0.4 nmol·mg−1·min−1, respectively (Fig. 2, I and J). Similarly, 18:0/15-HETE-PE (hydroxy 18O) was synthesized from 2–15-HETE-LPE (hydroxy 18O) (Fig. 2B), and the specific activity of the acyltransferase reaction was 1.2 nmol·mg−1·min−1 (Fig. 2K). Accurate mass analysis revealed the m/z of 18:0/20:4-PC-d9 was 819.6547 (calculated m/z is 819.6565, Δ = 2 ppm), the m/z of 18:0/15-HETE-PC-d9 was 835.6525 (calculated m/z = 835.6514, Δ = 1 ppm), and the m/z of 18:0/15-HETE-PE (hydroxy 18O) was 786.5538 (calculated m/z = 786.5530, Δ = 1 ppm) (Fig. 2, C, E, and G, respectively). To substantiate the identities of these three reaction products, MS2 analysis was performed. The predominant fragment ion of 18:0/20:4-PC-d9 was 193.1292, which is d9-phosphocholine ([C5H15NO4P-d9]+ calculated m/z = 193.1296, Δ = 2 ppm) (Fig. 2D). In contrast, the predominant fragment ion of 18:0/15-HETE-PC-d9 was 817.6399, which results from the neutral loss of water from the parent ion (calculated m/z = 817.6409, Δ = 1 ppm) (Fig. 2F). The predominant fragment ion of 18:0/15-HETE-PE (hydroxy 18O) was 766.5383, which results from the neutral loss of H218O from the parent ion (calculated m/z = 766.5389, Δ = 1 ppm) (Fig. 2H).
Figure 2.

Synthesis of phospholipid/oxidized phospholipid from 2-arachidonoyl-lysophospholipid or 2-eicosanoid lysophospholipid by sn-1 acyltransferase. A, extracted ion chromatograms of PC/oxidized PC synthesized by murine hepatic microsomal sn-1 acyltransferase activity(ies) utilizing 2-AA-LPC or 2–15-HETE-LPC as substrate. Microsomal homogenates isolated from mouse liver were incubated with the indicated substrates for 5 min at 37 °C in 75 mm sodium phosphate buffer (pH 7.4). After incubation, 14:1-PC was added as an internal standard. The lipids were extracted and analyzed by LC-MS. The extracted ion chromatograms (with a 5-ppm mass window) of the metabolic products 18:0/20:4-PC-d9 (m/z 819.6565) and 18:0/15-HETE-PC-d9 (m/z 835.6514) are shown. B, extracted ion chromatograms of oxidized PE synthesized by murine hepatic microsomal sn-1 acyltransferase activity(ies) utilizing 2–15-HETE-LPE as described above. After incubation, 16:1-PE was added as an internal standard. The extracted ion chromatograms (with a 5-ppm mass window) of the metabolic product 18:0/15-HETE-PE (hydroxy 18O) (m/z 786.5530) is shown. C and D, MS1 spectrum (C) and MS2 spectrum (D) of 18:0/20:4-PC-d9. E and F, MS1 spectrum (E) and MS2 spectrum (F) of 18:0/15-HETE-PC-d9. G and H, MS1 spectrum (G) and MS2 spectrum (H) of 18:0/15-HETE-PE (hydroxy 18O). I–K, specific activities of sn-1 acyltransferase–mediated production of 18:0/20:4-PC-d9 (I), 18:0/15-HETE-PC-d9 (J), and 18:0/15-HETE-PE (hydroxy 18O) (K) from 2-AA-LPC-d9 (10 μm), 2–15-HETE-LPC-d9 (10 μm), and 2–15-HETE-LPE (hydroxy 18O), respectively, in the presence of 18:0-CoA (10 μm). Values are the average of four independent preparations. Error bars represent S.D.
Stereoselective synthesis of the nonhydrolyzable ether analog of 2-AA-LPC
To unambiguously establish the importance of this putative pathway and the stereospecificity of sn-1 acyltransferase activity in mammalian tissues, we developed a chemical synthesis for nonhydrolyzable 2-AA-ether-LPC (Fig. 3). Starting with commercially available 2-AA-glyceryl ether, we stereoselectively acetylated the sn-1 position in methyl acetate using porcine pancreatic lipase absorbed on Celite as described previously (22). This afforded the l-conformation at the sn-2 carbon because this transacylase reaction highly selectively acetylates the hydroxy group on the sn-1 carbon. To synthesize the d-enantiomer, we protected the sn-3 hydroxyl with a trityl group and subsequently hydrolyzed the more labile acetyl group using tetrabutylammonium hydroxide. Thus, two different protected chiral intermediates were obtained.
Figure 3.

Stereoselective synthesis of l-2-AA-ether-LPC and d-2-AA-ether-LPC. a, lipase from porcine pancreas absorbed on Celite, anhydrous methyl acetate, 3-Å molecular sieves, room temperature (rt). b, (i) Et3N, POCl3, anhydrous CH2Cl2, 0 °C to rt; (ii) choline tosylate, pyridine, anhydrous CH2Cl2, rt. c, Bu4NOH, Et2O, rt. d, TrCl, dimethylaminopyridine, Et3N, anhydrous CH2Cl2, rt. e, Bu4NOH, Et2O, rt. f, (i) Et3N, POCl3, anhydrous CH2Cl2, 0 °C to rt; (ii) choline tosylate, pyridine, anhydrous CH2Cl2, rt. g, TFA, anhydrous CH2Cl2, rt. Details of reactions are found under “Experimental procedures.”
To validate the optical purity of these critical intermediates, both were transformed to their respective diastereomers by derivatization with (R)-(+)-α-methylbenzyl isocyanate. The carbamates produced from the two chiral intermediates were analyzed by straight-phase HPLC, and their chromatograms were compared with racemic standards. As shown in Fig. 4A, derivatized 1-Ac-2-AA-ether-rac-glycerol has two resolvable chromatographic peaks of similar heights at 31.5 and 33.5 min, which were the two predicted (R)-(+)-α-methylbenzyl isocyanate–derivatized enantiomers (d- and l-) of 1-Ac-2-AA-ether-rac-glycerol. In contrast, the derivatized 1-Ac-2-AA-ether-sn-glycerol from the enzymatic reaction with pancreatic lipase has only one major peak at 33.5 min, which suggests that the product is optically active. By comparing the area of the peaks at 31.5 and 33.5 min in Fig. 1B, the optical purity of 1-Ac-2-AA-ether-sn-glycerol obtained from the lipase-catalyzed acetylation reaction was calculated to be 92%. Similarly, the optical purity of 2-AA-ether-3-trityl-sn-glycerol was 91% as calculated by comparison of the areas of the two peaks of the (R)-(+)-α-methylbenzyl–derivatized enantiomers at 18.5 and 20.0 min in Fig. 4B.
Figure 4.

HPLC analysis of R-MBIC–derivatized racemic standards and optically active products from the enzymatic reaction. Chemical derivatization of the enantiomers of diacylglycerol standards/products with R-MBIC generates the corresponding diastereomers, which can be separated utilizing a Si HPLC column. The resultant chromatograms show racemic standards with two peaks of equal intensity representing the two R-MBIC-diacylglycerol enantiomers, whereas the products from the enzymatic reaction have only one major peak, thereby indicating that the products are enantiomeric products. A, chromatogram of R-MBIC–derivatized racemic 1-Ac-2-AA-ether-glycerol (top) and R-MBIC–derivatized optically active 1-Ac-2-AA-ether-sn-glycerol (bottom). B, chromatogram of R-MBIC–derivatized racemic 2-AA-ether-3-trityl-glycerol (top) and R-MBIC–derivatized 2-AA-ether-3-trityl-sn-glycerol (bottom).
Next, a phosphocholine group was attached to each intermediate by sequential reactions with POCl3 and choline tosylate as described (23). After attachment of the phosphocholine group, the acetyl group of 1-Ac-2-AA-ether-sn-glycerol-3-phosphocholine was removed by tetrabutylammonium hydroxide, yielding l-2-AA-ether-LPC. The polar headgroup phosphocholine was attached to 2-AA-ether-3-trityl-sn-glycerol by the same series of reactions. Next, the trityl group was subsequently removed by TFA, yielding d-2-AA-ether-LPC. The structural identities of both final products and all intermediates were confirmed by multistage high-resolution MS and proton NMR as described in Figs. S1–S5.
Tandem mass spectrometric analyses of l-2-AA-ether-LPC and d-2-AA-ether-LPC demonstrated the anticipated m/z of l-2-AA-ether-LPC as 552.3420 ([C28H52NO6P + Na]+ calculated m/z = 552.3424, Δ = 0.7 ppm) (Fig. 5). Similarly, the m/z of d-2-AA-ether-LPC was 552.3416 ([C28H52NO6P + Na]+ calculated m/z = 552.3424, Δ = 1.4 ppm) (Fig. 5). The MS2 and MS3 spectra of l-2-AA-ether-LPC and d-2-AA-ether-LPC are indistinguishable. In MS2 spectra, the fragmentation of the sodium adducts of l- and d-2-AA-ether-LPC produce a fragment ion with an m/z of 493.2677 that is due to the neutral loss of trimethylamine (m/z 59) from each precursor ion. In MS3 spectra, fragmentation of the ion at m/z 493.2677 produces two major product ions with m/z 449.2416 and m/z 369.2752 that result from the neutral loss of CH2CH2O and the neutral loss of CH2CH2PO4H, respectively. The MS1, MS2, and MS3 spectra unambiguously prove the structure of the desired optically active intermediates.
Figure 5.

Tandem mass spectrometric analysis of l-2-AA-ether-LPC, d-2-AA-ether-LPC, and l-2-AA-LPC. Stereoselectively synthesized l-2-AA-ether-LPC and d-2-AA-ether-LPC as well as l-2-AA-LPC were separated on a C18 HPLC column and analyzed by MS. Tandem MS was performed using an LTQ ion trap with collision energy of 25 eV for MS2 and 30 eV for MS3. The resultant fragment ions were detected in an Orbitrap mass spectrometer with a mass resolution of 30,000 at m/z = 400 and a mass accuracy within 5 ppm.
Tandem mass spectrometric analysis of the 2-AA-LPC standard demonstrated fragmentation of the sodium adduct, generating a product ion with neutral loss of m/z 59. However, in contrast to the ether analogs, MS3 fragmentation of the ion at m/z 507.2462 produced only a fragment ion with an m/z of 383.2538 that results from the neutral loss of CH2CH2PO4H. The product ion resulting from the neutral loss of CH2CH2O is not present in the MS3 spectrum of 2-AA-LPC, further substantiating the differences between lysolipids and their nonhydrolyzable ether analogs.
Acylation of l-2-AA-ether-LPC by different subcellular fractions from murine liver
To determine whether l-2-AA-ether-LPC could be acylated by an acyl-CoA–dependent sn-1 acyltransferase in different subcellular fractions of liver, 10 μm l-2-AA-ether-LPC was incubated with sonicated mitochondria, cytosol, or microsomes in the presence or absence of 10 μm stearoyl-CoA. The results demonstrate that highest specific activity for acylation of l-2-AA-ether-LPC is present in the microsomal fraction (Table 1). The formation of 1-stearoyl-2-arachidonyl-ether-sn-PC from l-2-AA-ether-LPC is minimal in the cytosolic fraction and highest in microsomes. Because preparations of mitochondria inevitably contain some microsomal components, the sn-1 acylation activity within the mitochondrial fraction is likely due to a small amount of microsomal contamination in the mitochondrial fraction. For the microsomal fraction, the formation of 1-stearoyl-2-arachidonyl-ether-sn-PC with 10 μm stearoyl-CoA is ∼4-fold higher than in the absence of fatty acyl-CoA. These results highlight the requirement of acyl-CoA for PC synthesis from l-2-AA-ether-LPC and rule out significant transacylation effects.
Table 1.
sn-1 acyltransferase activity of murine hepatic subcellular fractions
Subcellular fractions from C57 mouse liver were isolated by differential centrifugation as described under “Experimental procedures”. Cytosolic, mitochondrial, and microsomal proteins were incubated with the indicated concentrations of l-2-AA-ether-LPC and 18:0-CoA at 37 °C for 5 min in 75 mm sodium phosphate buffer (pH 7.4). The 1-stearoyl-2-arachidonyl-ether-sn-glycero-3-phosphocholine produced in the reaction was extracted in the presence of dimyristoleoylphosphatidylcholine (14:1-PC) internal standard and quantified by LC-MS. Values are the average of four independent preparations ±S.D.
| 1-Stearoyl-2-arachidonyl-ether-sn-PC production |
|||
|---|---|---|---|
| Cytosol | Mitochondria | Microsomes | |
| nmol·mg−1·min−1 | |||
| Control | 0 ± 0 | 0.001 ± 0.001 | 0.002 ± 0.001 |
| 10 μm l-2-AA-ether-LPC | 0 ± 0 | 0.01 ± 0.01 | 0.01 ± 0.004 |
| 10 μm l-2-AA-ether-LPC + 10 μm 18:0-CoA | 0.006 ± 0.002 | 0.03 ± 00.01 | 0.13 ± 0.03 |
The acyl-CoA dependence of the sn-1 acyltransferase reaction is enantioselective for the l configuration
To determine whether the sn-1 acyltransferase activity was enantioselective, we compared the initial rates of acyl-CoA–dependent acylation of d- and l-2-AA-ether-LPCs. Incubation of 10 μm l- or d-2-AA-ether-LPC with murine hepatic microsomes in the presence of 10 μm stearoyl-CoA demonstrated that the formation of PC from l-2-AA-ether-LPC was approximately 3 times higher than that from d-2-AA-ether-LPC (Fig. 6). These results demonstrate that the acyl-CoA–dependent sn-1 acyltransferase reaction catalyzed by hepatic microsomal acyltransferase(s) is stereoselective for the naturally occurring form of lysophospholipids (i.e. l-lysophosphatidylcholine).
Figure 6.

Stereoselectivity of the mouse liver microsomal sn-1 acyltransferase reaction to produce 1-stearoyl-2-AA-ether-PC. Microsomal homogenates isolated from mouse liver were incubated with either 10 μm l-2-AA-ether-LPC or 10 μm d-2-AA-ether-LPC in the presence of 10 μm 18:0-CoA for 0, 2, 5, 9, or 15 min at 37 °C in 75 mm sodium phosphate buffer (pH 7.4). The reactions were terminated by adding chloroform/methanol (1:1, v/v) and vortexed. The chloroform phase was isolated and dried under a nitrogen stream. The dried residues were redissolved in water/methanol (1:4), and the sn-1 acyltransferase product, 1-stearoyl-2-AA-ether-PC, was analyzed and quantitated by LC-MS in the positive ion mode. Values are the average of four independent preparations. Error bars represent S.D.
The effect of 2-AA-ether-LPC and fatty acyl-CoA concentrations on the sn-1 acyltransferase reaction
Previously, it has been shown that the acyl-CoA acyltransferase reaction at the sn-2 position of LPC depends on the concentrations of sn-1 LPC and fatty acyl-CoA substrates. Accordingly, we examined the concentration dependence of each substrate on initial reaction velocity. Selected concentrations of these two substrates were incubated with the microsomal fraction isolated from mouse liver. The LPC sn-1 acyltransferase reaction increases linearly up to 50 μm 2-AA-ether-LPC in the presence of 10 μm exogenous stearoyl-CoA (Fig. 7A). Approximately 10 μm stearoyl-CoA saturated the sn-1 acyltransferase reaction in the presence of 10 μm 2-AA-ether-LPC (Fig. 7B).
Figure 7.

Effect of LPC and acyl-CoA concentrations on microsomal sn-1 acyltransferase activity. A, microsomal homogenates isolated from mouse liver were incubated with 10 μm 18:0-CoA in the presence of increasing concentrations of l-2-AA-ether-LPC for 1 min at 37 °C in 75 mm sodium phosphate buffer (pH 7.4). The reactions were terminated by adding chloroform/methanol (1:1, v/v). Di-14:1-PC was added as an internal standard, and the extraction mixture was vortexed. The chloroform layer was collected and dried under a nitrogen stream. The dried residues were redissolved in water/methanol (1:4), and the sn-1 acyltransferase product, 1-stearoyl-2-AA-ether-PC, was analyzed and quantitated by LC-MS in the positive ion mode. B, microsomal homogenates isolated from mouse liver were incubated with 10 μm l-2-AA-ether-LPC in the presence of increasing concentrations of 18:0-CoA for 2 min at 37 °C in 75 mm sodium phosphate buffer (pH 7.4). The resulting 1-stearoyl-2-AA-ether-PC was extracted and analyzed as described above. Values are the average of four independent preparations. Error bars represent S.E.
Acyl-CoA specificity of the microsomal sn-1 LPC acyltransferase reaction
Years ago, Lands (24) and Lands and Merkl (25) demonstrated that polyunsaturated fatty acids are preferentially incorporated at the sn-2 hydroxyl of sn-1 LPC by cellular acyl-CoA–dependent acyltransferases in a process now known as the Lands cycle. Because naturally occurring PC predominantly contains saturated fatty acids at the sn-1 position, we reasoned that palmitate and stearate should be preferentially incorporated at the sn-1 hydroxyl of sn-2 LPC by microsomal sn-1 LPC acyltransferase activity in parallel with the regiospecificity of nearly all of the polyunsaturated phospholipids in mammalian tissues. Accordingly, we compared the initial reaction velocities using selected concentrations of saturated and unsaturated fatty acyl-CoAs in the presence of l-2-AA-ether-LPC and hepatic microsomes. Saturated fatty acids (i.e. palmitoyl-CoA and stearoyl-CoA) were preferentially incorporated at the sn-1 position of sn-2-AA-LPC compared with their unsaturated fatty acyl counterparts (Fig. 8).
Figure 8.
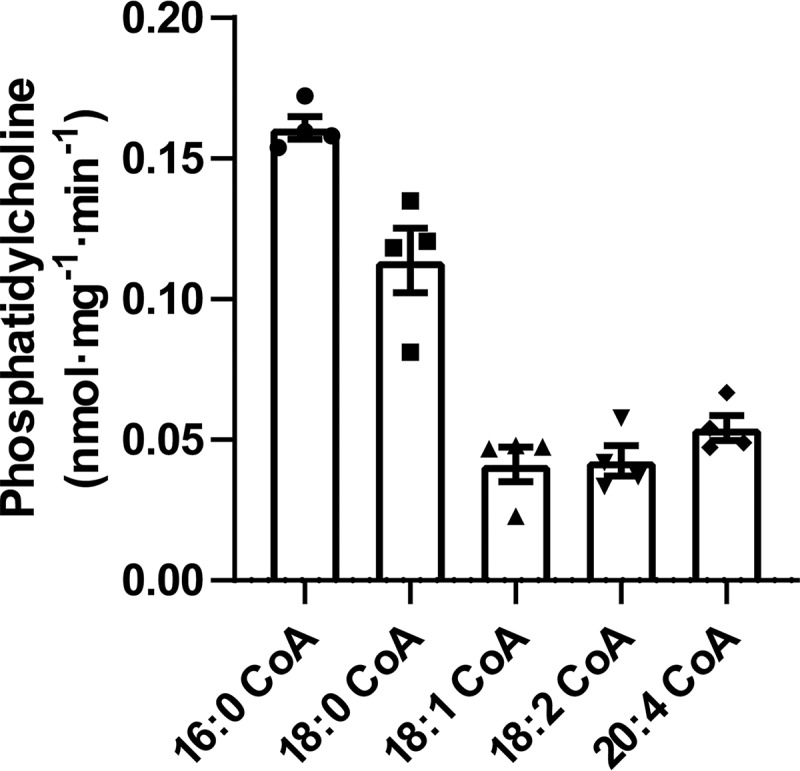
Substrate specificities of the microsomal sn-1 acyltransferase reaction. Microsomal homogenates isolated from mouse liver were incubated with 10 μm l-2-AA-ether-LPC in the presence of selected saturated and unsaturated molecular species of acyl-CoAs for 5 min at 37 °C in 75 mm sodium phosphate buffer (pH 7.4). The reactions were terminated by addition of chloroform/methanol (1:1, v/v) and vortexed. The chloroform layer was isolated and dried under a nitrogen stream. The dried residues were redissolved in water/methanol (1:4), and the sn-1 acyltransferase product, 1-acyl-2-AA-ether-PC, was analyzed and quantitated by LC-MS in the positive ion mode. Values are the average of four independent preparations. Error bars represent S.D.
Oxidation of the 2-AA-LPC ether analog by 15-LOX
Previously, we have demonstrated that 2-AA-LPC can be directly oxidized by oxygenases such as COX-2 and 15-LOX to produce the corresponding oxidized LPCs (19). Considering the structural similarity between 2-AA-LPC and 2-AA-ether-LPC, we anticipated that the 2-AA-ether-LPC would also be oxidized by 15-LOX. To test this possibility, purified human recombinant 15-LOX was incubated with either l-2-AA-ether-LPC or d-2-AA-ether-LPC. The resulting products were extracted and analyzed by LC-MS and LC-MS/MS. The results demonstrated that both l- and d-2-AA-ether-LPC were efficiently oxidized by 15-lipoxygenase with no significant differences in the specific activity between these stereoisomers (Fig. 9).
Figure 9.

Oxidation of d and l stereoisomers of 2-AA-ether-LPC by 15-lipoxygenase. Purified recombinant human 15-lipoxygenase was incubated with either 10 μm l-2-AA-ether-LPC or 10 μm d-2-AA-ether-LPC for 0, 2.5, 5, 10, or 15 min at 37 °C in 50 mm Tris-Cl buffer (pH 7.2). The reactions were terminated by addition of 2 volumes of chloroform/methanol (1:1) containing 0.1% acetic acid and internal standards (17:1-LPC). The chloroform phase was separated and dried by nitrogen. The resulting oxidized LPC was dissolved in methanol and analyzed by LC-MS. Values are the average of four independent preparations. Error bars represent S.E.
Production of oxidized PC from oxidized 2-AA-ether-LPC by microsomal fatty acyl-CoA–dependent sn-1 acyltransferase
Previous work has suggested that the production of oxidized phospholipids occurs mainly through a four-step process initiated by the release of AA followed by its oxidation by various oxidases, thioesterification by acyl-CoA synthetase, and incorporation of the oxidized acyl-CoAs into lysophospholipids by acyl-CoA acyltransferases. Considering our observation of the relative abundance of oxidized lysolipids in biological systems, we were interested in determining whether oxidized phospholipids could be synthesized through the direct acylation of oxidized lysophospholipids by the sn-1–selective acyl-CoA–dependent acyltransferase activity we discovered. To determine whether oxidized lysophospholipids could serve as substrates for the sn-1 acyltransferase, 2–15-HETE-ether-LPC was prepared by 15-LOX–mediated oxidation of 2-AA-ether-LPC and incubated with hepatic microsomes in the presence of stearoyl-CoA. The resultant reaction products were extracted and analyzed by LC-MS. Notably, 1-stearoyl-2-15-HETE-ether-PC was rapidly generated by the microsomal sn-1 acyltransferase activity, which incorporated stearic acid from stearoyl-CoA into 2–15-HETE-ether-LPC (Fig. 10A). To substantiate the identification of 1-stearoyl-2–15-HETE-ether-PC, the accurate mass of the predominant precursor ion in MS1 (Fig. 10B) was compared with its theoretical value (m/z 812.6165), revealing a 1-ppm difference in mass. Fragmentation of the precursor 1-stearoyl-2–15-HETE-ether-PC ion at m/z 812.6178 yielded a product ion at m/z 794.6045 resulting from the neutral loss of water, thereby substantiating the existence of a hydroxy group, likely in the arachidonoyl acyl chain (Fig. 10C). Collectively, these results demonstrate separate and distinct biochemical pathways for the production of oxidized phospholipids in biologic systems.
Figure 10.

Synthesis of 1-stearoyl-2–15-HETE-ether-PC from stearoyl-CoA and 2–15-HETE-ether-LPC by murine hepatic microsomal homogenates. Microsomal homogenates isolated from mouse liver were incubated with 10 μm l-2–15-HETE-ether-LPC and 10 μm stearoyl-CoA for 0, 1, 2.5, and 5 min at 37 °C in 75 mm sodium phosphate buffer (pH 7.4). The reactions were terminated by addition of chloroform/methanol (1:1, v/v) containing di-14:1-PC internal standard. The mixture was vortexed, and the chloroform layer was separated and dried under a nitrogen stream. The dried residues were redissolved in water/methanol (1:4), and the resultant 1-stearoyl-2–15-HETE-ether-PC was chromatographed on a C18 HPLC column and analyzed by MS. Fragmentations were performed in an LTQ ion trap with a collision energy of 35 eV, and the resultant fragment ion was detected in an Orbitrap mass spectrometer with a mass resolution of 30,000 at m/z = 400 and a mass accuracy within 5 ppm. A, production of 1-stearoyl-2–15-HETE-ether-PC from stearoyl-CoA and 2–15-HETE-ether-LPC incubated with murine hepatic microsomal homogenates. B, MS1 spectrum and accurate mass of 1-stearoyl-2–15-HETE-ether-PC. C, MS2 spectrum of 1-stearoyl-2–15-HETE-ether-PC. Values are the average of three independent preparations. Error bars represent S.D.
Synthesis of oxidized phosphatidylcholine from oxidized 2-AA-ether-LPC by platelets and comparison with the synthesis of oxidized phosphatidylcholine from incorporation of oxidized nonesterified fatty acid
To estimate the relative contribution of each pathway to the synthesis of oxidized phosphatidylcholine in murine platelets after thrombin stimulation, we compared the initial rates of oxidized PC synthesis from oxidized fatty acids with that of oxidized lysophosphatidylcholine. To this end, we incubated 8 μm 2–15-HETE-ether-LPC and 8 μm 15-HETE-d8 with murine platelets and activated the platelets with 1 unit/ml thrombin for 30 min. The lipids were extracted and analyzed by LC-MS as described under “Experimental procedures”. Similar amounts of oxidized PC were synthesized from both oxidized fatty acid and oxidized LPC using palmitoyl-CoA, whereas ∼⅓ the amount was synthesized using stearoyl-CoA (Fig. 11). Thus, dual pathways exist for the synthesis of oxidized phospholipids in activated platelets that are likely dependent on the spatial, temporal, and subcellular location of the participating enzymes and their preferred substrates.
Figure 11.
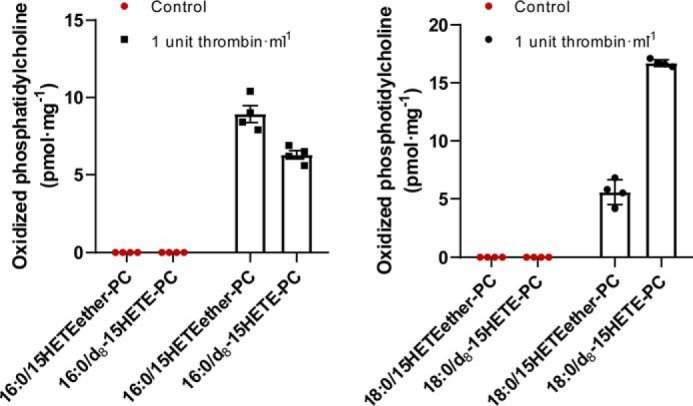
Synthesis of oxidized phosphatidylcholine from oxidized fatty acid (deuterated 15-HETE) or oxidized (15-HETE-ether–linked) sn-2 lysophosphatidylcholine by activated platelets. Murine blood in the presence of 3.8% sodium citrate was centrifuged at 150 × g for 10 min. The platelet-rich plasma (upper layer) was collected and centrifuged again at 1500 × g for 10 min. The supernatant was discarded, and the platelet pellet was suspended in 200 μl of Tyrode buffer. The protein concentration was determined by a Bradford assay. 2–15-HETE-ether-LPC and 15-HETE-d8 were added to the isolated platelets, each with a final concentration of 10 μm. The platelets were activated by addition of 1 unit·ml−1 thrombin and incubated at 37 °C for 30 min. The reactions were terminated by addition of chloroform/methanol (1:1, v/v) containing 0.1 nmol of di-14:1-PC as an internal standard and vortexed. The chloroform layer was collected and dried under a nitrogen stream. The dried residues were redissolved in water/methanol (1:4), and the resultant oxidized phosphatidylcholines were analyzed and quantitated by LC-MS in the positive ion mode. Values are the average of four independent preparations. Error bars represent S.D.
Discussion
In this study, we describe a novel pathway for the synthesis of enzymatically generated oxidized phospholipids resulting from sn-1 deacylation/reacylation cycling and the direct enzymatic oxidation of arachidonoyl-LPC and arachidonoyl-LPE. This discovery was mechanistically substantiated by development of a facile synthesis of enantiomerically pure nonhydrolyzable ether analogs of 2-AA-LPC. Through the use of these enabling reagents, this novel pathway was shown to be activated in thrombin-stimulated platelets, contributing to the production of oxidized phospholipids. Additionally, the stereospecificity and substrate preferences of the major murine hepatic microsomal sn-1 acyltransferase activity were determined. This activity was selective for the naturally occurring lysolipid enantiomer (l > d) as well as favoring incorporation of saturated aliphatic chains as normally found at the sn-1 position of cellular phosphatidylcholines.
The canonical pathway of oxidized phospholipid synthesis is a four-step process initiated by the sn-2 hydrolysis of 1-palmitoyl-2-arachidonoyl-sn-PC by a PLA2, the oxidization of the released nonesterified arachidonic acid to various eicosanoids, the activation of the resultant eicosanoid by acyl-CoA synthetase, and the esterification of the eicosanoid-CoA to the sn-2 position of 1-palmitoyl (or 1-stearoyl)-LPC. In the present study, we demonstrated the existence of a previously unknown pathway that contributes to the enzymatic production of oxidized phospholipids in platelets. This pathway uses a three-step sequential process initiated by an sn-1 phospholipase (PLA1) activity to generate 2-AA-LPC and direct oxidation of 2-AA-LPC to 2–15-HETE-LPC by 15-LOX followed by the acylation of the eicosanoid-LPC by palmitoyl-CoA or stearoyl-CoA catalyzed by sn-1 LPC acyltransferase. This new pathway is separate and distinct from the canonical pathway for oxidized phospholipid synthesis in platelets and other cells.
The sn-2 lysophospholipid acyltransferase reaction has been thoroughly characterized primarily as it relates to phospholipid remodeling (24, 25). In this process, now known as the Lands cycle, the sequential deacylation/reacylation is a primary determinant of the fatty acyl chain composition at the sn-2 position. De novo synthesized phospholipids are often produced with a mixture of fatty acyl chains not typically found under steady-state conditions. Through exchange/remodeling inherent in the Lands cycle by the sequential actions of PLA2s and sn-2 lysophospholipid acyltransferases, the mature phospholipid composition can be obtained. The sn-2 acyltransferase reaction is critical to this process because it is highly selective for the use of unsaturated fatty acyl-CoAs that are channeled into the corresponding sn-2 position of lysophospholipids, which typically contain a saturated fatty acyl chain at the sn-1 position. The importance of remodeling is underscored in several disease states, including Barth syndrome, cardiolipin depletion in diabetic cardiomyopathy, and neuromuscular diseases (26–29). Lysophospholipid acyltransferases also play important roles in other biological processes, including phospholipid trafficking, cell differentiation, and cancer chemoresistance (30–33). Compared with the sn-2 acyltransferase, the sn-1 acyltransferase reaction has been much less thoroughly investigated because 2-acyl-lysophospholipids readily undergo α-hydroxy migration due to the thermodynamically favored 1-acyl-lysophospholipids. Thus, discrimination of the specificity of bona fide sn-1 versus sn-2 lysolipid acyltransferase activities has remained difficult to precisely define. To overcome this obstacle, the use of synthetic optically active nonhydrolyzable 2-AA-ether-LPC as the acyl acceptor in this study unambiguously defined the stereospecificity and fatty acyl selectivity of the sn-1 acyltransferase activity by preventing fatty acid α-hydroxy migration or hydrolysis during the reaction.
Although oxidized phospholipids have many significant biological effects, the pathways through which oxidized phospholipids are synthesized have not been unambiguously identified. In human platelets and neutrophils, Thomas and co-workers (2, 34) have suggested that oxidized phospholipids are generated through PLA2 hydrolysis of a nonoxidized phospholipid to release a polyunsaturated fatty acid, which is then oxidized by cyclooxygenases or lipoxygenase(s), activated by ligation to CoASH, and finally re-esterified into acceptor lysophospholipids by sn-2 acyltransferase. Maskrey et al. (35) have proposed that, in activated human monocytes, oxidized phospholipids are synthesized through direct oxidation of the phospholipid by lipoxygenases.
Previously, we reported that 2-arachidonoyl-lysophospholipids could be directly oxidized by both COX-1 and COX-2 as well as by 15-LOX, leading to the formation of 2-eicosanoid lysophospholipids (19). In addition, we have demonstrated that 2-arachidonoyl-lysophospholipids can be produced through oxidative cleavage of the vinyl ether linkage in plasmalogens catalyzed by cytochrome c (36) or selective sn-1 hydrolysis of phospholipids by phospholipases such as iPLA2γ (17) and group XV phospholipase A2 among other sn-1 phospholipases (37, 38). Using stable isotope experiments, when 15-HETE and 15-HETE-ether-LPC are present in equal concentrations, we demonstrated that ∼40% of palmitate is directed to the sn-1 position to generate oxidized PC, whereas ∼30% of stearate is directed to the synthesis of oxidized PC by this previously unknown pathway. This result shows that utilization of 2-eicosanoid-LPC for eicosanoid-PC synthesis constitutes a substantial portion of total oxidized PC synthesis. Precise determination of the relative importance of this noncanonical pathway is difficult because the enzymes and substrate targets are not necessarily present in the same subcellular compartment, and the potential presence of metabolic channeling to generate a lipid synthetic metabolon makes definitive relative rates of each pathway difficult to determine.
In conclusion, the present work demonstrates a facile enantiomeric synthesis of both stereoisomers of nonhydrolyzable 2-AA-ether-LPC that were employed to study their oxidation by 15-LOX and subsequent acylation by microsomal sn-1 lysophospholipid acyltransferase(s). The results establish the existence of a previously unknown pathway in platelets capable of generating oxidized phospholipids in activated platelets and likely by analogy in other cells. These results raise many intriguing questions for further investigation of this pathway regarding the generation of other enzymatically produced oxidized phospholipids, their mechanism of formation, and their roles in signaling in different cell types and subcellular compartments in a spatial and context-dependent manner.
Experimental procedures
Materials
2-Arachidonyl glycerol ether (2-AA-glycerol ether) was purchased from Cayman Chemical (Ann Arbor, MI). Palmitoyl-CoA, stearoyl-CoA, linoleoyl-CoA, oleoyl-CoA, arachidonoyl-CoA, 1-(1Z-octadecenyl)-2-arachidonoyl-sn-glycero-3-phosphoethanolamine (plasmenyl-SAPE), and 1-(1Z-octadecenyl)-2-arachidonoyl-sn-glycero-3-phosphocholine (plasmenyl-SAPC) were obtained from Avanti Polar Lipids (Alabaster, AL). Kinetex 5-μm EVO C18 column (250 × 4.6 mm) and Kinetex 2.7-μm EVO C18 column (150 × 2.1 mm) were purchased from Phenomenex (Torrance, CA). Discovery DSC-NH2 SPE cartridges (500 mg/6 ml) and C18 SPE cartridges (2 g, 12 ml) were purchased from Supelco (Bellefonte, PA). Silica gel for column chromatography (60 Å, 35–75 μm) and high-performance TLC plates (HPTLC-HLF, UV254, 150 μm) were purchased from Analtech (Newark, DE). Celite Hyflo Supercel (diatomaceous earth) was purchase from EMD Millipore (Billerica, MA). Lipase from porcine pancreas (type II) was purchased from Sigma-Aldrich. LC-MS–grade acetonitrile and water were obtained from Fisher Scientific. LC-MS–grade methanol and isopropanol were purchased from Burdick & Jackson (Muskegon, MI). HPLC-grade methanol and acetonitrile were purchased from Fisher Scientific. All other chemicals were purchased from Sigma-Aldrich.
General animal studies
Animal protocols were conducted in strict accordance with the National Institutes of Health guidelines for humane treatment of animals and were reviewed and approved by the Animal Studies Committee of Washington University.
NMR analysis
1H NMR spectra were recorded on a Varian spectrometer (400 MHz) and are referenced relative to tetramethylsilane proton signals at δ 0 ppm.
Synthesis of 1-Ac-2-AA-ether-glycerol
Lipase immobilized on Celite was prepared as described (22). Briefly, 8 g of crude porcine pancreas lipase was dissolved in 80 ml of 18 mm sodium phosphate buffer (pH 8.0). The lipase solution was stirred for 20 min at room temperature and then centrifuged at 10,000 × g for 10 min. The supernatant was collected and cooled to 4 °C. 20 g of Celite Hyflo Supercel was slowly added to the supernatant to form a cloudy suspension that was stirred for 10 min at 4 °C. Cold acetone (150 ml) was added dropwise to the suspension over 20 min followed by 30 min of stirring at 4 °C. The suspension was filtered by vacuum, and the solid filtride was washed with 100 ml of cold acetone. The filtride was dried by vacuum until it became a fine and loose powder and was stored at 4 °C.
90 mg (0.25 mmol) of 2-AA-glycerol ether, 360 mg of Celite-immobilized porcine pancreas lipase, 0.2 g of 4-Å molecular sieve beads, and 18 ml of methyl acetate were mixed and stirred under nitrogen at room temperature. After 4.5 h, the solid was filtered out. The filtrate was dried by vacuum and purified by silica-gel column chromatography (hexane/ethyl acetate, 2:1, to ethyl acetate). Approximately 18 mg of 1-Ac-2-AA-ether-glycerol was obtained at this step of the synthesis. The unreacted 2-AA-glycerol ether was eluted by ethyl acetate and recycled.
Synthesis of 2-AA-ether-3-Tr-glycerol
9 mg (0.022 mmol) of 1-Ac-2-AA-ether-glycerol, 0.13 mg (0.0011 mmol) of dimethylaminopyridine, and 6.6 μl (0.047 mmol) of triethylamine were dissolved in 2 ml of dichloromethane. 12 mg (0.043 mmol) of trityl chloride was then added and stirred under nitrogen at room temperature overnight. The solvent was dried by vacuum, and the product was purified by silica-gel column chromatography (hexane to hexane/ethyl acetate, 10:1)
The 1-Ac-2-AA-3-Tr-glycerol obtained was then dissolved in 1 ml of ethyl ether. 30 μl of 40% tetrabutylammonium hydroxide in methanol was added, and the solution was stirred at room temperature for 1.5 h. The solvent was dried by vacuum, and the product was purified by column chromatography (hexane/ethyl acetate, 10:1, to hexane/ethyl acetate, 2:1). The yield of 2-AA-ether-3-Tr-glycerol was 5.5 mg.
Synthesis of l-2-AA-ether-LPC
25 μl (0.27 mmol) of POCl3 and 200 μl (1.4 mmol) of triethylamine were dissolved in 1 ml of dichloromethane at 0 °C and placed under nitrogen. 20 mg (0.05 mmol) of 1-Ac-2-AA-ether-glycerol was dissolved in 1 ml of anhydrous dichloromethane and added dropwise. Next, the reaction was allowed to warm to room temperature with continuous stirring for 1 h. Then 125 mg (0.45 mmol) of choline tosylate and 500 μl of pyridine were added, and the reaction was stirred at room temperature under nitrogen for 24 h. The reaction was quenched by addition of 1.08 ml of 1 m NaOH solution and stirred for 1 h followed by addition of 1.08 ml of methanol. The dichloromethane phase was collected, and the aqueous phase was extracted twice with 1 ml of chloroform. The organic phases were combined, dried over Na2SO4, and loaded onto a 500-mg Discovery DSC-NH2 SPE cartridge that had been equilibrated with 6 ml of chloroform. The column was washed with 6 ml of chloroform, and the 1-Ac-2-AA-ether-PC was eluted in 8 ml of methanol. The methanol eluent was dried under nitrogen and dissolved in 1 ml of ethyl ether. 50 μl of 40% tetrabutylammonium hydroxide in methanol was added, and the reaction was then stirred for 1 h. The reaction was stopped by adding 0.5 ml of saturated ammonium chloride solution and 0.5 ml of water. The mixture was vortexed, and the ether phase was collected. 1 ml of chloroform and 1 ml of methanol were added into the aqueous phase and vortexed. The chloroform phase was collected. The organic phases were combined and dried by nitrogen. The dried product was dissolved in 250 μl of methanol followed by addition of 750 μl of water after which it was loaded onto a 2-g C18 SPE column that had been prewashed with 12 ml of methanol and equilibrated with 12 ml of methanol/water (1:4). The column was then washed by 12 ml of methanol/water (2:3), and the product was eluted by 24 ml of methanol. The methanol solution was dried under a nitrogen stream for HPLC purification.
Synthesis of d-2-AA-ether-LPC
25 μl (0.27 mmol) of POCl3 and 50 μl (0.36 mmol) of triethylamine were dissolved in 1 ml of dichloromethane at 0 °C under a nitrogen atmosphere. 3 mg (0.005 mmol) of 2-AA-ether-3-trityl-glycerol was dissolved in 1 ml of anhydrous dichloromethane and added dropwise to the POCl3/triethylamine solution. The reaction was warmed to room temperature and stirred for 1 h. Next, 137 mg (0.50 mmol) of choline tosylate and 60 μl of pyridine were added. The reaction was stirred at room temperature under nitrogen for 24 h. The reaction was quenched by addition of 0.94 ml of 1 m NaOH solution and stirred for 1 h followed by addition of 1.06 ml of methanol and 1 ml of chloroform. The mixture was vortexed, and the organic phase was collected. The aqueous phase was re-extracted by 1 ml of chloroform. The organic phases were combined and dried under a nitrogen stream. The dried product was dissolved in 0.5 ml of chloroform, and 0.1 ml of TFA was added. After 20 min of stirring, the reaction was quenched by 0.65 ml of 1 m Na2CO3 followed by addition of 0.35 ml of water and 1 ml of methanol. The mixture was vortexed, and the organic phase was collected. The aqueous phase was re-extracted with 1 ml of chloroform. The organic phases were combined and dried under a nitrogen stream. The product was purified using a C18 SPE column as described above and was ready for HPLC purification.
Purification of 2-AA-ether-LPC by reversed-phase HPLC
A Phenomenex Kinetex EVO C18 column (5 μm, 250 × 4.6 mm) was used for HPLC purification. Solvent A was acetonitrile/methanol/water (2:1:1), solvent B was methanol, and solvent C was water. A linear gradient was used as follows with a flow rate of 1 ml/min: 0 min, 70% A, 0% B, 30% C; 2 min, 70% A, 0% B, 30% C; 8 min, 100% A; 15 min, 100% A; 15.1 min, 100% B; 20 min, 100% B; 20.1 min, 70% A, 0% B, 30% C; 30 min, 70% A, 0% B, 30% C. 2-AA-ether-LPC was eluted at 14 min. The purified 2-AA-ether-LPC was extracted with chloroform/methanol/water (1:1:1).
(R)-(+)-α-Methylbenzyl isocyanate derivatization and optical purity determination of 1-Ac-2-AA-ether-glycerol and 2-AA-ether-3-Tr-glycerol
0.5 mg of 1-Ac-2-AA-ether-glycerol-lipase was dissolved in 100 μl of (R)-(+)-α-methylbenzyl isocyanate (R-MBIC) and incubated at 37 °C under nitrogen for 24 h. After 24-h incubation, the reaction mixture was diluted with 1 ml of hexane and loaded onto a 300-mg silica-gel column packed in hexane. The column was washed with 2 ml of hexane, and the product was eluted in 2 ml of ethyl acetate. The solvent was dried under nitrogen, and the product was dissolved in heptane. The precipitate was filtered using glass wool, and the filtrate was used for HPLC analysis. The 2-AA-ether-3-Tr-glycerol was derivatized and purified in the same manner.
A Phenomenex Luna Silica column (3 μm, 250 × 4.6 mm) was used for the analysis of (R)-(+)-α-methylbenzyl isocyanate–derivatized product. Solvent A was heptane, and solvent B was ethanol. For (R)-(+)-α-methylbenzyl isocyanate–derivatized 1-Ac-2-AA-ether-glycerol, a linear gradient with a flow rate of 1.5 ml/min was used as follows: 0 min, 0.2% B; 40 min, 0.2% B; 40.1 min, 20% B; 45 min, 20% B; 45.1 min, 0.2% B; 57 min, 0.2% B. For (R)-(+)-α-methylbenzyl isocyanate–derivatized 2-AA-ether-3-Tr-glycerol, a different linear gradient with a flow rate of 1.5 ml/min was used as follows: 0 min, 0.4% B; 30 min, 0.4% B; 30.1 min, 20% B; 35 min, 20% B; 35.1 min, 0.4% B; 47 min, 0.4% B. Blank injections were performed before analyzing each sample.
Synthesis and purification of 2-AA-LPC-d9 and 2-AA-LPE
Plasmenyl-SAPC-d9 was synthesized from plasmenyl-SAPE as described (39). Briefly, 5 mg of plasmenyl-SAPE and 1.6 mg of benzyltriethylammonium chloride were dissolved in 0.2 ml of chloroform. 125 μl of 0.6 m Na2CO3 and 80 μl of CD3I were then added. The reaction was stirred at room temperature in the dark overnight. 0.8 ml of chloroform, 0.5 ml of saturated NaCl solution, 0.3 ml of water, and 1 ml of methanol were added to the reaction mixture. The mixture was vortexed and centrifuged at 800 × g for 5 min. The chloroform phase was collected and dried under nitrogen flow. The resulting plasmenyl-SAPC-d9 was dissolved in 0.8 ml of methanol. 0.2 ml of 0.4 m sulfuric acid was added, and the reaction mixture was incubated at 70 °C for 5 min. The reaction mixture was quickly cooled on ice after the incubation. 0.6 ml of water and 0.8 ml of chloroform were then added. The extraction mixture was vortexed and centrifuged at 800 × g for 5 min. The chloroform phase was collected and dried under nitrogen flow. The resulting 2-AA-LPC-d9 was purified by HPLC as described under “Purification of 2-AA-ether-LPC by reversed-phase HPLC.”
2 mg of plasmenyl-SAPE was dissolved in 0.8 ml of methanol. 0.2 ml of 0.4 m sulfuric acid was added, and the reaction mixture was incubated at 70 °C for 5 min. The resulting 2-AA-LPE was extracted and purified in the same manner as 2-AA-LPE-d9.
Subcellular fractionation and incubation of different fractions with lysophosphatidylcholine
C57 mice were purchased from The Jackson Laboratory. Following euthanasia of the mice, livers were removed and washed in cold isolation buffer (10 mm sodium phosphate, 0.25 m sucrose, 1 mm EDTA, 1 mm DTT (pH 7.4)) and then homogenized at 4 °C with a 15-ml Teflon pestle tissue grinder (eight strokes at a speed of 15). The homogenates were first centrifuged at 700 × g for 10 min to pellet nuclei and cellular debris. The supernatants were centrifuged at 10,000 × g for 10 min to pellet mitochondria. The mitochondrial pellet was resuspended in isolation buffer, and the supernatants were then centrifuged at 100,000 × g for 60 min to pellet the microsomal fraction. The supernatant after ultracentrifugation was used as cytosol, and the pellet was resuspended in isolation buffer and used as the microsomal fraction.
2-AA-ether-LPC or 2-AA-LPC-d9 was resuspended in 75 mm sodium phosphate buffer (pH 7.4) by vortexing and brief sonication (15 × 1 s), with a final concentration of 10 μm. Stearoyl-CoA was added into the buffer as indicated, with a final concentration of 10 μm. 5 μg of protein from different subcellular fractions was added to 200 μl of buffer containing the lipid substrate, and the mixture was incubated at 37 °C for the indicated times. The reaction was stopped by addition of 0.3 ml of water and 1 ml of chloroform/methanol (1:1) containing 0.1% acetic acid. 0.2 nmol of di-14:0-PC was added as internal standard. The mixture was vortexed and centrifuged at 3000 × g for 10 min. The chloroform phase was collected, and the aqueous phase was extracted again by 1 volume of chloroform. Chloroform phases were combined and dried under nitrogen stream. The dried residue was suspended in 200 μl of methanol and ready for LC-MS/MS analysis.
Oxidation of LPC and LPE by 15-lipoxygenase
2-AA-ether-LPC was resuspended in 50 mm Tris-HCl (pH 7.4) by vortexing, with a final concentration of 100 μm. Human recombinant 15-lipoxygenase-2 was added at a concentration of 5 μg/ml, and the reaction was incubated at 37 °C for the indicated times. The reaction was quenched by addition of 2 volumes of chloroform/methanol (1:1) containing 0.1% acetic acid. After vortexing and 10 min of centrifugation at 3000 × g, the chloroform phase was collected, and the aqueous phase was re-extracted with 1 volume of chloroform. The chloroform phases were combined and dried under a nitrogen stream. The obtained oxidized LPC was analyzed by LC-MS as described above. To prepare the 2–15-HETE-ether-LPC for acyltransferase activity assay, the dried residue was dissolved in 50–100 μl of methanol containing 1 mg/ml triphenylphosphine, which reduced the hydroperoxide to hydroxy. The methanol solution was injected into the HPLC system, and the 2–15-HETE-ether-LPC was purified as described under “Purification of 2-AA-ether-LPC by reversed-phase HPLC.” The oxidized LPC was eluted at 9 min.
2-AA-LPC-d9 was resuspended in 50 mm Tris-HCl, 100 mm NaCl, 0.5 mm EDTA (pH 7.0) by vortexing, with a final concentration of 100 μm. Then the 2-AA-LPC-d9 was oxidized and purified in the same manner as 2-AA-ether-LPC.
200 nmol of 2-AA-LPE was dissolved in 10 μl of DMSO followed by adding 2 ml of reaction buffer (50 mm Tris-HCl, 100 mm NaCl, 0.5 mm EDTA (pH 7.0)) that was preconditioned by 18O2. The product was extracted and reduced. The resulting 2–15-HETE-LPC (hydroxy 18O) was purified as described above.
Synthesis of oxidized phosphatidylcholine from oxidized 2-AA-ether-LPC by microsomal acyltransferase
2–15-HETE-ether-LPC was resuspended in 75 mm sodium phosphate buffer (pH 7.4) by vortexing and brief sonication, with a final concentration of 10 μm. Stearoyl-CoA was added into the buffer to a concentration of 10 μm. 5 μg of microsomal protein was added into 200 μl of buffer containing LPC and acyl-CoA, and the mixture was incubated at 37 °C for the indicated amount of time. The reaction was extracted as described above and ready for LC-MS/MS analysis.
Platelet isolation and incubation with oxidized 2-AA-ether-LPC
Blood was obtained by intracardiac puncture from euthanized mice. About 0.8 ml of blood was drawn into a syringe containing 0.15 ml of 3.8% sodium citrate to prevent platelet activation. The blood was then centrifuged at 150 × g for 10 min, and the platelet-rich plasma was subsequently centrifuged at 200 × g for 5 min to remove residual red blood cells. The purified platelet-rich plasma was spun at 1500 × g for 10 min, and the supernatant was discarded. The pellet was resuspended into Tyrode buffer. The protein concentration was measured using a Bradford protein assay (Bio-Rad).
2–15-HETE-ether-LPC (8 μm) and 15-HETE-d8 (8 μm) were resuspended in Tyrode buffer by vortexing and brief sonication (15 × 1 s). 30 μg of platelet protein was added into 200 μl of buffer, and the reaction was initiated by addition of 0.2 unit of thrombin to each sample. The mixture was incubated at 37 °C for the indicated times. The reaction was extracted as described above and was utilized for LC-MS/MS analyses.
LC-MS/MS analysis
LC-MS/MS analysis was performed using an LTQ Orbitrap mass spectrometer connected to a Waters Acquity UPLC system. Lipids were separated using a C18 reversed-phase column (Kinetex EVO C18, 2.7 μm, 150 × 2.1 mm) at 22 °C with a flow rate of 200 μl/min. A linear gradient of solvent A (10 mm ammonium acetate and 0.1% acetic acid (v/v) in water and solvent B (isopropanol) was used as follows: 0 min, 25% B; 5 min, 25% B; 20 min, 95% B; 25 min, 95% B; 25.1 min, 25% B; 35 min, 25% B. The autosampler tray temperature was set at 4 °C. The spray voltage in electrospray ionization source was 4.1 kV. The sheath gas flow rate was 40 (arbitrary unit). The capillary temperature was 270 °C.
Statistical analyses
Results are expressed as averages ± S.E. (or S.D.).
Author contributions
G.-Y. L. and R. W. G. conceptualization; G.-Y. L., S. H. M., C. M. J., H. F. S., and R. W. G. data curation; G.-Y. L., S. H. M., C. M. J., and R. W. G. formal analysis; G.-Y. L. and H. F. S. validation; G.-Y. L., H. F. S., and S. G. investigation; G.-Y. L. visualization; G.-Y. L. methodology; G.-Y. L. and R. W. G. writing-original draft; G.-Y. L., S. H. M., C. M. J., and R. W. G. writing-review and editing; H. F. S. and S. G. resources.
Supplementary Material
This work was supported by National Institutes of Health Grants R01HL118639 and R01HL133178. R. W. G. has a financial relationship with Platomics. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5.
- cPLA2α
- cytosolic phospholipase A2α
- HETE
- hydroxyeicosatetraenoic acid
- 2-AA-LPC
- 2-arachidonoyl-lysophosphatidylcholine
- 2-AA-LPE
- 2-arachidonoyl-lysophosphatidylethanolamine
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- COX
- cyclooxygenase
- 15-LOX
- 15-lipoxygenase
- iPLA2γ
- calcium-independent phospholipase A2γ
- Ac
- acetyl
- Tr
- trityl
- plasmenyl-SAPE
- 1-(1Z-octadecenyl)-2-arachidonoyl-sn-glycero-3-phosphoethanolamine
- plasmenyl-SAPC
- 1-(1Z-octadecenyl)-2-arachidonoyl-sn-glycero-3-phosphocholine
- SPE
- solid-phase extraction
- R-MBIC
- (R)-(+)-α-methylbenzyl isocyanate
- 18:0/20:4-PC
- 1-stearoyl-2-arachidonoyl-sn-PC
- rt
- room temperature.
References
- 1. Aldrovandi M., Hammond V. J., Podmore H., Hornshaw M., Clark S. R., Marnett L. J., Slatter D. A., Murphy R. C., Collins P. W., and O'Donnell V. B. (2013) Human platelets generate phospholipid-esterified prostaglandins via cyclooxygenase-1 that are inhibited by low dose aspirin supplementation. J. Lipid Res. 54, 3085–3097 10.1194/jlr.M041533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clark S. R., Guy C. J., Scurr M. J., Taylor P. R., Kift-Morgan A. P., Hammond V. J., Thomas C. P., Coles B., Roberts G. W., Eberl M., Jones S. A., Topley N., Kotecha S., and O'Donnell V. B. (2011) Esterified eicosanoids are acutely generated by 5-lipoxygenase in primary human neutrophils and in human and murine infection. Blood 117, 2033–2043 10.1182/blood-2010-04-278887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bochkov V. N., Oskolkova O. V., Birukov K. G., Levonen A. L., Binder C. J., and Stöckl J. (2010) Generation and biological activities of oxidized phospholipids. Antioxid. Redox Signal. 12, 1009–1059 10.1089/ars.2009.2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O'Donnell V. B., and Murphy R. C. (2012) New families of bioactive oxidized phospholipids generated by immune cells: identification and signaling actions. Blood 120, 1985–1992 10.1182/blood-2012-04-402826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang X., Liu B., Wei Y., Beyea R., Yan H., and Olson S. T. (2017) Lipid oxidation inactivates the anticoagulant function of protein Z-dependent protease inhibitor (ZPI). J. Biol. Chem. 292, 14625–14635 10.1074/jbc.M117.793901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fruhwirth G. O., Loidl A., and Hermetter A. (2007) Oxidized phospholipids: from molecular properties to disease. Biochim. Biophys. Acta 1772, 718–736 10.1016/j.bbadis.2007.04.009 [DOI] [PubMed] [Google Scholar]
- 7. Kagan V. E., Mao G., Qu F., Angeli J. P., Doll S., Croix C. S., Dar H. H., Liu B., Tyurin V. A., Ritov V. B., Kapralov A. A., Amoscato A. A., Jiang J., Anthonymuthu T., Mohammadyani D., et al. (2017) Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 10.1038/nchembio.2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chu L. H., Indramohan M., Ratsimandresy R. A., Gangopadhyay A., Morris E. P., Monack D. M., Dorfleutner A., and Stehlik C. (2018) The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat. Commun. 9, 996 10.1038/s41467-018-03409-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuosmanen S. M., Kansanen E., Kaikkonen M. U., Sihvola V., Pulkkinen K., Jyrkkanen H. K., Tuoresmaki P., Hartikainen J., Hippelainen M., Kokki H., Tavi P., Heikkinen S., and Levonen A. L. (2018) NRF2 regulates endothelial glycolysis and proliferation with miR-93 and mediates the effects of oxidized phospholipids on endothelial activation. Nucleic Acids Res. 46, 1124–1138 10.1093/nar/gkx1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zanoni I., Tan Y., Di Gioia M., Broggi A., Ruan J., Shi J., Donado C. A., Shao F., Wu H., Springstead J. R., and Kagan J. C. (2016) An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352, 1232–1236 10.1126/science.aaf3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oehler B., Kistner K., Martin C., Schiller J., Mayer R., Mohammadi M., Sauer R. S., Filipovic M. R., Nieto F. R., Kloka J., Pflücke D., Hill K., Schaefer M., Malcangio M., Reeh P. W., et al. (2017) Inflammatory pain control by blocking oxidized phospholipid-mediated TRP channel activation. Sci. Rep. 7, 5447 10.1038/s41598-017-05348-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morgan A. H., Dioszeghy V., Maskrey B. H., Thomas C. P., Clark S. R., Mathie S. A., Lloyd C. M., Kühn H., Topley N., Coles B. C., Taylor P. R., Jones S. A., and O'Donnell V. B. (2009) Phosphatidylethanolamine-esterified eicosanoids in the mouse: tissue localization and inflammation-dependent formation in Th-2 disease. J. Biol. Chem. 284, 21185–21191 10.1074/jbc.M109.021634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Slatter D. A., Aldrovandi M., O'Connor A., Allen S. M., Brasher C. J., Murphy R. C., Mecklemann S., Ravi S., Darley-Usmar V., and O'Donnell V. B. (2016) Mapping the human platelet lipidome reveals cytosolic phospholipase A2 as a regulator of mitochondrial bioenergetics during activation. Cell Metab. 23, 930–944 10.1016/j.cmet.2016.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hutchins P. M., and Murphy R. C. (2012) Cholesteryl ester acyl oxidation and remodeling in murine macrophages: formation of oxidized phosphatidylcholine. J. Lipid Res. 53, 1588–1597 10.1194/jlr.M026799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mancuso D. J., Jenkins C. M., and Gross R. W. (2000) The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium-independent phospholipase A2. J. Biol. Chem. 275, 9937–9945 10.1074/jbc.275.14.9937 [DOI] [PubMed] [Google Scholar]
- 16. Mancuso D. J., Jenkins C. M., Sims H. F., Cohen J. M., Yang J., and Gross R. W. (2004) Complex transcriptional and translational regulation of iPLAγ resulting in multiple gene products containing dual competing sites for mitochondrial or peroxisomal localization. Eur. J. Biochem. 271, 4709–4724 10.1111/j.1432-1033.2004.04435.x [DOI] [PubMed] [Google Scholar]
- 17. Yan W., Jenkins C. M., Han X., Mancuso D. J., Sims H. F., Yang K., and Gross R. W. (2005) The highly selective production of 2-arachidonoyl lysophosphatidylcholine catalyzed by purified calcium-independent phospholipase A2γ: identification of a novel enzymatic mediator for the generation of a key branch point intermediate in eicosanoid signaling. J. Biol. Chem. 280, 26669–26679 10.1074/jbc.M502358200 [DOI] [PubMed] [Google Scholar]
- 18. Moon S. H., Jenkins C. M., Liu X., Guan S., Mancuso D. J., and Gross R. W. (2012) Activation of mitochondrial calcium-independent phospholipase A2γ (iPLA2γ) by divalent cations mediating arachidonate release and production of downstream eicosanoids. J. Biol. Chem. 287, 14880–14895 10.1074/jbc.M111.336776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu X., Moon S. H., Jenkins C. M., Sims H. F., and Gross R. W. (2016) Cyclooxygenase-2 mediated oxidation of 2-arachidonoyl-lysophospholipids identifies unknown lipid signaling pathways. Cell Chem. Biol. 23, 1217–1227 10.1016/j.chembiol.2016.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pete M. J., and Exton J. H. (1996) Purification of a lysophospholipase from bovine brain that selectively deacylates arachidonoyl-substituted lysophosphatidylcholine. J. Biol. Chem. 271, 18114–18121 10.1074/jbc.271.30.18114 [DOI] [PubMed] [Google Scholar]
- 21. Snyder J. M., and Scholfield C. R. (1982) cis-trans isomerization of unsaturated fatty acids with p-toluenesulfinic acid. J. Am. Oil Chem. Soc. 59, 469–470 10.1007/BF02636144 [DOI] [Google Scholar]
- 22. Tombo G. M. R., Schär H. P., i Busquets X. F., and Ghisalba O. (1986) Synthesis of both enantiomeric forms of 2-substituted 1,3-propanediol monoacetates starting from a common prochiral precursor, using enzymatic transformations in aqueous and in organic media. Tetrahedron Lett. 27, 5707–5710 10.1016/S0040-4039(00)85306-X [DOI] [Google Scholar]
- 23. Huang Z., Guo X., Li W., MacKay J. A., and Szoka F. C. Jr. (2006) Acid-triggered transformation of diortho ester phosphocholine liposome. J. Am. Chem. Soc. 128, 60–61 10.1021/ja057024w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lands W. E. (1960) Metabolism of glycerolipids. 2. The enzymatic acylation of lysolecithin. J. Biol. Chem. 235, 2233–2237 [PubMed] [Google Scholar]
- 25. Lands W. E., and Merkl I. (1963) Metabolism of glycerolipids. III. Reactivity of various acyl esters of coenzyme A with α′-acylglycerophosphorylcholine, and positional specificities in lecithin synthesis. J. Biol. Chem. 238, 898–904 [PubMed] [Google Scholar]
- 26. Schlame M., and Ren M. (2006) Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 580, 5450–5455 10.1016/j.febslet.2006.07.022 [DOI] [PubMed] [Google Scholar]
- 27. Xu Y., Phoon C. K., Berno B., D'Souza K., Hoedt E., Zhang G., Neubert T. A., Epand R. M., Ren M., and Schlame M. (2016) Loss of protein association causes cardiolipin degradation in Barth syndrome. Nat. Chem. Biol. 12, 641–647 10.1038/nchembio.2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gaspard G. J., and McMaster C. R. (2015) Cardiolipin metabolism and its causal role in the etiology of the inherited cardiomyopathy Barth syndrome. Chem. Phys. Lipids 193, 1–10 10.1016/j.chemphyslip.2015.09.005 [DOI] [PubMed] [Google Scholar]
- 29. Cowling B. S., Toussaint A., Muller J., and Laporte J. (2012) Defective membrane remodeling in neuromuscular diseases: insights from animal models. PLoS Genet. 8, e1002595 10.1371/journal.pgen.1002595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lin S., Ikegami M., Moon C., Naren A. P., and Shannon J. M. (2015) Lysophosphatidylcholine acyltransferase 1 (LPCAT1) specifically interacts with phospholipid transfer protein StarD10 to facilitate surfactant phospholipid trafficking in alveolar type II cells. J. Biol. Chem. 290, 18559–18574 10.1074/jbc.M115.666701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tabe S., Hikiji H., Ariyoshi W., Hashidate-Yoshida T., Shindou H., Shimizu T., Okinaga T., Seta Y., Tominaga K., and Nishihara T. (2017) Lysophosphatidylcholine acyltransferase 4 is involved in chondrogenic differentiation of ATDC5 cells. Sci. Rep. 7, 16701 10.1038/s41598-017-16902-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cotte A. K., Aires V., Fredon M., Limagne E., Derangère V., Thibaudin M., Humblin E., Scagliarini A., de Barros J. P., Hillon P., Ghiringhelli F., and Delmas D. (2018) Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat. Commun. 9, 322 10.1038/s41467-017-02732-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eto M., Shindou H., Koeberle A., Harayama T., Yanagida K., and Shimizu T. (2012) Lysophosphatidylcholine acyltransferase 3 is the key enzyme for incorporating arachidonic acid into glycerophospholipids during adipocyte differentiation. Int. J. Mol. Sci. 13, 16267–16280 10.3390/ijms131216267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thomas C. P., Morgan L. T., Maskrey B. H., Murphy R. C., Kühn H., Hazen S. L., Goodall A. H., Hamali H. A., Collins P. W., and O'Donnell V. B. (2010) Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependent thrombin generation. J. Biol. Chem. 285, 6891–6903 10.1074/jbc.M109.078428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maskrey B. H., Bermúdez-Fajardo A., Morgan A. H., Stewart-Jones E., Dioszeghy V., Taylor G. W., Baker P. R., Coles B., Coffey M. J., Kühn H., and O'Donnell V. B. (2007) Activated platelets and monocytes generate four hydroxyphosphatidylethanolamines via lipoxygenase. J. Biol. Chem. 282, 20151–20163 10.1074/jbc.M611776200 [DOI] [PubMed] [Google Scholar]
- 36. Jenkins C. M., Yang K., Liu G., Moon S. H., Dilthey B. G., and Gross R. W. (2018) Cytochrome c is an oxidative stress-activated plasmalogenase that cleaves plasmenylcholine and plasmenylethanolamine at the sn-1 vinyl ether linkage. J. Biol. Chem. 293, 8693–8709 10.1074/jbc.RA117.001629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Abe A., Hiraoka M., and Shayman J. A. (2006) Positional specificity of lysosomal phospholipase A2. J. Lipid Res. 47, 2268–2279 10.1194/jlr.M600183-JLR200 [DOI] [PubMed] [Google Scholar]
- 38. Shayman J. A., Kelly R., Kollmeyer J., He Y., and Abe A. (2011) Group XV phospholipase A2, a lysosomal phospholipase A2. Prog. Lipid Res. 50, 1–13 10.1016/j.plipres.2010.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ayanoglu E., Wegmann A., Pilet O., Marbury G. D., Hass J. R., and Djerassi C. (1984) Mass spectrometry of phospholipids. Some applications of desorption chemical ionization and fast atom bombardment. J. Am. Chem. Soc. 106, 5246–5251 10.1021/ja00330a035 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.