

Abstract
Dysfunction of human phenylalanine hydroxylase (hPAH, EC 1.14.16.1) is the primary cause of phenylketonuria, the most common inborn error of amino acid metabolism. The dynamic domain rearrangements of this multimeric protein have thwarted structural study of the full-length form for decades, until now. In this study, a tractable C29S variant of hPAH (C29S) yielded a 3.06 Å resolution crystal structure of the tetrameric resting-state conformation. We used size-exclusion chromatography in line with small-angle X-ray scattering (SEC–SAXS) to analyze the full-length hPAH solution structure both in the presence and absence of Phe, which serves as both substrate and allosteric activators. Allosteric Phe binding favors accumulation of an activated PAH tetramer conformation, which is biophysically distinct in solution. Protein characterization with enzyme kinetics and intrinsic fluorescence revealed that the C29S variant and hPAH are otherwise equivalent in their response to Phe, further supported by their behavior on various chromatography resins and by analytical ultracentrifugation. Modeling of resting-state and activated forms of C29S against SAXS data with available structural data created and evaluated several new models for the transition between the architecturally distinct conformations of PAH and highlighted unique intra- and inter-subunit interactions. Three best-fitting alternative models all placed the allosteric Phe-binding module 8–10 Å farther from the tetramer center than do all previous models. The structural insights into allosteric activation of hPAH reported here may help inform ongoing efforts to treat phenylketonuria with novel therapeutic approaches.
Keywords: allosteric regulation, small-angle X-ray scattering (SAXS), X-ray crystallography, protein conformation, inborn error of metabolism, structural biology, ACT domain, phenylalanine hydroxylase, phenylketonuria, resting-state structure, structural modeling
Introduction
Human phenylalanine hydroxylase (hPAH,6 EC 1.14.16.1) dysfunction is the primary cause of phenylketonuria (PKU, ∼1:10,000 births global average), which results in neurotoxic concentrations of the essential amino acid l-phenylalanine (Phe) (1). hPAH catalyzes the conversion of Phe to tyrosine (Tyr) using tetrahydrobiopterin (BH4) and molecular oxygen as co-substrates and a nonheme iron. PKU is a highly heterogeneous disorder with nearly 1,000 different disease-associated alleles (1). hPAH is a 452-amino acid multidomain protein that contains an N-terminal regulatory domain, composed of an autoregulatory segment and an ACT domain (Fig. 1A). ACT domains are a family of structurally homologous elements that serve as sensors for small molecules, often amino acids, and are named for the first three discovered examples (2–4). Following the ACT domain, hPAH contains a large catalytic domain, and a C-terminal multimerization domain (Fig. 1A).
Figure 1.

PAH structure. A, refined description of the domain structure of hPAH. The N-terminal 32 amino acids have been called an autoregulatory sequence, which together with the ACT subdomain (residues 33–110) constitute the regulatory domain (residues 1 to ∼117). The N-terminal ∼20 amino acids are disordered in all PAH crystal structures; residues 111–117 constitute a loop that connects the ACT subdomain to the rest of the protein. The residues between 118 and ∼410 have been defined as the catalytic domain. The region between 118 and 127 contains a tryptophan, whose intrinsic fluorescence changes upon enzyme activation (43), residues 128–148 constitute the active-site lid, and residues 137–141 are disordered in all full-length PAH structures in the RS-PAH conformation. Residues 411–453 have been designated as the multimerization domain based on truncation analysis. However, the crystal structure suggests that the β-hairpin at 411–424 might best be considered part of the catalytic domain. Residues 425 and 426 constitute a connection between this expanded catalytic domain and a long C-terminal α-helix (residues 427–452). The C-terminal helices form a 4-helix bundle that secures the tetramer (see B). The most C-terminal residues are disordered in all full-length mammalian PAH structures. B, illustrated is the highest-resolution structure of full-length rPAH in the RS-PAH conformation (7). The regulatory domains of subunit B (red) and subunit C (blue) are in bolder tones. Subunits A and D are colored gray. The interaction between the auto-regulatory region and the catalytic domain, which partially occludes the active site in the RS-PAH conformation, is stabilized by a 2.7 Å hydrogen bond between Ser-29 and Asp-112 (inset). C, top is a schematic of the RS-PAH ⇔ A-PAH conformational interchange using coloring as in B. Bottom is the crystal structure of the ACT-domain dimer of hPAH with allosteric Phe bound (shown as spheres) (6). The repositioned regulatory domain in the A-PAH conformation releases active-site occlusion (not shown).
PAH populates architecturally distinct multimeric conformations, including a low-activity resting-state PAH (RS-PAH) conformation, first determined for rat PAH (rPAH) (Fig. 1B) and an activated PAH (A-PAH) conformation with high activity (5). The ACT domain of PAH binds the allosteric Phe (at one per ACT domain), which secures an ACT–ACT dimer, shown in Fig. 1C, which is present only in the A-PAH conformation (6). The equilibrium between RS-PAH and A-PAH serves to maintain Phe levels sufficient for normal metabolism but below neurotoxic levels (5). As Phe levels rise, the conformational equilibrium shifts toward A-PAH; the subsequent metabolism of Phe is accompanied by a conformational shift back toward RS-PAH. Because Phe is both substrate and allosteric activator, the kinetic behavior of hPAH depends upon the order of addition of reaction components; preincubation with Phe results in a significant increase in the initial velocity of Tyr production, relative to hPAH that has not been preincubated.
Our understanding of hPAH recently advanced with the determination of the crystal structure of the highly homologous rPAH in a resting-state conformation (PDB codes 5DEN and 5FGJ (7, 8)) (Fig. 1B), along with the structure of the ACT domain of WT hPAH in its activated form (PDB code 5FII (6)) (Fig. 1C). The latter reveals Phe bound to an A-PAH–specific protein–protein interface, a conformation supported by NMR (9, 10) and SAXS (7, 8). Recently, in silico work has advanced a conformational selection mechanism (11) for the allosteric binding of Phe. Together, these data support the hypothesis that 1) activation of mammalian PAH minimally involves substantial movement of the N-terminal regulatory domain, and 2) the A-PAH conformation is stabilized by Phe binding allosterically to a multimer-specific binding site at the interface of two ACT domains (12). Unfortunately, crystals of rPAH grown in the presence of Phe have yet to provide a structure in the A-PAH conformation (8).
rPAH has long-served as a surrogate for the biochemical, biophysical, and structural study of hPAH due to its favorable in vitro properties (12–14), with a wealth of data now available. Although the hPAH and rPAH sequences are highly homologous, the proteins are sufficiently different in their in vitro properties to justify pursuit of structures of full-length hPAH for a better understanding of PKU. One clue to the basis for the resistance of hPAH to structural studies is a lower-fold activation of hPAH (∼3) relative to rPAH (∼5) when comparing assays done with and without preincubation with 1 mm Phe (15). This difference suggests that hPAH has a higher propensity to sample the A-PAH conformation in the absence of Phe, making it more heterogeneous under Phe-free conditions, relative to rPAH. Understanding that the mole fractions of the RS-PAH and A-PAH conformations reflect an equilibrium, one approach to increasing the uniformity of hPAH is to further stabilize the RS-PAH conformation, which we have done here by design.
We describe an hPAH variant where cysteine at position 29 is replaced with serine (C29S). Inspection of the highest-resolution crystal structure of full-length rPAH (PDB code 5DEN; RS-PAH conformation) allowed identification of a species-specific stabilizing intra-subunit hydrogen bond between Ser-29 in the autoregulatory region and Asp-112 near the hinge region between the regulatory and catalytic domains (Fig. 1B). This substitution was predicted to introduce an RS-PAH–stabilizing interaction by similarity to the rPAH sequence. Indeed, the crystal structure of C29S reveals the hPAH in an RS-PAH conformation. Additional biochemical and biophysical analysis demonstrates that the solution behavior of C29S mirrors that of hPAH both in its resting and activated forms. Consistent with the hypothesis that Ser-29 more highly favors the RS-PAH conformation relative to Cys-29 is a prior report that the S29C variant of rPAH is activated relative to WT rPAH (16).
Results
Purification of hPAH and C29S
A classic affinity purification of mammalian PAH exploits the different physical properties of the alternative conformations of mammalian PAH (17). At high Phe concentrations, the protein has high affinity for phenyl-Sepharose resin, whereas removal of Phe from the elution buffer causes PAH to dissociate. Relative to the high yield and purity of rPAH prepared using this method, the yields and purity of hPAH are reproducibly lower (12). In contrast, C29S could be obtained in good yield and high purity with this method and retained full catalytic activity when stored at modest ionic strength (150 mm KCl). Analytical characterization of hPAH, C29S, and rPAH using phenyl-Sepharose suggests that hPAH experiences a slower conversion to the RS-PAH conformation following Phe removal from the elution buffer (Fig. S1). These observations are consistent with a prior report that the S29C variant of rPAH impaired purification using phenyl-Sepharose (16).
hPAH and C29S are comparably active
The initial velocity of the PAH-catalyzed reaction is higher if the protein is incubated with Phe prior to being introduced to the assay mixture (12). The magnitude of the difference (+/− Phe preincubation) is a function of the concentration of Phe and of the initial position of the conformational equilibrium (RS-PAH ⇔ A-PAH) in the enzyme sample. For this reason, PAH activity assays performed using a preincubation of PAH with versus without Phe (often set to 1 mm) have become a standard approach to determining the ability of a given PAH (or disease-associated PAH variant) to become “activated.”
Table 1 reports kinetic parameters derived from activity assays performed with and without preincubation with Phe, using Phe concentrations in the range of 10 μm to 1 mm. The preincubation is carried out at the same Phe concentration as is used in the assay. We find comparable KM values for hPAH relative to C29S, both with and without preincubation with Phe and regardless of whether or not the protein was cleaved from an affinity purification tag. We find hPAH undergoes an ∼3-fold activation due to preincubation with 1 mm Phe, whereas the activation for C29S is ∼5-fold. These observations support the design criterion for C29S, which was to stabilize the RS-PAH conformation of hPAH. Consistent with this idea is the observation that C29S exhibits a lower Vmax (−Phe) relative to hPAH.
Table 1.
Measured kinetic parameters for the hPAH and C29S catalyzed production of Tyr from Phe
Experiments were performed with 75 μm BH4 at 25 °C, without and with a 1-min preincubation with Phe. Kinetic data were fitted to the Hill equation using SigmaPlot and reporting standard errors.
| Enzyme or condition | KM for Phe | Hill coefficient | Vmax | Fold activation | |||
|---|---|---|---|---|---|---|---|
| μm | h | nmol of Tyr·min−1·mg−1 | |||||
| Phe preincubation | − | + | − | + | − | + | (Vmax+Phe ÷ Vmax−Phe) |
| hPAHa,b | 303 ± 54 | 144 ± 10 | 1.1 ± 0.1 | 2.4 ± 0.3 | 1749 ± 140 | 5179 ± 1 | 3.0 |
| C29Sc,d | 217 ± 8 | 180 ± 4 | 1.9 ± 0.1 | 2.6 ± 0.1 | 837 ± 17 | 4077 ± 6 | 4.9 |
| C29Sb,e | 391 ± 45 | 153 ± 10 | 1.7 ± 0.2 | 2.5 ± 0.3 | 1367 ± 101 | 6919 ± 209 | 5.1 |
a Data were on PAH cleaved from a His6–SUMO construct and affinity-purified with Tris buffers.
b [Phe] was at 10, 50, 100, 200, 300, 600, 800, and 1,000 μm.
c Native protein was purified using the Shiman method (17).
d [Phe] was at 10, 50, 100, 200, 300, and 1,000 μm.
e Data were on PAH cleaved from a His6–TEV construct, affinity purified with Tris buffers.
Phe stabilizes both hPAH and C29S in an A-PAH conformation
PAH's intrinsic fluorescence is routinely used to monitor activation and was one of the first indications that the A-PAH conformation was significantly different from the RS-PAH conformation (18, 19). The intrinsic protein fluorescence for both hPAH and C29S (Fig. 2A), both in the absence and presence of activating concentrations of Phe, is identical to each other and reproduces a well-documented red shift upon addition of 1 mm Phe (20).
Figure 2.

Characterization of hPAH and C29S. A, intrinsic fluorescence of WT hPAH and C29S in the absence or presence of activating concentrations of Phe. B, activation by Phe promotes oligomerization of C29S. Shown is a linearized plot of S.E. data from a single rotor speed (10,000 rpm) for 2.5 μm C29S in the absence (gray circles) and presence of 1 mm Phe (open circles) at 4 °C. The slopes are proportional to M̄w at the given value of r2. Shown as dashed lines are the calculated lines expected for a monomer, dimer, and tetramer, respectively. See Fig. S2 for global fits to three rotor speeds and two concentrations in each state. C, SEC profiles of C29S in the absence and presence of saturating Phe. Black arrows indicate the elution volume (Ve) for the following standard proteins: 1, blue dextran (2000 kDa); 2, ferritin (440 kDa); 3, catalase (232 kDa); 4, aldolase (158 kDa); 5, BSA (67 kDa); and 6, bovine serum ovalbumin (43 kDa).
Analytical ultracentrifugation of C29S confirms that Phe stabilizes tetramers
Sedimentation equilibrium (SE–AUC) analysis of C29S in the absence and presence of 1 mm Phe provides additional information about its self-association equilibrium. Both linearized plots (Fig. 2B) and global fits (Fig. S2) of the radial distributions show M̄w values as those expected for PAH in its tetrameric form in the presence of Phe, and intermediate values between dimer and tetramer in the absence of Phe. In the absence of Phe, the apparent KD value for the dimer ⇔ tetramer equilibrium at 4 °C is 18.8 ± 0.91 μm, whereas in the presence of Phe the fit yields an uncertain value of 0.46 ± 0.82 μm, which indicates the virtual absence of dimer under these experimental conditions (Fig. S2). In all cases, attempts to model species larger than tetramer failed to yield reasonable fits to the experimental data. These data are consistent with a wealth of literature supporting the notion that Phe stabilizes mammalian PAH tetramers relative to dimers (12, 21).
Crystal structure of C29S
The overall structure of C29S in the resting state resembles that of rPAH (7, 8), including the extensive buried surfaces that compose the dimer and tetramer interfaces. Each of the four ACT domains are situated in a characteristic RS-PAH conformation, where they do not interact with each other. The asymmetric unit is a tetramer assembled as a dimer–of–dimers that form an irregular quadrilateral shape (Fig. S3). The shortest sides of the tetramer contain two dimers that are twisted about a point that is centered on the 4-helix bundle composed of each subunit's C-terminal α-helix (see Fig. 1B and Fig. S3). Each of the four active sites of C29S are not significantly different from those observed in any of the 13 documented crystal structures of the truncated hPAH catalytic domain (residues 117–424) or within the full-length rPAH structure (PDB code 5DEN). Each active site contains one iron ion (with an average occupancy of 90%), and first coordination sphere water molecules can be modeled for all chains.
The overall quaternary architecture of C29S in the tetrameric RS-PAH conformation is like that of rPAH, yet there is a more pronounced difference between the orientations of the dimer pairs. Alignment with the catalytic domain of one chain shows that the four chains of C29S segregate into two conformations that differ at the hinge that connects the C-terminal helix with the rest of the protein (Fig. 3A, top). Although a similar asymmetry is observed within rPAH, alignment of the C-terminal helices, instead of the catalytic domain, shows the more pronounced differences between the hinge positions of each dimer pair for C29S relative to rPAH (Fig. 3A, bottom).
Figure 3.

Flexibility within regulatory and multimerization domains of C29S. A, both C29S (left) and rPAH (PDB code 5DEN, right) are aligned on their respective chain A at catalytic domain residues 144–410 (top) and on their respective chain A C-terminal helices (residues 426–447, bottom). The overlay of helices was created by orienting the models such that Thr-427 is pointing out toward the reader to highlight the different spacing between the BC and AD pairs at the hinge that precedes the C-terminal helix (see top). B, secondary structure elements of the ACT subdomain of hPAH are defined (top), highlighting the connecting loops (L1–L5), and noting different degrees of disorder among the four subunits of tetrameric C29S. Differences in sequence between hPAH and rPAH are denoted with an underlined letter; the two PAH proteins are 89% sequence identical through residue 111. The RMSD after alignment of C29S with rPAH on their regulatory domains (i.e. through residue 111) is 0.76 Å. Colored magenta (bottom) are the loops that contain unmodeled, disordered segments in at least one subunit of the structure of C29S. C, four structures are overlaid on their ACT domains (from the N-terminal most residue through residue 111), and L1 is at center. Only one chain of PDB code 5FII (isolated human PAH ACT domains in an A-PAH conformation, with Phe bound, but not shown) and one chain of PDB entry 5DEN (the full-length rPAH structure in the RS-PAH conformation) are shown because the comparison is identical when using any other chain in those structures. Similarly, only two chains are shown for PDB code 5EGQ; chains B and D are identical to A at this position, whereas chain C is different from any other rPAH structure at this position.
The structure of C29S shows regions of apparent disorder. Like the rPAH structure, disorder at the N and C termini, as well as at the active-site lid, is observed, and the extent of the disorder is different in each of the four chains of the asymmetric unit. The disorder is most notable within the ACT domains, where electron density for known atomic inventory is not observed (see Table 2). The overall degree of disorder is greater for this structure relative to rPAH (PDB code 5DEN), despite similarities in resolution (3.06 versus 2.9 Å, respectively). Unlike the structure of rPAH, the structure of C29S contains additional disorder within the ACT subdomains and at the hinge between the auto-regulatory sequence and the ACT subdomain (Fig. 3B). We surmise that sequence differences between rPAH and hPAH may partially underlie the disorder of the missing inventory within the ACT domain. There are nine residues that differ between the two species; these differences occur at residues 60 at loop L2; 73 at L3; 82 at the β3–L4 junction; 88 and 90 at L4; and 92, 93, 97, and 100 all at α2.
Table 2.
Data collection and refinement statistics for the X-ray crystal structure of full-length hPAH C29S
Values in parentheses are for the highest-resolution shell.
| PDB code | 6N1K | |||
|---|---|---|---|---|
| Data collection | ||||
| Beamline | BNL-NSLS II 17-ID-1 (AMX) | |||
| Wavelength (Å) | 1.008 | |||
| Space group | P1211 | |||
| Unit cell a, b, c (Å); b (°) | 72.4, 202.6, 72.6; 90, 90.3, 90 | |||
| No. of monomers in asymmetric unit | 4 | |||
| Resolution range (Å) | 36.3–3.06 (3.17–3.06) | |||
| Total no. of observations | 153,999 (15,045) | |||
| No. of unique reflections | 39,316 (3,885) | |||
| Completeness (%) | 99.54 (98.16) | |||
| Multiplicity | 3.9 (3.9) | |||
| 〈I/σ(I)〉 | 11.5 (1.7) | |||
| R-merge§ | 0.078 (0.66) | |||
| R-meas+ | 0.091 (0.76) | |||
| CC1/2 | 0.998 (0.63) | |||
| R-pim | 0.045 (0.38) | |||
| Wilson B-factor (Å2) | 93.6 | |||
| Refinement | ||||
| No. of nonhydrogen atoms | 13,689 | |||
| Chains | A | B | C | D |
| Residues modeled | 21–28; 31–136; 142–446 | 20–29; 32–93; 86–102; 105–137; 141–448 | 22–136; 141–448 | 20–29; 33–59; 63–102; 105–137; 141–446 |
| No. of waters | 2 | 2 | 1 | 2 |
| No. of Fe ions | 1 | 1 | 1 | 1 |
| R-work/R-free* | 0.228/0.276 | |||
| Twin law | l, −k, h | |||
| No. of reflections in working set | 39,316 (3,885) | |||
| No. of reflections in test set | 1,980 (203) | |||
| Mean B-factor (Å2) | 101.2 | |||
| RMS deviations | ||||
| Bonds (Å) | 0.003 | |||
| Angles (°) | 0.54 | |||
| Ramachandran plot (%) | ||||
| Preferred | 95.4 | |||
| Allowed | 4.6 | |||
| Outliers | 0 |
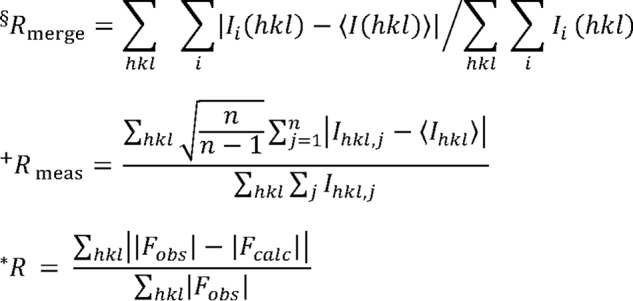
R-free is the R factor based on 5% of the data that were excluded from refinement.
Evidence for flexibility in the regulatory domains of C29S
The unexpected amount of disorder within the regulatory domain, as evidenced by missing electron density, suggests plasticity in the mobile loops of the ACT domain that may be involved in allosteric regulation (11, 22). To probe the scope of this plasticity, the final crystal structure was subjected to molecular dynamics simulations restrained by the experimental diffraction data as implemented within phenix.ensemble_refinement (23). The results, when inspected as an ensemble and relative to the rPAH structure, provide an indication of the greater mobility of the C29S ACT subdomain and the C-terminal helices of the multimerization domain (residues ∼426-end) versus the same regions in rPAH. The analysis shows that despite the two structures possessing comparable coordinate error (0.47 Å for rPAH and 0.5 Å for C29S), and even though the structure of C29S contains a lower-phase error (28.7° versus 36.8° for rPAH), the C29S ensemble appears to sample more accessible space as shown by larger RMSD at more positions throughout the tetrameric assembly of C29S relative to rPAH (Fig. 4).
Figure 4.

C29S demonstrates considerable flexibility in the regulatory (ACT) domain. Molecular dynamics simulations of C29S (this work, left) and rPAH (PDB code 5DEN, right) using diffraction data as an experimental restraint in phenix.ensemble_refinement generated ensembles of compatible structures. Shown is the average structure of the ensemble of structures generated for each coordinate set, rendered as a cartoon putty. The thickness and color (warm is red) of the putty at any particular position is proportional to the RMSD at that position over the ensemble relative to the average structure.
The C29S structure was also superposed with all other PAH structures that contain the ACT domain, and the overlay was used to inspect the structure of C29S for regions of inferred flexibility. Comparison of the individual ACT domains of the Phe-bound hPAH ACT-domain dimer (PDB code 5FII (6), which contains four similar ACT-domain conformations representative of A-PAH), with the four ACT domains from each of the chains in the C29S tetramer, rPAH (PDB code 5DEN), and the Δ24-rPAH-R270K variant (PDB code 5EGQ), highlights the conformational space available to the ACT domain. The secondary structural elements are virtually identical between the RS-PAH conformations of the ACT domains of rPAH and C29S. However, there are several significant differences among the loops that connect the domain's helices and β-strands (see Fig. 4B). Fig. 3C highlights the flexibility of L1, a loop that interacts directly with the allosteric effector Phe, and is involved in the Phe-stabilized interface of hPAH ACT-domain dimers (6). Residues 41–47, which constitute L1 and are identical between hPAH and rPAH, are situated differently in the rPAH versus C29S structures. L1 of the four ACT domains of rPAH are in the same conformation, whereas L1 of the four ACT domains of the C29S tetramer vary from one another, and all are significantly different from those observed in the rPAH structure. Consistent with this observation, Ge et al. (11) report molecular dynamics simulations of the isolated hPAH ACT-domain dimer (A-PAH specific) indicating that Phe binding requires that L1 swing between open and closed positions, acting as a gate to expose the allosteric Phe-binding site, and then closing to stabilize Phe binding. Of all the various ACT-domain conformations seen in RS-PAH (rPAH and hPAH), only one resembles that seen in the Phe-bound A-PAH structure of an hPAH ACT-domain dimer (PDB code 5FII); this is chain C of the Δ24-rPAH-R270K variant (PDB code 5EGQ) (Fig. 3C).
The backbone conformation of L4 in chain B of C29S is different from that observed within any other documented structure that includes the PAH ACT domain. In contrast, the conformations of L3 and L5 are virtually the same among all multidomain PAH structures, but L3 has a different conformation in the isolated Phe-bound ACT-domain dimer. Specifically, L3 is the only loop within PDB code 5FII that is not identically placed at each of the four chains in the asymmetric unit (two dimers) of that structure, with one of the four chains lacking coordinates for several residues within L3. Molecular dynamics simulations of 5FII also reveal mobility within L3 (11), which, in this case as compared with the mobility observed in L1, is not essential to allosteric Phe binding.
SEC–SAXS analysis of C29S in the RS-PAH and A-PAH conformations
Small-angle X-ray scattering (SAXS) was next employed to test this crystal structure against its solution properties and to assess the extent of the conformational change that occurs in the Phe-stabilized transition from an RS-PAH to A-PAH conformation, as performed previously with rPAH (7, 8, 24). Because the reliable interpretation of SAXS data rests upon an intimate understanding of the concentration-dependent behavior and monodispersity of the material studied, we first assessed C29S using size-exclusion chromatography at room temperature.
At a modest injection concentration (∼1 mg/ml) and against known molecular weight markers, the broad elution profile of C29S in the absence of Phe is consistent with a distribution of distinct tetramer and dimer species at room temperature (Fig. 2C), in agreement with our observations in SE–AUC at 4 °C (Fig. 2B). Addition of saturating Phe results in loss of the dimeric component and a small shift in tetramer mobility (∼0.1 ml earlier), consistent with the shift in Kd values observed by SE–AUC and our previous observations with rPAH (12). The addition of saturating Phe to C29S also results in accumulation of larger molecular mass species. These larger assemblies, previously proposed to form through Phe-stabilized association of regulatory domains between tetramers (12), were shown to redistribute to tetramer upon removal of Phe (data not shown). The reversible appearance of higher-order multimers and the apparent polydispersity due to the concentration-dependent behavior motivated our application of size-exclusion chromatography in-line with SAXS (SEC–SAXS).
Synchrotron SAXS data were recorded continuously during the elution of an injected ∼100 μm bolus of C29S from a SEC column at room temperature in the presence and absence of Phe, providing a large dataset of sequential profiles in each state. The variation in Rg as a function of elution time provides a robust reading of the concentration-dependent behavior of the sample (25). In the absence of Phe, a single peak is observed when the forward scattering extrapolated to zero-angle (I(0)) is plotted as a function of frame number (Fig. S4A). The apparent radius of gyration (Rg) across this series of data is relatively constant across the peak, with apparent higher-order species preceding the main peak. In contrast, the Phe-stabilized activated protein yields a similar primary species with comparable Rg values, alongside a significant population of higher-order multimers (Fig. S5A).
These datasets were analyzed in a model-free fashion using singular value decomposition with evolving factor analysis (SVD–EFA) (8), allowing for the decomposition of the datasets into its minimal components with maximal redundancy; the details of these analyses are provided in Figs. S4A and S5A. Masses were calculated for the decomposed scattering profiles by two different methods (Qr and Porod (26, 27), see Table 3) and used to assign the profiles to an oligomeric form of PAH. Deconvoluted tetramer profiles for both RS-PAH and A-PAH conformations displayed linearity in the Guinier region of the scattering data and agreement of the I(0) and Rg values determined with inverse Fourier transform analysis (Table 3). As observed previously with the rat homolog (7, 8), a distinctive change in the primary data was observed, with the evolution of a peak feature near q = ∼0.13 Å−1 in the presence of Phe (Fig. 5A). These changes in the primary data correlate with a redistribution in interatomic vectors by shape distribution (P(r)) analysis, consistent with the rearrangement of structural domains (Fig. 5B).
Table 3.
Parameters derived from SVD–EFA SEC–SAXS analysis of C29S
| Sample | Guinier |
GNOM |
Pxb | Mass (kDa) |
||||
|---|---|---|---|---|---|---|---|---|
| qRg | Rga | Rg | Dmax | T.E. | By Qrc | By Porodd | ||
| Å | Å | Å | ||||||
| No Phe | 0.35 − 1.26 | 41.4 ± 0.6 | 41.0 | 135 | 0.85 | 3.9 | 191 | 207 |
| 1 mm Phe | 0.48 − 1.26 | 40.5 ± 5.2 | 41.2 | 131 | 0.93 | 4.0 | 218 | 234 |
a Errors reported reflect the uncertainty in the value for Rg determined using classical Guinier fitting.
b Px is Porod exponent. Values near ∼4 indicate compactness, whereas lower values between 2 and 3 indicate significant lack of compactness and increased volumes (28). These values were determined using the program ScÅtter (https://bl1231.als.lbl.gov/scatter/).
c Mass determinations using the Qr invariant (26) were determined using the program RAW.
d Mass determinations using the Porod volume (27) were determined using the program RAW.
Figure 5.

Small-angle scattering analysis of C29S in the absence and presence of Phe. A, data were obtained by singular value decomposition-evolving factor analysis (SVD–EFA (8)) of SEC–SAXS data obtained for C29S in the presence and absence of 1 mm Phe (see under “Experimental procedures” and Figs. S2 and S3). Shown in A is a comparison of deconvoluted SAXS data for C29S without (blue) and with (red) incubation with 1 mm Phe. Data are shown as the superposed log–log plots of intensity as a function of q. Shown below this panel is a ratio plot where the discrepancy between the two profiles is highlighted as a function of 1. Identical regions have a value of 1, whereas higher discrepancies will deviate from unity. B, shape distribution function analysis for C29S in the absence (blue, resting state) and presence of 1 mm Phe (red, activated). Lower panel, ΔP(r) is shown, highlighting the change in interatomic vectors that occur upon activation of C29S with 1 mm Phe. C, complete atomistic model of C29S in the resting state derived from the crystal structure reported herein was created using NAMD, with missing inventory simulated as compacted random coil is shown in Fig. 6 (left). This model showed strong correlation with its solution scatter (χ2 = 0.99 using the program CRYSOL).
For RS-PAH and A-PAH conformations, both Kratky plot analysis and the Porod exponent (Table 3) indicate the presence of compact particles in solution with no detectable flexibility or disorder, motivating the use of single-structural models to best describe the experimental data (28). In the absence of Phe, the solution data closely matches the Rg and the maximum dimension (Dmax) of the full-length C29S crystal structure (Fig. 6, left). As was the case in our prior examinations of the rPAH tetramer in the absence of Phe (7), modeling of the missing atomic inventory was necessary for the most accurate reconciliation with the solution data at the higher-scattering angles. An all-atom model of C29S was derived from its crystal structure using molecular dynamics to simulate a compact coil-like structure for missing inventory (Fig. 6, left). Without additional manipulation, this model shows strong correlation with the SAXS data (Fig. 5C). Together, these results allow us to conclude that the crystal structure of C29S in the RS-PAH conformation very closely resembles its solution form.
Figure 6.

Structures of human PAH. A, X-ray crystal structure of hPAH in the RS-PAH conformation is illustrated (PDB code 6N1K). As in Fig. 1, the regulatory domains of subunit B (red) and subunit C (blue) are in bolder tones. Subunits A and D are colored gray. The disordered regions (missing inventory) are shown as balls and were modeled using NAMD to reconcile the crystal structure with the solution structure obtained from SAXS analysis. B, model of the A-PAH conformation, which contains a repositioned regulatory domain that no longer occludes the active site and contains an ACT-domain dimer. This model, the optimization of which is extensively discussed herein, employs the crystal structure of the truncated ACT domain of human PAH (residues 34–111) with allosteric Phe bound (PDB code 5FII (6)). The illustrated conformation of the entire autoregulatory region is shown as balls, as modeled by NAMD. The best-fit model contains the ACT-domain dimers at 8–10 Å farther from the tetramer center of mass than had previously been considered (see Fig. 7).
Using SAXS to refine our understanding of the A-PAH conformation
The year 2013 marked the realization that activation of mammalian PAH by Phe could require significant movement of the entire PAH regulatory domain to facilitate formation of an allosteric Phe-binding site at the interface of two ACT subdomains (12). Although then state-of-the-art models of tetrameric PAH did not include close proximity between ACT domains (29), it was already established that most ACT domains in the PDB exist as dimers and that the dimer interface can form a binding site for ligands such as amino acids (2, 3); for many years, PAH was considered an interesting exception to this rule (30). By 2011, Fitzpatrick and co-workers (9) had established that the isolated rPAH ACT domain would spontaneously form dimers and that conformational changes ensued in the presence of Phe. Herein, we address three uncertainties in the quaternary model of A-PAH containing a Phe-stabilized ACT-domain dimer. The first is the architecture of the ACT-domain dimer; the second is the extent of translation and rotation of the ACT-domain dimers relative to the catalytic and multimerization domains of the tetramer; and the third is the extent (in number of residues) of the N-terminal region that is repositioned in the RS-PAH to A-PAH transition.
ACT domain architecture
Multiple ACT-domain dimers observed in the PDB can be described as bringing the two monomers together in a “palms-down” conformation. In this form, the dimer contains an eight-stranded β-sheet, most often proximal to the rest of the protein, whereas the four α-helices of the ACT-domain dimer are positioned outward toward the solvent. This was the architecture of the 2013 model we developed (12). There is also precedent for a “palm–to–palm” architecture in which the ACT dimer interface forms through interactions between two four-stranded β-sheets, one from each ACT domain (4, 31). These alternative ACT dimers are shown in Fig. 7A. Although the recently determined human PAH ACT-domain dimer crystal structure (6) is consistent with the palms-down geometry, as had been determined for the regulatory domain of tyrosine hydroxylase (32), herein we address whether SAXS can corroborate the palms-down conformation in the context of the intact tetramer and further distinguish between the palms-down and palm–to–palm models.
Figure 7.

Modeling the activated form of hPAH. A, two models for the ACT-domain dimer are considered. Shown in red is the hPAH ACT-domain dimer determined by X-ray crystallography that features an eight-stranded β-sheet (PDB 5FII (6), palms-down). Shown in orange is an ACT-domain dimer model derived from the PDB 3PG9 structure (31), where the dimer interface is composed of two four-stranded β-sheets (palm–to–palm). B, all-atom models were constructed where the rotation and translation of the ACT-domain dimer (dark gray) was systematically sampled in 5° and 1 Å increments, respectively, in such a way that preserved the 2-fold symmetry plane across the xz plane. Linker regions with no known atomic structure were generated ab initio to complete the atomic inventory (see “Experimental procedures”). The remainder of the hPAH tetramer (light gray) was templated from the X-ray structure presented herein. These operations yielded an ensemble of theoretical models that could be tested against the experimental data. C, alternative possibilities for the hPAH subunit structure in the A-PAH conformation are shown. On the left (purple) is the traditional, proximal, or trans-connectivity, in which the position of residues 117–142 (spheres) is not substantially different relative to RS-PAH. For this model, the RS-PAH to A-PAH transition includes an ∼90° backbone rotation at residue ∼117 and a repositioning/restructuring of the autoregulatory region. On the right (green) is the newly proposed, reaching, or cis connectivity model, in which the repositioning/restructuring additionally involves residues 117–128 and the regulatory domain swings out and is transposed to the other side of the PAH tetramer. For both images, the backbone of residues 117–128 is colored white, Trp-120 (space-filling) is red, and Cys-237 (space-filling) is orange. D, calculated scattering profiles for each model in the ensemble with the palms-down–activated conformation were tested against the experimental data, and the best matches were identified using the χ2CRYSOL metric as a function of rotation and translation to derive a contour plot. The best model selected is denoted with an asterisk. E, log–log plot of the calculated scattering intensity of the best model (red line) is plotted against the experimental SAXS data (χ2CRYSOL = 0.698). F, orthogonal views of the best atomistic model identified by this approach is illustrated. Highlighted in red is the modeled ACT domains, with the ab initio–generated linkers (residues 1–30, 111:142) shown as red C-α spheres. G, contour plot for the ensemble of models modeled with the palm–to–palm conformation is shown. The best model selected is denoted with an asterisk. H, log–log plot of the calculated scattering intensity of the best model (orange line) is plotted against the experimental SAXS data (χ2CRYSOL = 0.627). I, orthogonal views of the best atomistic model identified is shown. The ACT-domain dimer and linkers are highlighted in orange, and the ab initio–generated linkers (residues 1–30, 111:142) are shown as C-α spheres. J, contour plot for the modeling results testing the palms down conformation is shown, but with an alternative topology of interdomain linkers (C). The best model selected is denoted with an asterisk. K, log–log plot of the calculated scattering intensity of the best model (green line) is plotted against the experimental SAXS data (χ2CRYSOL = 0.623). L, orthogonal views of the best atomistic model identified is shown, with the ACT-domain dimer and linkers highlighted in green and the ab initio-generated linkers (residues 1–30, 111:142) shown as C-α spheres.
We had previously reported that our 2013 model for full-length hPAH in an A-PAH conformation does not provide an outstanding fit to the SAXS data of activated rPAH without manipulation (7). In this model, the modeled center-of-mass (COM) to COM distance between the ACT-domain dimer and the rest of the catalytic tetramer is 31.7 Å. Here, we also investigate alternative models for the A-PAH conformation, wherein we incrementally adjust both the rotation (in 5° increments) and translation (in 1 Å increments) of the ACT-domain dimer relative to the catalytic and multimerization domains of the tetramer (Fig. 7B).
To directly reconcile the available atomic inventory against the solution data and these considerations, we constructed all-atom models of the A-PAH conformation of C29S, using the recently available PDB 5FII ACT-domain dimer conformation or a homology model dimer derived from DAH7P synthase (PDB 3PG9), connected to the catalytic and multimerization domain crystal structure determined here for C29S in the RS-PAH conformation. From these starting models, an ensemble of test structures was systematically created to probe parameter space by varying the rotation and translation of the ACT-domain dimers relative to the center-of-mass (Fig. 7B). Because two ACT-domain dimers are presumed present in an A-PAH tetramer, the operations were performed as to constrain the 2-fold symmetry relationship across the xz plane of the catalytic domain tetramer. For each model structure generated, its calculated scattering profile and fit to the experimental data were generated, and the best fit to the experimental data was identified by contour plots of χ2CRYSOL (Fig. 7, D–E for the palms-down model, and G and H for the palm–to–palm model). Using this approach, a model was derived that optimized the correlation with the solution scatter in each case (Fig. 7, F and I), failing to unambiguously discriminate a preferred dimer conformation. In these test ensembles, the best matches to the features of these experimental data (e.g. the peak feature at q = ∼0.13 Å−1) correlated most strongly with the translation of the ACT-domain dimer such that the COM–COM distance between the ACT dimer and remaining catalytic tetramer was ∼41 Å; there was a far less stringent correlation with the ACT dimer rotation.
Linker connectivity between the catalytic and regulatory domains
In building the original model for activated PAH (12), the simplest model involved an ∼90° backbone rotation at residue ∼118 to reposition the entire regulatory domain and form an ACT-domain dimer. The proposed allosteric mechanism included tetramer dissociation based on the reported morpheein-like characteristics of PAH (33). A morpheein has been described as a protein that can reversibly populate alternative assemblies as a consequence of conformational changes in the dissociated state (34). For PAH, where a tetramer–dimer equilibrium is well-established to occur in the RS-PAH conformation (e.g. Fig. 2B and Fig. S2), conformational change in a dissociated state is possible. Nevertheless, it is not obvious why a rotation at a hinge between the regulatory and catalytic domains would require tetramer dissociation. Here, we consider whether the RS-PAH to A-PAH transition involves repositioning an additional ∼10-amino acid region to include all residues N-terminal to the active site lid (residues 1–127), wherein the mobile element swings to the opposite face of the tetramer. The alternative structures of a monomer (trans versus cis) are shown in Fig. 7C. The large molecular motion required to accommodate the cis possibility would necessitate tetramer dissociation, as is more consistent with the biophysical and kinetic behavior of PAH from which we first suggested that it might fall into the category of shape-shifting proteins that have been called morpheeins (33, 34) or transformers (35).
To test whether SAXS could discriminate between the trans versus cis architectures, a similar all-atom palms-down model was prepared with the monomers in the cis conformation (Fig. 7, J–L). An ensemble of test structures was again generated by the systematic rotation and translation of the ACT-domain dimers (Fig. 7J). The results are shown in Fig. 7, K and L. Although an optimal model was identified using this approach, it could not be unambiguously distinguished from the other two structural models derived, including χ2CRYSOL and the COM–COM distance. In summary, solution scattering cannot resolve the three considered features of the quaternary structure of the C29S tetramer in the A-PAH conformation. However, optimal fits suggest a significantly further translation of the ACT-domain dimers, about 9–10 Å farther from the catalytic domains relative to the originally published A-PAH model (12) or about 8 Å relative to an arbitrary starting model (Fig. 7, F, I, and L).
Discussion
The X-ray crystal structure and SAXS analysis of C29S establish that rPAH and hPAH sample the same structural space, establishing that the bulk of the extensive published data (10, 12, 20, 36–38) on the more tractable rPAH protein is likely applicable to understanding hPAH and human PKU. Design of the C29S variant provided sufficient stabilization for the RS-PAH conformation to provide a 3 Å crystal structure of an hPAH whose only notable difference from rPAH is increased sampling of alternative conformations within the ACT domain and the C-terminal helices. The implied molecular motions within the monomeric ACT domains of the RS-PAH conformation are consistent with recent molecular dynamics calculations (11), although the bulk of that study was on the ACT-domain dimer found in the A-PAH conformation. The implied motions within the C-terminal helices are consistent with the observed dissociation of the hPAH tetramer (e.g. Fig. S2 for C29S). Unlike the other aromatic amino acid hydroxylases, PAH does not contain a classic leucine zipper motif in the C-terminal helices that secure the tetramers of this class of proteins (39).
The most significant aspect of this study is our improved understanding of the structure of the A-PAH conformation through extensive modeling relative to the SEC–SAXS data on the activated PAH tetramer. Here, we establish that SAXS cannot discriminate between different precedented ACT-domain dimer assemblies (Fig. 7A), although the crystal structure of the Phe-bound hPAH ACT domain (PDB code 5FII) confirms palms-down dimerization. More significantly, we conclude that the SAXS data are consistent with an A-PAH conformation for which the ACT-domain dimers are ∼8–10 Å more distant from the catalytic and multimerization domains (Fig. 6, right) than had previously been considered (6–8, 12, 24). The unexpectedly large space between the ACT-domain dimers and the rest of the protein provides potential for repositioning the autoregulatory region and/or a large binding cavity for pharmacological chaperones that might selectively stabilize A-PAH. We have previously discussed the therapeutic potential of such molecules either alone or in concert with BH4 (Kuvan®), which is posited to stabilize the RS-PAH conformation (5). Most significantly, however, we present an unprecedented alternative “cis” conformation for the monomer of activated protein (Fig. 7C), the formation of which would require tetramer dissociation as a component of PAH allostery. PAH was originally identified as a putative morpheein due, in part, to the slow nature of its allosteric activation (12). A similar slow equilibration of alternative quaternary structure assemblies has been established to proceed via a rate-determining conformational change in the dissociated state (40). Required tetramer dissociation as a component of PAH allostery would dictate the mixing of disease-associated variants in compound heterozygous individuals living with PKU. This provides a structural rationale for the interallelic complementation that has been reported for PKU (41, 42).
Although future experimentation is required to discriminate between the monomer structure possibilities illustrated in Fig. 7, they correspond to significant differences in the environments of Trp-120 and Cys-237, for which biochemical and biophysical studies have determined increased solvent accessibility upon PAH activation. Trp-120 is the residue most responsible for the change in intrinsic fluorescence upon activation (43), where the red shift implies increased solvent accessibility (see Fig. 2A). Cys-237 has been shown to be susceptible to chemical modification only when PAH is activated, again implying increased solvent accessibility in the A-PAH conformation (44, 45).
This study has potential for simplifying characterization of disease-associated hPAH variants. Sophisticated contemporary studies of such variants use a fusion protein with a maltose-binding protein on the N terminus (46); for these studies, kinetic data are routinely reported for the fusion protein, whereas stability studies employ hPAH from which the fusion protein has been cleaved. We established herein that C29S is a tractable variant of hPAH, which suggests that it may be useful for characterization of disease-associated variants.
Experimental procedures
Materials
All chemicals were purchased from Sigma unless otherwise noted. Enzymes used in cloning were purchased from Agilent and New England Biolabs.
Quantification of Phe, BH4, and Tyr stock concentrations
Phe and Tyr were dissolved in water to ∼150 and ∼2.5 mm, respectively. The Phe concentration was determined at 257.5 nm in 0.1 m HCl using a molar extinction coefficient (ϵ257.5 nm) of 195 cm−1 m−1 (47). The Tyr concentration was determined in 0.1 m HCl using ϵ274.5 nm of 1,400 cm−1 m−1 (48). BH4 was dissolved in 10 mm DTT to give ∼100 mm, and concentration was determined in 0.1 m HCl using ϵ264 nm of 13,750 cm−1 m−1 (49).
Construction of hPAH expression vectors
Expression plasmid pMRH160 encoding His6–SUMO–hPAH was constructed by subcloning the hPAH gene from plasmid pEPAH1 (13) into plasmid pETHSUL with the ligation-independent cloning method (50). The C29S mutation was introduced into this construct by QuikChange mutagenesis to yield expression plasmid pMRH161. Plasmid pAHhum-C29S encoding hPAH C29S variant was created with the QuikChange method using plasmid pEPAH1 (13). pMRH173 was created by using the QuikChange method to replace a Gly codon in TEV recognition site Ser codon in plasmid pETHT (gift from the Patrick Loll laboratory, Drexel University). The expression plasmid pMRH174 encodes for His6–TEV–C29S, where digestion of the fusion protein with TEV protease results in C29S where the first N-terminal amino acid in the matured protein to be Ser-02. The construct was created by the subcloning of the PCR product from pMRH161 into BsaI-digested pMRH173 via blunt-end ligation. The PAH gene sequences of all plasmids were verified by standard sequencing (GeneWiz). All primers used in the creation of these plasmids are provided in Table S1.
Protein expression and purification
The WT hPAH and C29S proteins were expressed in E. coli as fusion proteins with an N-terminal His6–SUMO tag that is cleaved such that no non-native amino acids remain at the N terminus (50). The N terminus of C29S and hPAH is Ser-02, omitting the initiator methionine codon. This is anticipated from similarity with mouse PAH (UniProtKB: P16331), whereby LC-MS determined that the initiating methionine is cleaved from the mature protein. The details of the expression and purification of all proteins used in this study are provided in supporting Methods.
The hPAH and C29S used for kinetic characterization were derived from proteins with no fusion partners and free of cloning artifacts that were purified using the classic Shiman method (17), or from fusion protein constructs purified using Tris-HCl, pH 7.4, for the nickel column procedures.
The theoretical ϵ280 nm of 49,280 m−1 cm−1 (51) was used to calculate the concentration of the purified hPAH and C29S proteins. Purified protein was flash-frozen in liquid N2 and stored at −80 °C.
Steady-state kinetics
PAH activity was measured as reported previously (11).
Intrinsic protein fluorescence
PAH intrinsic fluorescence was measured at 25.0 °C at an excitation wavelength of 295 nm and an emission spectrum of 305–400 nm with a PTI fluorescence system spectrophotometer equipped with a USHIO Xenon Short Arc Lamp and FelixTM for Windows software. Protein (0.5 μm monomer) was analyzed in 30 mm Tris-HCl, pH 7.4, 150 mm KCl with and without 1 mm Phe. Samples were preheated at 25.0 °C for 15 min before each measurement.
SEC
Analyses were performed using a calibrated Superdex 200 10/300 GL gel-filtration column (GE Healthcare) in 30 mm Tris, pH 7.4, 150 mm KCl with and without 1.5 mm Phe with a flow-rate of 0.5 ml/min at 22 °C. The isocratic elution of ∼1 mg of injected protein was monitored continuously using UV detection at 280 nm.
Sedimentation equilibrium analysis (SE–AUC)
Analytical ultracentrifugation experiments were performed with an XL-A analytical ultracentrifuge (Beckman-Coulter) and a TiAn60 rotor with six-channel charcoal-filled Epon centerpieces and quartz windows. S.E. data were collected at 4 °C with detection at 280 nm for two sample concentrations in the presence and absence of 1 mm Phe at 2.5 and 5 μm protein concentrations in 30 mm Tris, pH 7.4, and 100 mm KCl. Analyses were carried out using global fits to data acquired at multiple speeds for each concentration with strict mass conservation using the program SEDPHAT (52). Error estimates for equilibrium constants were determined from a 1,000-iteration Monte Carlo simulation. The partial specific volume (v̄), solvent density (ρ), and viscosity (η) were derived from chemical composition by SEDNTERP (53).
Protein crystallization
Crystals were grown from C29S (16 mg/ml) in 2 mm Tris-HCl, pH 7.4, mixed with reservoir solutions in a protein/reservoir ratio equal to 1:1.25 and equilibrated using hanging-drop vapor diffusion. The 1-ml reservoir solution initially contained 16% PEG 3,350, 100 mm bis-tris-propane, pH 7.5, and 200 mm sodium sulfate. Crystals appeared at 22 °C within 1 day.
Structure determination
Crystals were looped for several seconds through a solution composed of 1 part mother liquor and 1 part of a 1:1 mixture of 50% sucrose in H2O and 100% ethylene glycol (Hampton Research) before flash-freezing in liquid N2. Data from a single crystal was collected at NSLS-II, 17-ID-1 beamline at Brookhaven National Laboratories. Table 2 reports the statistics for data collection, processing, and model building. Data were processed using XDS (54), and the phases were solved using molecular replacement using the approach described by Korasick et al. (55). Search models were generated by BALBES (56). The two top-scoring models were derived from PDB entries 1PHZ (57) (residues 1–429), and 1J8U (58) (residues 103–427). Ab initio model building as implemented within PHENIX version 1.12 rc1–2807 (59) generated an initial model that was 58.9% complete. Further iterative manual model building and local refinement was done using Coot version 0.8.8 (60). PHENIX was used for further refinement, restraint generation, and map calculations that were used for manual model building. NCS restraints (torsion-angle), conformation-dependent rotamer libraries, and automatic optimization of atomic displacement factor and stereochemistry weights were used throughout refinement, as well as restraints for iron coordination by waters and amino acid residues at the active site. The L-test analysis with Xtriage as implemented within Phenix indicated a possible twin operator of (l, −k, h) and a twin fraction of 0.245. However, application of twin operators early in the refinement worsened R-free by ∼1.3% and a widened the gap between R-work and R-free by ∼12%. Therefore, during subsequent refinements, the twin law was not applied. In the latest stages of refinement, application of the twin operator significantly improved the overall refinement statistics (e.g. improvement of R-free by ∼3.3% and a decrease in the R-free–R-work gap by ∼1.2%.) in the final model. A single TLS group per chain, Ramachandran restraints, and riding hydrogens were applied at later stages of refinement. An iterative-built composite omit map (61) was generated toward the end of refinement and was used to make adjustments to backbone within regions of uncertainty. The final coordinate and phase errors are reported by Phenix to be 0.5 Å and 28.7, respectively. All molecular structure alignments (using the all-atom align tool) were performed, and illustrations were created using the PyMOL version 2.0 (Schrödinger, LLC). All measurements related to protein interfaces were performed using PDBePISA (62). PDB_redo (63) was used at a near final stage to inspect the model and inform minor adjustments to rotamers.
SAXS data collection
SEC-SAXS data were collected at beamline 16-ID (LiX) of the National Synchrotron Light Source II (Upton, NY) (64). Data were collected at a wavelength of 1.0 Å in a three-camera configuration and yielded accessible scattering angle where 0.006 < q < 3.0 Å−1, where q is the momentum transfer, defined as q = 4π sin(θ)/λ, where λ is the X-ray wavelength and 2θ is the scattering angle; data to q <0.5 Å−1 were used in subsequent analyses. 100 μl of 5 mg/ml C29S was injected and eluted isocratically at 0.5 ml/min from a Superdex 200 10/300 sizing column (GE Healthcare) equilibrated in 30 mm Tris/HCl, pH 7.4, 100 mm KCl, with or without 1 mm Phe, at room temperature. Eluent from the column flowed into a 1-mm capillary for subsequent X-ray exposures at 1-s intervals. Plots of intensity from the forward scatter closely correlated to in-line UV and refractive index measurements. C29S was activated by the addition of a 100 mm Phe stock to the protein sample followed by incubation at room temperature for 30 min and subsequent high-speed centrifugation in a table-top centrifuge before injection.
SAXS analysis
SVD–EFA analysis of the SEC–SAXS datasets were performed as described previously (8) and as implemented in the program RAW (65). Buffer-subtracted profiles were analyzed by SVD, and the ranges of overlapping peak data were determined using EFA (66). The determined peak windows were used to identify the basis vectors for each component, and the corresponding SAXS profiles were calculated (see Fig. S4 and S5 for additional details of the deconvolution process). When fitting manually, the maximum diameter of the particle (Dmax) was incrementally adjusted in GNOM (67) to maximize the goodness-of-fit parameter, to minimize the discrepancy between the fit and the experimental data, and to optimize the visual qualities of the distribution profile. The theoretical SAXS profiles for atomic models were created using the CRYSOL program (68). To facilitate comparison of atomic models to small-angle scattering data, atomistic models representing the complete composition of the protein were constructed, and the resulting model was gradually relaxed by energy minimization in a box of water by the programs VMD (69) and NAMD (70), using CHARMM42 force fields. The models were rendered using the program PYMOL (71).
Author contributions
E. C. A. and E. K. J. conceptualization; E. C. A., K. G., and M. R. H. data curation; E. C. A., K. G., M. R. H., E. B., and E. K. J. formal analysis; E. C. A., K. G., M. R. H., and E. B. investigation; E. C. A., K. G., and E. K. J. writing-original draft; E. C. A., K. G., and E. K. J. writing-review and editing; E. K. J. supervision; E. K. J. funding acquisition; E. K. J. project administration.
Supplementary Material
Acknowledgments
The crystallographic data were obtained at the 17-ID-1 beamline, and the SEC–SAXS data were obtained at 16-ID (LIX) at the National Synchrotron Light Source II, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. We also acknowledge the intellectual input from Prof. Patrick J. Loll on various aspects of the crystallographic analysis of the structure of C29S. We acknowledge the contribution of Dr. Paul Emsley for guidance on model building and for providing the code to generate the structures presented in Fig. 4. We are grateful to Prof. Paula Leandro for sharing her complementary studies on several His-tagged hPAH variants, including C29S, which were designed to stabilize the protein for therapeutic purposes.
This work was supported by National Institutes of Health Grant 5R01-NS100081 (to E. K. J. and K. G.) and a grant from the National PKU Alliance (to E. K. J.) and in part by the National Institutes of Health NCI Cancer Center Support Grant P30 CA006927. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5, Table S1, supporting Methods, and supporting Refs. 1–8.
The atomic coordinates and structure factors (code 6N1K) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- hPAH
- human PAH
- PAH
- phenylalanine hydroxylase
- rPAH
- rat PAH
- A-PAH
- activated PAH
- RS-PAH
- resting-state PAH
- PKU
- phenylketonuria
- SEC
- size-exclusion chromatography
- SAXS
- small-angle X-ray scattering
- SE-AUC
- sedimentation equilibrium analytical ultracentrifugation
- PDB
- Protein Data Bank
- RMSD
- root mean square deviation
- BH4
- tetrahydrobiopterin
- TEV
- tobacco etch virus
- SVD–EFA
- singular value decomposition with evolving factor analysis
- COM
- center-of-mass.
References
- 1. Blau N. (2016) Genetics of phenylketonuria: then and now. Hum. Mutat. 37, 508–515 10.1002/humu.22980 [DOI] [PubMed] [Google Scholar]
- 2. Chipman D. M., and Shaanan B. (2001) The ACT domain family. Curr. Opin. Struct. Biol. 11, 694–700 10.1016/S0959-440X(01)00272-X [DOI] [PubMed] [Google Scholar]
- 3. Grant G. A. (2006) The ACT domain: a small molecule binding domain and its role as a common regulatory element. J. Biol. Chem. 281, 33825–33829 10.1074/jbc.R600024200 [DOI] [PubMed] [Google Scholar]
- 4. Lang E. J., Cross P. J., Mittelstädt G., Jameson G. B., and Parker E. J. (2014) Allosteric ACTion: the varied ACT domains regulating enzymes of amino-acid metabolism. Curr. Opin. Struct. Biol. 29, 102–111 10.1016/j.sbi.2014.10.007 [DOI] [PubMed] [Google Scholar]
- 5. Jaffe E. K. (2017) New protein structures provide an updated understanding of phenylketonuria. Mol. Genet. Metab. 121, 289–296 10.1016/j.ymgme.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Patel D., Kopec J., Fitzpatrick F., McCorvie T. J., and Yue W. W. (2016) Structural basis for ligand-dependent dimerization of phenylalanine hydroxylase regulatory domain. Sci. Rep. 6, 23748 10.1038/srep23748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arturo E. C., Gupta K., Héroux A., Stith L., Cross P. J., Parker E. J., Loll P. J., and Jaffe E. K. (2016) First structure of full-length mammalian phenylalanine hydroxylase reveals the architecture of an autoinhibited tetramer. Proc. Natl. Acad. Sci. U.S.A. 113, 2394–2399 10.1073/pnas.1516967113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meisburger S. P., Taylor A. B., Khan C. A., Zhang S., Fitzpatrick P. F., and Ando N. (2016) Domain movements upon activation of phenylalanine hydroxylase characterized by crystallography and chromatography-coupled small-angle X-ray scattering. J. Am. Chem. Soc. 138, 6506–6516 10.1021/jacs.6b01563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li J., Ilangovan U., Daubner S. C., Hinck A. P., and Fitzpatrick P. F. (2011) Direct evidence for a phenylalanine site in the regulatory domain of phenylalanine hydroxylase. Arch. Biochem. Biophys. 505, 250–255 10.1016/j.abb.2010.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang S., and Fitzpatrick P. F. (2016) Identification of the allosteric site for phenylalanine in rat phenylalanine hydroxylase. J. Biol. Chem. 291, 7418–7425 10.1074/jbc.M115.709998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ge Y., Borne E., Stewart S., Hansen M. R., Arturo E. C., Jaffe E. K., and Voelz V. A. (2018) Simulations of the regulatory ACT domain of human phenylalanine hydroxylase (PAH) unveil its mechanism of phenylalanine binding. J. Biol. Chem. 293, 19532–19543 10.1074/jbc.RA118.004909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jaffe E. K., Stith L., Lawrence S. H., Andrake M., and Dunbrack R. L. Jr. (2013) A new model for allosteric regulation of phenylalanine hydroxylase: implications for disease and therapeutics. Arch. Biochem. Biophys. 530, 73–82 10.1016/j.abb.2012.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Daubner S. C., Hillas P. J., and Fitzpatrick P. F. (1997) Expression and characterization of the catalytic domain of human phenylalanine hydroxylase. Arch. Biochem. Biophys. 348, 295–302 10.1006/abbi.1997.0435 [DOI] [PubMed] [Google Scholar]
- 14. Ledley F. D., Grenett H. E., and Woo S. L. (1987) Biochemical characterization of recombinant human phenylalanine hydroxylase produced in Escherichia coli. J. Biol. Chem. 262, 2228–2233 [PubMed] [Google Scholar]
- 15. Kaufman S. (1993) The phenylalanine hydroxylating system. Adv. Enzymol. Relat. Areas Mol. Biol. 67, 77–264 [DOI] [PubMed] [Google Scholar]
- 16. Wang G. A., Gu P., and Kaufman S. (2001) Mutagenesis of the regulatory domain of phenylalanine hydroxylase. Proc. Natl. Acad. Sci. U.S.A. 98, 1537–1542 10.1073/pnas.98.4.1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shiman R., Gray D. W., and Pater A. (1979) A simple purification of phenylalanine hydroxylase by substrate-induced hydrophobic chromatography. J. Biol. Chem. 254, 11300–11306 [PubMed] [Google Scholar]
- 18. Phillips R. S., Parniak M. A., and Kaufman S. (1984) Spectroscopic investigation of ligand interaction with hepatic phenylalanine hydroxylase: evidence for a conformational change associated with activation. Biochemistry 23, 3836–3842 10.1021/bi00312a007 [DOI] [PubMed] [Google Scholar]
- 19. Parniak M. A., Davis M. D., and Kaufman S. (1988) Effect of alkaline pH on the activity of rat liver phenylalanine hydroxylase. J. Biol. Chem. 263, 1223–1230 [PubMed] [Google Scholar]
- 20. Zhang S., Hinck A. P., and Fitzpatrick P. F. (2015) The amino acid specificity for activation of phenylalanine hydroxylase matches the specificity for stabilization of regulatory domain dimers. Biochemistry 54, 5167–5174 10.1021/acs.biochem.5b00616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martinez A., Knappskog P. M., Olafsdottir S., Døskeland A. P., Eiken H. G., Svebak R. M., Bozzini M., Apold J., and Flatmark T. (1995) Expression of recombinant human phenylalanine hydroxylase as fusion protein in Escherichia coli circumvents proteolytic degradation by host cell proteases. Isolation and characterization of the wild-type enzyme. Biochem. J. 306, 589–597 10.1042/bj3060589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carluccio C., Fraternali F., Salvatore F., Fornili A., and Zagari A. (2013) Structural features of the regulatory ACT domain of phenylalanine hydroxylase. PLoS ONE 8, e79482 10.1371/journal.pone.0079482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burnley B. T., Afonine P. V., Adams P. D., and Gros P. (2012) Modelling dynamics in protein crystal structures by ensemble refinement. Elife 1, e00311 10.7554/eLife.00311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khan C. A., Meisburger S. P., Ando N., and Fitzpatrick P. F. (2019) The phenylketonuria-associated substitution R68S converts phenylalanine hydroxylase to a constitutively active enzyme but reduces its stability. J. Biol. Chem. 294, 4359–4367 10.1074/jbc.RA118.006477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Malaby A. W., Chakravarthy S., Irving T. C., Kathuria S. V., Bilsel O., and Lambright D. G. (2015) Methods for analysis of size-exclusion chromatography–small-angle X-ray scattering and reconstruction of protein scattering. J. Appl. Crystallogr. 48, 1102–1113 10.1107/S1600576715010420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rambo R. P., and Tainer J. A. (2013) Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496, 477–481 10.1038/nature12070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 10.1107/S0021889812007662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rambo R. P., and Tainer J. A. (2011) Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye Law. Biopolymers 95, 559–571 10.1002/bip.21638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Erlandsen H., and Stevens R. C. (1999) The structural basis of phenylketonuria. Mol. Genet. Metab. 68, 103–125 10.1006/mgme.1999.2922 [DOI] [PubMed] [Google Scholar]
- 30. Liberles J. S., Thórólfsson M., and Martínez A. (2005) Allosteric mechanisms in ACT domain containing enzymes involved in amino acid metabolism. Amino Acids 28, 1–12 10.1007/s00726-004-0152-y [DOI] [PubMed] [Google Scholar]
- 31. Cross P. J., Dobson R. C., Patchett M. L., and Parker E. J. (2011) Tyrosine latching of a regulatory gate affords allosteric control of aromatic amino acid biosynthesis. J. Biol. Chem. 286, 10216–10224 10.1074/jbc.M110.209924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang S., Huang T., Ilangovan U., Hinck A. P., and Fitzpatrick P. F. (2014) The solution structure of the regulatory domain of tyrosine hydroxylase. J. Mol. Biol. 426, 1483–1497 10.1016/j.jmb.2013.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Selwood T., and Jaffe E. K. (2012) Dynamic dissociating homo-oligomers and the control of protein function. Arch. Biochem. Biophys. 519, 131–143 10.1016/j.abb.2011.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jaffe E. K. (2005) Morpheeins–a new structural paradigm for allosteric regulation. Trends Biochem. Sci. 30, 490–497 10.1016/j.tibs.2005.07.003 [DOI] [PubMed] [Google Scholar]
- 35. Wasserman H., and Saphire E. O. (2016) More than meets the eye: hidden structures in the proteome. Annu. Rev. Virol. 3, 373–386 10.1146/annurev-virology-100114-054923 [DOI] [PubMed] [Google Scholar]
- 36. Khan C. A., and Fitzpatrick P. F. (2018) Phosphorylation of phenylalanine hydroxylase increases the rate constant for formation of the activated conformation of the enzyme. Biochemistry 57, 6274–6277 10.1021/acs.biochem.8b00919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li J., and Fitzpatrick P. F. (2013) Regulation of phenylalanine hydroxylase: conformational changes upon phosphorylation detected by H/D exchange and mass spectrometry. Arch. Biochem. Biophys. 535, 115–119 10.1016/j.abb.2013.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fitzpatrick P. F. (2012) Allosteric regulation of phenylalanine hydroxylase. Arch. Biochem. Biophys. 519, 194–201 10.1016/j.abb.2011.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fitzpatrick P. F. (2015) Structural insights into the regulation of aromatic amino acid hydroxylation. Curr. Opin. Struct. Biol. 35, 1–6 10.1016/j.sbi.2015.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Selwood T., Tang L., Lawrence S. H., Anokhina Y., and Jaffe E. K. (2008) Kinetics and thermodynamics of the interchange of the morpheein forms of human porphobilinogen synthase. Biochemistry 47, 3245–3257 10.1021/bi702113z [DOI] [PubMed] [Google Scholar]
- 41. Djordjevic M., Klaassen K., Sarajlija A., Tosic N., Zukic B., Kecman B., Ugrin M., Spasovski V., Pavlovic S., and Stojiljkovic M. (2013) Molecular genetics and genotype-based estimation of BH4-responsiveness in Serbian PKU patients: spotlight on phenotypic implications of p.L48S. JIMD Rep. 9, 49–58 10.1007/8904_2012_178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shen N., Heintz C., Thiel C., Okun J. G., Hoffmann G. F., and Blau N. (2016) Co-expression of phenylalanine hydroxylase variants and effects of interallelic complementation on in vitro enzyme activity and genotype-phenotype correlation. Mol. Genet. Metab. 117, 328–335 10.1016/j.ymgme.2016.01.004 [DOI] [PubMed] [Google Scholar]
- 43. Knappskog P. M., and Haavik J. (1995) Tryptophan fluorescence of human phenylalanine hydroxylase produced in Escherichia coli. Biochemistry 34, 11790–11799 10.1021/bi00037a017 [DOI] [PubMed] [Google Scholar]
- 44. Gibbs B. S., and Benkovic S. J. (1991) Affinity labeling of the active site and the reactive sulfhydryl associated with activation of rat liver phenylalanine hydroxylase. Biochemistry 30, 6795–6802 10.1021/bi00241a024 [DOI] [PubMed] [Google Scholar]
- 45. Parniak M. A., and Kaufman S. (1981) Rat liver phenylalanine hydroxylase. Activation by sulfhydryl modification. J. Biol. Chem. 256, 6876–6882 [PubMed] [Google Scholar]
- 46. Staudigl M., Gersting S. W., Danecka M. K., Messing D. D., Woidy M., Pinkas D., Kemter K. F., Blau N., and Muntau A. C. (2011) The interplay between genotype, metabolic state and cofactor treatment governs phenylalanine hydroxylase function and drug response. Hum. Mol. Genet. 20, 2628–2641 10.1093/hmg/ddr165 [DOI] [PubMed] [Google Scholar]
- 47. Beaven G. H., and Holiday E. R. (1952) Ultraviolet absorption spectra of proteins and amino acids. Adv. Protein Chem. 7, 319–386 10.1016/S0065-3233(08)60022-4 [DOI] [PubMed] [Google Scholar]
- 48. Edelhoch H., and Lippoldt R. E. (1962) The properties of thyroglobulin. IX. The molecular properties of iodinated thyroglobulin. J. Biol. Chem. 237, 2788–2794 [PubMed] [Google Scholar]
- 49. Stone K. J. (1976) The role of tetrahydrofolate dehydrogenase in the hepatic supply of tetrahydrobiopterin in rats. Biochem. J. 157, 105–109 10.1042/bj1570105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weeks S. D., Drinker M., and Loll P. J. (2007) Ligation independent cloning vectors for expression of SUMO fusions. Protein Expr. Purif. 53, 40–50 10.1016/j.pep.2006.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pace C. N., Vajdos F., Fee L., Grimsley G., and Gray T. (1995) How to measure and predict the molar absorption-coefficient of a protein. Protein Sci. 4, 2411–2423 10.1002/pro.5560041120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vistica J., Dam J., Balbo A., Yikilmaz E., Mariuzza R. A., Rouault T. A., and Schuck P. (2004) Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 326, 234–256 10.1016/j.ab.2003.12.014 [DOI] [PubMed] [Google Scholar]
- 53. Laue T. M., Shah B. D., Ridgeway T. M., and Pelletier S. L. (1992) Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S., and Rowe A., eds) pp. 90–125, The Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 54. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Korasick D. A., Gamage T. T., Christgen S., Stiers K. M., Beamer L. J., Henzl M. T., Becker D. F., and Tanner J. J. (2017) Structure and characterization of a class 3B proline utilization A: ligand-induced dimerization and importance of the C-terminal domain for catalysis. J. Biol. Chem. 292, 9652–9665 10.1074/jbc.M117.786855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Long F., Vagin A. A., Young P., and Murshudov G. N. (2008) BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 10.1107/S0907444907050172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kobe B., Jennings I. G., House C. M., Michell B. J., Goodwill K. E., Santarsiero B. D., Stevens R. C., Cotton R. G., and Kemp B. E. (1999) Structural basis of autoregulation of phenylalanine hydroxylase. Nat. Struct. Biol. 6, 442–448 10.1038/8247 [DOI] [PubMed] [Google Scholar]
- 58. Andersen O. A., Flatmark T., and Hough E. (2001) High resolution crystal structures of the catalytic domain of human phenylalanine hydroxylase in its catalytically active Fe(II) form and binary complex with tetrahydrobiopterin. J. Mol. Biol. 314, 279–291 10.1006/jmbi.2001.5061 [DOI] [PubMed] [Google Scholar]
- 59. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L. W., Read R. J., and Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 10.1107/S090744490705024X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Paxman J. J., and Heras B. (2017) Bioinformatics tools and resources for analyzing protein structures. Methods Mol. Biol. 1549, 209–220 10.1007/978-1-4939-6740-7_16 [DOI] [PubMed] [Google Scholar]
- 63. Joosten R. P., Salzemann J., Bloch V., Stockinger H., Berglund A. C., Blanchet C., Bongcam-Rudloff E., Combet C., Da Costa A. L., Deleage G., Diarena M., Fabbretti R., Fettahi G., Flegel V., Gisel A., et al. (2009) PDB_REDO: automated re-refinement of X-ray structure models in the PDB. J. Appl. Crystallogr. 42, 376–384 10.1107/S0021889809008784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. DiFabio J., Chodankar S., Pjerov S., Jakoncic J., Lucas M., Krywka C., Graziano V., and Yang L. (2016) in Proceedings of the 12th International Conference on Synchrotron Radiation Instrumentation (Sri2015) (Shen Q., and Nelson C., eds) Vol. 1741, p. 30049, American Institute of Physics (AIP), New York [Google Scholar]
- 65. Hopkins J. B., Gillilan R. E., and Skou S. (2017) BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Crystallogr. 50, 1545–1553 10.1107/S1600576717011438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Maeder M. (1987) Evolving factor-analysis for the resolution of overlapping chromatographic peaks. Anal. Chem. 59, 527–530 10.1021/ac00130a035 [DOI] [Google Scholar]
- 67. Semenyuk A. V., and Svergun D. I. (1991) Gnom–a program package for small-angle scattering data-processing. J. Appl. Crystallogr. 24, 537–540 10.1107/S002188989100081X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Svergun D., Barberato C., and Koch M. H. J. (1995) CRYSOL–a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 10.1107/S0021889895007047 [DOI] [Google Scholar]
- 69. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 70. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. DeLano W. L. (2004) Use of PYMOL as a communications tool for molecular science. Abstracts of Papers of the American Chemical Society 228, U313–U314 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.