

Abstract
The epithelial sodium channel (ENaC) mediates Na+ transport in several epithelia, including the aldosterone-sensitive distal nephron, distal colon, and biliary epithelium. Numerous factors regulate ENaC activity, including extracellular ligands, post-translational modifications, and membrane-resident lipids. However, ENaC regulation by bile acids and conjugated bilirubin, metabolites that are abundant in the biliary tree and intestinal tract and are sometimes elevated in the urine of individuals with advanced liver disease, remains poorly understood. Here, using a Xenopus oocyte-based system to express and functionally study ENaC, we found that, depending on the bile acid used, bile acids both activate and inhibit mouse ENaC. Whether bile acids were activating or inhibiting was contingent on the position and orientation of specific bile acid moieties. For example, a hydroxyl group at the 12-position and facing the hydrophilic side (12α-OH) was activating. Taurine-conjugated bile acids, which have reduced membrane permeability, affected ENaC activity more strongly than did their more membrane-permeant unconjugated counterparts, suggesting that bile acids regulate ENaC extracellularly. Bile acid–dependent activation was enhanced by amino acid substitutions in ENaC that depress open probability and was precluded by proteolytic cleavage that increases open probability, consistent with an effect of bile acids on ENaC open probability. Bile acids also regulated ENaC in a cortical collecting duct cell line, mirroring the results in Xenopus oocytes. We also show that bilirubin conjugates activate ENaC. These results indicate that ENaC responds to compounds abundant in bile and that their ability to regulate this channel depends on the presence of specific functional groups.
Keywords: epithelial sodium channel (ENaC), bile acid, protein palmitoylation, allosteric regulation, proteolysis, renal physiology, bililrubin, cholic acid, degenerin family, kidney tubule, physico-chemical properties, trimeric cation channel
Introduction
Epithelial Na+ channel (ENaC)2-mediated Na+ transport is rate-limiting for Na+ reabsorption in principal cells in the aldosterone-sensitive distal nephron, where the channel plays a key role regulating total extracellular fluid volume (1). ENaC expression in the epithelia of the airways, colon, and biliary tree has also been implicated in regulating total luminal Na+ and fluid volume (2–4). The channel's activity is directly regulated by several environmental factors, including extracellular ions (e.g. H+, Na+, and Cl−), mechanical forces, and proteases (5). It has recently emerged that amphipathic compounds, including bile acids, also regulate ENaC-mediated currents (6–8).
Primary bile acids are synthesized in hepatocytes and are ultimately secreted into the duodenum to emulsify dietary lipids and facilitate excretion of toxic metabolites (9). Gut microbes metabolize these and generate secondary bile acids by modifying key functional groups (10). Approximately 95% of bile acids are reabsorbed in the ileum and are transported back to the liver via the portal vein, completing the enterohepatic loop. Bile is also composed of conjugated bilirubin (c-bilirubin), an end product of heme catabolism that allows for its aqueous phase excretion (11). Under physiologic conditions, high concentrations of bile acids and c-bilirubin are restricted to the bile ducts, gall bladder, and gut. Blood and urine concentrations of both increase in liver disease or injury, whereupon urine becomes a major vehicle for their elimination (12–16). Whether increased biliary factor levels contribute to the pathology of liver disease remains unclear.
Bile acids influence numerous physiologic processes through the nuclear farnesoid X receptor and the G protein–coupled receptor TGR5 (17–20). Recent reports have shown that bile acids regulate several members of the ENaC/degenerin family (6, 7, 21, 22). ENaC belongs to a family of trimeric cation channels, which include acid-sensing ion channels (ASICs) and the bile acid–sensitive ion channel (BASIC) (23, 24). ENaC is a heterotrimer composed of α, β, and γ subunits. Each subunit contributes two transmembrane helices to a single pore, with the bulk of each subunit contained in the extracellular domains (25, 26). Gating regulation by extracellular factors is emblematic of this protein family. Numerous studies have identified sites and structures key to regulation by extracellular factors (27).
Here we examined the molecular determinants of ENaC regulation by amphipathic compounds found in bile. We found that specific bile acids regulate mouse ENaC, both heterologously expressed in Xenopus oocytes and endogenously expressed in a mouse cortical collecting duct cell line (mpkCCDc14). When systematically screening bile acids, we found that both channel activation and inhibition were associated with specific moieties and that neither activation nor inhibition depended on membrane permeability. We also found that conjugates of bilirubin activated ENaC. Other known modes of ENaC regulation, including proteolytic processing, influenced bile acid activation of the channel. Deoxycholic acid (DCA) robustly activated uncleaved channels, which have a low basal open probability (Po), whereas DCA and taurocholic acid (t-CA) failed to activate fully cleaved channels, which have a high Po. Our data suggest that biliary factors may be natural ligands for the channel and bind to the extracellular side of the channel to influence its Po.
Results
Biliary factors regulate ENaC expressed in Xenopus oocytes
It has previously been reported that bile acids activate human and rat ENaC, ASIC1, and BASIC expressed in Xenopus oocytes (6, 7, 21, 22). Here, we investigated bile acid regulation of mouse ENaC, which has been extensively studied and is relevant in key model systems. We expressed WT mouse α, β, and γ subunits in oocytes and measured the effect of 1 mm DCA, cholic acid (CA), chenodeoxycholic acid (CDCA), and hyodeoxycholic acid (HDCA) (Fig. 1A) on whole-cell currents by two-electrode voltage clamp (TEVC). We found that bile acids enhanced ENaC currents in the order DCA > CA > CDCA and that HDCA inhibited ENaC currents (Fig. 1B). Bile acid regulation of ENaC currents occasionally demonstrated oocyte batch dependence (note the bimodal distribution of CA data), suggesting that differences in protein biosynthesis may influence bile acid regulation. DCA activation was dose-dependent, with evident activation at 10 μm DCA and more than half-maximal activation at 100 μm DCA (Fig. 1C).
Figure 1.
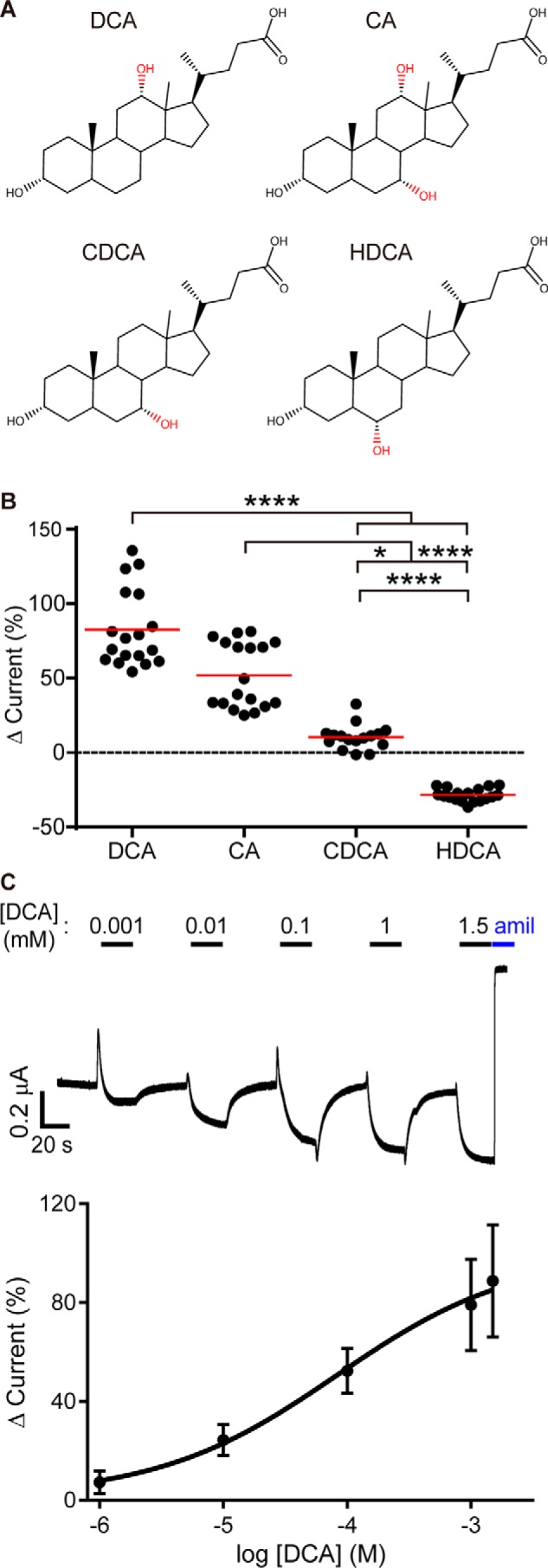
Specific Bile acids activate mouse ENaC in Xenopus oocytes. Oocytes were injected with cRNA encoding WT mouse ENaC subunits. Currents were measured the following day using TEVC. A, Oocytes were treated with bile acids (1 mm) for 40 s, followed by 10 μm amiloride. B, the effect of bile acid addition in each experiment was determined as a percentage of the baseline amiloride-sensitive current. The basal amiloride-sensitive current was −3.8 ± 2.8 μA (n = 72). Individual experiments are plotted with the means indicated by a horizontal bar. Each bile acid treatment resulted in a significant change in current (p = 0.002 for CDCA and p < 0.0001 for all others by paired Student's t test). Treatments were compared using a Kruskal–Wallis test followed by Dunn's multiple comparison test: *, p < 0.05; ****, p < 0.0001. C, dose-dependent activation of ENaC by DCA. Top, representative current trace of ENaC activated by successive increasing concentrations of DCA. Bottom, summary of DCA concentration–dependent activation on ENaC, presented as mean ± S.D. (n = 15). Data were fit to the Hill equation using nonlinear regression (logEC50 = −4.1 (78.5 μm) ± 0.26, Hill coefficient = 0.6).
Because bilirubin conjugated to glucuronic acid (c-bilirubin) is also a major bile component, we determined whether c-bilirubin activates ENaC (Fig. 2A). Bilirubin conjugation allows for aqueous phase excretion of the heme catabolite into bile. We found that c-bilirubin reversibly activated ENaC and that activation was dose-dependent (Fig. 2, B and C). ENaC activation by c-bilirubin required higher doses than DCA for equivalent increases in current and did not saturate even at 5 mm c-bilirubin (Figs. 1C and 2C). To test whether ENaC activation depended on the opened porphyrin ring of bilirubin or the glucuronide moieties introduced by conjugation, we tested the ability of taurine-conjugated bilirubin to activate the channel (t-bilirubin; Fig. 2A). We found that t-bilirubin activated ENaC more potently than c-bilirubin did (Fig. 2, B and C). These data suggest that bilirubin itself mediates activation of ENaC by c-bilirubin and t-bilirubin. Taken together, our data show that specific biliary factors activate mouse ENaC expressed in Xenopus oocytes.
Figure 2.
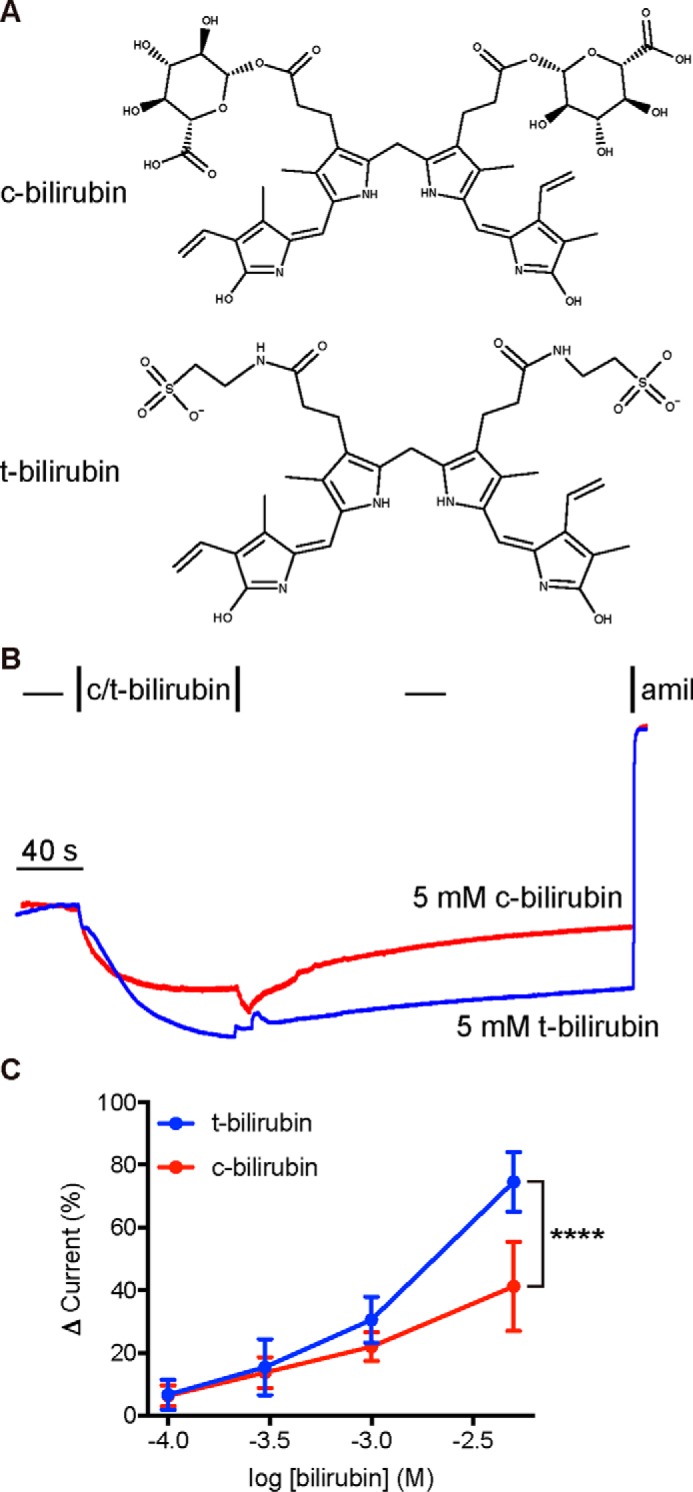
Conjugated bilirubin activates mouse ENaC in Xenopus oocytes. A and B, oocytes expressing WT ENaC were perfused with 5 mm bilirubin conjugated with either glucuronic acid (c-bilirubin) or taurine (t-bilirubin) and then washed out before the addition of 10 μm amiloride. C, concentration-dependent activation of ENaC by bilirubin conjugates. The basal amiloride-sensitive current was −2.7 ± 1.9 μA (n = 14). Data are mean ± S.D. (n = 5–9). Lack of saturation suggests an EC50 > 1 mm. ****, p < 0.0001 by two-way ANOVA with Sidak's multiple-comparison post hoc test.
Preventing ENaC palmitoylation enhances activation by DCA
Schmidt et al. (28) proposed that bile acids activate BASIC by facilitating interactions between a cytosolic amphipathic helix and the plasma membrane. Palmitoylation (post-translational modification by hydrophobic palmitate) of the ENaC β and γ subunits has been proposed to activate ENaC through an analogous mechanism (29, 30). ENaC β and γ subunits are palmitoylated at specific intracellular cysteine residues (βCys-43, βCys-557, γCys-33, and γCys-41), and mutating these sites to alanine prevents palmitoylation. We hypothesized that DCA could activate ENaC through a similar mechanism and that there might be overlap between the two modes of regulation. We expressed ENaC subunits with all four palmitoylation sites mutated to alanine (ENaCΔP) in oocytes and tested whether mutation affected activation by DCA (Fig. 3). We found that removing all four palmitoylation sites potentiated DCA activation of ENaC currents by 7.5-fold (Fig. 3C). We also tested ENaCs lacking palmitoylation sites in only the β (βΔP) or γ (γΔP) subunits (Fig. 3). We found that γΔP recapitulated the effect of removing all four sites, whereas βΔP had an intermediate effect. When we mutated individual palmitoylation sites, we found the greatest effects at the N-terminal palmitoylation sites flanking a highly conserved His-Gly sequence. These results suggest that palmitoylation affects ENaC regulation by DCA.
Figure 3.
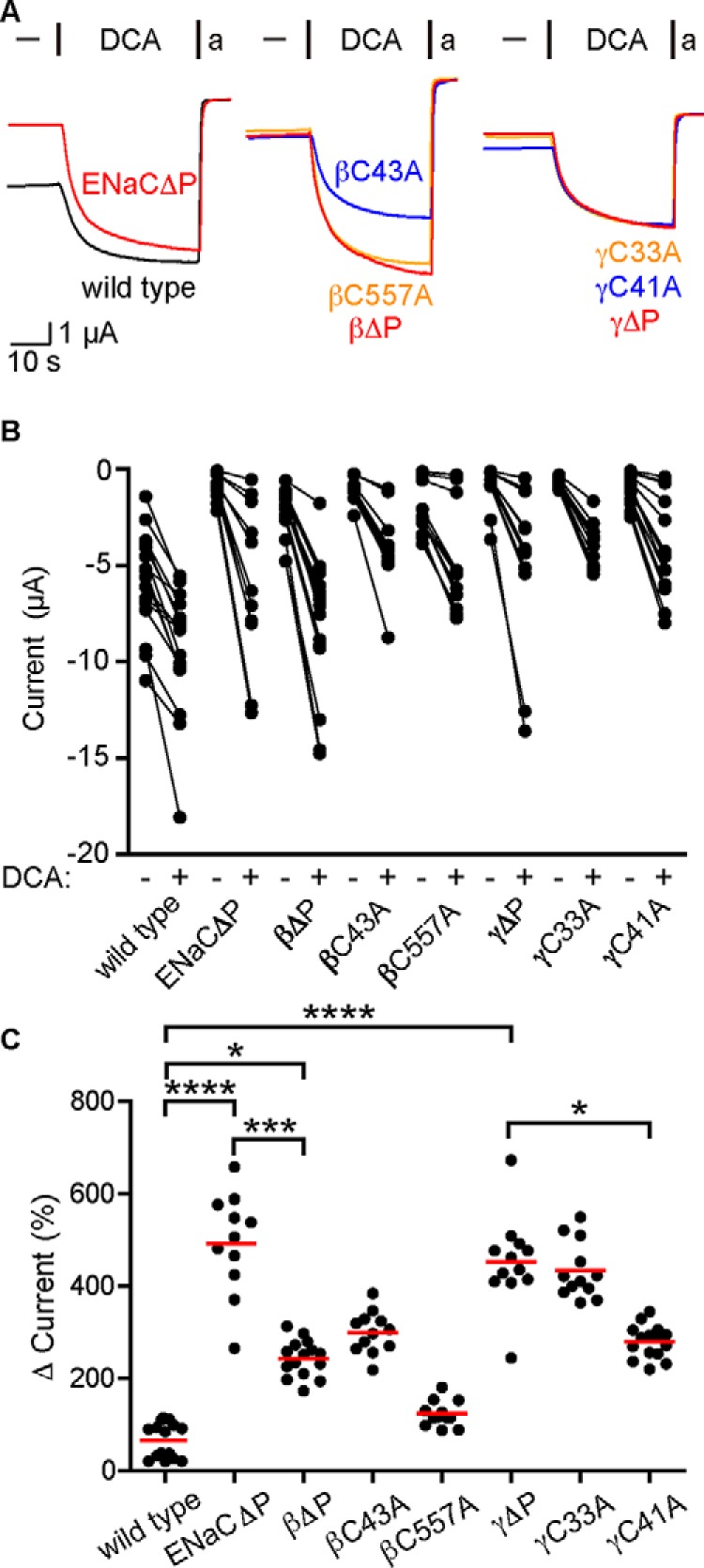
Removal of palmitoylation sites increases ENaC activation by DCA. Oocytes were injected with cRNA encoding β or γ subunits with palmitoylation site cysteines mutated to alanine along with complementary WT ENaC subunits. The effect of 1 mm DCA on amiloride (a)-sensitive currents was measured the following day. A, representative current traces showing the effect of DCA on currents from WT ENaC or various palmitoylation-deficient mutants, where all sites have been mutated (ENaCΔP), both sites in a single subunit have been mutated (βΔP and γΔP), or individual sites have been mutated (βC43A, βC557A, γC33A, and γC41A). Oocytes were clamped at a holding potential of −70 mV due to high WT basal currents. B, amiloride-sensitive currents are shown before and after exposure to DCA for each oocyte. Baseline currents were −6.1 ± 2.4 μA for WT ENaC, −0.98 ± 0.65 μA for ENaCΔP, −2.4 ± 1.2 μA for βΔP, −1.1 ± 0.6 μA for βC43A, −2.0 ± 1.4 μA for βC557A, −1.0 ± 1.1 μA for γΔP, −0.8 ± 0.2 μA for γC33A, and −1.2 ± 0.7 μA for γC41A (n = 11–15 for all groups). All groups showed significant increases in current upon DCA addition (p < 0.001 by Wilcoxon matched-pairs signed rank test). C, DCA activation of ENaC currents was determined as a percentage of the amiloride-sensitive baseline current. Individual experiments are shown with bars indicating the mean. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001 by Kruskal–Wallis test followed by Dunn's multiple-comparison test.
Trypsin treatment precludes ENaC activation by DCA
Preventing ENaC palmitoylation may have affected DCA activation through overlapping mechanisms of activation (i.e. by enhancing membrane association of key intracellular structures). Alternatively, preventing palmitoylation may simply reduce the channel's basal Po, leaving greater potential for activation (note the lower basal currents in Fig. 3B). We tested the ability of DCA to activate channels forced into a low-Po state through an independent mechanism: by preventing ENaC cleavage. ENaC α and γ subunits each undergo double cleavage that releases autoinhibitory tracts (31–33). Cleavage occurs in the periphery of the extracellular domains, far from the palmitoylation sites (25, 34–37). Uncleaved channels are “nearly silent” and have a very low Po (38, 39). Mutating the furin sites in the α and the γ subunits (ENaCΔF: αR205A,R231AβγR143A) prevents cleavage by furin but leaves channels susceptible to cleavage by less stringent proteases like trypsin (33, 40). We found that DCA robustly increased ENaCΔF currents (Fig. 4, A and B). We also found that preventing cleavage greatly increased activation by DCA as compared with WT (Fig. 4C), similar to the effect of preventing palmitoylation. We then measured the effect of DCA activation before and after trypsin activation (Fig. 5A). As before, DCA activated ENaC currents, and the effect was reversible (Fig. 5A, DCA #1). Trypsin addition (2 μg/ml) irreversibly activated ENaC currents, consistent with cleavage. However, subsequent DCA addition (DCA #2) failed to activate currents further. Notably, trypsin activation was comparable with the first DCA activation. We confirmed these results by testing channels lacking autoinhibitory tracts in the α and γ subunits (ENaCΔI: αΔ206–231βγΔ144–186). Channels lacking both tracts have a high open probability (39) and were not further activated by DCA (Fig. 5B). Taken together, our data suggest that preventing palmitoylation or cleavage indirectly enhanced DCA activation by suppressing activity and that trypsin treatment prevented DCA activation by “locking” the channel open.
Figure 4.
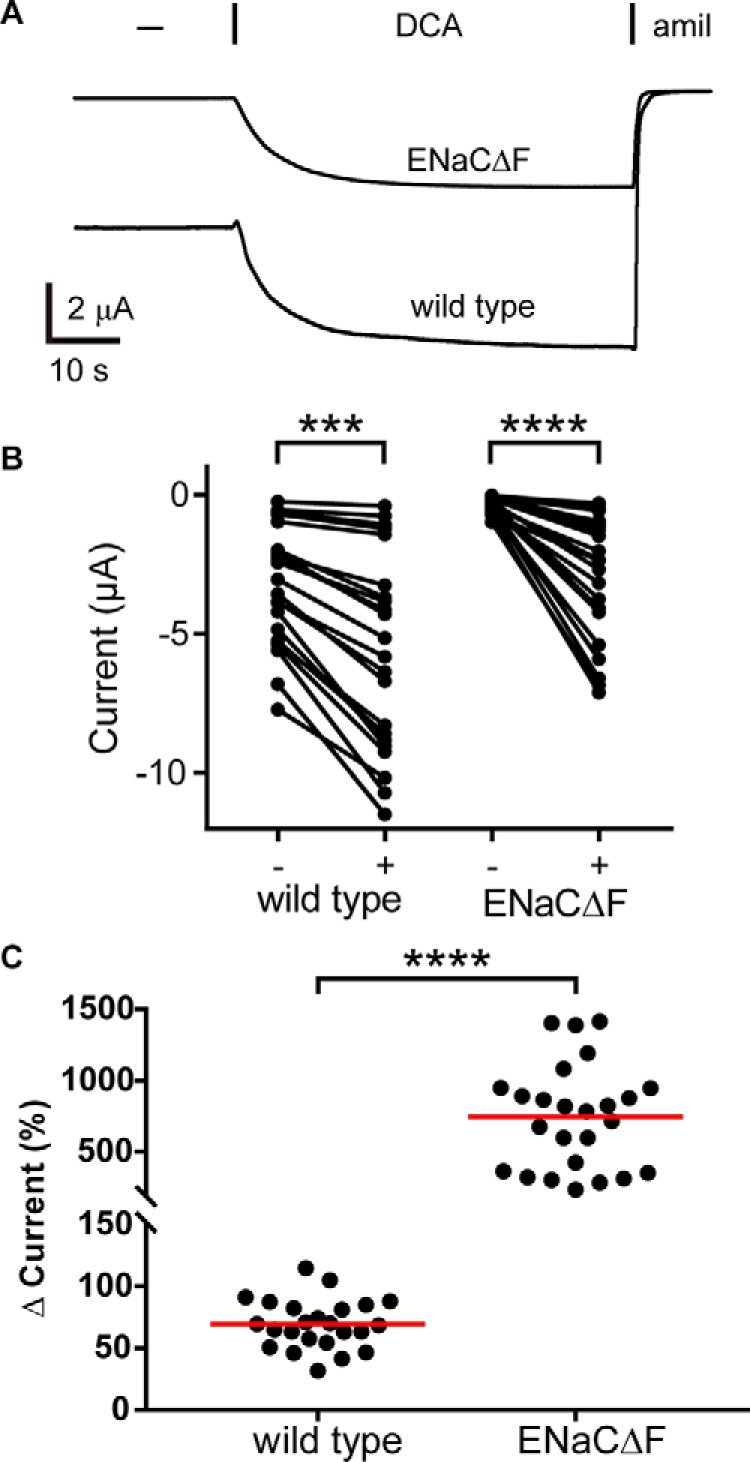
Removal of ENaC furin cleavage sites increases DCA activation of the channel. A, oocytes were injected with cRNA encoding WT ENaC subunits or ENaC subunits lacking the furin cleavage sites in the α and γ subunits (ENaCΔF: αR205A,R231AβγR143A). Whole-cell currents were measured by TEVC at −100 mV. Representative current traces of oocytes perfused with 1 mm DCA for 40 s, followed by 10 μm amiloride (amil) are shown. B, amiloride-sensitive currents are shown before and after exposure to DCA for each oocyte. Baseline amiloride-sensitive currents were −4.6 ± 1.21 μA for WT ENaC (n = 11) and −0.6 ± 0.3 μA for ENaCΔF (n = 19). ***, p < 0.001; ****, p < 0.0001 by Wilcoxon matched-pairs signed rank test. C, DCA activation of ENaC current was determined as a percentage of the amiloride-sensitive baseline current. Individual experiments are shown with bars indicating the mean. ****, p < 0.0001 by Mann–Whitney U test.
Figure 5.
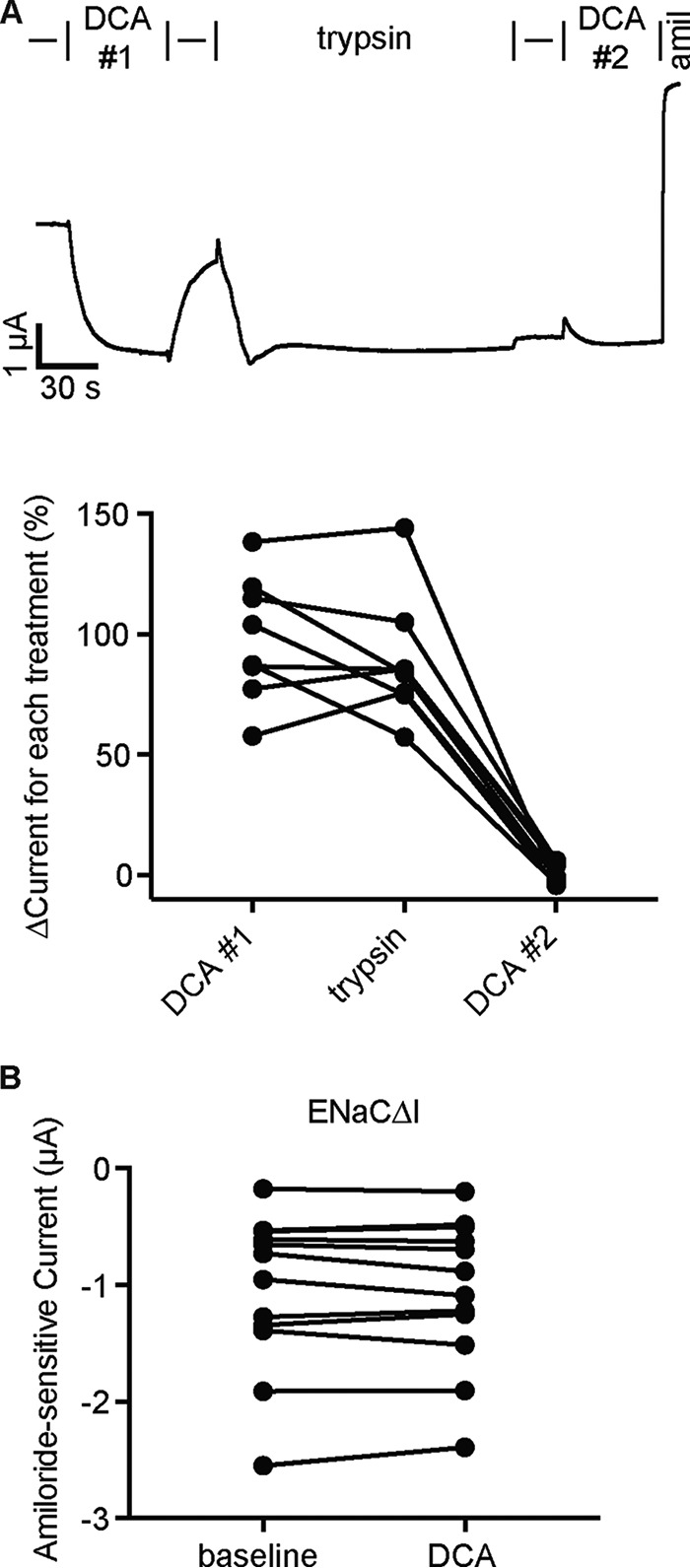
Trypsin cleavage of ENaC precludes DCA activation of the channel. A, representative whole-cell current trace of an oocyte expressing WT ENaC sequentially treated with DCA, trypsin, and then DCA again. Cells clamped at −100 mV were treated with 1 mm DCA (DCA #1) for 60 s. Following washout, oocytes were then treated with 2 μg/ml trypsin for 2 min. After removing trypsin from the bath, a second perfusion of 1 mm DCA was applied (DCA #2), followed by 10 μm amiloride (amil). Changes in current were determined by comparing steady-state currents during DCA or trypsin treatment to amiloride-sensitive steady-state currents just prior to treatment. Individual experiments are plotted with bars indicating the mean of each group (n = 8). ****, p < 0.0001 by repeated measures one-way ANOVA with the Greenhouse–Geisser correction and Tukey's post hoc analysis. B, oocytes expressing ENaC subunits lacking inhibitory tracts (ENaCΔI: αΔ206–231βγΔ144–186) were clamped at −100 mV and exposed to 1 mm DCA in the bath solution, followed by 10 μm amiloride. No effect was detected upon DCA addition (n = 11; p = not significant by paired Student's t test).
DCA activates ENaC independent of purinergic signaling
Purinergic signaling regulates ENaC activity on a time scale similar to DCA-dependent activation (41). ATP inhibits ENaC through purinergic receptor signaling that ultimately depletes phosphoinositide 4,5-bisphosphate necessary for ENaC activity (42). DCA could inhibit purinergic signaling to activate ENaC. However, purinergic receptor-dependent ENaC regulation has not been demonstrated in Xenopus oocytes, and deep proteomics of Xenopus eggs did not detect P2Y receptors (43). To test this idea, we used apyrase at concentrations sufficient to activate ENaC 3-fold in other cells (4) to scavenge extracellular ATP and reduce purinergic signaling in our cells. When we perfused 5 units/ml apyrase to the cell bath, whole-cell currents increased by 13 ± 6% (Fig. 6), consistent with modest ATP inhibition of ENaC. When we added 1 mm DCA in the continued presence of apyrase, amiloride-sensitive currents nevertheless increased. The magnitude of current increase was similar to DCA-dependent activation in the absence of apyrase (see Figs. 1 and 7). These data suggest that purinergic signaling has a modest effect on ENaC currents in oocytes and that DCA activates ENaC independent of purinergic signaling.
Figure 6.
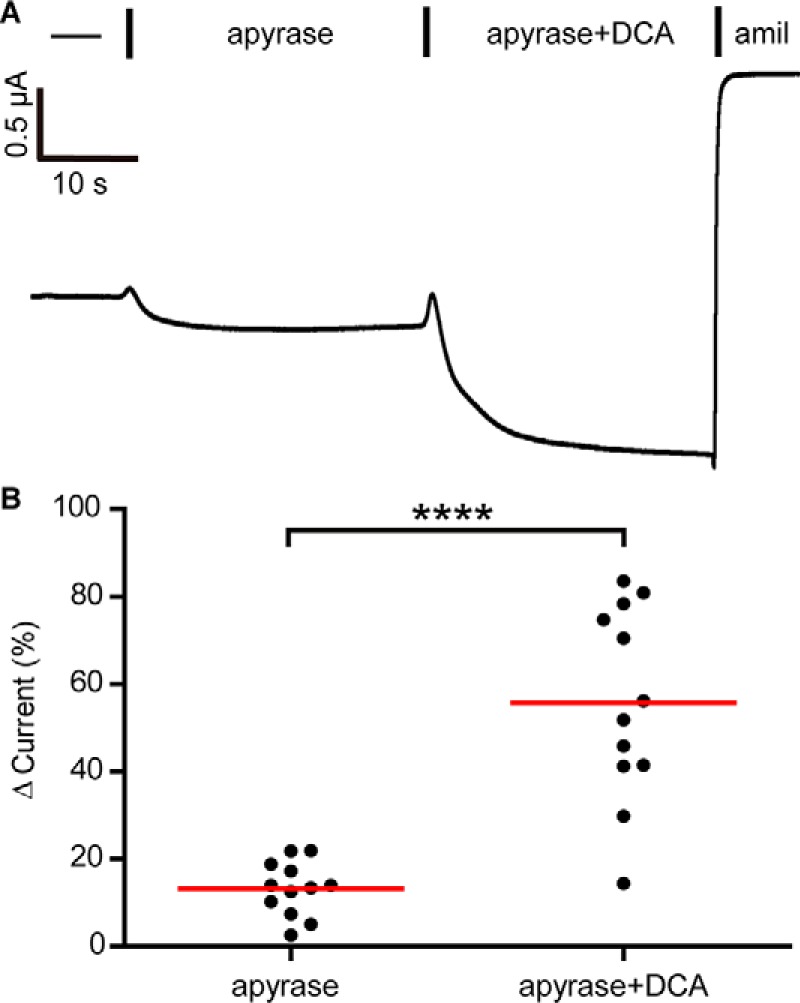
DCA activates ENaC independent of ATP. A, representative whole-cell recording of oocytes expressing WT ENaC. Bath solutions were supplemented with 5 units/ml apyrase, 1 mm DCA, and 10 μm amiloride (amil) as indicated. B, changes in current were determined as a percentage of the amiloride-sensitive baseline current. Individual experiments are plotted with bars indicating the mean of each group (n = 12). ****, p < 0.0001 by paired Student's t test.
Figure 7.
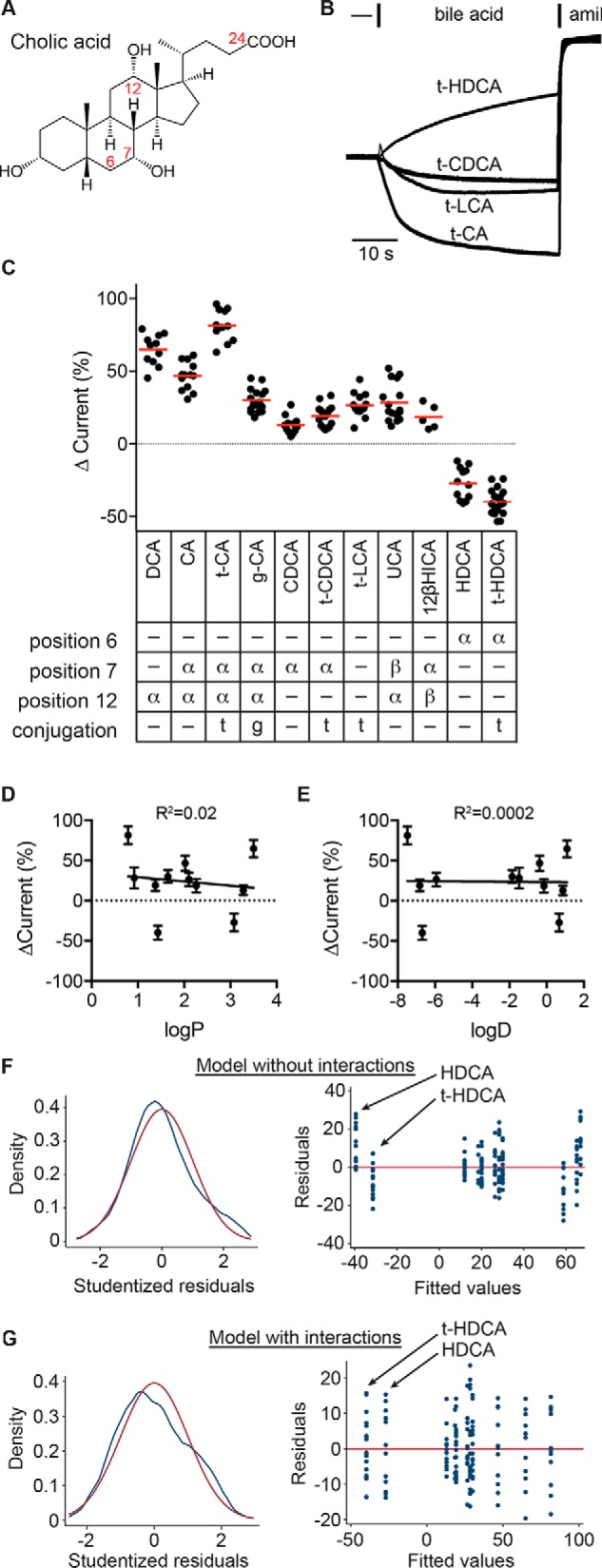
ENaC-activating bile acids possess specific moieties. A, schematic of cholic acid structure. Solid wedges connect groups facing the hydrophobic side of the molecule, termed the β side. Dashed wedges connect groups facing the hydrophilic side of the molecule, termed the α side. Bile acids can be conjugated with taurine or glycine at position 24. B, representative recordings of the effect of various taurine-conjugated bile acids at 1 mm, followed by 10 μm amiloride (amil). C, bile acid–driven changes in current were determined as a percentage of the amiloride-sensitive baseline current. Individual experiments are shown with bars indicating the mean. The position and orientation (facing the α or β side) of hydroxyl groups in the tested bile acids and the presence of taurine or glycine conjugation at position 24 are summarized. All bile acids tested had a 3α-OH. Data were analyzed by one-way ANOVA with Tukey's multiple-comparison test, with results summarized in Table 1. D and E, the effect on ENaC currents for each bile acid was plotted against the bile acid's logP and logD values (see Table 1). Neither correlation was significant. Data were also analyzed by multiple linear regression, with functional groups as independent factors. Models with no interactions between functional groups (F) or with interactions between functional groups (G; see Table 2) were evaluated. F and G (left), kernel density plot of Studentized residuals (blue line) to evaluate distribution of residuals, with normal density (red line) overlaid. Both models gave normally distributed residuals, as determined by the Shapiro–Wilk W test (p = not significant). F and G (right), residuals versus fitted values plot to evaluate heteroscedasticity in models. White's test for heteroscedasticity gave p = 0.0003 for the model without interactions and p = 0.049 for the model with interactions.
ENaC activation by bile acids depends on specific moieties, but not on membrane permeability
CA and CDCA are synthesized from cholesterol in the liver. These primary bile acids may then be conjugated to taurine or glycine in the liver and further modified into secondary bile acids (e.g. DCA) and/or deconjugated in the gut (10). Bile acids are differentiated based on the presence and orientation (facing the hydrophilic α side or hydrophobic β side) of hydroxyl groups on the steroid rings and whether they are conjugated at position 24 (Fig. 7A). Data in Fig. 1 suggested that ENaC activation depends on the presence of specific functional groups. We tested this idea by screening conjugated and unconjugated bile acids that varied by the presence and orientation of hydroxyl groups at positions 6, 7, and 12. These bile acids varied in the lipophilicity of the nonionized form, indicated by the partition coefficient (logP; Table 1) and the propensity to cross the membrane at pH 7.4, indicated by the distribution coefficient (logD; Table 1). We applied each bile acid at 1 mm and determined the effect on amiloride-sensitive currents in oocytes (Fig. 7, B and C). Although bile acids had varied effects on ENaC currents (Fig. 7C and Table 2), neither the bile acid's partition coefficient nor distribution coefficient was predictive of that effect (Fig. 7, D and E).
Table 1.
Physico-chemical properties of bile acids
Distribution coefficients were calculated using the formula, logD = logP + log (1 + 10pH-pKa).
Table 2.
Adjusted p values for each pair in Fig. 7C using one-way ANOVA with Tukey's multiple-comparison test
| t-HDCA | HDCA | 12βHICA | UCA | t-LCA | t-CDCA | CDCA | g-CA | t-CA | CA | |
|---|---|---|---|---|---|---|---|---|---|---|
| DCA | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.003 | 0.0004 |
| CA | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.0003 | <0.0001 | |
| t-CA | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | ||
| g-CA | <0.0001 | <0.0001 | NSa | NS | NS | NS | 0.0003 | |||
| CDCA | <0.0001 | <0.0001 | NS | 0.002 | 0.02 | NS | ||||
| t-CDCA | <0.0001 | <0.0001 | NS | NS | NS | |||||
| t-LCA | <0.0001 | <0.0001 | NS | NS | ||||||
| UCA | <0.0001 | <0.0001 | NS | |||||||
| 12βHICA | <0.0001 | <0.0001 | ||||||||
| HDCA | 0.02 |
a NS, not significant.
To determine the contribution of the various functional groups to the effect on ENaC current, we performed a multiple linear regression analysis of our data in Fig. 7C. Each functional group (with either α or β orientation) was included in the linear regression model as an independent factor (Table 3, “Main effects”). A model without interactions between functional groups fit the data poorly and was characterized by strongly heteroscedastic residuals (Fig. 7F). For example, the fitted values at −40 that represent the HDCA data had largely positive residuals, whereas the fitted values at −33 that represent the t-HDCA data had largely negative residuals (Fig. 7F, right). A model including interactions between functional groups fit the data well-and had smaller residuals that were normally distributed and nearly homoscedastic (Fig. 7G) (e.g. the residuals for each bile acid were symmetrically distributed about the red model line at zero) (Fig. 7G, right). In this model, the HDCA and t-HDCA data were fit with values approximating the averages for their groups. Although all possible interactions were considered, most were automatically omitted due to collinearity with other factors in the model. Our linear regression model fit the data with R2 = 0.93, with parameters reported in Table 3.
Table 3.
Linear regression model parameters of Fig. 7 data (143 data points)
Functional groups at various sites on the lithocholic acid backbone were included as independent variables in the model. Interactions between functional groups were also included. Several interactions were automatically omitted due to collinearity. R2 was 0.93 with a root mean squared error of 9.5.
| Coefficient | p value | |
|---|---|---|
| Main effects | ||
| 6α-OH | −47 ± 0.5 | <0.0001 |
| 7α-OH | −7 ± 0.3 | 0.01 |
| 7β-OH | −36 ± 0.5 | <0.0001 |
| 12α-OH | 44 ± 0.5 | <0.0001 |
| 12β-OH | 6 ± 0.4 | 0.2 |
| Glycine conjugation | −16 ± 0.3 | <0.0001 |
| Taurine conjugation | 6 ± 0.3 | 0.01 |
| Model intercept | 20 ± 0.3 | <0.0001 |
| Interactions | ||
| 7α-OH–12α-OH | −11 ± 0.5 | 0.04 |
| 6α-OH–taurine conjugation | −19 ± 0.5 | <0.0001 |
| 12α-OH–taurine conjugation | 29 ± 0.5 | <0.0001 |
With an intercept of 20, our model predicts that a bile acid lacking all of the functional groups considered (i.e. lithocholic acid (LCA)) would moderately activate ENaC. We could not test LCA due to its low aqueous solubility; however, taurolithocholic acid (t-LCA) increased ENaC currents by 27 ± 8%. The presence of the 12α-OH group was strongly associated with an increase in ENaC activity in the model (Table 3). Indeed, compounds that possessed a 12α-OH (DCA, CA, t-CA, glycocholic acid (g-CA), and ursocholic acid (UCA)) generally activated ENaC more than other compounds (Fig. 7C and Table 2). Both 6α-OH and 7β-OH were associated with inhibition in the model. Our data show that 6α-OH–bearing HDCA and t-HDCA both decreased ENaC currents. UCA, which bears both 12α-OH and 7β-OH had weaker activation compared with DCA, which only bears 12α-OH. Both 7α-OH and taurine conjugation had mild main effects but interacted with other functional groups in our model. Our data show that adding a 7α-OH to t-LCA, creating t-CDCA, had little effect (not significant). However, adding 7α-OH to 12α-OH–bearing DCA, making CA, decreased activation by 28% (p = 0.0004). Interestingly, taurine conjugation interacted with 12α-OH to enhance its channel-activating effect (compare CA versus t-CA) and with the 6α-OH to enhance its channel-inhibitory effect (compare HDCA versus t-HDCA). Notably, glycine conjugation, which also increases hydrophilicity and decreases membrane permeability, had a negative impact on channel activation (compare CA and g-CA). Our results suggest that specific moieties influence the ENaC-regulating properties of bile acids. Furthermore, taurine conjugation enhanced the effect of the underlying bile acid, whether that bile acid activated or inhibited ENaC. Because taurine conjugation lowers the pKa of bile acids to <2 (44), our data suggest that bile acid regulation of ENaC does not require membrane permeability.
Bile acids regulate ENaC endogenously expressed in mpkCCDc14 cells
ENaCs expressed in Xenopus oocytes have recapitulated many of the channel's functional properties observed in other cells (e.g. ion selectivity, blocker sensitivity, and proteolytic activation). Because ENaC regulation by bile acids has only been reported in oocytes, we determined whether bile acids activate ENaC in a mouse cortical collecting duct cell line (mpkCCDc14), which endogenously expresses ENaC (45, 46). We cultured cells on permeable supports for 5 days and measured ENaC-mediated short-circuit currents (ISC) in Ussing chambers. Experiments with DCA failed due to irreversible loss of electrical resistance across the monolayer. We then tried t-CA due to its low membrane permeability. Our initial experiments showed no effect with t-CA, which robustly activated ENaC expressed in oocytes (see Fig. 7). We hypothesized that ENaC subunits expressed in mpkCCDc14 cells may be cleaved, leaving the channel “locked open” and insensitive to channel modulators (see Fig. 5 and below). Indeed, observing trypsin activation of ENaC currents in cultured cells has required inhibiting endogenous proteases to circumvent this issue (32, 47). Nevertheless, there is strong evidence that cleavage of ENaC subunits is dynamically regulated in vivo and that channels at the luminal surface of the distal nephron are not fully cleaved (48–54). We therefore incubated cells overnight with the serine protease inhibitors aprotinin and camostat before measuring the effect of t-CA on ENaC-mediated ISC. We found that t-CA added to the apical side in the continued presence of camostat activated amiloride-sensitive ISC in a dose-dependent manner, with 1 mm t-CA activating by 54 ± 14% (Fig. 8). The t-CA dose–response curve from mpkCCDc14 cells was similar to the t-CA dose–response curve in oocytes (Fig. 8B).
Figure 8.
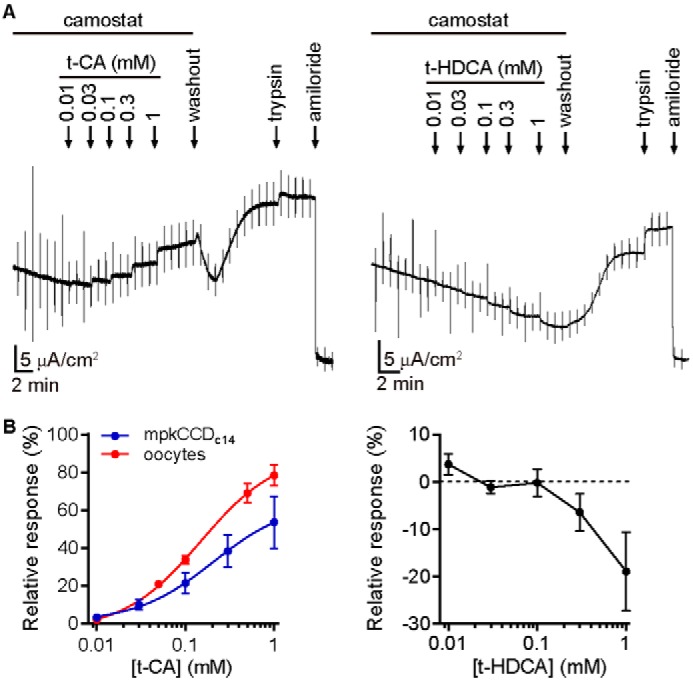
Bile acids regulate ENaC in mpkCCDc14 cells. Polarized mpkCCDc14 cells grown on permeable supports were mounted in an Ussing chamber for ISC measurements. A, representative recordings shown. Increasing concentrations of t-CA or t-HDCA were added to Ringer's buffer supplemented with 10 μm camostat on the apical side of the chamber, as indicated. After the last bile acid addition, bile acids were removed by replacing the solution on the apical side with Ringer's buffer lacking camostat. 2 μm trypsin was then added, followed by 10 μm amiloride. Baseline amiloride-sensitive ISC was 16 ± 9 μA/cm2. B, changes in current in response to t-CA (n = 6) or t-HDCA (n = 6) were determined as a percentage of the amiloride-sensitive baseline current, while correcting for time-dependent changes in current. Linear estimates of the time-dependent change in current under each condition were determined using a 30-s window during the steady-state period just before each bile acid addition and were used to account for run-down during each experiment. A dose–response experiment for t-CA activation of ENaC-expressing oocytes is overlaid for comparison (n = 5) and was performed analogously to the experiment in Fig. 1C. Data for t-CA were fit to the Hill equation, EC50 = 210 ± 100 μm in mpkCCDc14 cells and 148 ± 5 μm in oocytes.
To test the specificity of the effect, we also tested t-HDCA, which inhibited ENaC expressed in oocytes (Fig. 7). We found that t-HDCA inhibited amiloride-sensitive ISC in a dose-dependent manner with 1 mm t-HDCA inhibiting by 13 ± 6% (Fig. 8). The relative changes in current are smaller than those measured in oocytes in both cases. We also observed modest increases in ISC upon adding 2 μm trypsin. Before adding trypsin, we replaced the apical solution with a solution lacking camostat and waited until currents stabilized before proceeding. During this period, we observed a consistent increase in ISC even after reversal of t-CA stimulation. We speculate that this resulted from cleavage by endogenous proteases after removal of the protease inhibitor. Still, our data clearly show that t-CA and t-HDCA regulation of ENaC endogenously expressed in mpkCCDc14 cells is analogous to their regulation of ENaC heterologously expressed in Xenopus oocytes.
To confirm that locking the channel open precludes ENaC activation by t-CA in mpkCCDc14 cells, we tested the effect of t-CA on ENaC currents after trypsin treatment. One day before current measurements, cell culture medium was supplemented with protease inhibitors as before or with vehicle. For cells cultured with protease inhibitors, recording medium was supplemented with 5 μm camostat on the apical side. When we tested cells that had not been exposed to protease inhibitors, we observed no change in ISC after the addition of 2 μm trypsin or after the subsequent addition of 1 mm t-CA (Fig. 9). When we tested cells that were cultured with protease inhibitors, we observed a robust increase in ISC after trypsin addition in excess of camostat but no change in ISC after the subsequent addition of 1 mm t-CA. Taken together with data in Fig. 8, these data suggest that t-CA activates ENaC by increasing ENaC Po.
Figure 9.
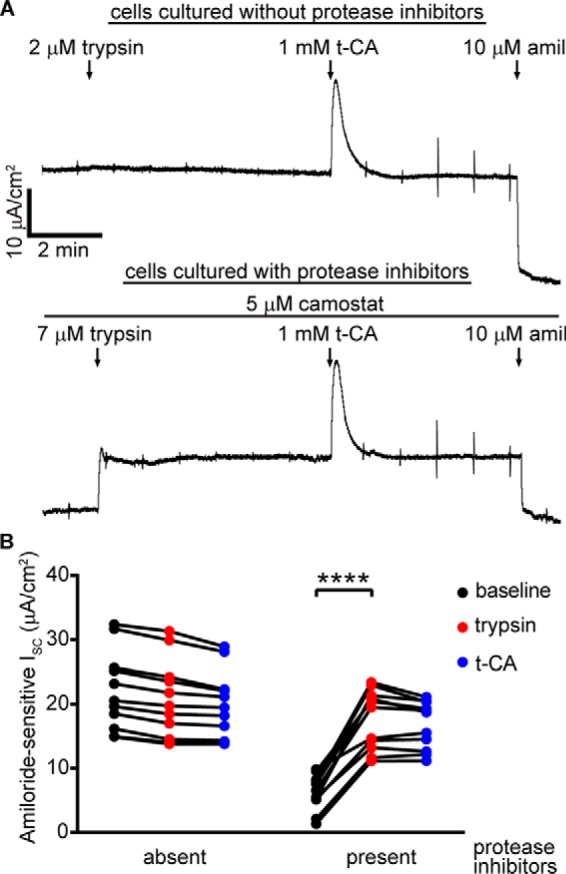
Trypsin precludes t-CA activation of amiloride-sensitive ISC in mpkCCDc14 cells. Cells were incubated with or without 500 μg/ml aprotinin and 20 μm camostat 24 h before recordings. A, representative recordings shown. 5 μm camostat was maintained on the apical side for the protease inhibitor–treated cells. After the currents stabilized, 7 μm trypsin was added, followed by 1 mm t-CA. For untreated cells, no camostat was present during the recording, during which 2 μm trypsin was added, followed by 1 mm t-CA. 10 μm amiloride was applied at the end of each recording. B, amiloride-sensitive ISC values are calculated based on currents measured just prior to the addition of the subsequent agent. Baseline amiloride-sensitive currents were 22.1 ± 6.2 μA/cm2 for untreated cells (n = 11) and 6.3 ± 2.8 μA/cm2 for protease inhibitor–treated cells (n = 11; p < 0.0001 versus untreated cells by repeated-measures two-way ANOVA with Sidak's multiple-comparison test). **, p < 0.01; ****, p < 0.0001 by repeated measures two-way ANOVA with Sidak's multiple-comparison test.
Discussion
Recent reports have shown that ENaC/degenerin family members are regulated by a variety of amphipathic molecules (6–8, 21, 22). In this study, we show that specific biliary factors regulate ENaC. Our results suggest that regulation by bile acids depends on specific moieties and that these compounds affect the channel Po. We also found that bilirubin conjugates regulate the channel and that bile acids regulate ENaC endogenously expressed in cultured polarized epithelial cells.
Bile acids are synthesized by hepatocytes and are modified by gut bacteria, giving rise to a family of compounds that vary with respect to hydroxyl groups at key positions. Our results show that bile acids with a 12α-OH activate ENaC, whereas bile acids with a 6α-OH inhibit it. Rat and human ENaCs were also activated by bile acids bearing 12α-OH (6, 7). Sensitivity to bile acid dosing was similar for mouse ENaC, rat ENaC, and human ASIC1a (6, 22). Our results also suggest that taurine conjugation had mixed effects on mouse ENaC. Whereas taurine conjugation had a mild independent stimulatory effect, it also amplified the effect of the underlying bile acid. The independent stimulatory effect of taurine conjugation appears to be larger for human ENaC (7). In contrast to mouse ENaC, glycine and taurine conjugation of CA had no effect on the activation of rat ENaC (6). Conjugation to taurine or glycine increases bile acid hydrophilicity and lowers the compound's pKa, resulting in reduced membrane permeability. Given the effect to enhance both activation and inhibition we observed with taurine conjugation, our data suggest that membrane permeability is not required for ENaC regulation by bile acids. Our data also show that bilirubin conjugates activated ENaC currents. As compared with DCA, bilirubin conjugates required much higher concentrations to activate ENaC to similar levels. Because both the glucuronic acid and taurine conjugates activated ENaC with similar dose dependences, our data suggest that regulation likely depends on the pyrrole moieties of bilirubin. As with bile acids, our data with taurine-conjugated bilirubin suggest that membrane permeability is not required for ENaC regulation.
Our bile acid data are consistent with regulation through effects on channel Po. Suppressing Po by preventing palmitoylation or furin processing markedly increased the response to DCA. In contrast, elevating Po through trypsin treatment or mutagenesis prevented a response to both DCA and t-CA. These results accord with previous studies that used trypsin to prevent bile acid activation and showed that bile acids increase ENaC Po in single-channel recordings (7). However, they contrast with earlier single-channel experiments suggesting that bile acids activate only furin-processed channels, and not uncleaved near-silent channels (6, 7). Notably, lower bile acid concentrations (250 μm t-DCA or 500 μm DCA) were used as compared with experiments presented here. Nonetheless, our data clearly show that bile acids robustly activate channels lacking furin processing.
Several lines of evidence suggest that bile acids regulate ENaC by directly binding to an extracellular site to affect the Po of the channel. Here we showed that bile acids likely regulate ENaC independent of purinergic signaling. The Wiemuth group (8) recently reported that while amphipathic compounds influenced membrane properties, no effect on membrane properties predicted effects on the function of ENaC/degenerin channels. We did not find that channel regulation correlated with differences in the biophysical properties of the bile acids tested. Instead, the presence of specific moieties accounted for differences between the effects of different bile acids. Our results with taurine conjugates limit the likely sites of interaction to the extracellular domains and the outer portion of the transmembrane helices. We showed that altering channel function through mutations at opposite ends of the channel profoundly affects ENaC regulation by bile acids, illustrating cross-talk between all three modes of regulation. The Haerteis group (7) recently proposed that bile acids bind ENaC near the degenerin site on the basis of functional analysis and molecular modeling. This region of the protein, long implicated in ENaC function, lies in the transmembrane helices of the pore and is consistent with these data.
ENaC is expressed in epithelia that are exposed to high concentrations of bile acids and c-bilirubin. ENaC is robustly expressed in the aldosterone-sensitive distal colon and was recently reported to be expressed in cholangiocytes, which line the bile ducts and gallbladder (4, 56). ENaC-mediated Na+ transport has been proposed to regulate the viscosity of bile and stool fluid content (4, 57). Additionally, cholestasis, cirrhosis, and hepatitis are often associated with elevated biliary factor levels in blood (13, 16, 58–60). Urinary bile acid concentrations are well-correlated with serum total bile acids and were elevated in cirrhotic patients (210 ± 130 μmol/g creatinine, n = 11 (13)). In the context of liver disease and disruption of the enterohepatic circulation, conjugated primary bile acids (t-CA, g-CA, t-CDCA, and g-CDCA) predominate (16). These bile acids either mildly or strongly increased ENaC currents (Fig. 7) (7). If sufficient concentrations are achieved in the distal nephron, direct ENaC activation by these compounds could contribute to the fluid retention and electrolyte imbalances often associated with advanced liver diseases (61–63). ENaC had affinities for DCA and t-CA of 80 and 150–200 μm, respectively. These values suggest that bile acids may be a physiologic ENaC regulator in the biliary tree and gut, where ENaC regulates luminal Na+, and a pathophysiologic ENaC regulator in the kidney tubule, where ENaC regulates systemic Na+ and extracellular fluid volume. ENaC regulation by c-bilirubin was much weaker by comparison. Bilirubin concentrations in bile are ∼1–6 mm (64), which suggests that bilirubin could regulate ENaC activity in the gallbladder epithelium. It is unclear whether bilirubin concentrations in the kidney tubule reach sufficient concentrations to affect ENaC in liver disease; however, the occurrence of bilirubin precipitates (e.g. urinary crystals and bile cast nephropathy) suggests that concentrations may be high in disease (65–67).
In summary, we found that bile acids and conjugated bilirubin are potent regulators of mouse ENaC function. Bile acid regulation of ENaC endogenously expressed in mpkCCDc14 cells mimicked regulation in Xenopus oocytes. Regulation depended on the presence of specific moieties in both bile acids and bilirubin. We propose that bile acids directly bind ENaC on the extracellular side of the channel to modulate its Po. Further investigation will be required to determine the mechanism of channel activation and its contribution to physiologic and pathophysiologic ENaC regulation.
Experimental procedures
Materials and reagents
The following reagents were purchased from commercial sources: CA, g-CDCA, and t-HDCA (Spectrum Chemical, New Brunswick, NJ); DCA, apyrase, aprotinin, trypsin, and camostat (Millipore-Sigma); CDCA, t-CA, t-CDCA, t-LCA, and t-bilirubin (Cayman Chemical, Ann Arbor, MI); g-CA (Frontier Scientific, Logan, UT); UCA (Astatech Inc., Bristol, PA); HDCA (Thermo Fisher Scientific); c-bilirubin (Lee BioSolutions, Maryland Heights, MO); and 12β-hydroxyisocholic acid (12βHICA; Toronto Research Chemicals Inc., North York, Canada). cDNAs for WT mouse α, β, and γ ENaC subunits and mutant subunits (βC43A, βC557A, βC43A,C557A, γC33A, γC41A, γC33A,C41A, αR205A,R231A, αδ206–231, γδ144–186, and γR143A) were described previously (30, 68–70). The corresponding cRNAs for oocyte injection were prepared using mMESSAGE mMACHINE kits (Thermo Fisher Scientific).
ENaC functional expression in Xenopus laevis oocytes
Oocytes from X. laevis were harvested and defolliculated using standard protocols. 1–4 ng of cRNA encoding each ENaC subunit was injected into stage V or VI oocytes using a Nanoject II (Drummond Scientific, Broomall, PA). After injection, oocytes were incubated at 18 °C for 20–24 h in modified Barth's saline (88 mm NaCl, 1 mm KCl, 2.4 mm NaHCO3, 15 mm HEPES, 0.3 mm Ca(NO3)2, 0.41 mm CaCl2, 0.82 mm MgSO4, 10 μg/ml sodium penicillin, 10 μg/ml streptomycin sulfate, 100 μg/ml gentamicin sulfate, pH 7.4) prior to current measurements. The protocol for harvesting oocytes from X. laevis was approved by the University of Pittsburgh's Institutional Animal Care and Use Committee.
Two-electrode voltage clamp (TEVC) recordings
TEVC was performed using an Axoclamp 900A voltage clamp amplifier and DigiData 1550A (Molecular Devices, Sunnyvale, CA) interfaced to a personal computer. Data acquisition and analyses were performed using pClamp 10.5 software (Molecular Devices). Pipettes for electrophysiological recordings were pulled from borosilicate glass capillaries (World Precision Instruments, Inc., Sarasota, FL) using a model p-87 micropipette puller (Sutter Instrument Co., Novato, CA) and had resistances of 0.5–5 milliohm when filled with 3 m KCl and inserted into the bath solution. Oocytes were maintained in a recording chamber (Automate Scientific, San Francisco, CA) with 20 μl of bath solution and continuously perfused with Na-110 buffer (110 mm NaCl, 2 mm KCl, 2 mm CaCl2, 10 mm HEPES, pH 7.4) at a flow rate of 5–8 ml/min. Various compounds were diluted into Na-110 solution from 100 mm stock solutions in water (t-CA, g-CA, DCA, HDCA, and t-HDCA) or DMSO (amiloride, CA, CDCA, t-CDCA, t-LCA, UCA, and 12βHICA). At the end of each experiment, currents were measured in 10 μm amiloride to determine the amiloride-sensitive component of the current. All experiments were performed at ambient temperatures (20–24 °C). The DMSO cosolvent used for some compounds at 1% final concentration did not significantly affect ENaC currents (ΔI = −10 ± 5%; n = 8; p = 0.08 by paired Student's t test).
mpkCCD14 cell culture
Cells were cultured in Dulbecco's modified Eagle's medium/F12 (Thermo Fisher Scientific) supplemented with 2% fetal calf serum, 50 nm dexamethasone, 60 nm selenium, 1 nm triiodothyronine, 5 μg/ml insulin, 5 μg/ml transferrin, 10 ng/ml epidermal growth factor, and 1% penicillin/streptomycin (Millipore-Sigma), as described previously (33). Cells were seeded at confluent density onto 1.2-cm diameter Costar Transwell inserts (0.4-μm pore size, Corning, NY). The subcultured cells were grown for at least 5 days. 24 h before ISC measurements, growth medium was replaced with unsupplemented Dulbecco's modified Eagle's medium/F12. At the same time, serine protease inhibitors (5 mg/ml aprotinin and 20 μm camostat mesylate) were added to both the upper and lower compartments of the transwells, as indicated.
Short-circuit current measurements
Transwell inserts covered by cell monolayers were mounted in modified Ussing chambers and continuously short-circuited using a VCC MC6 voltage clamp system (Physiologic Instruments, San Diego, CA) (33). Each hemi-chamber was bathed in 5 ml of Ringer's solution (110 mm NaCl, 25 mm NaHCO3, 5.8 mm KCl, 1.2 mm KH2PO4, 2 mm MgSO4, 2 mm CaCl2, and 11 mm glucose, pH 7.4). A gas mixture of 95% O2 and 5% CO2 at 37 °C was constantly infused into the bath solution. Transepithelial resistance was monitored throughout the recording by periodically applying a 1-mV pulse via an automated pulse generator. Recordings were digitized and analyzed using pClamp 10.5 software (Molecular Devices). To limit ENaC cleavage during recordings, camostat was added to the apical hemi-chamber, as indicated. The apical chamber was washed with a 6-fold volume of warmed Ringer's solution to remove bile acids and camostat prior to the trypsin addition where indicated. 10 μm amiloride was added at the end of each experiment to determine the amiloride-sensitive component of the ISC.
Statistical analyses
Prism 7 software (GraphPad, Inc., La Jolla, CA) was used to perform curve fitting and group comparisons (as indicated). Adjusted values of p < 0.05 were considered significant. Multiple linear regression and tests evaluating statistical models were performed using Stata 15.1 SE software (StataCorp LLC, College Station, TX).
Author contributions
X.-P. W., S. J. I., E. C. R., M. D. C., and O. B. K. conceptualized the study. X.-P. W., D. M. B., and S. J. I. performed oocyte experiments. X.-P. W. and N. M. performed Ussing chamber recordings. X.-P. W., S. J. I., and O. B. K. analyzed the data. X.-P. W. and O. B. K. wrote the manuscript. X.-P. W., N. M., E. C. R., M. D. C., and O. B. K. edited the manuscript. All authors discussed and approved the final manuscript.
Acknowledgment
The Pittsburgh Center for Kidney Research was supported by NIDDK, National Institutes of Health, Grant P30 DK079307.
This work was supported by NIDDK, National Institutes of Health, Grant R01 DK098204 (to O. B. K.) and a grant from the Pittsburgh Liver Research Center. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ENaC
- epithelial Na+ channel
- TEVC
- two-electrode voltage clamp
- DCA
- deoxycholic acid
- CA
- cholic acid
- CDCA
- chenodeoxycholic acid
- HDCA
- hyodeoxycholic acid
- c-bilirubin
- bilirubin diglucuronide
- t-bilirubin
- bilirubin ditaurate
- LCA
- lithocholic acid
- t-LCA
- taurolithocholic acid
- t-CA
- taurocholic acid
- g-CA
- glycocholic acid
- t-HDCA
- taurohyodeoxycholic acid
- UCA
- ursocholic acid
- t-CDCA
- taurochenodeoxycholic acid
- 12βHICA
- 12β-hydroxyisocholic acid
- ASIC
- acid-sensing ion channel
- BASIC
- bile acid–sensitive ion channel
- ANOVA
- analysis of variance
- cRNA
- complementary RNA.
References
- 1. Pearce D., Soundararajan R., Trimpert C., Kashlan O. B., Deen P. M., and Kohan D. E. (2015) Collecting duct principal cell transport processes and their regulation. Clin. J. Am. Soc. Nephrol. 10, 135–146 10.2215/CJN.05760513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ghosh A., Boucher R. C., and Tarran R. (2015) Airway hydration and COPD. Cell Mol. Life Sci. 72, 3637–3652 10.1007/s00018-015-1946-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kunzelmann K., and Mall M. (2002) Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol. Rev. 82, 245–289 10.1152/physrev.00026.2001 [DOI] [PubMed] [Google Scholar]
- 4. Li Q., Kresge C., Bugde A., Lamphere M., Park J. Y., and Feranchak A. P. (2016) Regulation of mechanosensitive biliary epithelial transport by the epithelial Na+ channel. Hepatology 63, 538–549 10.1002/hep.28301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kleyman T. R., Kashlan O. B., and Hughey R. P. (2018) Epithelial Na+ channel regulation by extracellular and intracellular factors. Annu. Rev. Physiol. 80, 263–281 10.1146/annurev-physiol-021317-121143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wiemuth D., Lefèvre C. M., Heidtmann H., and Gründer S. (2014) Bile acids increase the activity of the epithelial Na+ channel. Pflugers Arch. 466, 1725–1733 10.1007/s00424-013-1403-0 [DOI] [PubMed] [Google Scholar]
- 7. Ilyaskin A. V., Diakov A., Korbmacher C., and Haerteis S. (2016) Activation of the human epithelial sodium channel (ENaC) by bile acids involves the degenerin site. J. Biol. Chem. 291, 19835–19847 10.1074/jbc.M116.726471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmidt A., Alsop R. J., Rimal R., Lenzig P., Joussen S., Gervasi N. N., Khondker A., Gründer S., Rheinstädter M. C., and Wiemuth D. (2018) Modulation of DEG/ENaCs by amphiphiles suggests sensitivity to membrane alterations. Biophys. J. 114, 1321–1335 10.1016/j.bpj.2018.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chiang J. Y. (2013) Bile acid metabolism and signaling. Compr. Physiol. 3, 1191–1212 10.1002/cphy.c120023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Enright E. F., Joyce S. A., Gahan C. G., and Griffin B. T. (2017) Impact of gut microbiota-mediated bile acid metabolism on the solubilization capacity of bile salt micelles and drug solubility. Mol. Pharm. 14, 1251–1263 10.1021/acs.molpharmaceut.6b01155 [DOI] [PubMed] [Google Scholar]
- 11. Memon N., Weinberger B. I., Hegyi T., and Aleksunes L. M. (2016) Inherited disorders of bilirubin clearance. Pediatr. Res. 79, 378–386 10.1038/pr.2015.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Simko V., and Michael S. (1998) Urinary bile acids in population screening for inapparent liver disease. Hepatogastroenterology 45, 1706–1714 [PubMed] [Google Scholar]
- 13. Miura R., Tanaka A., and Takikawa H. (2011) Urinary bile acid sulfate levels in patients with primary biliary cirrhosis. Hepatol. Res. 41, 358–363 10.1111/j.1872-034X.2011.00779.x [DOI] [PubMed] [Google Scholar]
- 14. Schlattjan J. H., Winter C., and Greven J. (2003) Regulation of renal tubular bile acid transport in the early phase of an obstructive cholestasis in the rat. Nephron Physiol. 95, p49–56 10.1159/000074330 [DOI] [PubMed] [Google Scholar]
- 15. Jiao N., Baker S. S., Chapa-Rodriguez A., Liu W., Nugent C. A., Tsompana M., Mastrandrea L., Buck M. J., Baker R. D., Genco R. J., Zhu R., and Zhu L. (2018) Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 67, 1881–1891 10.1136/gutjnl-2017-314307 [DOI] [PubMed] [Google Scholar]
- 16. Brandl K., Hartmann P., Jih L. J., Pizzo D. P., Argemi J., Ventura-Cots M., Coulter S., Liddle C., Ling L., Rossi S. J., DePaoli A. M., Loomba R., Mehal W. Z., Fouts D. E., Lucey M. R., et al. (2018) Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J. Hepatol. 69, 396–405 10.1016/j.jhep.2018.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parks D. J., Blanchard S. G., Bledsoe R. K., Chandra G., Consler T. G., Kliewer S. A., Stimmel J. B., Willson T. M., Zavacki A. M., Moore D. D., and Lehmann J. M. (1999) Bile acids: natural ligands for an orphan nuclear receptor. Science 284, 1365–1368 10.1126/science.284.5418.1365 [DOI] [PubMed] [Google Scholar]
- 18. Wang H., Chen J., Hollister K., Sowers L. C., and Forman B. M. (1999) Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 3, 543–553 10.1016/S1097-2765(00)80348-2 [DOI] [PubMed] [Google Scholar]
- 19. Makishima M., Okamoto A. Y., Repa J. J., Tu H., Learned R. M., Luk A., Hull M. V., Lustig K. D., Mangelsdorf D. J., and Shan B. (1999) Identification of a nuclear receptor for bile acids. Science 284, 1362–1365 10.1126/science.284.5418.1362 [DOI] [PubMed] [Google Scholar]
- 20. Kawamata Y., Fujii R., Hosoya M., Harada M., Yoshida H., Miwa M., Fukusumi S., Habata Y., Itoh T., Shintani Y., Hinuma S., Fujisawa Y., and Fujino M. (2003) A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 278, 9435–9440 10.1074/jbc.M209706200 [DOI] [PubMed] [Google Scholar]
- 21. Wiemuth D., Sahin H., Falkenburger B. H., Lefèvre C. M., Wasmuth H. E., and Gründer S. (2012) BASIC—a bile acid-sensitive ion channel highly expressed in bile ducts. FASEB J. 26, 4122–4130 10.1096/fj.12-207043 [DOI] [PubMed] [Google Scholar]
- 22. Ilyaskin A. V., Diakov A., Korbmacher C., and Haerteis S. (2017) Bile acids potentiate proton-activated currents in Xenopus laevis oocytes expressing human acid-sensing ion channel (ASIC1a). Physiol. Rep. 5, e13132 10.14814/phy2.13132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boscardin E., Alijevic O., Hummler E., Frateschi S., and Kellenberger S. (2016) The function and regulation of acid-sensing ion channels (ASICs) and the epithelial Na+ channel (ENaC): IUPHAR Review 19. Br. J. Pharmacol. 173, 2671–2701 10.1111/bph.13533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wiemuth D., Assmann M., and Gründer S. (2014) The bile acid-sensitive ion channel (BASIC), the ignored cousin of ASICs and ENaC. Channels 8, 29–34 10.4161/chan.27493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Noreng S., Bharadwaj A., Posert R., Yoshioka C., and Baconguis I. (2018) Structure of the human epithelial sodium channel by cryo-electron microscopy. Elife 7, e39340 10.7554/eLife.39340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Snyder P. M., McDonald F. J., Stokes J. B., and Welsh M. J. (1994) Membrane topology of the amiloride-sensitive epithelial sodium channel. J. Biol. Chem. 269, 24379–24383 [PubMed] [Google Scholar]
- 27. Kashlan O. B., and Kleyman T. R. (2011) ENaC structure and function in the wake of a resolved structure of a family member. Am. J. Physiol. Renal Physiol. 301, F684–F696 10.1152/ajprenal.00259.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schmidt A., Löhrer D., Alsop R. J., Lenzig P., Oslender-Bujotzek A., Wirtz M., Rheinstädter M. C., Gründer S., and Wiemuth D. (2016) A cytosolic amphiphilic α-helix controls the activity of the bile acid-sensitive ion channel (BASIC). J. Biol. Chem. 291, 24551–24565 10.1074/jbc.M116.756437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mueller G. M., Maarouf A. B., Kinlough C. L., Sheng N., Kashlan O. B., Okumura S., Luthy S., Kleyman T. R., and Hughey R. P. (2010) Cys palmitoylation of the β subunit modulates gating of the epithelial sodium channel. J. Biol. Chem. 285, 30453–30462 10.1074/jbc.M110.151845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mukherjee A., Mueller G. M., Kinlough C. L., Sheng N., Wang Z., Mustafa S. A., Kashlan O. B., Kleyman T. R., and Hughey R. P. (2014) Cysteine palmitoylation of the γ subunit has a dominant role in modulating activity of the epithelial sodium channel. J. Biol. Chem. 289, 14351–14359 10.1074/jbc.M113.526020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hughey R. P., Mueller G. M., Bruns J. B., Kinlough C. L., Poland P. A., Harkleroad K. L., Carattino M. D., and Kleyman T. R. (2003) Maturation of the epithelial Na+ channel involves proteolytic processing of the α- and γ-subunits. J. Biol. Chem. 278, 37073–37082 10.1074/jbc.M307003200 [DOI] [PubMed] [Google Scholar]
- 32. Hughey R. P., Bruns J. B., Kinlough C. L., Harkleroad K. L., Tong Q., Carattino M. D., Johnson J. P., Stockand J. D., and Kleyman T. R. (2004) Epithelial sodium channels are activated by furin-dependent proteolysis. J. Biol. Chem. 279, 18111–18114 10.1074/jbc.C400080200 [DOI] [PubMed] [Google Scholar]
- 33. Carattino M. D., Sheng S., Bruns J. B., Pilewski J. M., Hughey R. P., and Kleyman T. R. (2006) The epithelial Na+ channel is inhibited by a peptide derived from proteolytic processing of its a subunit. J. Biol. Chem. 281, 18901–18907 10.1074/jbc.M604109200 [DOI] [PubMed] [Google Scholar]
- 34. Kashlan O. B., Adelman J. L., Okumura S., Blobner B. M., Zuzek Z., Hughey R. P., Kleyman T. R., and Grabe M. (2011) Constraint-based, homology model of the extracellular domain of the epithelial Na+ channel a subunit reveals a mechanism of channel activation by proteases. J. Biol. Chem. 286, 649–660 10.1074/jbc.M110.167098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kashlan O. B., Boyd C. R., Argyropoulos C., Okumura S., Hughey R. P., Grabe M., and Kleyman T. R. (2010) Allosteric inhibition of the epithelial Na+ channel (ENaC) through peptide binding at peripheral finger and thumb domains. J. Biol. Chem. 285, 35216–35223 10.1074/jbc.M110.167064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Balchak D. M., Thompson R. N., and Kashlan O. B. (2018) The epithelial Na+ channel γ subunit autoinhibitory tract suppresses channel activity by binding the gamma subunit's finger-thumb domain interface. J. Biol. Chem. 293, 16217–16225 10.1074/jbc.RA118.004362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kashlan O. B., Blobner B. M., Zuzek Z., Carattino M. D., and Kleyman T. R. (2012) Inhibitory tract traps the epithelial Na+ channel in a low activity conformation. J. Biol. Chem. 287, 20720–20726 10.1074/jbc.M112.358218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Caldwell R. A., Boucher R. C., and Stutts M. J. (2004) Serine protease activation of near-silent epithelial Na+ channels. Am. J. Physiol. Cell Physiol. 286, C190–C194 10.1152/ajpcell.00342.2003 [DOI] [PubMed] [Google Scholar]
- 39. Carattino M. D., Hughey R. P., and Kleyman T. R. (2008) Proteolytic processing of the epithelial sodium channel g subunit has a dominant role in channel activation. J. Biol. Chem. 283, 25290–25295 10.1074/jbc.M803931200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kabra R., Knight K. K., Zhou R., and Snyder P. M. (2008) Nedd4-2 induces endocytosis and degradation of proteolytically cleaved epithelial Na+ channels. J. Biol. Chem. 283, 6033–6039 10.1074/jbc.M708555200 [DOI] [PubMed] [Google Scholar]
- 41. Stockand J. D., Mironova E., Bugaj V., Rieg T., Insel P. A., Vallon V., Peti-Peterdi J., and Pochynyuk O. (2010) Purinergic inhibition of ENaC produces aldosterone escape. J. Am. Soc. Nephrol. 21, 1903–1911 10.1681/ASN.2010040377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Toney G. M., Vallon V., and Stockand J. D. (2012) Intrinsic control of sodium excretion in the distal nephron by inhibitory purinergic regulation of the epithelial Na+ channel. Curr. Opin. Nephrol. Hypertens. 21, 52–60 10.1097/MNH.0b013e32834db4a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wühr M., Freeman R. M. Jr., Presler M., Horb M. E., Peshkin L., Gygi S., and Kirschner M. W. (2014) Deep proteomics of the Xenopus laevis egg using an mRNA-derived reference database. Curr. Biol. 24, 1467–1475 10.1016/j.cub.2014.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roda A., Minutello A., Angellotti M. A., and Fini A. (1990) Bile acid structure-activity relationship: evaluation of bile acid lipophilicity using 1-octanol/water partition coefficient and reverse phase HPLC. J. Lipid Res. 31, 1433–1443 [PubMed] [Google Scholar]
- 45. Rossier B. C., Pradervand S., Schild L., and Hummler E. (2002) Epithelial sodium channel and the control of sodium balance: interaction between genetic and environmental factors. Annu. Rev. Physiol. 64, 877–897 10.1146/annurev.physiol.64.082101.143243 [DOI] [PubMed] [Google Scholar]
- 46. Butterworth M. B., Frizzell R. A., Johnson J. P., Peters K. W., and Edinger R. S. (2005) PKA-dependent ENaC trafficking requires the SNARE-binding protein complexin. Am. J. Physiol. Renal Physiol. 289, F969–F977 10.1152/ajprenal.00390.2003 [DOI] [PubMed] [Google Scholar]
- 47. Vallet V., Chraibi A., Gaeggeler H. P., Horisberger J. D., and Rossier B. C. (1997) An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature 389, 607–610 10.1038/39329 [DOI] [PubMed] [Google Scholar]
- 48. Yang L., Xu S., Guo X., Uchida S., Weinstein A. M., Wang T., and Palmer L. G. (2018) Regulation of renal Na transporters in response to dietary K. Am. J. Physiol. Renal Physiol. 315, F1032–F1041 10.1152/ajprenal.00117.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Frindt G., Yang L., Bamberg K., and Palmer L. G. (2018) Na restriction activates epithelial Na channels in rat kidney through two mechanisms and decreases distal Na(+) delivery. J. Physiol. 596, 3585–3602 10.1113/JP275988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Frindt G., Yang L., Uchida S., Weinstein A. M., and Palmer L. G. (2017) Responses of distal nephron Na+ transporters to acute volume depletion and hyperkalemia. Am. J. Physiol. Renal Physiol. 313, F62–F73 10.1152/ajprenal.00668.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Frindt G., and Palmer L. G. (2015) Acute effects of aldosterone on the epithelial Na channel in rat kidney. Am. J. Physiol. Renal Physiol. 308, F572–F578 10.1152/ajprenal.00585.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bohnert B. N., Menacher M., Janessa A., Wörn M., Schork A., Daiminger S., Kalbacher H., Häring H. U., Daniel C., Amann K., Sure F., Bertog M., Haerteis S., Korbmacher C., and Artunc F. (2018) Aprotinin prevents proteolytic epithelial sodium channel (ENaC) activation and volume retention in nephrotic syndrome. Kidney Int. 93, 159–172 10.1016/j.kint.2017.07.023 [DOI] [PubMed] [Google Scholar]
- 53. Morimoto T., Liu W., Woda C., Carattino M. D., Wei Y., Hughey R. P., Apodaca G., Satlin L. M., and Kleyman T. R. (2006) Mechanism underlying flow stimulation of sodium absorption in the mammalian collecting duct. Am. J. Physiol. Renal Physiol. 291, F663–F669 10.1152/ajprenal.00514.2005 [DOI] [PubMed] [Google Scholar]
- 54. Nesterov V., Dahlmann A., Bertog M., and Korbmacher C. (2008) Trypsin can activate the epithelial sodium channel (ENaC) in microdissected mouse distal nephron. Am. J. Physiol. Renal Physiol. 295, F1052–F1062 10.1152/ajprenal.00031.2008 [DOI] [PubMed] [Google Scholar]
- 55. Tetko I. V., Gasteiger J., Todeschini R., Mauri A., Livingstone D., Ertl P., Palyulin V. A., Radchenko E. V., Zefirov N. S., Makarenko A. S., Tanchuk V. Y., and Prokopenko V. V. (2005) Virtual computational chemistry laboratory—design and description. J. Comput. Aided Mol. Des. 19, 453–463 10.1007/s10822-005-8694-y [DOI] [PubMed] [Google Scholar]
- 56. Canessa C. M., Schild L., Buell G., Thorens B., Gautschi I., Horisberger J. D., and Rossier B. C. (1994) Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367, 463–467 10.1038/367463a0 [DOI] [PubMed] [Google Scholar]
- 57. Koyama K., Sasaki I., Naito H., Funayama Y., Fukushima K., Unno M., Matsuno S., Hayashi H., and Suzuki Y. (1999) Induction of epithelial Na+ channel in rat ileum after proctocolectomy. Am. J. Physiol. 276, G975–G984 10.1152/ajpgi.1999.276.4.G975 [DOI] [PubMed] [Google Scholar]
- 58. Sullivan J. I., and Rockey D. C. (2017) Diagnosis and evaluation of hyperbilirubinemia. Curr. Opin. Gastroenterol. 33, 164–170 10.1097/MOG.0000000000000354 [DOI] [PubMed] [Google Scholar]
- 59. Hernaez R., Solà E., Moreau R., and Ginès P. (2017) Acute-on-chronic liver failure: an update. Gut 66, 541–553 10.1136/gutjnl-2016-312670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Singal A. K., Bataller R., Ahn J., Kamath P. S., and Shah V. H. (2018) ACG clinical guideline: alcoholic liver disease. Am. J. Gastroenterol. 113, 175–194 10.1038/ajg.2017.469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Soler N. G., Jain S., James H., and Paton A. (1976) Potassium status of patients with cirrhosis. Gut 17, 152–157 10.1136/gut.17.2.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wong F., Massie D., Hsu P., and Dudley F. (1994) Renal response to a saline load in well-compensated alcoholic cirrhosis. Hepatology 20, 873–881 10.1002/hep.1840200415 [DOI] [PubMed] [Google Scholar]
- 63. Wensing G., Lotterer E., Link I., Hahn E. G., and Fleig W. E. (1997) Urinary sodium balance in patients with cirrhosis: relationship to quantitative parameters of liver function. Hepatology 26, 1149–1155 10.1002/hep.510260510 [DOI] [PubMed] [Google Scholar]
- 64. Lapidus A., Akerlund J. E., and Einarsson C. (2006) Gallbladder bile composition in patients with Crohn's disease. World J. Gastroenterol. 12, 70–74 10.3748/wjg.v12.i1.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Patel J., Walayat S., Kalva N., Palmer-Hill S., and Dhillon S. (2016) Bile cast nephropathy: a case report and review of the literature. World J. Gastroenterol. 22, 6328–6334 10.3748/wjg.v22.i27.6328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sequeira A., and Gu X. (2015) Bile cast nephropathy: an often forgotten diagnosis. Hemodial. Int. 19, 132–135 10.1111/hdi.12169 [DOI] [PubMed] [Google Scholar]
- 67. Cavanaugh C., and Perazella M. A. (2019) Urine sediment examination in the diagnosis and management of kidney disease: core curriculum 2019. Am. J. Kidney Dis. 73, 258–272 10.1053/j.ajkd.2018.07.012 [DOI] [PubMed] [Google Scholar]
- 68. Mukherjee A., Wang Z., Kinlough C. L., Poland P. A., Marciszyn A. L., Montalbetti N., Carattino M. D., Butterworth M. B., Kleyman T. R., and Hughey R. P. (2017) Specific palmitoyltransferases associate with and activate the epithelial sodium channel. J. Biol. Chem. 292, 4152–4163 10.1074/jbc.M117.776146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Passero C. J., Carattino M. D., Kashlan O. B., Myerburg M. M., Hughey R. P., and Kleyman T. R. (2010) Defining an inhibitory domain in the gamma subunit of the epithelial sodium channel. Am. J. Physiol. Renal Physiol. 299, F854–F861 10.1152/ajprenal.00316.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ahn Y. J., Brooker D. R., Kosari F., Harte B. J., Li J., Mackler S. A., and Kleyman T. R. (1999) Cloning and functional expression of the mouse epithelial sodium channel. Am. J. Physiol. 277, F121–F129 10.1152/ajprenal.1999.277.1.F121 [DOI] [PubMed] [Google Scholar]