

Abstract
Proper cell division relies on the coordinated regulation between a structural component, the mitotic spindle, and a regulatory component, anaphase-promoting complex/cyclosome (APC/C). Hematopoietic PBX-interacting protein (HPIP) is a microtubule-associated protein that plays a pivotal role in cell proliferation, cell migration, and tumor metastasis. Here, using HEK293T and HeLa cells, along with immunoprecipitation and immunoblotting, live-cell imaging, and protein-stability assays, we report that HPIP expression oscillates throughout the cell cycle and that its depletion delays cell division. We noted that by utilizing its D box and IR domain, HPIP plays a dual role both as a substrate and inhibitor, respectively, of the APC/C complex. We observed that HPIP enhances the G2/M transition of the cell cycle by transiently stabilizing cyclin B1 by preventing APC/C–Cdc20–mediated degradation, thereby ensuring timely mitotic entry. We also uncovered that HPIP associates with the mitotic spindle and that its depletion leads to the formation of multiple mitotic spindles and chromosomal abnormalities, results in defects in cytokinesis, and delays mitotic exit. Our findings uncover HPIP as both a substrate and an inhibitor of APC/C–Cdc20 that maintains the temporal stability of cyclin B1 during the G2/M transition and thereby controls mitosis and cell division.
Keywords: mitosis, cell cycle, cell biology, molecular cell biology, cell division, cytokinesis, APC-C complex, chromosome segregation, Hematopoietic PBX-interacting protein (HPIP)/PBX homeobox interacting protein 1 (PBXIP1), post-translational modification, spindle assembly checkpoint (SAC)
Introduction
Accurate chromosome segregation is essential for proper cell division in normal cells. Two important post-translational modifications, phosphorylation, and ubiquitin-mediated proteolysis during mitosis, play crucial roles in this process (1). The anaphase promoting complex/cyclosome (APC/C),3 a ubiquitin ligase, controls the cell division by regulating mitosis through ubiquitin-directed proteolysis of key substrates in an ordered fashion to direct progression through the mitotic exit, chromosome segregation, and cytokinesis (2–4). The activity of APC/C is coordinated by two regulatory proteins, Cdc20 and Cdh1, through phase-specific interactions during the cell cycle and promotes cell division with precision and accuracy (5). For example, APC/C–Cdc20 degrades Securin and cyclin B1 at anaphase onset. This ensures Separase activation and proteolysis of Cohesin, which holds pair of sister chromatids together during early mitosis. The spindle assembly checkpoint, which depends on multiprotein complexes including Mad2, BubR1, and Bub3, delays APC/C–Cdc20 activation until all chromosomes are properly aligned at the metaphase plate (6, 7). Perturbation of this checkpoint results in chromosomal abnormality (8).
Entry into mitosis is coordinated by cyclin B1–dependent activation of cyclin-dependent kinase 1 (CDK1) during G2 phase and form a Cdk1–cyclin B1 complex also known as maturation-promoting factor, which is crucial for G2/M transition (9). Cyclin B1 accumulates in the nucleus as the cells progress to mitosis, although the activation of CDK1–cyclin B1 is initiated at the cytoplasm (10). Cyclin B1 binding triggers a conformational change in Wee1 phosphorylated and inactive CDK1, restoring the activity in CDK1 (11). The activated CDK1–cyclin B1 complex triggers initiation of chromosome condensation, nuclear envelope breakdown, and mitotic spindle assembly through phosphorylation of its substrates (10). It also phosphorylates APC/C–Cdc20 for its complete activation, but later during mitosis cyclin B1 is degraded by APC/C–Cdc20 (12, 13). Abolishing the degradation of cyclin B1 leads to arrest of cells in mitosis, suggesting timely degradation of cyclin B1 by APC/C, is important for proper cell cycle progression (14, 15). Although transcriptional up-regulation of cyclin B1 and its increased stability of mRNA during the G2 phase has been described before (16), the role of proteasomal pathway in increased cyclin B1 levels is largely unknown.
Hematopoietic PBX-interacting protein (HPIP, also known as PBXIP1) is a protooncoprotein that has been shown to be overexpressed in several cancer types including infiltrative ductal carcinoma (17), hepatocellular carcinoma (18), glioma (19), and ovarian cancer (20). Previous reports have shown that HPIP promotes cell proliferation by modulating the expression of cyclins during the cell cycle (21, 22). However, the precise mechanism by which HPIP regulates cell cycle progression remains elusive. Here, we demonstrated that HPIP protein levels oscillate during various stages of cell cycle and HPIP is degraded during mitosis by APC/C–Cdc20 but not by APC/C–Cdh1. Further, we also found that HPIP inhibits APC/C–Cdc20–mediated cyclin B1 degradation during early mitosis perhaps to facilitate the G2/M transition. Moreover, this study also demonstrates that HPIP associates with mitotic spindle, and its depletion leads to the formation of multiple spindle poles, chromosomal abnormality, and delayed mitotic exit. These studies suggest a dual role for HPIP as substrate and an inhibitor of APC/C–Cdc20 during cell cycle progression.
Results
Loss of HPIP expression delays cell division
To determine the role of HPIP in cell proliferation, we silenced endogenous HPIP expression in HeLa cells by lentiviral-mediated shRNA silencing approach. After confirming HPIP silencing in HeLa cells (Fig. 1A), the cell proliferation rate was determined. We found a significant decrease in cell growth upon depletion of HPIP in HeLa cells (Fig. 1B). Next, cell division dynamics were monitored by time-lapse microscopy. Depletion of HPIP significantly delayed the cell division in HeLa cells by an average of ∼3.4 h as compared with control cells (shCtrl versus shHPIP: 14.2 ± 0.6 h versus 17.6 ± 1.0 h) (Fig. 1, C and D, Videos S1 and S2). To assess which phase of the cell cycle might be influenced by HPIP, cell cycle progression was analyzed by flow cytometry (FACS). The FACS analysis indicated a significant accumulation of cells at the G2/M phase upon HPIP silencing as compared with control cells (Fig. 1E and Fig. S1). To explore the role of HPIP during cell-cycle progression more precisely, we depleted HPIP in HeLa cells expressing H2B–EGFP and α-tubulin–mCherry and synchronized them in the S phase by double thymidine (DT) block. We subsequently measured the time between the S and M phases in HeLa–H2B/tubulin cells by time-lapse microscopy following release from a DT block and found a significant delay in mitotic entry in HPIP knockdown cells as compared with control siRNA-treated cells (shCtrl versus shHPIP: 12.9 ± 1.9 versus 18.7 ± 2.5 h) (Fig. 1, F and G; and Videos S3 and S4). Moreover, HPIP depletion markedly decreased the mitotic index, which is measured by FACS based histone H3Ser-10 staining, in HeLa cells as compared with control cells (shCtrl versus shHPIP, 2.4 ± 0.2% versus 1.4 ± 0.1%) (Fig. 1H). Together, these results indicated that HPIP expression is required for normal cell division, and HPIP may act as a G2/M transition regulator.
Figure 1.

Loss of HPIP expression delays cell division. A, HPIP knockdown by various HPIP-specific shRNAs (shHPIP-1, shHPIP-2, and shHPIP-3) in HeLa cells was analyzed by Western blotting. B, cell proliferation upon HPIP knockdown in HeLa cells was analyzed by WST-1 assay. C, representative time-lapse live cell images of HPIP-depleted HeLa cells (magnification, 20×). D, quantification data of C. A total of 20 cells were analyzed for each sample (n = 20). E, flow cytometry (FACS) analysis showing HeLa cells at various stages of cell cycle (percentage) upon HPIP knockdown. F, representative time-lapse live cell fluorescent images of either siCtrl or siHPIP-treated HeLa–H2B/tubulin cells that are synchronized by DT block at the S phase followed by release into fresh medium and captured at indicated time points. Green, H2B–EGFP; red, α-Tubulin–mCherry (magnification, 20×). EGFP, enhanced green fluorescence protein. G, quantification data of F. A total of 13 cells were analyzed for each sample (n = 13). H, HeLa cells transfected with either shCtrl or shHPIP were fixed and stained with H3Pho-S10 antibody and propidium iodide and then subjected to flow cytometry analysis. The mitotic cells were boxed. The mitotic indices are indicated as percentages of total cell population. The quantified results are presented as means ± S.D. using Student's t test. **, p < 0.001; ***, p < 0.0001 were considered significant. Ctrl, control; sh, short hairpin; Rel. OD, relative optical density.
HPIP protein level oscillates during cell cycle
To decipher the underlying mechanism of HPIP-mediated cell division, we examined the expression dynamics of HPIP in different stages of the cell cycle. HeLa cells synchronized at G1/S boundary by DT block were released and harvested at different time points, which are confirmed by FACS analysis (Fig. S2A), and HPIP protein levels were analyzed by Western blotting. HPIP protein levels oscillated to varying degree throughout the cell cycle, specifically a sharp decline at the time period between hours 9 and 10 when mitosis took place (Fig. 2, A and B). The degradation of HPIP was similar to cyclin A, which is a substrate of APC/C–Cdc20. But mRNA levels of HPIP remain unchanged (Fig. S2B). We next studied the protein dynamics of HPIP during mitosis by thymidine–nocodazole block/release approach. We observed that HPIP levels were decreased as mitosis progresses, suggesting a possibility of proteasomal degradation of HPIP in the midmitosis (Fig. 2, C and D; and Fig. S2C). Taken together, our data indicated that HPIP might be a possible substrate of APC/C during mitosis.
Figure 2.

HPIP protein levels oscillate across the cell cycle progression. A, HeLa cells synchronized by double thymidine block were release into fresh medium. The cells collected at indicated time points were analyzed by Western blotting (upper panel). (Note: Protein samples from lanes 1–9 and 10–12 were run on two different gels.) The percentage of cells at various stages of the cell cycle indicated was derived from FACS analysis (lower panel). B, bar graph showing the quantification of protein bands (relative to glyceraldehyde-3-phosphate dehydrogenas) from A (upper panel). C, HeLa cells synchronized by thymidine–nocodazole block were released into fresh medium at indicated time points, and cell lysates were analyzed by Western blotting. D, bar graph showing the quantification of protein bands (relative to glyceraldehyde-3-phosphate dehydrogenase) from C. Protein band intensities was determined using ImageJ software. MW, molecular weight.
HPIP is a substrate of APC/C–Cdc20 but not APC/C–Cdh1
Next, we investigated whether HPIP is degraded by APC/C ubiquitin-mediated pathway. The APC/C complex utilizes either Cdc20 or Cdh1 as coactivators to degrade its substrates during cell cycle progression (5). To prove which coactivator is likely to be involved in HPIP proteolysis during mitosis, we co-transfected T7–HPIP and either HA–Cdc20 or HA–Cdh1 in HEK293T cells, and protein levels were analyzed by Western blotting. HA–Cdc20 co-transfected cells showed reduced T7–HPIP protein level (Fig. 3A, lane 5), but MG132 treatment restored it (Fig. 3A, lane 6). However, there is no change in the levels of T7–HPIP upon overexpression of HA–Cdh1 (Fig. 3A, lane 3). We next monitored the dosage effect of Cdc20 or Cdh1 on HPIP in HEK293T cells. As shown in Fig. 3B, we observed a gradual decrease of T7–HPIP upon dose-dependent increase of HA–Cdc20 but not with HA–Cdh1 (Fig. 3C). Conversely, knockdown of Cdc20 substantially increased HPIP levels in HeLa cells (Fig. 3D, lane 2), whereas Cdh1 knockdown did not alter it (Fig. 3D, lane 3). Furthermore, HPIP protein dynamics were altered upon Cdc20 knockdown (although it was ∼50%) and remained almost unchanged from 6 to 12 h in synchronized HeLa cells as compared with control cells, suggesting that Cdc20-mediated degradation of HPIP is cell cycle–specific (Fig. 3E). Together, these data indicated that HPIP is a possible substrate of APC/C complex utilizing Cdc20 as a coactivator but not Cdh1.
Figure 3.

Cdc20 but not Cdh1 stimulates HPIP degradation. A, HEK293T cells were co-transfected with T7–HPIP and HA–Cdc20 or HA–Cdh1 plasmid constructs. Following MG132 (10 μm) treatment, cell lysates were blotted as indicated. B and C, HEK293T cells were co-transfected with T7–HPIP and increasing concentrations of either HA–Cdc20 (B) or HA–Cdh1 (C) (1–5 μg) plasmid constructs. After 8 h of MG132 treatment, cell lysates were blotted as indicated. D, HeLa cells were transfected with control shRNA, Cdc20 shRNA, or Cdh1shRNA, and cell lysates were analyzed by Western blotting as indicated. E, Cdc20 shRNA-treated cells were synchronized and released at indicated time points and blotted as indicated (upper panel). The bar graph showing the quantification of HPIP protein band intensity (lower panel) (n = 2). Ctrl, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; sh, short hairpin; MW, molecular weight.
Role of D box in HPIP degradation by APC/Cdc20 during mitosis
APC/C complex recognizes its substrates through a specific degron motif (23, 24). For instance, APC/C–Cdc20 utilizes either D box (RXXL, where R is arginine, L is leucine, and X is any amino acid) or KEN motifs in the substrates for their interaction and degradation, whereas APC/C–Cdh1 utilizes a KEN box. We analyzed the HPIP protein sequence and found seven putative D box motifs, which are located at different regions of HPIP and one KEN motif (277–279 amino acids) at the N-terminal region of HPIP (Fig. 4A). Of seven D box motifs, only four are conserved in human and mouse (Fig. 4B). To determine whether these motifs are important for proteolysis, we generated mutants for all the conserved degron motifs of HPIP. First, HEK293T cells were transfected with these mutants with or without the HA–Cdc20 expression construct. The protein levels of all mutants except D4 mutant were reduced upon HA–Cdc20 ectopic expression (Fig. 4C, lanes 8 and 9). Next, we performed co-immunoprecipitation (co-IP) analysis using protein lysates prepared from HEK293T cells that were co-transfected with either T7–HPIP or mtHPIP–D4 and with or without the GFP–Cdc20 expression to check whether loss of a D box motif (D4 in HPIP) abrogates APC/C–Cdc20–mediated ubiquitination. As expected, T7–HPIP readily ubiquitinates in Cdc20-overexpressing cells, but D4 mutation abrogated Cdc20-mediated ubiquitination (Fig. 4D). Consistent with these data, mtHPIP–D4 was stable during mitosis in synchronized HeLa cells unlike T7–HPIP (Fig. 4E). Together, the compelling evidence suggests that D4 domain is responsible for HPIP proteolysis by APC/C–Cdc20.
Figure 4.

D box but not KEN motif is required for HPIP degradation by APC-C/Cdc20. A, physical map of HPIP displaying the location of various D boxes and the KEN box present in it. B, HPIP domain conservation in human (h) and mouse (m). Conserved arginine and leucine residues at respective D box were replaced with alanine. Similarly, conserved lysine, glutamic acid, and asparagine residues at KEN box were replaced with alanine. C, HEK293T cells were co-transfected with various HPIP (T7 tag) mutant plasmid constructs and HA–Cdc20. 48 h post-transfection, the cell lysates were blotted as indicated. D, HEK293T cells were co-transfected with HA–ubiquitin and various combinations of plasmid constructs including wtHPIP or mtHPIP–D4 (T7 tag) and with or without GFP–Cdc20 constructs. 48 h post-transfection, T7 immunoprecipitates were blotted as indicated. E, T7–HPIP or mtHPIP–D4 transfected HeLa cells were synchronized by thymidine–nocodazole (Thy-Noc) block and released into fresh medium as indicated and blotted. IP, immunoprecipitation; MW, molecular weight; Vec, vector.
Cdc20 interacts and ubiquitinates HPIP at lysine 274 during early mitosis
We hypothesized that HPIP proteolysis by Cdc20 depends on their physical interaction. To test this, HEK293T cells were co-transfected with either T7–HPIP or mtHPIP–D4 (expressed with a T7 tag) and HA–Cdc20, and cell lysates were subjected to co-IP using T7 antibody. T7–HPIP, but not mtHPIP–D4 or control IgG, was readily associated with HA–Cdc20 (Fig. 5A). Furthermore, immunoprecipitation analysis of lysates prepared from synchronized HeLa cells demonstrated a strong interaction between HPIP and Cdc20 at the beginning of mitosis (at 0–1 h after release) but almost lost at 2 h after release, a time point where cells will undergo anaphase and cytokinesis (Fig. 5B). Together, these results indicate that HPIP and Cdc20 are specifically associated during early to midmitosis.
Figure 5.

Cdc20 interacts with HPIP during mitosis and ubiquitinates at lysine 274. A, HEK293T cells were co-transfected with either wtHPIP or mtHPIP–D4 (T7 tag) and HA–Cdc20 plasmid constructs. 48 h post-transfection, cell lysates were immunoprecipitated using T7 antibody and blotted as indicated. B, thymidine–nocodazole (Thy-Noc) synchronized HeLa cells were released into fresh medium and lysed, and cell lysates were immunoprecipitated using HPIP antibody and blotted as indicated. C, HPIP protein sequence at indicated region from human, mouse, and rat showing conservation of lysines at 274 and 634. D, HEK293T cells co-transfected with T7–HPIP, mtHPIP-K274R, or mtHPIP-K634R and HA–Cdc20 were harvested 48 h post-transfection, and cell lysates were blotted as indicated. E, HEK293T cells were co-transfected with either T7–HPIP or mtHPIP-K274R with or without GFP–Cdc20 and HA–Ub constructs. 48 h post-transfection, cell lysates were T7-immunoprecipitated and blotted with indicated antibodies. F, WST-1 assay demonstrating the effect of HPIP silencing followed by ectopic expression of T7–HPIP, mtHPIP–D4, or mtHPIP-K274R on HeLa cell proliferation. G, Western blotting demonstrating the knockdown of HPIP (lanes 2–5) and ectopic expression (rescue) of various HPIP mutants (lanes 3–5) in HeLa cells. The quantified results are presented as means ± S.D. using analysis of variance. **, p < 0.001; ***, p < 0.0001 were considered significant. Ctrl, control; IP, immunoprecipitation; sh, short hairpin; MW, molecular weight; WB, Western blot.
Because there are several putative ubiquitination sites on HPIP, we considered only two potential lysines i.e. Lys-274 and Lys-634, which have been shown to undergo ubiquitination by whole proteome analysis (25). Therefore, these two conserved lysines were mutated (Fig. 5C). These mutants were then checked to verify whether APC/C–Cdc20 mediates ubiquitination at Lys-274 or Lys-634 sites, by co-transfecting T7–HPIP (wt), mtHPIP-K274R, or mtHPIP-K634R constructs along with HA–Cdc20. Interestingly, protein levels of both T7—HPIP, as well as mtHPIP-K634R were reduced upon ectopic expression of HA–Cdc20 (Fig. 5D, lanes 2 and 6), although the expression levels of these proteins are different (Fig. 5D, lanes 1, 3, and 5), whereas mutation at Lys-274 abrogated it (Fig. 5D, lane 4). Consistent with these data, T7–HPIP but not mtHPIP-K274R was readily ubiquitinated, supporting the fact that lysine 274 of HPIP undergoes APC/C–Cdc20–mediated ubiquitination perhaps during mitosis (Fig. 5E).
Having demonstrated that HPIP modulates the G2/M phase of cell cycle, we next examined the functional significance of the D box motif and lysine 274 of HPIP in this process. To test this, cell proliferation was measured by the WST-1 assay, which showed that neither mtHPIP–D4 nor mtHPIP-K274R could rescue the cell proliferation similar to T7–HPIP (Fig. 5, F and G). Together, these results indicate that D4 domain and Lys-274 are critical for HPIP in the regulation of the G2/M transition.
HPIP stabilizes cyclin B1 to facilitate timely mitotic entry
During the normal cell cycle, activated CDK1–cyclin B1 complex promotes the progression of cells from G2 to M phase, but its diminished activity renders G2 phase arrest (26). In accordance with the previous reports, we have shown that HPIP promotes cell proliferation by enhancing the G2/M transition during the cell cycle (Fig. 1) (18). We next focused on investigating the molecular mechanism by which HPIP enhances G2/M phase of the cell cycle. Depletion of HPIP in HeLa cells resulted in a significant loss of cyclin B1 expression as compared with control cells (Fig. 6A, lanes 1 and 2), but it was restored upon treatment with MG132, a proteasome inhibitor, suggesting a possible proteasomal degradation of cyclin B1 (Fig. 6A, lane 3). Furthermore, ectopic expression of T7–HPIP increased cyclin B1 levels in HEK293T cells where HPIP abundance is very low (Fig. 6B, lane 2). Although ectopic expression of HPIP increased cyclin B1 expression, coexpression of HA–Cdc20 markedly reduced it, further reinforcing the possibility of HPIP involvement in blocking Cdc20-mediated cyclin B1 degradation (Fig. 6B). To further strengthen these data, a chasing experiment was carried out using cycloheximide, a de novo protein synthesis inhibitor, in synchronized HeLa cells. As shown in (Fig. 6, C and D), stability of the cyclin B1 was significantly decreased upon HPIP silencing (t½ = 41 ± 10 min) as compared with control cells (t½ = 72 ± 15 min). We reasoned that increased cyclin B1 upon HPIP ectopic expression could be due to decreased ubiquitination of cyclin B1 by APC/C–Cdc20. Hence, we co-transfected GFP–cyclin B1, HA–Cdc20, and control vector or T7–HPIP into HEK293T cells, and then cyclin B1 ubiquitination was determined. Ectopic expression of HA–Cdc20 ensured cyclin B1 ubiquitination as reported earlier (27) (Fig. 6E, lane 3); however, T7–HPIP coexpression markedly reduced it (Fig. 6E, lane 4). We next analyzed the levels of pH3Ser-10, a mitosis marker, in HPIP-depleted HeLa cells that are synchronized with thymidine block followed by treatment with nocodazole. As shown Fig. 6F, in control cells pH3Ser-10 levels appeared at 10 h with a gradual increase and peaked at 14 h, whereas in HPIP-depleted cells, although pH3Ser-10 levels appeared at 10 h but with at lower levels than in control cells. Together, these results indicate that HPIP stabilizes cyclin B1 to ensure timely mitotic entry.
Figure 6.

HPIP stabilizes cyclin B1 during G2/M transition. A, HeLa cells transfected with shCtrl (lane 1) and shHPIP (lanes 2 and 3) were treated with MG132 (lane 3) and blotted as indicated. B, HEK293T cells were transfected with T7–HPIP alone or in combination of T7–HPIP and HA–Cdc20, and cell lysates were blotted as indicated. C, stably knocked down HeLa cells with indicated shRNAs were synchronized by thymidine–nocodazole block and treated with cycloheximide (20 μg/ml) for the indicated time points. Cell lysates were then analyzed by Western blotting. D, the line graph represents the quantification of cyclin B1 protein bands from C. E, HEK293T cells were co-transfected with GFP–cyclin B1 or HA–Cdc20 and with or without T7–HPIP. 48 h post-transfection, cell lysates were T7-immunoprecipitated and blotted as indicated. F, double thymidine block synchronized HPIP-depleted HeLa cells were released in presence of nocodazole (100 ng/ml) at the indicated time points and analyzed by Western blotting as indicated. Ctrl, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IP, immunoprecipitation; sh, short hairpin; MW, molecular weight; Vec, vector.
HPIP antagonizes APC/C–Cdc20 activity during cell cycle progression
We wondered how HPIP stabilizes cyclin B1. We ascertained whether HPIP inhibits APC/C–Cdc20 to stabilize cyclin B1. To test this possibility, we analyzed the interaction between APC3, a core component of APC/C complex, and Cdc20 in HPIP-depleted cells. Immunoprecipitation analysis showed an increased interaction between Cdc20 and APC3 upon HPIP depletion (Fig. 7A). Because Cdc20 is also a component of mitotic checkpoint complex (MCC), we examined whether loss of HPIP expression could enhance interaction of MAD2 and Cdc20. However, the interaction between MAD2, a component of MCC, and Cdc20 is unaltered, suggesting that HPIP does not interfere with the interaction of Cdc20 with Mad2 (Fig. 7B). It has been demonstrated that Cdc20 interacts with APC/C complex through IR (where I is isoleucine, and R is arginine) motif, and thus the loss of IR motif in Cdc20 hampers its interaction with APC/C and associated functions (28–30). Inspired by these prior reports, we analyzed HPIP protein sequence and found a conserved IR motif in the N-terminal region, between amino acids 282 and 283 (Fig. 7C). We mutated IR motif in HPIP by replacing with alanine residues (IR to AA), and co-IP was performed to determine its interaction ability with APC3. As shown in Fig. 7D, IR mutation in HPIP abrogated its interaction with APC3, whereas wtHPIP, as well as D4 mutant, could bind to it efficiently.
Figure 7.

HPIP binds and inhibits APC/C–Cdc20 activity through IR motif. A and B, HeLa cells stably transfected with either shCtrl or shHPIP were lysed, and cell lysates were subjected to co-IP by APC3 (A) or MAD2 (B) followed by Western blotting as indicated. C, conservation of IR motif in HPIP among various species. D, HeLa cells transfected with wtHPIP, mtHPIP–D4, or mtHPIP–IR were subjected to co-IP with APC3 antibody followed by Western blotting as indicated. E, HeLa cells transfected with wtHPIP, mtHPIP–D4, or mtHPIP–IR were subjected to double thymidine synchronization followed by release at the indicated time points and blotted as indicated. An asterisk denotes expression of the indicated proteins at peak in the specified time period. F, representative time-lapse live cell fluorescent images of GFP–HPIP, GFP–mtHPIP–D4, or GFP–mtHPIP–IR transfected HeLa–H2B–mCherry (red) cells that are synchronized by DT block at the S phase followed by release into fresh medium and captured at indicated time points (magnification, 20×). G, quantification data of F. The quantified results are presented as means ± S.D. using Student's t test. *, p < 0.01; **, p < 0.001 were considered significant. Ctrl, control; IP, immunoprecipitation; MW, molecular weight; sh, small hairpin.
Next, we analyzed the effect of IR mutation in HPIP on cyclin B1, Securin dynamics. HeLa cells were transfected with T7–HPIP (wt), mtHPIP–D4, or mtHPIP–IR followed by double thymidine block and then released at various time points. We observed that mtHPIP–IR protein dynamics were similar to T7–HPIP, being low at the time period between hours 9 and 10 during which cells are in mitosis, whereas D4 mutation ensured elevated levels of HPIP and sustained up to hour 12, indicating that D4 domain but not IR domain is involved in HPIP stability during mitosis (Fig. 7E). Histone H3pSer-10 was also analyzed to study the mitotic index among these mutants. H3pSer-10 signal started appearing at hour 9 in both mutants as well as in wtHPIP. In T7–HPIP (WT), cyclin B1 accumulation started at hour 7 (G2 phase) and continued until hour 9, at which cells are in mitosis, and then declined. However, in mtHPIP–IR cells, cyclin B1 accumulation started at hour 7 as similar to wtHPIP but sustained only for a short period of 1 h (hours 7 to 8) and declined at the onset of mitosis (from hour 9). Interestingly, in mtHPIP–D4 cells, cyclin B1 accumulation started at hour 9, i.e. at the onset of mitosis; peaked at hour 10, and then declined at the later time points (Fig. 7E). We also analyzed Securin dynamics, an indicator of metaphase to anaphase shift, in all the mutants. All the mutants as well as T7–HPIP cells had elevated levels of Securin in the time period between hours 7 and 10 and declined at later time points. To further support these findings, we measured the time between the S and M phases in these mutants. We transfected either wtHPIP or its mutants (mtHPIP–D4 or mtHPIP–IR) in HeLa cells expressing H2B-mCherry and synchronized them in S phase by DT block. Subsequently we measured the time between the S and M phases by time-lapse microscopy following release from a DT block. In support of biochemical data, we found that mitotic entry was significantly delayed in D4 mutant of HPIP, i.e. mtHPIP–D4 as compared with wtHPIP and mtHPIP–IR cells (wtHPIP, mtHPIP-D, and mtHPIP–IR: 11.9 ± 1.5, 16.0 ± 2.8, and 11.0 ± 1.7 h, respectively) (Fig. 7, F and G; and Video S5–S7). Together these data suggests that D4 and IR motifs of HPIP function differently, because D4 domain is primarily involved in HPIP stability, whereas IR domain participates in the inhibition of APC–Cdc20 activity and therefore the altered cyclin B1 dynamics.
HPIP associates with mitotic spindle during mitosis, and its loss of expression delays mitosis exit
Microtubules are integral part of mitotic spindle that aid in chromosome segregation during mitosis (31). Although earlier studies established that HPIP is a microtubule-binding protein (32, 33), its role in mitotic spindle function is unexplored. Therefore, we hypothesized that HPIP may associate with mitotic spindle and thereby control its function. Confocal imaging indeed showed a marked co-localization of HPIP with mitotic spindle during mitosis (Fig. 8A). Furthermore, loss of HPIP expression leads to formation of multiple spindle poles (Fig. 8B). Next we measured the time taken for mitotic exit upon HPIP depletion in HeLa cells. Live cell imaging of G2/M synchronized HeLa cells showed delayed mitotic exit upon HPIP knockdown (n = 60 cells; shCtrl versus shHPIP: 74.6 ± 24.2 versus 90.5 ± 29.7 min; p = 0.001) (Fig. 8, C and D; and Videos S8 and S9). To further support our findings, we next monitored Clover-Geminin (1–110) levels, a M/G1 transition marker, during mitosis (34). We depleted HPIP in HeLa cells transfected with Clover-Geminin (1–110) and synchronized them at the early M phase by nocodazole block. We subsequently monitored the Clover-Geminin disappearance, which is an indicator of mitosis exit, by time-lapse fluorescence microscopy. In control siRNA-treated cells, Geminin disappeared by approximately 60 min after nocodazole release. By contrast, Geminin levels persisted up to ∼90 min in HPIP-depleted cells, suggesting that loss of HPIP delays mitotic exit (Fig. 8, E and F; and Videos S10 and S11). These data suggest that a loss of HPIP delays mitotic exit.
Figure 8.

HPIP associate with mitotic spindle during mitosis. A, confocal images representing the localization of indicated proteins at various stages of mitosis in HeLa cells. Green, HPIP; red, α-tubulin; blue, DNA (DAPI) (magnification, 60×). B, confocal images representing the spindle morphology in control shRNA or HPIP shRNA-treated HeLa cells (magnification, 60×). C, representative time-lapse live cell images of shCtrl or shHPIP-treated HeLa cells. 48 h post-transfection cells were synchronized by aphidicolin–RO-3306 followed by released into fresh medium and time-lapse imaging and then monitoring of the mitotic exit (magnification, 20×). D, scatter plot representing the quantification of division time. A total of 60 cells were analyzed for each sample (n = 60). The quantified results are presented as means ± S.D. using Student's t test. **, p < 0.001 was considered significant. E, HeLa cells co-transfected with either siCtrl or siHPIP and Clover-Geminin (1–110) were synchronized at the early M phase by nocodazole block. After nocodazole release of the cells, Clover-Geminin was monitored by time-lapse fluorescence microscopy (magnification, 20×). F, schematic diagram depicts the pattern of Clover-Geminin (green) during cell cycle progression. BF, brightfield; Ctrl, control; Noc., nocodazole; sh, small hairpin.
HPIP has spindle check point function
To further delineate the functions of HPIP in mitosis, we set forth to examine the morphology of the chromosomes during metaphase. As shown in Fig. 9, A and C, HPIP-depleted cells shows a significantly higher percentage of metaphase cells having premature segregated chromosomes and also higher chromosome breaks per cell (Fig. 9, D and F). MCC or SAC complex prevents the errors in chromosome segregation, aneuploidy, tumor progression, and cell death when it is activated. Hence premature segregation can happen only when SAC is not activated (35, 36). Previous reports have demonstrated that suppression of MAD2, a SAC component, and Securin, which inhibits Separase-mediated proteolysis of Cohesin that tether chromatids, leads to premature chromosomal segregation. We observed a marked down-regulation of MAD2 and Securin expression in HPIP-depleted cells (Fig. 9G). However, Securin, but not MAD2, levels were restored upon treatment with MG132 (Fig. 9G, lane 3), suggesting a possible transcriptional regulation of MAD2. Conversely, ectopic expression of HPIP led to a marked increase in MAD2 levels (Fig. 9H). We also determined the duration of metaphase (min) upon HPIP depletion in G2/M synchronized HeLa cells by time-lapse imaging. We found that HPIP depletion resulted in a faster metaphase-to-anaphase transition as compared with control cells (shCtrl versus shHPIP: 39.6 ± 7.0 versus 26.7 ± 5.3 min) (Fig. 9, I and J; and Videos S12 and S13). Together, these data indicate that loss of HPIP expression accelerates metaphase to anaphase transition, which could cause chromosomal presegregation and breaks.
Figure 9.

Loss of HPIP expression leads to chromosomal presegregation and predisposes for chromosomal breaks. A, representative images of HeLa metaphase spread showing a normal metaphase in siCtrl cells but premature sister chromatid segregation (arrowheads) in siHPIP-treated cells. A total of ∼50 chromosome spreads were analyzed for each sample (magnification, 60×). B, number of metaphase cells from A. C–F, quantitative analysis of premature segregation (C), chromosome breaks per cell (E), and percentage of chromosomal breaks (F) from D. Arrowheads indicate chromosome breaks in D. G, effect of HPIP knockdown on MAD2 and Securin levels in HeLa cells was analyzed by Western blotting. As indicated, MG132 (10 μm) was treated for 8 h. H, HeLa cells were transfected with either vector alone or T7–HPIP. 48 h post-transfection, the cells were harvested, and lysates were analyzed by Western blotting as indicated. I, HeLa cells transfected with either siCtrl or siHPIP were synchronized at prometaphase by nocodazole, and cells were time-lapse imaged after nocodazole release (magnification, 20×). J, the duration (min) of metaphase in control and HPIP knockdown cells was determined from I. The data represent the average duration from the time when all of the chromosomes were aligned at the metaphase plate to the onset of anaphase. The number of cells in each experiment is indicated. The error bars represent S.E. ****, p < 0.0001 was considered significant. Ctrl, control; MW, molecular weight; si, small interfering; Vec, vector.
Loss of HPIP expression leads to defects in cytokinesis
Cytokinesis, a process involving the division of the cytoplasm to generate two daughter cells, overlaps with the final stages of mitosis. It may start in either anaphase or telophase (66). We ascertained that the delayed mitotic exit, whereas faster metaphase to anaphase transition could be due to defects in cytokinesis. To test this hypothesis, first we examined the localization of HPIP during telophase and cytokinesis by confocal microscopy. We found that HPIP localize to the midbody, which appeared in telophase and persisted up to abscission (Fig. 10A). Next, we analyzed the morphology of midbody upon HPIP depletion in HeLa cells. Interestingly, we found significant defects in midbody formation in HPIP-depleted HeLa cells (Fig. 10B). Together, these results indicate a plausible role for HPIP in cytokinesis.
Figure 10.
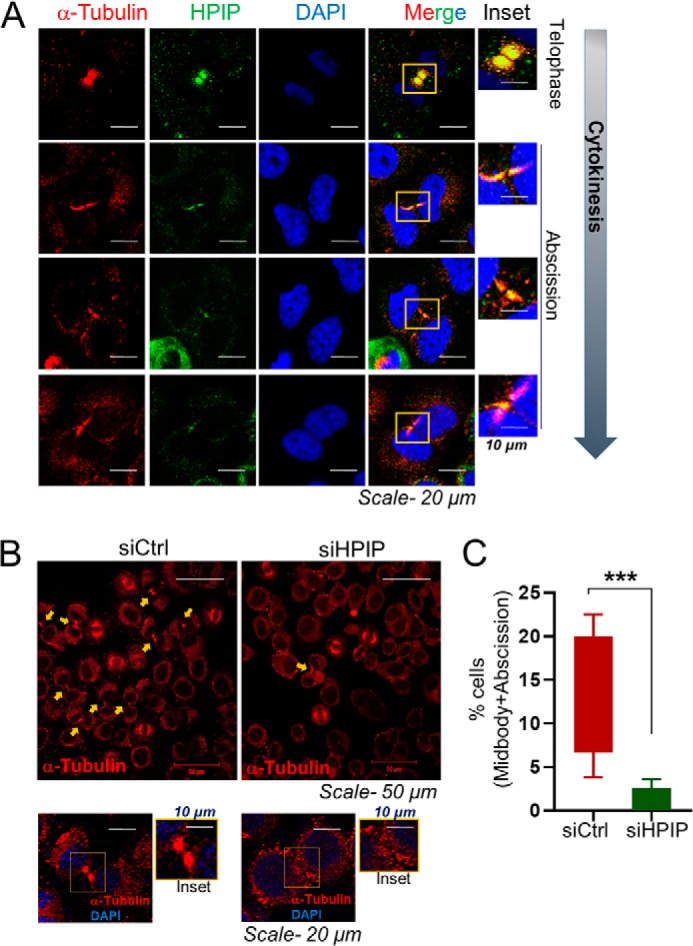
HPIP localizes to midbody during cytokinesis and thus loss of HPIP expression leads to defects in cytokinesis. A, confocal images representing the localization of indicated proteins at telophase and cytokinesis in HeLa cells. Green, HPIP; red, α-tubulin; blue, DNA (DAPI) (magnification, 60×). Scale bar, 20 μm. B, confocal images representing the localization of tubulin in siCtrl or siHPIP transfected HeLa cells. Upper panels, magnification, 20×. Lower panels, magnification, 60×). C, data represent percentages of cells having midbody. ***, p < 0.001 was considered significant.
Discussion
Our study demonstrates that HPIP is a critical regulator of G2/M transition by regulating temporal stability of cyclin B1 via inhibition of APC/C–Cdc20 activity. We show that HPIP and APC/C–Cdc20 antagonizes each other as HPIP inhibits APC/C–Cdc20, and in turn APC/C–Cdc20 degrades HPIP. Reciprocal regulation of HPIP and APC/C–Cdc20 represents a unique mechanism in control of mitotic entry and progression.
HPIP as a G2/M transition regulator
Although earlier studies demonstrated a role for HPIP in cell proliferation, yet the molecular mechanism that underlie in this function remain elusive (21, 22). Consistent with these reports, we provide the mechanistic evidence that HPIP promotes cell proliferation by enhancing G2/M transition. Time-lapse live cell imaging and cell cycle analysis revealed that HPIP expression is required for normal cell division. The delay in cell division is due to accumulation of cells at G2/M transition. Cyclin B1–Cdk1 complex is essential for G2/M transition because its diminished activity renders G2 phase arrest (26). Recent knockout studies reiterated that cyclin B1 knockout mouse embryos indeed arrest in G2 phase (37). Furthermore, accumulation of cyclin B1 is a prerequisite for timely mitotic entry because a loss of cyclin B1 expression delays it (9). Based on these previous reports, we argued that HPIP could regulate G2/M transition by controlling cyclin B1 levels. In concordance with these reports, loss-of-function and gain-of-function studies demonstrated that HPIP stabilizes cyclin B1. Further ubiquitination studies supported that HPIP stabilizes cyclin B1 by precluding APC/Cdc20-mediated ubiquitination and subsequent proteasomal degradation. Together, these findings point to the critical role of HPIP in G2/M transition function that partly occurs by controlling cyclin B1 stability.
HPIP and cyclin B1 are two crucial regulators involved in cell cycle regulation. Multiple lines of evidence indicate that HPIP and cyclin B1 are overexpressed in a variety of human tumors, and increased expression of HPIP and cyclin B1 has been correlated with malignant behavior of tumors (17, 18, 21, 38, 39). Because elevated levels of cyclin B1 were associated with increased ploidy (40), HPIP overexpression observed in several cancers might result in increased cyclin B1 levels and thus polyploidy. This could partly explain that HPIP overexpression may be a driving force for cellular transformation via stabilization of cyclin B1, which can potentially derail the cell division.
HPIP antagonizes APC/C–Cdc20 function during G2/M transition
To decipher the mechanism that underlies in HPIP-mediated cyclin B1 stability, we uncovered that HPIP antagonizes APC/C–Cdc20 activity. Earlier studies established that APC3 acts as a receptor for IR motif, and perhaps, Cdc20 utilizes this domain to interact with APC3 (29). HPIP contains one such conserved IR motif similar to Cdc20. Based on the following evidence, we argue that HPIP renders temporal and spatial stability to cyclin B1 during G2/M transition utilizing its IR domain, through which HPIP competitively inhibits the interaction between APC3 and Cdc20 and thus its activity. First, loss of IR domain abrogated HPIP interaction with APC3, whereas the D box 4 mutant could still bind APC3. Second, in the absence of HPIP, APC3 interaction with Cdc20 is potentiated (Fig. 7A). Third, in mtHPIP–IR cells, cyclin B1 accumulation occurred in a narrow window of time (hours 7–8) before mitosis in contrast to wtHPIP cells where cyclin B1 was stable from hours 7 to 9 (Fig. 7E). This could be due to the loss of inhibitory binding of HPIP, as a result of IR mutation, toward APC3, whereas Cdc20 is accessible to APC3. Co-IP data from Figs. 5 (A and B) and 7D together suggest that D4 and IR domains in HPIP mediate the interaction with Cdc20 and APC3, respectively. Considering the competitive interaction between HPIP and Cdc20 toward APC3 based on the co-IP data, we propose the existence of four possible protein complexes during G2/M transition including HPIP/Cdc20, APC/Cdc20, APC/HPIP, and APC/Cdc20/HPIP (Fig. 11A). Because either Cdc20 or Cdc20/mtHPIP–IR complex are freely accessible to bind APC3, mtHPIP–IR could be a prior substrate for APC/Cdc20 complex and thus degraded faster as observed in Fig. 7E. Because of this, cyclin B1 accumulation is confined to a narrow window of hours 7 and 8 in mtHPIP–IR cells. Whereas in D4 mutants where interaction between HPIP and Cdc20 is lost, as a consequence, APC3 could have preferred Cdc20-bound cyclin B1 over mtHPIP–IR and subsequently degraded it. Therefore, cyclin B1 accumulation is delayed (9–11 h) in mt-HPIP-D4 cells in contrast to wtHPIP where it occurred in the time window between hours 7 and 9. There are several known cellular inhibitors for APC/Cdc20, which include Mad3p, Acm1, RASSF1A, and Emi1 (41–44). Similar to Mes1 (45), RASSF1A (41), and SMURF2 (46), HPIP also acts as both an inhibitor and a substrate for APC/C complex. However, among these, HPIPs stand out as a different category of proteins that display antagonistic activity utilizing IR motif while serving as a substrate using D4 domain toward APC/Cdc20. Together, these results imply that D box and IR domains enable a sort of “facultative molecular bridge” between HPIP and APC/C–Cdc20 to facilitate a temporal and spatial stability to cyclin B1 and thus timely mitotic entry and cell cycle progression.
Figure 11.

Model represents the role of HPIP during cell cycle progression. A, schematic illustration showing the mechanistic role of HPIP in G2/M transition during mitosis. I, II, II, IV, and V are the possible protein complexes formed in a combinatorial fashion involving HPIP, APC, and Cdc20. HPIP promotes G2/M transition by stabilizing cyclin B1 during late G2 phase and itself is subjected to degradation during mitosis. Degradation of cyclin B1 and then Securin follows soon after the HPIP destruction by APC/Cdc20. D4 and IR motifs of HPIP function differently, because the D4 domain is primarily involved in HPIP stability, whereas the IR domain participates in inhibition of APC–Cdc20 activity. B, schematic illustration showing the role of HPIP in spindle checkpoint function and cytokinesis. In the presence of HPIP, the SAC is on, and cells proceed to cytokinesis with proper midbody/abscission and generate daughter cells with normal phenotype. In contrast, loss of HPIP results in SAC being off, defects in midbody formation, and thus abnormal cells.
HPIP is a substrate of APC/C–Cdc20 complex
This is the first report to show that HPIP is a substrate of APC/C–Cdc20 during mitosis. We show that HPIP protein levels oscillate during cell cycle stages and are subjected to proteasomal degradation during mitosis. The protein profile of HPIP during cell cycle stages was similar to that of cyclin A. cyclin A destruction occurs prior to mitotic entry (47, 48). Similar to cyclin A, Nek2A also undergoes APC/C–Cdc20-mediated ubiquitination and proteasomal degradation (29, 49). We found that APC/C–Cdc20, but not APC/C–Cdh1, mediates HPIP destruction during early mitosis. From the co-IP data (Fig. 5B), it infers that HPIP interact with Cdc20 during early/midmitosis (0 and 1 h), although the interaction is substantially decreased at a later time point (2 h). As reported earlier, Cdc20 undergoes proteasomal degradation by APC–Cdh1 during mitosis exit (50, 51). The reduced levels of Cdc20 at 2 h could be due to its proteasomal degradation by APC–Cdh1. That Cdc20 has also undergone post-translational modification during mitosis exit; therefore we cannot rule out its effect on interaction with HPIP. Further, we could also map the ubiquitination site as lysine at 274 in HPIP. Cdc20 interacts with APC/C substrates through various destruction motifs such as KEN or D box (52). Although HPIP contains four putative conserved D box motifs and one KEN domain, it utilizes D box 4 for interaction with Cdc20. D box 4 mutation rendered HPIP resistant to APC/Cdc20–mediated destruction during mitosis. Together, these findings point to the importance of Lys-274 and D box 4 in HPIP stability during mitosis. Substrate destruction by APC/C complex follows a temporal and spatial pattern that may depend on phosphorylation status of the substrates (53). Interestingly, HPIP appears to undergo phosphorylation during mitosis by an unknown kinase (54). Thus, we propose that ubiquitination of HPIP by APC/C–Cdc20 may be dependent on its post-translational modification such that phosphorylation might trigger HPIP destruction by APC/C–Cdc20. Further studies are warranted to investigate this aspect.
Role of HPIP in mitotic checkpoint function
This study also reports that HPIP is a spindle checkpoint regulator. At prophase, the spindle checkpoint proteins Bub1, BubR1, and Mad2 prevent premature separation of sister chromatids by specifically inhibiting APC/Cdc20 (55). Therefore, a lack of SAC function results in premature segregation of chromosomes occurs. We show that HPIP depletion causes chromosomal presegregation and breaks and accelerated metaphase to anaphase transition, a phenotype that was displayed upon silencing of SAC proteins (55) (Fig. 11B). We partly attribute this phenotype to the impaired MAD2 expression in the absence of HPIP. Although the precise mechanism that underlie in MAD2 expression by HPIP is unknown, it is possible that HPIP may regulate MAD2 transcription via Myc. It is based on the earlier observations that Myc is the transcriptional regulator of MAD2 gene (56), whereas HPIP is known to cause Myc up-regulation via the mTOR pathway (57). Loss of SAC function also results in early degradation of Securin, an inhibitor of Separase whose activation is required for Cohesin destruction and subsequently chromosome separation (52). In support of this, we also found that HPIP depletion results in early degradation of Securin and thus premature segregation chromosomes. Together, it implies that HPIP may indirectly regulate SAC by controlling MAD2 expression and proper segregation of chromosomes during mitosis.
Whereas microtubules are the principle components of the mitotic spindle and essential for accurate chromosome segregation during cell division, it is not surprising that a number of microtubule-associated proteins act predominantly during spindle assembly (58). Earlier we reported that HPIP is a microtubule-associated protein (30). In light of these earlier reports, we found that HPIP associates with mitotic spindle, and thus loss of HPIP results in the formation of multiple spindle poles. However, the role of HPIP in mitotic spindle assembly and function need to be further explored. Intriguingly, we also observed that HPIP not only localizes to mitotic spindle but also to midbody. Midbody is formed by central spindle microtubules during cytokinesis and is associated with cell fate and differentiation (59). In view of this, we also observed defects in midbody formation in HPIP-depleted cells. This partly explains the delay in mitotic exit upon loss of HPIP (Fig. 11B). However, the mechanistic role of HPIP in midbody formation and cytokinesis remains to be determined. In conclusion, our study provided compelling evidence to support that HPIP plays an essential role as a substrate as well as an inhibitor of APC/C–Cdc20 to maintain the temporal stability of cyclin B1 during G2/M transition to ensure timely mitotic entry and progression.
Experimental procedures
Molecular biology
pcDNA3.1-HPIP (T7–HPIP) was made as previously described (32). The D box mutants of HPIP, i.e. mtHPIP-D3, mtHPIP–D4, mtHPIP-D6, and mtHPIP-D7, in the T7–HPIP backbone were generated by replacing arginine and leucine with alanine at respective positions of the D boxes through site-directed mutagenesis using specific primers as listed in Table S1 (17). Similarly lysine mutations in HPIP at Lys-274 and Lys-634 were generated by replacing with arginine, and the clones generated are denoted as mtHPIP-K274R and mtHPIP-K634R, respectively. mtHPIP–IR mutant was generated by replacing isoleucine and arginine at positions 282 and 283 with alanine, respectively. wtHPIP, mtHPIP–D4, and mtHPIP–IR were PCR-amplified and subcloned into pEGFP-C1 vector. GFP–Cdc20 clone was generated by PCR using Cdc20-specific primers listed in Table S1. PCR-amplified Cdc20 was digested with BamHI and XhoI and cloned into pEGFP-C1 vector at the same sites. HA–Cdc20, HA–Cdh1 (Provided by Marc Krishner, Harvard Medical School through Addgene), GFP–cyclin B1 (Provided by Prof. Jonathon Pines through Addgene), H2B-mCherry (kind gift from Dr. Robert Benezra), and Clover-Geminin (1–110) (kind gift from Dr. Michael Lin) were purchased from Addgene. pCMV–Ub and H2B–GFP plasmids were kind gifts from Dr. Maddika Subbareddy (Center for DNA Fingerprinting Diagnosis, Hyderabad, India).
Cell culture, transfection, and treatment
The cells were maintained in standard conditions, and transfection was done using Lipofectamine 2000 (Invitrogen) based on manufacturer's protocols. Detailed cell culture techniques are described in supporting “Materials and methods.”
Lentiviral transduction was carried out as described previously (60). In brief, HEK293T cells were grown and co-transfected with shRNA plasmid (Cdc20 or HPIP) with packaging plasmids (pVsVg, pΔR, and pREV). Viral soups were harvested for every 24, 48, and 72 h by replacing with fresh growth medium to the cells. These viral soups were subsequently added to the HeLa cells by mixing with complete DMEM (1:1) containing protamine sulfate (5 μg/ml). 48 h post-transduction, the cells were treated continuously with puromycin (1 μg-10 μg/ml) for 7 days for stable selection. Knockdown of gene of interest was confirmed by Western blotting.
Cell proliferation assay
Cell proliferation assay was done by using WST-1 reagent according to the manufacturer's instructions, and details are described in the supporting “Materials and methods.”
Cell synchronization and cycle analysis
The cells were synchronized by double thymidine treatment procedure as described previously (61). The cells were then released into various time points to enter through various cell cycle phases and fixed the cells with 70% ethanol followed by RNase A (200 μg/ml) treatment to remove RNA. After fixing, the cells were stained with propidium iodide (5 μg/ml) (Sigma–Aldrich) and were processed by flow cytometry (FACS-Aria; Becton Dickinson). Subsequently, FCS Express software was used to analyze the data.
To determine the mitotic index, trypsinized cells were washed with PBS twice and fixed with 1% paraformaldehyde at 37 °C for 15 min. The cells were then permeabilized with 70% of methanol and stored at −20 °C overnight. After washing with PBS, the fixed cells were blocked with blocking buffer (0.5% BSA in PBS) for 10 min. The cells were then stained with rabbit anti-phosphohistone H3 (Ser-10) antibody at 1:50 dilution in blocking buffer for 1 h at room temperature followed by incubation with FITC-conjugated anti-rabbit secondary antibody at 1:100 dilutions for 1 h. The cells were then washed and incubated with 5 μg/ml propidium iodide and 250 μg/ml RNase A in PBS. Approximately 10,000 cells were analyzed by flow cytometry.
Thymidine–nocadazole treatment and release
For synchronization into the G2/M phase of cells cycle, HeLa cells (40% confluence) were treated with 2 mm thymidine for 24 h in cell culture incubator at 37 °C. Following three washes of cells with 1× PBS, the cells were incubated for 3 h with fresh DMEM, and subsequently, the cells were again treated with nocodazole (100 ng/ml) for 12 h. The cells were released in prewarmed fresh DMEM after washing three times with 1× PBS and collected at different time points for further analysis.
Live cell imaging for cell cycle progression
Synchronized cells with DT procedure were released into fresh Flurobrite DMEM (A1896701; Thermo Fisher Scientific), and live cell imaging was carried out using fluorescence microscope (Olympus IX83 inverted microscope with Andor Zyla 4.2 sCMOS camera, F-UBW for GFP, U-FGW for mCherry, Oko lab Uno live cell chamber, and Retiga 6000 monochrome detector; Singapore) connected with CO2 for ∼24 h. Synchronization of cells at the G2/M transition was carried out by arresting the cells first at G1/S transition using aphidicolin (9 μm) for 4 h followed treating with CDK1 inhibitor RO-3306 (35 nm) for 7 h or nocodazole (100 ng/ml) for 12 h. The cells were then released into fresh medium followed by the capturing of images with a regular intervals of 5 min, and the videos were generated using ImageJ software (Wayne Rasband).
Immunoprecipitation and Western blotting
The cell lysates were collected and subjected to immunoprecipitation and Western blotting as described previously (17), and the details are described in supporting “Materials and methods.”
RT-PCR
Real-time PCR analysis was done as described previously (62), and the details are described in supporting “Materials and methods.”
Ubiquitination assays
For in vivo ubiquitination of HPIP or cyclin B1, the cells were transfected with respective plasmid constructs using Lipofectamine 2000 and then treated with MG132 (10 μm) for 4 h before harvesting. 48 h post-transfection, the cell extracts were immunoprecipitated using protein-specific antibody or tag-specific antibody (GFP/HA/T7) followed by protein A/G bead incubation for 1 h at 4 °C. The protein A/G beads were collected by centrifugation and washed three times in Nonidet P-40 buffer. The samples were then analyzed by SDS-PAGE and Western blotting analysis using protein-specific antibodies (Table S2).
Protein stability and chasing experiments
Synchronized cells were treated with cycloheximide (25 μg/ml) followed by chasing for various time points, and cell extracts were subjected to Western blotting analysis using protein-specific antibodies (Table S2). The protein half-life of was determined as described previously (63, 64).
Confocal microscopy
The cells grown on coverslips were fixed with 4% paraformaldehyde and permeabilized by prechilled acetone and methanol (1:3). After blocking with 3% BSA followed by primary antibodies at 4 °C overnight, fluorescently labeled secondary antibodies were added and incubated at room temperature for 1 h. After thorough washing, coverslips were mounted on glass slide using DAPI (Thermo Scientific). Fluorescent images were captured by a confocal microscope (model NLO710; Carl Zeiss).
Metaphase spread preparation
Metaphase preparation of chromosomes was carried out as described previously (65). The cells were first arrested in metaphase by the addition of 1 μg/ml colcemid (Thermo Scientific) for 1 h. Then they were trypsinized and washed with PBS. After washing, the cells were treated with a hypotonic solution (0.56% KCl) at 37 °C for 6 min followed by the addition of three drops of freshly prepared ice-cold methanol/acetic acid (3:1) to stop the reaction. After brief centrifugation, the cells were resuspended with dropwise addition of methanol/acetic acid fixative followed by vortexing at a very low speed. This step was repeated again before storing the samples at −20 °C for further analysis. To get metaphase spreads, the cells were dropped onto a precleaned glass slide and dried overnight. Metaphases were then stained with DAPI and visualized under a microscope (Olympus, Singapore).
Statistical analysis
Statistical analysis was performed using GraphPad prism. All the data are presented as means ± S.D., with two or three independent experiments performed. Comparison between groups was performed using either Student's t test or analysis of variance. p < 0.05 was considered as significant.
Author contributions
S. S. K., V. S. K. S., V. P., A. K., and B. M. conceptualization; S. S. K., V. S. K. S., V. P., A. K., S. R. K., and B. M. data curation; S. S. K., V. S. K. S., V. P., and B. M. formal analysis; S. S. K., V. S. K. S., L. R. V., A. M., and B. M. validation; S. S. K., A. K., P.S.K., L. R. V., S. R. K., A. M., and B. M. methodology; S. S. K., V. P., and B. M. writing-original draft; S. S. K., V. P., and B. M. writing-review and editing; P.S.K., A. M., and B. M. resources; L. R. V., A. M., and B. M. visualization; B. M. supervision; B. M. funding acquisition; B. M. investigation; B. M. project administration.
Supplementary Material
Acknowledgments
We acknowledge University Grants Commission (UGC)-Departmental Research Support (DRS), Department of Science and Technology (DST)–India-Promotion of University Research and Scientific Excellence (PURSE) for providing research facilities at the University of Hyderabad. Experiments in Tata Institute of Fundamental Research, Hyderabad (TIFRH) were supported by intramural funds from Tata Institute of Fundamental Research (TIFR).
This work was supported by Department of Biotechnology, India Grants BT/PR7672/BRB/10/1173/2013, BT/Med/30/SP11273/2015, and BT/PR8764/MED/97/104/2013 and Department of Science and Technology, India Grant SB/SO/BB/013/2013 (to B. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supporting “Materials and methods,” Tables S1 and S2, Figs. S1 and S2, and Videos S1–S13.
- APC/C
- anaphase-promoting complex/cyclosome
- HPIP
- hematopoietic PBX-interacting protein
- CDK
- cyclin-dependent kinase
- DT
- double thymidine
- DMEM
- Dulbecco's modified Eagle's medium
- DAPI
- 4′,6′-diamino-2-phenylindole
- SAC
- spindle assembly checkpoint
- MCC
- mitotic checkpoint complex.
References
- 1. Gilberto S., and Peter M. (2017) Dynamic ubiquitin signaling in cell cycle regulation. J. Cell Biol. 216, 2259–2271 10.1083/jcb.201703170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pines J. (2011) Cubism and the cell cycle: the many faces of the APC/C. Nat. Rev. Mol. Cell Biol. 12, 427–438 10.1038/nrm3132 [DOI] [PubMed] [Google Scholar]
- 3. Primorac I., and Musacchio A. (2013) Panta rhei: the APC/C at steady state. J. Cell Biol. 201, 177–189 10.1083/jcb.201301130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Leuken R., Clijsters L., and Wolthuis R. (2008) To cell cycle, swing the APC/C. Biochim. Biophys. Acta 1786, 49–59 [DOI] [PubMed] [Google Scholar]
- 5. Sivakumar S., and Gorbsky G. J. (2015) Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat. Rev. Mol. Cell Biol. 16, 82–94 10.1038/nrm3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Musacchio A., and Hardwick K. G. (2002) The spindle checkpoint: structural insights into dynamic signalling. Nat. Rev. Mol. Cell Biol. 3, 731–741 10.1038/nrm929 [DOI] [PubMed] [Google Scholar]
- 7. Bharadwaj R., and Yu H. (2004) The spindle checkpoint, aneuploidy, and cancer. Oncogene 23, 2016–2027 10.1038/sj.onc.1207374 [DOI] [PubMed] [Google Scholar]
- 8. Malmanche N., Maia A., and Sunkel C. E. (2006) The spindle assembly checkpoint: preventing chromosome mis-segregation during mitosis and meiosis. FEBS Lett. 580, 2888–2895 10.1016/j.febslet.2006.03.081 [DOI] [PubMed] [Google Scholar]
- 9. Champion L., Linder M. I., and Kutay U. (2017) Cellular reorganization during mitotic entry. Trends Cell Biol. 27, 26–41 10.1016/j.tcb.2016.07.004 [DOI] [PubMed] [Google Scholar]
- 10. Gavet O., and Pines J. (2010) Activation of cyclin B1–Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J. Cell Biol. 189, 247–259 10.1083/jcb.200909144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matheson C. J., Backos D. S., and Reigan P. (2016) Targeting WEE1 kinase in cancer. Trends Pharmacol. Sci. 37, 872–881 10.1016/j.tips.2016.06.006 [DOI] [PubMed] [Google Scholar]
- 12. Fujimitsu K., Grimaldi M., and Yamano H. (2016) Cyclin-dependent kinase 1-dependent activation of APC/C ubiquitin ligase. Science 352, 1121–1124 10.1126/science.aad3925 [DOI] [PubMed] [Google Scholar]
- 13. Qiao R., Weissmann F., Yamaguchi M., Brown N. G., VanderLinden R., Imre R., Jarvis M. A., Brunner M. R., Davidson I. F., Litos G., Haselbach D., Mechtler K., Stark H., Schulman B. A., and Peters J. M. (2016) Mechanism of APC/CCDC20 activation by mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 113, E2570–E2578 10.1073/pnas.1604929113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fang G., Yu H., and Kirschner M. W. (1998) The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 12, 1871–1883 10.1101/gad.12.12.1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kallio M., Weinstein J., Daum J. R., Burke D. J., and Gorbsky G. J. (1998) Mammalian p55CDC mediates association of the spindle checkpoint protein Mad2 with the cyclosome/anaphase-promoting complex, and is involved in regulating anaphase onset and late mitotic events. J. Cell Biol. 141, 1393–1406 10.1083/jcb.141.6.1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hwang A., Maity A., McKenna W. G., and Muschel R. J. (1995) Cell cycle-dependent regulation of the cyclin B1 promoter. J. Biol. Chem. 270, 28419–28424 10.1074/jbc.270.47.28419 [DOI] [PubMed] [Google Scholar]
- 17. Bugide S., David D., Nair A., Kannan N., Samanthapudi V. S., Prabhakar J., and Manavathi B. (2015) Hematopoietic PBX-interacting protein (HPIP) is over expressed in breast infiltrative ductal carcinoma and regulates cell adhesion and migration through modulation of focal adhesion dynamics. Oncogene 34, 4601–4612 10.1038/onc.2014.389 [DOI] [PubMed] [Google Scholar]
- 18. Xu X., Jiang C., Wang S., Tai Y., Wang T., Kang L., Fan Z., Li S., Li L., Fu J., Liu J., Ji Q., Wang X., Wei L., and Ye Q. (2013) HPIP is upregulated in liver cancer and promotes hepatoma cell proliferation via activation of G2/M transition. IUBMB Life 65, 873–882 [DOI] [PubMed] [Google Scholar]
- 19. Wang D., Wang L., Zhou Y., Zhao X., and Xiong H. (2016) The involvement of hematopoietic pre-B cell leukemia transcription factor-interacting protein in regulating epithelial-mesenchymal transition of human spinal glioblastoma. Tumour Biol. 37, 5897–5903 10.1007/s13277-015-4453-4 [DOI] [PubMed] [Google Scholar]
- 20. Bugide S., Gonugunta V. K., Penugurti V., Malisetty V. L., Vadlamudi R. K., and Manavathi B. (2017) HPIP promotes epithelial-mesenchymal transition and cisplatin resistance in ovarian cancer cells through PI3K/AKT pathway activation. Cell. Oncol. (Dordr.) 40, 133–144 10.1007/s13402-016-0308-2 [DOI] [PubMed] [Google Scholar]
- 21. Chen B., Zhao J., Zhang S., Zhang Y., and Huang Z. (2016) HPIP promotes gastric cancer cell proliferation through activation of cap-dependent translation. Oncol. Rep. 36, 3664–3672 10.3892/or.2016.5157 [DOI] [PubMed] [Google Scholar]
- 22. Wang X., Yang Z., Zhang H., Ding L., Li X., Zhu C., Zheng Y., and Ye Q. (2008) The estrogen receptor-interacting protein HPIP increases estrogen-responsive gene expression through activation of MAPK and AKT. Biochim. Biophys. Acta 1783, 1220–1228 10.1016/j.bbamcr.2008.01.026 [DOI] [PubMed] [Google Scholar]
- 23. da Fonseca P. C., Kong E. H., Zhang Z., Schreiber A., Williams M. A., Morris E. P., and Barford D. (2011) Structures of APC/C(Cdh1) with substrates identify Cdh1 and Apc10 as the D-box co-receptor. Nature 470, 274–278 10.1038/nature09625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tian W., Li B., Warrington R., Tomchick D. R., Yu H., and Luo X. (2012) Structural analysis of human Cdc20 supports multisite degron recognition by APC/C. Proc. Natl. Acad. Sci. U.S.A. 109, 18419–18424 10.1073/pnas.1213438109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim W., Bennett E. J., Huttlin E. L., Guo A., Li J., Possemato A., Sowa M. E., Rad R., Rush J., Comb M. J., Harper J. W., and Gygi S. P. (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 10.1016/j.molcel.2011.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Choi H. J., Fukui M., and Zhu B. T. (2011) Role of cyclin B1/Cdc2 up-regulation in the development of mitotic prometaphase arrest in human breast cancer cells treated with nocodazole. PLoS One 6, e24312 10.1371/journal.pone.0024312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Song M. S., Song S. J., Ayad N. G., Chang J. S., Lee J. H., Hong H. K., Lee H., Choi N., Kim J., Kim H., Kim J. W., Choi E. J., Kirschner M. W., and Lim D. S. (2004) The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC–Cdc20 complex. Nat. Cell Biol. 6, 129–137 10.1038/ncb1091 [DOI] [PubMed] [Google Scholar]
- 28. Hein J. B., and Nilsson J. (2014) Stable MCC binding to the APC/C is required for a functional spindle assembly checkpoint. EMBO Reports 15, 264–272 10.1002/embr.201337496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sedgwick G. G., Hayward D. G., Di Fiore B., Pardo M., Yu L., Pines J., and Nilsson J. (2013) Mechanisms controlling the temporal degradation of Nek2A and Kif18A by the APC/C–Cdc20 complex. EMBO J. 32, 303–314 10.1038/emboj.2012.335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang S., Chang L., Alfieri C., Zhang Z., Yang J., Maslen S., Skehel M., and Barford D. (2016) Molecular mechanism of APC/C activation by mitotic phosphorylation. Nature 533, 260–264 10.1038/nature17973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bertalan Z., Budrikis Z., La Porta C. A., and Zapperi S. (2015) Role of the number of microtubules in chromosome segregation during cell division. PLoS One 10, e0141305 10.1371/journal.pone.0141305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Manavathi B., Acconcia F., Rayala S. K., and Kumar R. (2006) An inherent role of microtubule network in the action of nuclear receptor. Proc. Natl. Acad. Sci. U.S.A. 103, 15981–15986 10.1073/pnas.0607445103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abramovich C., Chavez E. A., Lansdorp P. M., and Humphries R. K. (2002) Functional characterization of multiple domains involved in the subcellular localization of the hematopoietic Pbx interacting protein (HPIP). Oncogene 21, 6766–6771 10.1038/sj.onc.1205784 [DOI] [PubMed] [Google Scholar]
- 34. Bajar B. T., Lam A. J., Badiee R. K., Oh Y. H., Chu J., Zhou X. X., Kim N., Kim B. B., Chung M., Yablonovitch A. L., Cruz B. F., Kulalert K., Tao J. J., Meyer T., Su X. D., et al. (2016) Fluorescent indicators for simultaneous reporting of all four cell cycle phases. Nat. Methods 13, 993–996 10.1038/nmeth.4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wassmann K., and Benezra R. (2001) Mitotic checkpoints: from yeast to cancer. Curr. Opin. Genet. Dev. 11, 83–90 10.1016/S0959-437X(00)00161-1 [DOI] [PubMed] [Google Scholar]
- 36. Howell B. J., McEwen B. F., Canman J. C., Hoffman D. B., Farrar E. M., Rieder C. L., and Salmon E. D. (2001) Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J. Cell Biol. 155, 1159–1172 10.1083/jcb.200105093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strauss B., Harrison A., Coelho P. A., Yata K., Zernicka-Goetz M., and Pines J. (2018) Cyclin B1 is essential for mitosis in mouse embryos, and its nuclear export sets the time for mitosis. J. Cell Biol. 217, 179–193 10.1083/jcb.201612147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pan J., Qin Y., and Zhang M. (2016) HPIP promotes non-small cell lung cancer cell proliferation, migration and invasion through regulation of the Sonic hedgehog signaling pathway. Biomed. Pharmacother. 77, 176–181 10.1016/j.biopha.2015.12.012 [DOI] [PubMed] [Google Scholar]
- 39. Song Y., Zhao C., Dong L., Fu M., Xue L., Huang Z., Tong T., Zhou Z., Chen A., Yang Z., Lu N., and Zhan Q. (2008) Overexpression of cyclin B1 in human esophageal squamous cell carcinoma cells induces tumor cell invasive growth and metastasis. Carcinogenesis 29, 307–315 10.1093/carcin/bgm269 [DOI] [PubMed] [Google Scholar]
- 40. Roh M., Song C., Kim J., and Abdulkadir S. A. (2005) Chromosomal instability induced by Pim-1 is passage-dependent and associated with dysregulation of cyclin B1. J. Biol. Chem. 280, 40568–40577 10.1074/jbc.M509369200 [DOI] [PubMed] [Google Scholar]
- 41. Chow C., Wong N., Pagano M., Lun S. W., Nakayama K. I., Nakayama K., and Lo K. W. (2012) Regulation of APC/CCdc20 activity by RASSF1A-APC/CCdc20 circuitry. Oncogene 31, 1975–1987 10.1038/onc.2011.372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Burton J. L., and Solomon M. J. (2007) Mad3p, a pseudosubstrate inhibitor of APCCdc20 in the spindle assembly checkpoint. Genes Dev. 21, 655–667 10.1101/gad.1511107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Enquist-Newman M., Sullivan M., and Morgan D. O. (2008) Modulation of the mitotic regulatory network by APC-dependent destruction of the Cdh1 inhibitor Acm1. Mol. Cell 30, 437–446 10.1016/j.molcel.2008.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fry A. M., and Yamano H. (2006) APC/C-mediated degradation in early mitosis: how to avoid spindle assembly checkpoint inhibition. Cell Cycle 5, 1487–1491 10.4161/cc.5.14.3003 [DOI] [PubMed] [Google Scholar]
- 45. Kimata Y., Trickey M., Izawa D., Gannon J., Yamamoto M., and Yamano H. (2008) A mutual inhibition between APC/C and its substrate Mes1 required for meiotic progression in fission yeast. Dev. Cell 14, 446–454 10.1016/j.devcel.2007.12.010 [DOI] [PubMed] [Google Scholar]
- 46. Osmundson E. C., Ray D., Moore F. E., Gao Q., Thomsen G. H., and Kiyokawa H. (2008) The HECT E3 ligase Smurf2 is required for Mad2-dependent spindle assembly checkpoint. J. Cell Biol. 183, 267–277 10.1083/jcb.200801049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. den Elzen N., and Pines J. (2001) Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J. Cell Biol. 153, 121–136 10.1083/jcb.153.1.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kabeche L., and Compton D. A. (2013) Cyclin A regulates kinetochore microtubules to promote faithful chromosome segregation. Nature 502, 110–113 10.1038/nature12507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boekhout M., and Wolthuis R. (2015) Nek2A destruction marks APC/C activation at the prophase-to-prometaphase transition by spindle-checkpoint-restricted Cdc20. J. Cell Sci. 128, 1639–1653 10.1242/jcs.163279 [DOI] [PubMed] [Google Scholar]
- 50. Pfleger C. M., and Kirschner M. W. (2000) The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 14, 655–665 [PMC free article] [PubMed] [Google Scholar]
- 51. Robbins J. A., and Cross F. R. (2010) Regulated degradation of the APC coactivator Cdc20. Cell Div. 5, 23 10.1186/1747-1028-5-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Peters J. M. (2006) The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol. 7, 644–656 10.1038/nrm1988 [DOI] [PubMed] [Google Scholar]
- 53. Barford D. (2011) Structural insights into anaphase-promoting complex function and mechanism. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 3605–3624 10.1098/rstb.2011.0069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., and Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3 [DOI] [PubMed] [Google Scholar]
- 55. Lischetti T., and Nilsson J. (2015) Regulation of mitotic progression by the spindle assembly checkpoint. Mol. Cell. Oncol. 2, e970484 10.4161/23723548.2014.970484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Menssen A., Epanchintsev A., Lodygin D., Rezaei N., Jung P., Verdoodt B., Diebold J., and Hermeking H. (2007) c-MYC delays prometaphase by direct transactivation of MAD2 and BubR1: identification of mechanisms underlying c-MYC-induced DNA damage and chromosomal instability. Cell Cycle 6, 339–352 10.4161/cc.6.3.3808 [DOI] [PubMed] [Google Scholar]
- 57. Reid R. O., Deb P., Howell B. L., and Shrank W. H. (2013) Association between Medicare Advantage plan star ratings and enrollment. JAMA 309, 267–274 10.1001/jama.2012.173925 [DOI] [PubMed] [Google Scholar]
- 58. Foley E. A., and Kapoor T. M. (2013) Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat. Rev. Mol. Cell Biol. 14, 25–37 10.1038/nrm3494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dionne L. K., Wang X. J., and Prekeris R. (2015) Midbody: from cellular junk to regulator of cell polarity and cell fate. Curr. Opin. Cell Biol. 35, 51–58 10.1016/j.ceb.2015.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Manavathi B., Lo D., Bugide S., Dey O., Imren S., Weiss M. J., and Humphries R. K. (2012) Functional regulation of pre–B-cell leukemia homeobox interacting protein 1 (PBXIP1/HPIP) in erythroid differentiation. J. Biol. Chem. 287, 5600–5614 10.1074/jbc.M111.289843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang X., Arceci A., Bird K., Mills C. A., Choudhury R., Kernan J. L., Zhou C., Bae-Jump V., Bowers A., and Emanuele M. J. (2017) VprBP/DCAF1 regulates the degradation and nonproteolytic activation of the cell cycle transcription factor FoxM1. Mol. Cell. Biol. 37, e00609–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gajulapalli V. N., Samanthapudi V. S., Pulaganti M., Khumukcham S. S., Malisetty V. L., Guruprasad L., Chitta S. K., and Manavathi B. (2016) A transcriptional repressive role for epithelial-specific ETS factor ELF3 on oestrogen receptor alpha in breast cancer cells. Biochem. J. 473, 1047–1061 10.1042/BCJ20160019 [DOI] [PubMed] [Google Scholar]
- 63. Gilkerson J., Tam R., Zhang A., Dreher K., and Callis J. (2016) Cycloheximide assays to measure protein degradation in vivo in plants. BioProtocol. 6, e1919 [Google Scholar]
- 64. Gilkerson J., Kelley D. R., Tam R., Estelle M., and Callis (2015) Lysine residues are not required for proteasome-mediated proteolysis of the auxin/indole acidic acid protein IAA1. Plant Physiol. 168, 708–720 10.1104/pp.15.00402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bhat A., Wu Z., Maher V. M., McCormick J. J., and Xiao W. (2015) Rev7/Mad2B plays a critical role in the assembly of a functional mitotic spindle. Cell Cycle 14, 3929–3938 10.1080/15384101.2015.1120922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Reece J. B., Urry L. A., Cain M. L., Wasserman S. A., Minorsky P. V., and Jackson R. B. (2011) The Cell Cycle, 9th Ed., Campbell Biology [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.