

Summary
Whole-genome doubling (WGD) is common early in tumorigenesis. WGD doubles ploidy and centrosome number. In the ensuing mitoses, excess centrosomes form a multipolar spindle, resulting in a lethal multipolar cell division. To survive, cells must cluster centrosomes to allow bipolar cell division. Cancer cells are often more proficient at centrosome clustering than untransformed cells, but the mechanism behind increased clustering ability is not well understood. Heterozygous missense mutations in PPP2R1A, which encodes the alpha isoform of the “scaffolding” subunit of PP2A (PP2A-Aα), positively correlate with WGD. We introduced a heterozygous hotspot mutation, P179R, into PPP2R1A in human RPE-1 cells. PP2A-AαP179R decreases PP2A assembly and intracellular targeting in mitosis. Strikingly, PP2A-AαP179R enhances centrosome clustering when centrosome number is increased either by cytokinesis failure or centrosome amplification, likely through PP2A-Aα loss of function. Thus cancer-associated mutations in PP2A-Aα may increase cellular fitness after WGD by enhancing centrosome clustering.
Subject Areas: Biological Sciences, Cell Biology, Cancer
Graphical Abstract
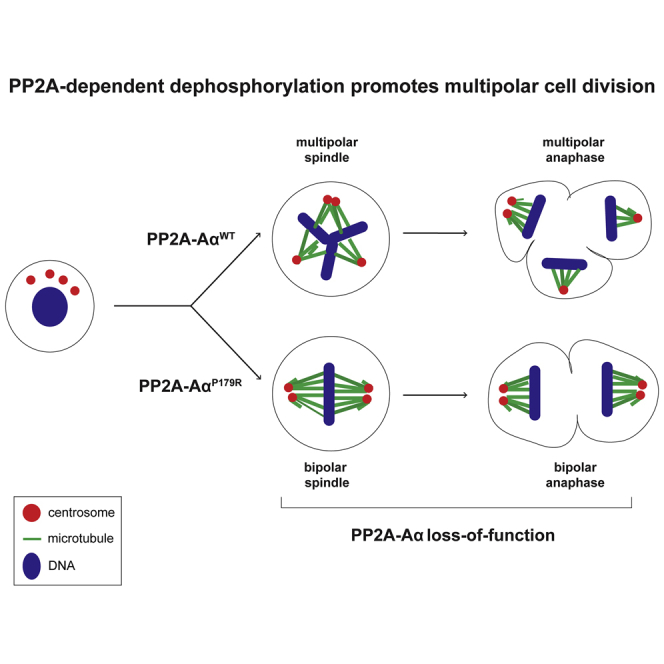
Highlights
-
•
A heterozygous P179R mutation in PP2A-Aα decreases PP2A phosphatase assembly
-
•
Intracellular targeting of PP2AB56α in mitosis is reduced in PP2A-AαP179R/+ cells
-
•
PP2A-AαP179R/+ cells are more proficient at centrosome clustering
-
•
Increased centrosome clustering is likely due to PP2A-Aα loss of function
Biological Sciences; Cell Biology; Cancer
Introduction
Most human tumors are aneuploid. In about one-third of tumors, aneuploidy is preceded by a whole-genome doubling (WGD) (Zack et al., 2013), an event that correlates with poor prognosis for patients (Bielski et al., 2018). After WGD, the newly tetraploid genome promotes the evolution of aneuploid karyotypes (Dewhurst et al., 2014), with tumor cells losing chromosomes and acquiring chromosomal rearrangements (Carter et al., 2012, Zack et al., 2013). However, WGD can be detrimental to subsequent mitoses because centrosome number doubles (from two to four) concomitantly with genome doubling. During mitosis, each centrosome nucleates microtubules (Ring et al., 1982), to form a multipolar spindle, which if uncorrected, can result in a lethal multipolar cell division (Ganem et al., 2009). Alternatively, cells can cluster supernumerary centrosomes into two spindle poles, to allow the formation of a pseudo-bipolar spindle (Quintyne et al., 2005), which permits bipolar cell division and survival. Given that centrosome amplification is common in cancers (Chan, 2011) and lethal multipolar cell divisions are expected to decrease the fitness of a tumor cell population, cancer cells likely experience selective pressures to efficiently cluster supernumerary centrosomes.
Human cells have an intrinsic ability to cluster centrosomes. This depends on proteins that directly or indirectly contribute to force generation during mitosis. Within the spindle, centrosome clustering arises from motor proteins located near spindle poles and centrosomes, such as dynein and KIFC1/HSET, as well as proteins localized at the kinetochore or centromere that control microtubule binding and spindle assembly checkpoint signaling (Drosopoulos et al., 2014, Kwon et al., 2008, Leber et al., 2010, Quintyne et al., 2005). Outside of the spindle, at the cell cortex, motor proteins associated with the cortical actin network, such as Myo10 and dynein, position centrosomes by generating force on astral microtubules (Kwon et al., 2015, Quintyne et al., 2005). Compared with non-transformed cells, centrosome clustering is more efficient in cancer cells (Ganem et al., 2009). Although centrosome clustering efficiency can be increased by depletion of the microtubule-binding protein NUMA (Quintyne et al., 2005) or the adhesion protein E-cadherin (Rhys et al., 2018), it remains largely unclear how cancer cells evolve to cluster centrosomes more proficiently.
Genomic analyses have identified genetic alterations that positively correlate with WGD (Bielski et al., 2018, Zack et al., 2013). Several changes (TP53 and RB1 mutation, CCNE1 amplification) enable cell proliferation after WGD by alleviating the G1 arrest triggered after cytokinesis failure (Andreassen et al., 2001). The molecular impact of other genetic changes that positively correlate with WGD, including alterations in protein phosphatase 2A (PP2A), is unknown. PP2A is a major source of serine/threonine phosphatase activity in eukaryotic cells. In the PP2A heterotrimer, a catalytic subunit (PP2A-Cα/β) and a “scaffolding” subunit (PP2A-Aα/β) are targeted to substrates by four evolutionarily conserved families of regulatory subunits. PP2A inactivation has been previously linked to tumorigenesis with the discovery that the SV40 small t antigen blocks the binding of PP2A-Aα/β to regulatory subunits (Pallas et al., 1990), leading to cellular transformation (Chen et al., 2004). Potentially similar perturbations in PP2A have been found to positively correlate with WGD in tumors. These include homozygous deletion of PPP2R2A, which encodes the B55α regulatory subunit, and heterozygous missense mutations in PPP2R1A, which encodes the α isoform of the scaffolding subunit PP2A-A, that accounts for ∼90% of total PP2A-A (Zhou et al., 2003). Hotspot mutations in PP2A-Aα are expected to prevent B55 and B56 regulatory subunit binding (Cho and Xu, 2007, Xu et al., 2006, Xu et al., 2008) and thus likely decrease the functionality of PP2A-Aα. Indeed, functional inactivation of PP2A by a recurrent P179R mutation in PPP2R1A has been recently implicated as a driver of tumorigenesis in high-grade endometrial carcinoma (Taylor et al., 2019). In other studies, over-expression of certain hotspot PP2A-Aα mutants in tissue culture cells has been observed to alter phospho-signaling (Haesen et al., 2016, Jeong et al., 2016). However, the impact of PP2A-Aα missense mutations with respect to WGD has not been examined.
Here we examine the impact of two prevalent hotspot mutations in PPP2R1A, P179R and R183W, on PP2A holoenzyme assembly and find that a reduction in protein-protein interactions predominates and is shared between the two mutants. We then focus on the P179R mutation in PPP2R1A, as this mutation is linked to tumorigenesis (Taylor et al., 2019). When introduced into one allele of endogenous PPP2R1A, the P179R mutation reduces PP2A holoenzyme assembly and intracellular targeting of PP2AB56 in mitosis. Strikingly, we find that these changes are sufficient to increase centrosome clustering after WGD, possibly in part through factors at the cell cortex that position centrosomes. Moreover, overexpression of wild-type PP2A-Aα partially rescues centrosome clustering, suggesting that this phenotype arises from a decrease in PP2A activity in mitosis.
Results
Characterization of Prevalent Cancer-Associated PP2A-Aα Missense Mutations
PPP2R1A is most frequently mutated in uterine cancers (Figure 1A), and to explore the cellular impact of the two most frequent PPP2R1A missense mutations (Figure 1B), we generated retinal pigment epithelial (RPE-1) hTERT cell lines expressing GFP-tagged PP2A-Aα wild-type (WT), P179R, or R183W. Each construct was expressed at 30%–40% of the level of endogenous PP2A-Aα/β (Figure 1C). Using quantitative mass spectrometry, we compared the composition of PP2A complexes isolated from stable isotope labeling by amino acids in cell culture (SILAC)-labeled cells by immunoprecipitation of WT or mutant GFP-PP2A-Aα. The P179R mutation significantly reduced PP2A-Aα binding to four B56 regulatory subunits (B56α/PPP2R5A, B56β/PPP2R5B, B56δ/PPP2R5D, and B56ε/PPP2R5E). Accordingly, the binding of proteins that associate with PP2A-Aα via B56 subunits including GEF-H1 (ARHGEF2), Liprinα1 (PPFIA1) (Hertz et al., 2016), and MTCL1 (Hyodo et al., 2016) was similarly reduced (Figure 1D). The P179R mutation also significantly reduced binding to the B55δ/PPP2R2D regulatory subunit (Figure 1D). The binding of STRN regulatory subunits (STRN, STRN3, and STRN4), a B″ regulatory subunit (PPP2R3A), and PP2A-C (PPP2CA, PPP2CB) was unaffected (Figure 1D). The R183W mutant had an overall similar decrease in protein-protein interactions (Figure 1E). Notably, although several proteins were observed to have increased binding to a PP2A-Aα mutant, none were shared between the two mutants (Figure 1F). Thus the major impact of both P179R and R183W mutations is to reduce protein-protein interactions.
Figure 1.

Quantitative Proteomic Characterization of Cancer-Associated PP2A-Aα Mutations
(A and B) Incidence of (A) PPP2R1A alterations and (B) missense mutations (Cerami et al., 2012, Gao et al., 2013).
(C) Western blot analysis of cells expressing GFP-PP2Aα-WT or indicated mutants. Solid line indicates intervening lanes have been removed.
(D and E) GFP immunoprecipitates from isotopically labeled RPE-1 cells expressing GFP-PP2A-Aα (WT, P179R, or R183W) were analyzed by mass spectrometry. Volcano plots with the mean log2 fold-change of proteins bound to mutant versus GFP-PP2Aα-WT against –log10 p value. 2-fold change (vertical dashed lines); p < 0.05 (horizontal dashed lines); red and blue circles indicate PP2A regulatory and catalytic subunits respectively.
(F) Heatmap of proteins with significant changes in association. Green to red gradient represents the mean log2 fold-change. X, protein not detected.
A Heterozygous P179R Mutation in PP2A-Aα Impacts PP2A Holoenzyme Assembly in Human Cells
To examine if a heterozygous PP2A-Aα missense mutation is sufficient to impact PP2A functionality, we introduced a P179R mutation into one allele of endogenous PPP2R1A in RPE-1 cells. The P179R mutation was selected because it is the most prevalent missense mutation in uterine tumors, which have the highest incidence of PP2A-Aα alterations (Cerami et al., 2012). We used adeno-associated virus-mediated gene targeting (Berdougo et al., 2009) to introduce a C to G mutation in exon five of PPP2R1A (Figure 2A) and isolated two independent heterozygous clones (Figure 2B). The mutation did not alter the levels of PP2A-Aα or PP2A-Aα/β (Figure 2C). Similarly, PP2A-Aα immunoprecipitates from WT and PP2A-AαP179R/+ cells had equivalent levels of PP2A-C (Figure S1A) and phosphatase activity (Figure S1B). By contrast, we observed near-2-fold reductions in PP2A-Aα association with B56γ, δ, and ε (Figures 2D, 2E, and S1C) and B55α (Figures S1D and S1E). Consistent with a decrease in PP2AB56 holoenzyme levels, intracellular targeting of both PP2A-Aα and B56α to the centromere or kinetochore was reduced in PP2A-AαP179R/+ cells (Figures 2F and 2G). Collectively, these results indicate that a heterozygous P179R mutation in PP2A-Aα is sufficient to alter the level of a subset of PP2A holoenzymes.
Figure 2.

A Heterozygous P179R Mutation in PP2A-Aα Decreases PP2AB56 Assembly and Targeting
(A) Schematic of PPP2R1A gene-targeting strategy. Exons, rectangles; loxP sites, triangles; ITR, AAV-specific inverted tandem repeats; homologous sequences, green and red dashed lines.
(B) Sanger sequencing the modified region of PPP2R1A in a mutant clone.
(C) Western blot analysis of lysates from WT (+/+) and independently derived PP2A-AαP179R/+ (P179R/+) clones. * Shows non-specific band.
(D and E) (D) Western blot analysis of lysates (lanes 1–2) and immunoprecipitations (IPs) of control IgG (lane 3) or PP2A-Aα IgG (lane 4–5) in indicated cell lines. (E) Plotted is the normalized mean ± SEM of the experiment in (D) performed three times.
(F–G) WT (+/+) and PP2A-AαP179R/+ (P179R/+) cells in nocodazole were fixed and processed for immunofluorescence. (F) Maximum intensity projection. Scale bar, 5 μm. (G) Normalized centromere or kinetochore signal is plotted. Circle, cell; line, mean.
Result is representative of three experiments. ∗∗∗∗p < 0.00005, ∗∗∗p < 0.0005; ns, not significant (p > 0.05), calculated from Student's t test.
See also Figure S1.
PP2A-AαP179R/+ Cells Suppress Multipolar Cell Division after Cytokinesis Failure
PPP2R1A mutations are enriched in tumors that experience WGD (Zack et al., 2013). Given that the levels of PP2AB55 and PP2AB56 holoenzymes were reduced in PP2A-AαP179R/+ cells, and that each of these holoenzyme families contribute to cell division (Craney et al., 2016, Espert et al., 2014, Foley et al., 2011, Kitajima et al., 2006, Lee et al., 2017, Nijenhuis et al., 2014, Schmitz et al., 2010), we considered the possibility that the P179R mutation in PPP2R1A might impact cells in mitosis following WGD. To test this, we induced one round of cytokinesis failure (Yang et al., 2008) by treating cells with cytochalasin D or B, inhibitors of actin polymerization (Cooper, 1987), or with blebbistatin, a myosin II inhibitor (Straight et al., 2003). Binucleate cells were imaged live during the ensuing mitosis (Figure 3A). To avoid a G1 cell-cycle arrest triggered by cytokinesis failure (Andreassen et al., 2001), we treated Tp53+/+ cells with p38 inhibitor SB 203580 (Cuenda et al., 1995) or utilized Tp53−/− cells. Almost 30% of WT cells treated with cytochalasin D underwent multipolar cell divisions. Strikingly, PP2A-AαP179R/+ cells exhibited 4-fold reduction in the frequency of multipolar cell divisions compared with WT cells (7% in clone a and 8% in clone b) (Figures 3B and 3C). PP2A-AαP179R/+ cells also underwent fewer multipolar cell divisions when cytokinesis failure was induced with cytochalasin B or blebbistatin (Figure 3C). Similar results were observed in Tp53−/− PP2A-AαP179R/+ cells (Figure 3D). Notably, whereas the expression of PP2A-Aα -P179R in WT cells had no impact on the outcome of cell division, the expression of GFP-PP2A-Aα-WT in PP2A-AαP179R/+ cells partially rescued the cell division phenotype (Figures 3E and 3F), suggesting that the observed phenotype is likely due to haploinsufficiency.
Figure 3.

The P179R Mutation in PP2A-Aα Suppresses Multipolar Cell Division after Cytokinesis Failure
(A) Schematic of cytokinesis failure assay.
(B) GFP-H2B (top) and differential interference contrast (bottom) time-lapse images of WT (+/+) and PP2A-AαP179R/+ (P179R/+) cells after cytochalasin D treatment. Time (minutes) relative to nuclear envelope breakdown is indicated. Scale bar, 5 μm.
(C) Quantification of cell division outcome of Tp53+/+ cells.
(D) Quantification of multipolar cell divisions in Tp53−/− cells treated as in (A).
(E and F) Cell lines of the indicated genotype expressing GFP-PP2A-Aα-WT or GFP-PP2A-Aα-P179R were (E) analyzed by western blot, where * shows non-specific band and ** shows GFP-PP2A-Aα truncation product, and (F) treated with cytochalasin D as in (A), imaged, and cell division outcome quantified.
(G) Mitotic duration of Tp53+/+ cells in (C) undergoing bipolar cell divisions is plotted. Bars, median; circles, cells.
Result is representative of three experiments. (C, D, and F) Mean ± SEM from three experiments is plotted. ***p < 0.0005, **p < 0.005, *p < 0.05; ns, not significant (p > 0.05) Student's t test.
See also Figure S2.
Given that PP2A-AαP179R/+ cells have a modest delay in mitosis (22 min in clone a and 21 min in clone b versus 18 min in WT cells) (Figure S2A) and increased time spent in mitosis suppresses multipolar mitoses (Kwon et al., 2008), we next compared the mitotic duration of binucleate cells that underwent a bipolar cell division. Both WT and PP2A-AαP179R/+ cells completed mitosis with indistinguishable kinetics (Figure 3G). We also examined if the cell division phenotype was caused by centrosome inactivation as observed in Drosophila (Basto et al., 2008). However, microtubule nucleation was observed at all centrosomes in WT and PP2A-AαP179R/+ cells (Figure S2B). Moreover, in PP2A-AαP179R/+ cells, centrosomes were always associated with spindle poles (Figure S2C) and were more likely to be clustered (Figure S2D). Our data suggest that enhanced bipolar cell division after WGD in PP2A-AαP179R/+ cells is likely due to more efficient centrosome clustering and not due to centrosome inactivation or lengthening of mitosis.
PP2A-AαP179R/+ Cells Cluster Supernumerary Centrosomes More Efficiently
To test the hypothesis that PP2A-AαP179R/+ cells may cluster supernumerary centrosomes more efficiently than WT cells, centrosome amplification was induced by over-expression of Plk4 kinase (Kleylein-Sohn et al., 2007, Peel et al., 2007) (Figure 4A). This increases centrosome number without altering ploidy. We introduced a doxycycline-inducible Plk4 into Tp53−/− WT and PP2A-AαP179R/+ cells. After doxycycline induction, immunofluorescence analyses of C-Nap1, a marker of functional centrioles (Wang et al., 2011), indicated that 64% of WT cells and 68% of PP2A-AαP179R/+ cells had centrosome amplification (Figures S2E and S2F). Immunofluorescence analysis of anaphase cells with extra centrosomes revealed that PP2A-AαP179R/+ cells clustered centrosomes into a bipolar spindle more efficiently than WT cells, with a 6-fold reduction in multipolar anaphases (Figures 4B and 4C). A similar result was observed by live imaging (Figures 4D and 4E), with no delay in mitosis (Figure 4F).
Figure 4.

PP2A-AαP179R/+ Cells Exhibit More Robust Centrosome Clustering
(A) Schematic of centrosome amplification induction by doxycycline (dox)-inducible Plk4 overexpression.
(B and C) Plk4-inducible cells were treated with dox and fixed and analyzed by immunofluorescence. (B) Representative bipolar (top) and multipolar (bottom) anaphases. (C) Multipolar anaphase incidence in cells with >4 centrin-1 foci is plotted.
(D–F) Plk4-inducible cells were treated with dox and imaged live (D) GFP-H2B (top) and differential interference contrast (bottom) montage. Time (minutes) relative to nuclear envelope breakdown is indicated. (E) Incidence of multipolar cell division is plotted. (F) A box-and-whisker plot of mitotic duration in dox-treated cells undergoing bipolar cell divisions. Whiskers indicate the 5th–95th percentile range, and circles indicate cells outside of this range. Result is representative of three experiments.
(G and H) Plk4-inducible cells were treated with dox and imaged on O- and Y-shaped fibronectin micro-patterns. (G) Representative images of interphase cells. (H) Incidence of multipolar anaphases. n, total cells analyzed.
(C and E) Mean ± SEM from three experiments. Scale bars, 5 μm. ***p < 0.0005, **p < 0.005; ns, not significant (p > 0.05) Student's t test.
See also Figure S2.
Next, to examine how PP2A-AαP179R/+ cells cluster centrosomes more efficiently, we asked if microtubule organization in mitosis is more robust in PP2A-AαP179R/+ cells. Although centrosomes are the major sites of microtubule nucleation, a bipolar spindle can assemble in the absence of one or both centrosomes, albeit with some delay (Khodjakov et al., 2000). We generated acentrosomal cells using centrinone, a small-molecule inhibitor of Plk4 (Wong et al., 2015) and measured the time between nuclear envelope breakdown and anaphase onset by live imaging. WT and PP2A-AαP179R/+ cells were similarly delayed upon centrinone treatment (Figures S2G and S2H), indicating that spindle assembly is not more robust in acentrosomal PP2A-AαP179R/+ cells. Spindle positioning and centrosome clustering in mitosis depends on cortical forces and interphase cell shape (Kwon et al., 2015, Rhys et al., 2018, Théry et al., 2005). To examine the contribution of cortical forces to spindle positioning, cells were plated on L-shaped fibronectin micro-patterned coverslips. Spindle positioning was not impaired in PP2A-AαP179R/+ cells with two centrosomes (Figures S2I–S2K). We next examined the contribution of cortical forces and interphase cell shape to centrosome clustering in PP2A-AαP179R/+ cells. Centrosome amplification was induced by Plk4 overexpression and cells plated onto O- or Y-shaped fibronectin micro-patterns (Figure 4G, S2L, and S2M). Both shapes bias cells with supernumerary centrosomes toward multipolar cell divisions (Kwon et al., 2008). Indeed, in WT cells these patterns resulted in 1.8-fold and 1.6-fold increases in the frequency of multipolar cell divisions on O- and Y- shaped patterns, respectively (Figure 4H). Interestingly, in PP2A-AαP179R/+ cells the frequency of multipolar cell divisions was increased by 2.6- and 3.9-fold on O- and Y- shaped patterns, respectively (Figure 4H). Thus, centrosome clustering efficiency in PP2A-AαP179R/+ cells is more sensitive to interphase cell shape compared with WT cells. However, the overall frequency of multipolar cell divisions in PP2A-AαP179R/+ cells was still reduced compared with WT cells (Figure 4H) suggesting that although cortical forces contribute to enhanced centrosome clustering in PP2A-AαP179R/+ cells, the phenotype likely also arises from mechanisms unrelated to cell shape.
Discussion
Human tumors frequently have centrosome amplification (Chan, 2011), and cancer cells tend to be more proficient at clustering supernumerary centrosomes (Ganem et al., 2009). Thus, in addition to relying on the intrinsic centrosome clustering ability of cells (Drosopoulos et al., 2014, Kwon et al., 2008, Kwon et al., 2015, Leber et al., 2010, Quintyne et al., 2005), cancer cells may acquire mutations that improve clustering efficiency. Here we have characterized a hotspot P179R mutation in PP2A-Aα that allows cells to cluster supernumerary centrosomes more efficiently. This finding is important because cells with supernumerary centrosomes only survive mitosis if they cluster centrosomes (Ganem et al., 2009). Our data suggest that a potential explanation for the enrichment of PPP2R1A mutations in tumors that experience WGD is to allow cells to survive the mitotic stress associated with centrosome amplification. Tolerance to WGD has been shown to confer chromosomal instability and promote tumor genome evolution and is linked to poor prognosis (Bielski et al., 2018, Dewhurst et al., 2014). Therefore, cancer-associated mutations in PP2A-Aα could also serve as a prognostic marker of tumor progression and is in agreement with PPP2R1A being annotated as a cancer driver in gynecological tumors (Bailey et al., 2018).
Our findings also highlight the importance of considering PP2A-Aα mutations in a context that closely resembles the genetic change in tumors and suggest that the enhanced centrosome clustering efficiency arises from haploinsufficiency in PP2A-Aα function. Interestingly, the reduction in PP2A functionality by the P179R mutation in PP2A-Aα has been identified as a driver of tumorigenesis in uterine cancers (Taylor et al., 2019) and is consistent with the known tumor suppressor role of PP2A (Pallas et al., 1990). Therefore, if the primary impact of the mutation is to reduce PP2A-Aα functionality, then it is perhaps unexpected that missense mutations dominate the PPP2R1A mutational landscape rather than truncations. Considering the heterotrimeric structure of PP2A holoenzymes, it is possible that several essential functions of PP2A may still be fulfilled by the mutant PP2A-Aα, including association with the catalytic subunit and association with STRN regulatory subunits, neither of which was altered by the P179R or R183W mutation. Thus the mutations may selectively impact the subset of PP2A holoenzymes with tumor suppressor functions. Given that small-molecule activators of PP2A have been shown to reduce the tumorigenicity associated with PP2A inactivation by the P179R mutation (Taylor et al., 2019), it will be important to further investigate the anti-tumor efficacies of pharmacologic activators of PP2A (Ramaswamy et al., 2015, Sangodkar et al., 2017) in human cancers with other oncogenic PP2A mutations.
Furthermore, our work reveals that decreasing PP2A functionality increases centrosome clustering. This observation together with the identification of a role for PP2A-B55/SUR-6 in regulating centrosome separation during mitotic entry (Boudreau et al., 2019) opens new avenues of exploration for a role of PP2A in the regulation of centrosome clustering. Our data also suggest that fine-tuning phosphoregulation, in the form of a heterozygous PP2A-Aα mutation, allows a human cell to become more proficient at centrosome clustering without compromising mitotic fidelity. Because the mutation only partially reduces PP2A-Aα function, and a WT allele remains present in the cell, we predict that the observed phenotype is due to small changes in phosphorylation of substrates that impact centrosome positioning and engagement during mitosis. Therefore, future work will focus on characterizing phosphorylation sites that control centrosome clustering in PP2A-AαP179R/+ cells.
Limitations of the Study
One limitation of this study is that we examined the impact of PP2A-Aα mutations in a non-cancer cell line. The advantage of this choice is that we could determine that the mutation is sufficient to alter centrosome clustering, whereas the limitation is that we did not examine the mutation in a transformed cell. The other limitation is that we did not perform gene replacement for all PP2A-Aα isoforms, and therefore further studies are needed to determine if this effect on centrosome clustering is shared among other recurrent mutations.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Veronica Rodriguez-Bravo and Sun Joo Lee for experimental assistance and Prasad Jallepalli and Bryan Tsou for helpful discussions. This work was supported by the Functional Genomics Initiative at Memorial Sloan Kettering (E.A.F. and A.K.), NIH/NIGMS GM125996 (E.A.F), and NCI CA214812 to A.K. This work was prepared while E.A.F. was employed at Memorial Sloan Kettering Cancer Center. The opinions expressed in this article are the author's own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the US government.
Author Contributions
Formal analysis and investigation, N.V.A., M.M.-O., and P.C.; Conceptualization, experiment design, and writing, N.V.A. and E.A.F.; Funding acquisition, E.A.F. and A. K.
Declaration of Interests
The authors declare no competing interests.
Published: September 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.07.018.
Supplemental Information
References
- Andreassen P.R., Lohez O.D., Lacroix F.B., Margolis R.L. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell. 2001;12:1315–1328. doi: 10.1091/mbc.12.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey M.H., Tokheim C., Porta-Pardo E., Sengupta S., Bertrand D., Weerasinghe A., Colaprico A., Wendl M.C., Kim J., Reardon B. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371–385.e18. doi: 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R., Brunk K., Vinadogrova T., Peel N., Franz A., Khodjakov A., Raff J.W. Centrosome amplification can initiate tumorigenesis in flies. Cell. 2008;133:1032–1042. doi: 10.1016/j.cell.2008.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdougo E., Terret M.-E., Jallepalli P.V. Functional dissection of mitotic regulators through gene targeting in human somatic cells. Methods Mol. Biol. 2009;545:21–37. doi: 10.1007/978-1-60327-993-2_2. [DOI] [PubMed] [Google Scholar]
- Bielski C.M., Zehir A., Penson A.V., Donoghue M.T.A., Chatila W., Armenia J., Chang M.T., Schram A.M., Jonsson P., Bandlamudi C. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018;50:1189–1195. doi: 10.1038/s41588-018-0165-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau V., Chen R., Edwards A., Sulaimain M., Maddox P.S. PP2A-B55/SUR-6 collaborates with the nuclear lamina for centrosome separation during mitotic entry. Mol. Biol. Cell. 2019;30:876–886. doi: 10.1091/mbc.E18-10-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter S.L., Cibulskis K., Helman E., McKenna A., Shen H., Zack T., Laird P.W., Onofrio R.C., Winckler W., Weir B.A. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.Y. A clinical overview of centrosome amplification in human cancers. Int. J. Biol. Sci. 2011;7:1122–1144. doi: 10.7150/ijbs.7.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Possemato R., Campbell K.T., Plattner C.A., Pallas D.C., Hahn W.C. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell. 2004;5:127–136. doi: 10.1016/s1535-6108(04)00026-1. [DOI] [PubMed] [Google Scholar]
- Cho U.S., Xu W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature. 2007;445:53–57. doi: 10.1038/nature05351. [DOI] [PubMed] [Google Scholar]
- Cooper J.A. Effects of cytochalasin and phalloidin on actin. J. Cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craney A., Kelly A., Jia L., Fedrigo I., Yu H., Rape M. Control of APC/C-dependent ubiquitin chain elongation by reversible phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 2016;113:1540–1545. doi: 10.1073/pnas.1522423113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenda A., Rouse J., Doza Y.N., Meier R., Cohen P., Gallagher T.F., Young P.R., Lee J.C. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- Dewhurst S.M., McGranahan N., Burrell R.A., Rowan A.J., Grönroos E., Endesfelder D., Joshi T., Mouradov D., Gibbs P., Ward R.L. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014;4:175–185. doi: 10.1158/2159-8290.CD-13-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosopoulos K., Tang C., Chao W.C.H., Linardopoulos S. APC/C is an essential regulator of centrosome clustering. Nat. Commun. 2014;5:3686. doi: 10.1038/ncomms4686. [DOI] [PubMed] [Google Scholar]
- Espert A., Uluocak P., Bastos R.N., Mangat D., Graab P., Gruneberg U. PP2A-B56 opposes Mps1 phosphorylation of Knl1 and thereby promotes spindle assembly checkpoint silencing. J. Cell Biol. 2014;206:833–842. doi: 10.1083/jcb.201406109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley E.A., Maldonado M., Kapoor T.M. Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat. Cell Biol. 2011;13:1265–1271. doi: 10.1038/ncb2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem N.J., Godinho S.A., Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haesen D., Abbasi Asbagh L., Derua R., Hubert A., Schrauwen S., Hoorne Y., Amant F., Waelkens E., Sablina A., Janssens V. Recurrent PPP2R1A mutations in uterine cancer act through a dominant-negative mechanism to promote malignant cell growth. Cancer Res. 2016;76:5719–5731. doi: 10.1158/0008-5472.CAN-15-3342. [DOI] [PubMed] [Google Scholar]
- Hertz E.P.T., Kruse T., Davey N.E., López-Méndez B., Sigurðsson J.O., Montoya G., Olsen J.V., Nilsson J. A Conserved motif provides binding specificity to the PP2A-B56 phosphatase. Mol. Cell. 2016;63:686–695. doi: 10.1016/j.molcel.2016.06.024. [DOI] [PubMed] [Google Scholar]
- Hyodo T., Ito S., Asano-Inami E., Chen D., Senga T. A regulatory subunit of protein phosphatase 2A, PPP2R5E, regulates the abundance of microtubule crosslinking factor 1. FEBS J. 2016;283:3662–3671. doi: 10.1111/febs.13835. [DOI] [PubMed] [Google Scholar]
- Jeong A.L., Han S., Lee S., Su Park J., Lu Y., Yu S., Li J., Chun K.-H., Mills G.B., Yang Y. Patient derived mutation W257G of PPP2R1A enhances cancer cell migration through SRC-JNK-c-Jun pathway. Sci. Rep. 2016;6:27391. doi: 10.1038/srep27391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodjakov A., Cole R.W., Oakley B.R., Rieder C.L. Centrosome-independent mitotic spindle formation in vertebrates. Curr. Biol. 2000;10:59–67. doi: 10.1016/s0960-9822(99)00276-6. [DOI] [PubMed] [Google Scholar]
- Kitajima T.S., Sakuno T., Ishiguro K., Iemura S., Natsume T., Kawashima S.A., Watanabe Y. Shugoshin collaborates with protein phosphatase 2A to protect cohesin. Nature. 2006;441:46–52. doi: 10.1038/nature04663. [DOI] [PubMed] [Google Scholar]
- Kleylein-Sohn J., Westendorf J., Le Clech M., Habedanck R., Stierhof Y.-D., Nigg E.A. Plk4-induced centriole biogenesis in human cells. Dev. Cell. 2007;13:190–202. doi: 10.1016/j.devcel.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Kwon M., Godinho S.A., Chandhok N.S., Ganem N.J., Azioune A., Thery M., Pellman D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–2203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon M., Bagonis M., Danuser G., Pellman D. Direct microtubule-binding by myosin-10 orients centrosomes toward retraction fibers and subcortical actin clouds. Dev. Cell. 2015;34:323–337. doi: 10.1016/j.devcel.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leber B., Maier B., Fuchs F., Chi J., Riffel P., Anderhub S., Wagner L., Ho A.D., Salisbury J.L., Boutros M. Proteins required for centrosome clustering in cancer cells. Sci. Transl. Med. 2010;2:33ra38. doi: 10.1126/scitranslmed.3000915. [DOI] [PubMed] [Google Scholar]
- Lee S.J., Rodriguez-Bravo V., Kim H., Datta S., Foley E.A. The PP2A B56 phosphatase promotes the association of Cdc20 with APC/C in mitosis. J. Cell Sci. 2017;130:1760–1771. doi: 10.1242/jcs.201608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijenhuis W., Vallardi G., Teixeira A., Kops G.J., Saurin A.T. Negative feedback at kinetochores underlies a responsive spindle checkpoint signal. Nat. Cell Biol. 2014;16:1257–1264. doi: 10.1038/ncb3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallas D.C., Shahrik L.K., Martin B.L., Jaspers S., Miller T.B., Brautigan D.L., Roberts T.M. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell. 1990;60:167–176. doi: 10.1016/0092-8674(90)90726-u. [DOI] [PubMed] [Google Scholar]
- Peel N., Stevens N.R., Basto R., Raff J.W. Overexpressing centriole-replication proteins in vivo induces centriole overduplication and de novo formation. Curr. Biol. 2007;17:834–843. doi: 10.1016/j.cub.2007.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintyne N.J., Reing J.E., Hoffelder D.R., Gollin S.M., Saunders W.S. Spindle multipolarity is prevented by centrosomal clustering. Science. 2005;307:127–129. doi: 10.1126/science.1104905. [DOI] [PubMed] [Google Scholar]
- Ramaswamy K., Spitzer B., Kentsis A. Therapeutic re-activation of protein phosphatase 2A in acute myeloid leukemia. Front. Oncol. 2015;5:16. doi: 10.3389/fonc.2015.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhys A.D., Monteiro P., Smith C., Vaghela M., Arnandis T., Kato T., Leitinger B., Sahai E., McAinsh A., Charras G. Loss of E-cadherin provides tolerance to centrosome amplification in epithelial cancer cells. J. Cell Biol. 2018;217:195–209. doi: 10.1083/jcb.201704102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring D., Hubble R., Kirschner M. Mitosis in a cell with multiple centrioles. J. Cell Biol. 1982;94:549–556. doi: 10.1083/jcb.94.3.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangodkar J., Perl A., Tohme R., Kiselar J., Kastrinsky D.B., Zaware N., Izadmehr S., Mazhar S., Wiredja D.D., O’Connor C.M. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J. Clin. Invest. 2017;127:2081–2090. doi: 10.1172/JCI89548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz M.H.A., Held M., Janssens V., Hutchins J.R.A., Hudecz O., Ivanova E., Goris J., Trinkle-Mulcahy L., Lamond A.I., Poser I. Live-cell imaging RNAi screen identifies PP2A–B55α and importin-β1 as key mitotic exit regulators in human cells. Nat. Cell Biol. 2010;12:886–893. doi: 10.1038/ncb2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight A.F., Cheung A., Limouze J., Chen I., Westwood N.J., Sellers J.R., Mitchison T.J. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Taylor Sarah E., O’Connor C.M., Wang Z., Shen G., Song H., Leonard D., Sangodkar J., LaVasseur C., Avril S., Waggoner S. The highly recurrent PP2A Aα-subunit mutation P179R alters protein structure and impairs PP2A enzyme function to promote endometrial tumorigenesis. Cancer Res. 2019 doi: 10.1158/0008-5472.CAN-19-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry M., Racine V., Pépin A., Piel M., Chen Y., Sibarita J.-B., Bornens M. The extracellular matrix guides the orientation of the cell division axis. Nat. Cell Biol. 2005;7:947–953. doi: 10.1038/ncb1307. [DOI] [PubMed] [Google Scholar]
- Wang W.J., Soni R.K., Uryu K., Tsou M.F.B. The conversion of centrioles to centrosomes: essential coupling of duplication with segregation. J. Cell Biol. 2011;193:727–739. doi: 10.1083/jcb.201101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong Y.L., Anzola J.V., Davis R.L., Yoon M., Motamedi A., Kroll A., Seo C.P., Hsia J.E., Kim S.K., Mitchell J.W. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science. 2015;348:1155–1160. doi: 10.1126/science.aaa5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Xing Y., Chen Y., Chao Y., Lin Z., Fan E., Yu J.W., Strack S., Jeffrey P.D., Shi Y. Structure of the protein phosphatase 2A holoenzyme. Cell. 2006;127:1239–1251. doi: 10.1016/j.cell.2006.11.033. [DOI] [PubMed] [Google Scholar]
- Xu Y., Chen Y., Zhang P., Jeffrey P.D., Shi Y. Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol. Cell. 2008;31:873–885. doi: 10.1016/j.molcel.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Lončarek J., Khodjakov A., Rieder C.L. Extra centrosomes and/or chromosomes prolong mitosis in human cells. Nat. Cell Biol. 2008;10:748–751. doi: 10.1038/ncb1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack T.I., Schumacher S.E., Carter S.L., Cherniack A.D., Saksena G., Tabak B., Lawrence M.S., Zhang C.-Z., Wala J., Mermel C.H. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Pham H.T., Ruediger R., Walter G. Characterization of the Aalpha and Abeta subunit isoforms of protein phosphatase 2A: differences in expression, subunit interaction, and evolution. Biochem. J. 2003;369:387–398. doi: 10.1042/BJ20021244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.