

Abstract
Introduction:
Numerous omics studies have been conducted to understand the molecular networks involved in Alzheimer’s disease (AD), but the pathophysiology is still not completely understood; new approaches that enable neuroscientists to better interpret the results of omics analysis are required.
Methods:
We have developed advanced methods to analyze and visualize publicly-available genomics and genetics data. The tools include a composite clinical-neuropathological score for defining AD, gene expression maps in the brain, and networks integrating omics data to understand the impact of polymorphisms on AD pathways.
Results:
We have analyzed over 50 public human gene expression data sets, spanning 19 different brain regions and encompassing three separate cohorts. We integrated genome-wide association studies with expression data to identify important genes in the pathophysiology of AD, which provides further insight into the calcium signaling and calcineurin pathways.
Discussion:
Biologists can use these freely-available tools to obtain a comprehensive, information-rich view of the pathways in AD.
Keywords: Genomics, Data visualization, Calcium signaling pathway, Calcineurin, Alzheimer’s disease, PTK2B, TOMM40
1. Introduction
Alzheimer’s disease (AD), the most common form of dementia, affects an estimated 5.7 million Americans and is the sixth leading cause of death in the United States [1]. To date, there are no disease-modifying therapies, and the failure of clinical trials can be attributed to multiple factors from inadequate study methodologies to targeting the wrong pathway, patient, or disease stage [2]. To address some of these challenges, AD researchers have recently advocated a paradigm shift from a syndromic diagnosis to a biologically based definition of the disease for interventional and observational studies [3]. In this framework, AD is defined either by postmortem pathologies or in vivo biomarkers of amyloid b deposition, pathologic tau, and neurodegeneration [3]. There are several underlying processes and putative risk factors that lead to these pathophysiologies, which are not completely understood, and an improved understanding of these will lead to better targeted therapies for AD.
Omics is one approach used by researchers to understand the biological complexity and heterogeneity of the disease. The largest such effort is the multiinstitution Accelerating Medicines Partnership in Alzheimer’s Disease (AMP-AD), a public-private open science initiative [4]. The AMP-AD consortium has generated genomics (microarray and RNA-Seq), epigenomics, and proteomics data from over 2000 human brain samples; the data are publicly available through the Synapse platform developed by Sage Bionetworks [4]. Several system biology analyses of these multiomics data have identified networks and pathways that may be implicated in AD [5–8]. Other efforts have combined genetic and transcriptomic data to identify novel pathways in AD [9,10]. More recently, the AMP-AD data integration was used to suggest the involvement of the human herpes virus 6A and human herpes virus 7 in AD [8].
A similar large-scale effort to understand the genetics of AD has led to the identification of genetic loci associated with the risk of developing AD that are thought to be involved in amyloid b precursor protein processing and tau pathways, inflammation, immune response, lipid transport, and endocytosis [11]. The epigenome and the effects of aging are other factors contributing to the molecular phenotypes observed in AD [12,13]. These studies and others have led to an understanding of how amyloid β and tau can lead to synapse degeneration, neuronal loss, and the eventual loss of cognitive abilities [14], but the complete mechanism and heterogeneity in AD are not fully understood.
The omics and systems biology efforts have uncovered several molecular networks in AD and taken strides to understand the pathophysiology of AD [5–7], but there have been no prior efforts to systematically analyze and evaluate the AMP-AD data from all the cohorts except the newly released Agora portal by Sage Bionetworks (https://agora.ampadportal.org/genes). We have developed a suite of tools to consistently process the AMP-AD transcriptomic data, combine it with genetic data, and visualize it using novel tools to explore the results. We have made our methods and visualization tools available to promote open and reproducible science research. As an example, our efforts validate previously implicated calcium-associated pathways and point to some novel mechanisms in the previously studied Aβ-mediated activation of the calcineurin pathway.
2. Methods
2.1. AMP-AD data
Gene expression data from AMP-AD was obtained from three centers: (1) The Religious Orders Study and Memory and Aging Project (ROSMAP); (2) The Mount Sinai Brain Bank (MSBB) study; and (3) The Mayo Clinic Brain Bank (MCBB).
The ROSMAP project contains two longitudinal cohort studies from Rush University. The Religious Order Study enrolls participants from 40 religious groups across the United States and the Memory Aging Project recruits older subjects with no cognitive deficits [15,16]. The ROSMAP study provides RNA-Seq data from the dorsolateral prefrontal cortex (PFC) of 724 subjects from the Religious Order Study and Memory Aging Project cohorts [7,17]. The ROSMAP study also provides microarray data from 490 samples (Myers data set) [18–20]. Data on clinical and demographic covariates including sex, race, age of death, apolipoprotein E (APOE) status, and clinical and neuropathological scores were provided for all subjects. We also used the genotype data provided for 1709 individuals using the Affymetrix GeneChip 6.0 for expression quantitative trait loci (eQTL) analysis [21].
The MSBB study has samples from the Mount Sinai/JJ Peters VA Medical Center Brain Bank which were sequenced from 301 individuals from PFC, inferior frontal gyrus, superior temporal gyrus, and parahippocampal gyrus (PHG). In addition, 19 brain regions were assayed with microarrays [6]. Covariates including sex, race, age of death, APOE status, and clinical and neuropathological scores were reported [22].
The MCBB study sequenced samples from 275 cerebellum and 276 temporal cortex brain regions from subjects with neuropathological diagnosis of AD, progressive supra-nuclear palsy, pathologic aging, or elderly controls without neurodegenerative diseases [23–25]. Data were processed, and normalized counts were provided [26]. We also downloaded the results of the eQTL analysis of cis-single-nucleotide polymorphisms (SNPs) affecting gene expression, which are provided by the Mayo Clinic [23].
With these three studies, a total of 27 data sets spanning 19 brain regions and including both microarrays and RNA-Seq were evaluated (Supplementary Table 1).
2.2. Differential gene expression analysis
Processed microarray and RNA-Seq data were downloaded from the AMP-AD portal [17,22,26]. All RNA-Seq Fragments Per Kilobase of transcript per Million mapped reads data were log transformed. RNA-Seq raw counts were normalized and transformed using the R edgeR [27] and voom [28] packages to prepare for linear modeling. Differential expression analysis was performed using the limma package in R [29]. In the microarray data, for each gene, we choose the probe with lowest P value. The false discovery rate for multiple comparisons was adjusted using the Benjamini-Hochberg method [30]. We analyzed the data using Braak [31,32], Consortium to Establish a Registry for Alzheimer’s Disease [32], and a composite clinical-neuropathological score (Fig. 1). Braak scores were divided into high (B3: V, VI), medium (B2: III, IV), and low (B1: 0, I, II) scores(Fig. 1A). A combined neuropathology score was developed using both Braak and Consortium to Establish a Registry for Alzheimer’s Disease scores (Fig. 1B) adapted from the NIA recommendations [33]. The final composite diagnosis (CpDx) is determined by combining the aforementioned neuropathological score with a clinical staging of dementia to define subjects with normal cognition, AD, preclinical AD, or dementia not AD (Fig. 1C). A subject is considered normal cognition if documented with a clinical dementia rating ≤ 0.5 or a Mini-Mental State Exam ≥ 26. These thresholds were chosen according to the study by Balsis et al. [34]. All scripts are available at https://github.com/ mindinformatics/CATS.
Fig. 1.
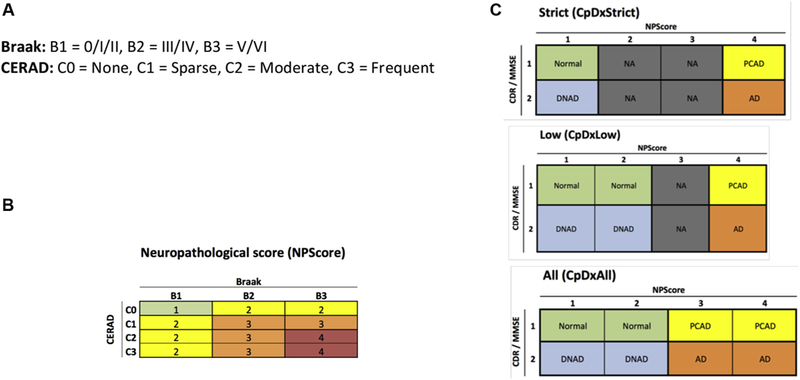
Defining analysis groups. (A) Braak scores were divided into high, medium, and low scores. (B) Combined neuropathological score was developed using Braak and CERAD. (C) Composite diagnosis score (CpDx) combines clinical and neuropathological measures. Comprehensive scoring CpDxAll applies to all categories with a neuropathological threshold NP score > 2. Low-scoring CpDxLow applies to low NP score values 1–2 and extreme value 4; cases with transitional NP score = 3 are ignored. Strict scoring CpDxStrict applies to extreme NP score values 1 and 4 only, and cases with transitional NP score values 2–3 are ignored. AD, Alzheimer’s disease; CDR, clinical dementiarating; CERAD, Consortiumto EstablishaRegistry forAlzheimer’sDisease; PCAD, preclinicalAD; DNAD, dementia not AD.
2.3. International Genomics of Alzheimer’s project
Genetics data were obtained from the International Genomics of Alzheimer’s Project (IGAP) stage 1 meta-analysis results of genome-wide association studies of 7,055,881 SNPs either genotyped or imputed in at least 40% of the AD cases and 40% of the control samples across all data sets for genome-wide association with AD (17,008 cases and 37,154 controls) in individuals of European ancestry. Gene-wide significance of SNPs was computed using the Gene-Wide Significance (GWiS) test that identifies the independent effects of SNPs within a gene using a greedy Bayesian approach and combines them to compute a stronger signal for each gene [35]. The GWiS P values for gene loci are computed using permutation tests.
2.4. Visualization
All visualizations were implemented using the JavaScript D3 libraries [36]. The bubble chart, scatter plot, and force-directed visualization methods were used. The input to the visualizations is provided as comma-separated values files. The plots are interactive, allowing the user to look up information or navigate to areas of further interest. The code is available at https://github.com/mindinformatics/CATS.
3. Results
3.1. Differential expression of AD pathway
We systematically analyzed all the various ways of defining AD and control subjects as described in the Methods section (Fig. 1) with all 25 data sets. The ROSMAP PFC and MSBB PHG RNA-Seq data sets contained the highest number of differentially expressed genes; similarly, for microarray data, it was the ROSMAP PFC and the MSBB HIPP (hippocampus) data sets. The dorsolateral PFC, PHG, and hippocampal (HIPP) brain regions are affected in AD [37–39]. For each of these four data sets, we ranked each gene by the 7 methods as applicable: Braak, Consortium to Establish a Registry for Alzheimer’s Disease, Clinical Diagnosis, Mini-Mental State Exam, CpDxAll, CpDxStrict, and CpDxLow. We summed the ranks of genes in the AD pathway from the Kyoto Encyclopedia of Genes and Genomes pathway database [40] for each data set. The CpDxLow method for analyzing AD versus control ranked among top 2 in the MSBB microarray and RNA-Seq data sets and top 3 in the ROSMAP data. Full results are available in the Supplementary Table 2. Several genes in the Kyoto Encyclopedia of Genes and Genomes AD pathway are differentially expressed using this method (Fig. 2). Such a combined clinical and neuropathological scoring was deemed superior and selected for the recent ROSMAP network analysis article [7]. Thus, we chose to use the CpDxLow scoring for the rest of our analyses. We also implemented a brain expression map for a set of genes (see example in Supplementary Fig. 1 and Supplementary Table 3).
Fig. 2.
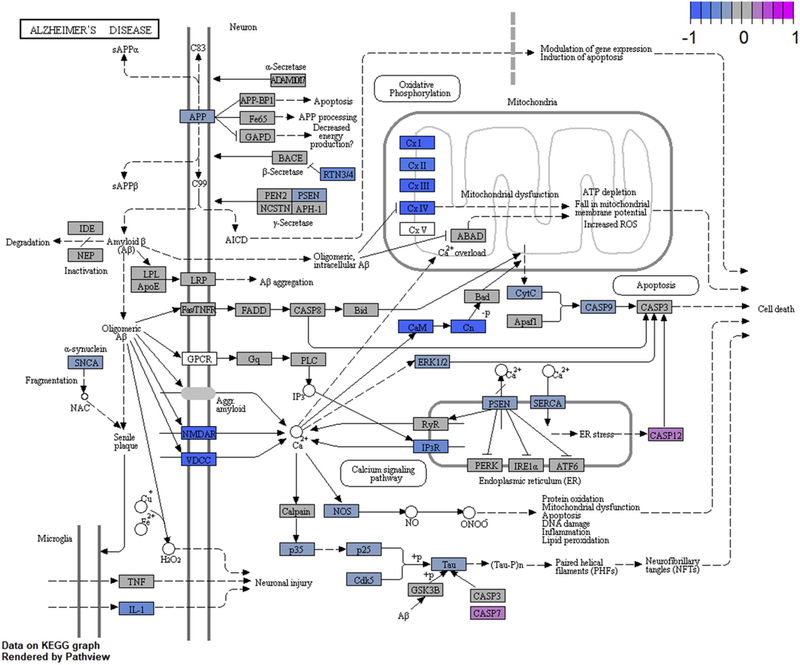
KEGG AD pathway. Differentially expressed genes are colored. AD, Alzheimer’s disease; KEGG, Kyoto Encyclopedia of Genes and Genomes; AICD, amyloid precursor protein intracellular domain; sAPP, soluble amyloid precursor protein; APP, amyloid precursor protein; ROS, reactive oxygen species; ATP, adenosine triphosphate; NAC, non-Aβ component.
3.2. Integration of genome-wide association studies and expression data
Then, we took the list of significantly differentially expressed genes in the ROSMAP PFC data set based on the composite CpDxLow score (p-adjusted < 0.01, 4044 genes) and combined it with the results from the IGAP stage 1 meta-analysis. For each gene, we selected the SNP with lowest P value within ± 1 Mbp of gene loci start and end. Although this method has the potential of selecting a gene that is not necessarily closest to the SNP, we wanted to look at the potential impacts on differential expression broadly. Fig. 3 demonstrates the fold change in the ROSMAP data set and the significance of the SNP in the IGAP stage 1 meta-analysis combined with the eQTL data from the MCBB study for these genes. We choose a threshold of IGAP P value < 1E-4 for further investigation (828 genes shown in blue and red).
Fig.3.
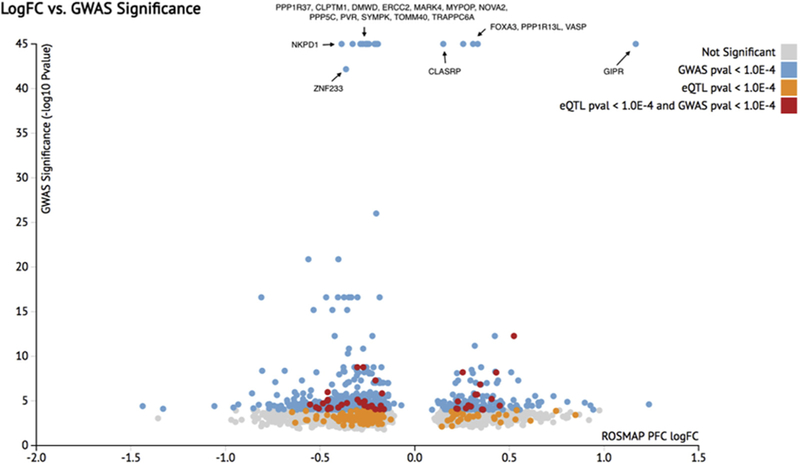
Plot of fold change in AD versus control of ROSMAP PFC using CpDxLow composite score vs. significance of SNP in IGAP meta-analysis that is within 61 Mbp of gene. All differentially expressed genes in the ROSMAP PFC data set that has IGAP data are shown. Each dot represents a gene and is colored by whether it is significant in the IGAP study, Mayo eQTL study, both, or none. GWAS, genome-wide association studies; AD, Alzheimer’s disease; IGAP, International Genomics of Alzheimer’s Project; ROSMAP, Religious Orders Study and Memory and Aging Project; PFC, prefrontal cortex.
Enrichment analysis with a hypergeometric test using the Molecular Signatures Database [41] identifies the calcium signaling pathway as the most significant one in this thresh-olded set of 828 differentially expressed genes, with a significant SNP within ± 1 Mbp of its locus (Table 1). The calcium signaling pathway and its involvement in AD has been well studied [42], and we chose to explore it in more detail. All the other pathways enriched such as chemokine signaling and mitogen-activated protein kinases signaling have also been implicated in AD [43].
Table 1.
Gene set enrichment using the MSigDB database.
| Gene Set Name [# Genes (K)] | Description | # Genes in Overlap (k) |
k/K | p-value |
FDR q-value |
|---|---|---|---|---|---|
| KEGG_CALCIUM_SIGNALING_PATHWAY [178] | Calcium signaling pathway | 18 | 4.42 e−10 | 8.21 e−8 | |
| KEGG_CHEMOKINE_SIGNALING_PATHWAY [190] | Chemokine signaling pathway | 16 | 5.56 e−8 | 5.17 e−6 | |
| KEGG_LONG_TERM_POTENTIATION [70] | Long-term potentiation | 8 | 1.29 e−5 | 8.02 e−4 | |
| KEGG_MAPK_SIGNALING_PATHWAY [267] | MAPK signaling pathway | 15 | 2.17 e−5 | 1.01 e−3 | |
| KEGG_HUNTINGTONS_DISEASE [185] | Huntington’s disease | 12 | 3.56 e−5 | 1.33 e−3 | |
| KEGG_ALZHEIMERS_DISEASE [169] | Alzheimer’s disease | 11 | 7.24 e−5 | 2.25 e−3 | |
| KEGG_NEUROACTIVE_LIGAND_RECEPTOR_INTER TERACTION [272] | Neuroactive ligand-receptor interaction | 14 | 1.03 e−4 | 2.74 e−3 | |
| KEGG_PHOSPHATIDYLINOSITOL_SIGNALING_SY_SYSTEM [76] | Phosphatidylinositol signaling system | 7 | 1.8 e−4 | 3.76 e−3 | |
| KEGG_GNRH_SIGNALING_PATHWAY [101] | GnRH signaling pathway | 8 | 1.82 e−4 | 3.76 e−3 | |
| KEGG_TIGHT_JUNCTION [134] | Tight Junction | 9 | 2.56 e−4 | 4.57 e−3 |
Abbreviations: MSigDB, Molecular Signatures Database; KEGG, Kyoto Encyclopedia of Genes and Genomes; MAPK, mitogen-activated protein kinases;GnRH, gonadotropin-releasing hormone; FDR, false discovery rate.
3.3. Calcium homeostasis dysregulation in AD
To look for potential effects of genotypes in AD net-works, we created a protein-protein and pathway interaction network of the Kyoto Encyclopedia of Genes and Genomes calcium signaling pathway (178 genes) and 9 additional genes that are significantly connected to this set using Gen-eMANIA [44]. The network of these 187 genes with their expression and gene locus significance, computed using GWiS test as described in the Methods section, is shown in Fig. 4. Each bubble is a gene and is colored by its fold change in AD versus normal in the ROSMAP dorsolateral prefrontal cortex RNA-Seq data. As seen in Fig. 4, the entire calcium pathway is downregulated in AD with the exception of 10 genes, even after correcting for neuronal loss using MAP2 and astrocyte and microglia activation using GFAP and CD68, respectively, as cell-specific markers. The most significant upregulated gene is the inositol trisphosphate 3-kinase B (ITPKB). ITPKB has previously been shown to be upregulated in mouse AD models and human brains and aggravates amyloid and tau pathologies [45]. The two genes with a high genetic risk in this network are TOMM40 and PTK2B.
Fig.4.
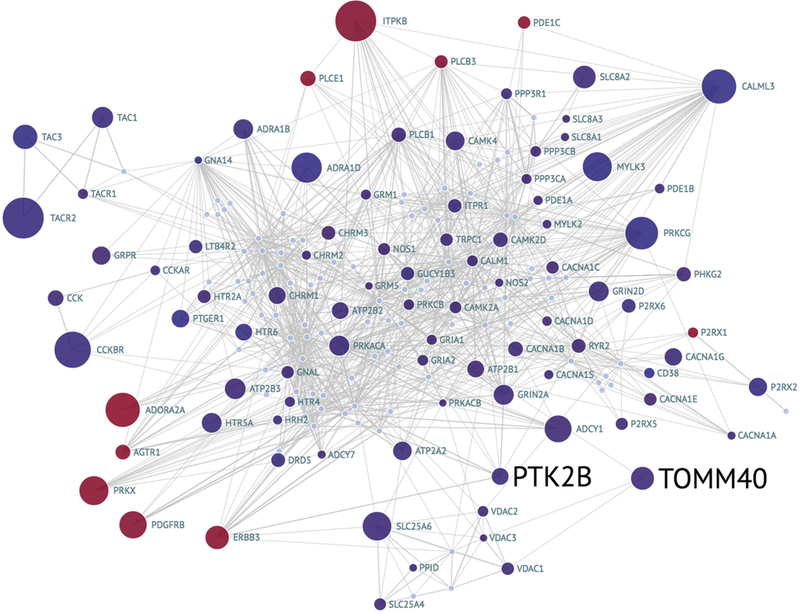
Network visualization of the calcium signaling pathway with expression and GWAS information. Each bubble is a gene and is colored by its fold change in differential expression analysis of AD versus normal defined using the composite clinical and neuropathological scores (CpDxLow) in the ROSMAP dorsolateral prefrontal cortex RNA-Seq data; the size indicates significance. Edges are protein or pathway interactions. The black text labels indicate significance of the locus computed using GWiS from the IGAP meta-analysis; the size indicates the magnitude of the significance log10 (P value). The blue labels are differentially expressed genes without significant GWiS results. GWAS, genome-wide association studies; AD, Alzheimer’s disease; IGAP, International Genomics of Alzheimer’s Project; ROSMAP, Religious Orders Study and Memory and Aging Project; GWiS, Gene-Wide Significance.
The TOMM40 gene and its genetic association with AD is a matter of debate. Some studies claim that the association of TOMM40 with AD is due to linkage disequilibrium [46], whereas others have found a significant association even after adjusting for the APOE e4 status [47]. From Fig. 4, TOMM40 interacts with the voltage-dependent anion channel [48], and both are located on the outer mitochondrial membrane, suggesting a potential role for TOMM40 in the calcium dysregulation of AD. Furthermore, eQTL analysis of the rs1160985 SNP, which was associated with a risk of AD [47], revealed 11 genes in the network (KCNA1, KCNA2, ATF4, CAMK2N1, PQBP1, CYFIP2, CLSTN1, SV2B, SNCB, TAGLN3, and OLFM1) that were differentially expressed in subjects with one or two copies of this SNP. Taken together, these results suggest a functional role of mutations in TOMM40 in the calcium signaling pathway. The role of PTK2B in calcium signaling and long-term potentiation has been previously studied [11]. Indeed, analysis of the rs7827965 SNP located within the PTK2B gene showed a differential expression of low-density lipoprotein receptor-related protein 6. Fig. 4 clearly illustrates disruption of calcium signaling in AD brains and suggests the role of genetic mutations in this network.
3.4. Synapse genes in the calcineurin pathway
Then, we investigated the hypothesis that the calcium/ calmodulin activated serine/threonine protein phosphatase, calcineurin (PPP3CA) pathway, which promotes neurodegeneration [49], and is altered in human brains. We took the 140 synapse genes from the set of ~ 1150 differentially expressed genes in mice HIPP neurons with constitutively active calcineurin [50]. We downloaded protein interactions and other significantly connected partners of these genes using GeneMANIA. Some of the genes (28) were not significantly connected and were discarded. The entire network of these 112 genes, visualized with the ROSMAP PFC differential expression and GWiS significance, is mostly downregulated even after adjusting for neuronal loss using the pan neuronal marker MAP2, which has robust expression and is downregulated in subjects with AD (Fig. 5). The genes in this network that have a genetic association (labeled in black and sized by significance)— and not necessarily downregulated themselves—are all membrane proteins or extracellular ones, suggesting a role of these mutations in calcium homeostasis in the calcium-calcineurin pathway. The gene GRIN2A (glutamate ionotropic receptor N-methyl-D-aspartate-type subunit 2A), which plays a critical role in long-term depression in AD [51], is significantly downregulated in AD brains (Fig. 5). GRIN2A interacts with DLG2 [52], which has an SNP associated with AD and is another membrane-associated guanylate kinase, and these 2 genes may play a role together in the synapse dysfunction of AD. Taken together, these data suggest the downregulation of the calcium-/calmodulin-activated calcineurin pathway in part modified by disease-associated polymorphisms. However, the SNPs in NRXN1, DLG2, and SDC2 do not have a significant eQTL association with the synapse genes of the calcineurin pathway.
Fig.5.
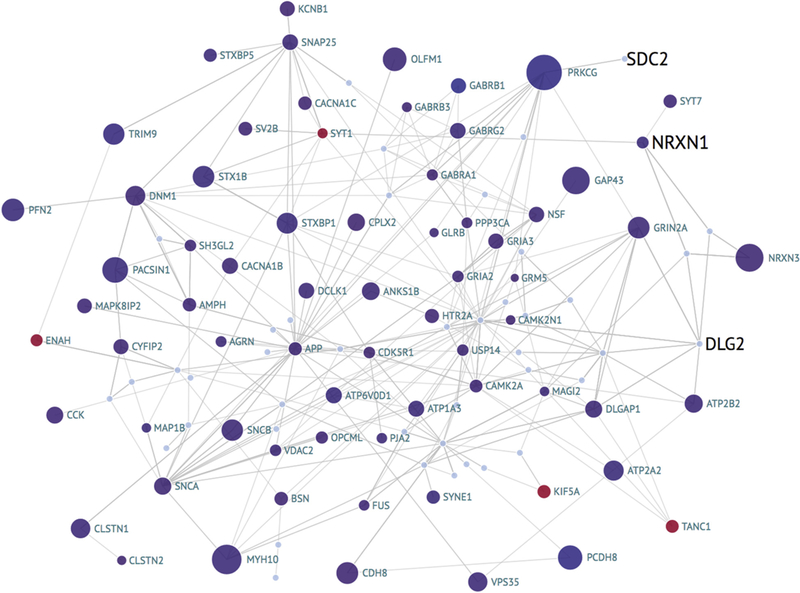
Network visualization of the synapse genes of the calcineurin pathway with expression and GWAS information. Each bubble is a gene and is colored by direction in differential expression analysis of AD versus normal defined using the composite clinical and neuropathological scores (CpDxLow) in the ROSMAP dorsolateral prefrontal cortex RNA-Seq data; the size indicates significance (-log10 P value). Edges are protein or pathway interactions. The black text labels indicate significance of the locus computed using GWiS from the IGAP meta-analysis; the size indicates the magnitude of the significance. The blue labels are differentially expressed genes without significant GWiS results. GWAS, genome-wide association studies; GWiS, Gene-Wide Significance; AD, Alzheimer’s disease; IGAP, International Genomics of Alzheimer’s Project; ROSMAP, Religious Orders Study and Memory and Aging Project.
4. Discussion
We used a combination of novel analytical and visualization methods to enable inquiries of extensive genetic and omics data. As one example, we test the hypothesis that the calcium signaling pathway, and in particular the calcium-/calmodulin-dependent calcineurin pathway, is activated in the pathogenesis of AD. The central claim of the calcium hypothesis is that oxidative stress and other mechanisms lead to disruption of homeostasis in early stages of the disease, which leads to synapse dysfunction, neuroinflammation, and other AD-associated phenomenon [42]. Although alterations in calcium homeostasis have been clearly demonstrated in animal models by, for example, multiphoton microscopy and calcium imaging in amyloid β precursor protein overexpressing mice [53,54], these approaches are not available in human samples, and so the implications of calcium dyshomeostasis in human AD remain uncertain; moreover, molecular mechanisms involved in these processes are not yet fully understood. We examined the possibility that transcriptomics can be used to further expand our understanding of calcium alterations in AD.
Previously, gene expression studies have shed light on upstream process in neurons that are vulnerable to chronic calcium dysregulation; stress and immune response genes are upregulated, whereas energy and signal transduction genes are downregulated [55]. In amyloid β precursor protein mice models with plaques, it has been shown that calcium overload within neurons causes abnormal morphologies through activation of the calcium-dependent calcineurin/nuclear factor of activated T-cells (NFAT) pathway [49,53]. Overexpression of calcineurin produces transcriptional suppression in mouse HIPP neurons [50]. Gene expression of calcium signaling genes in neuronal subcompartments, however, has not yet been fully explored in human brains. Here, we report a series of changes that are consistent with AD-related calcium dysregulation, including downregulation of calcium-signaling genes in neurons potentially leading to synaptic failure and abnormal neuronal function. These results match closely the predictions based on mouse data [50].
The dysregulation of calcium signaling in AD brains, however, is complex, and it is quite likely that there are several factors involved in how this pathway is downregulated. One possibility is the dephosphorylation and inactivation of the transcription factor CREB, which regulates several memory-associated genes via calcineurin [56]. Indeed, CREB transcriptional targets obtained from the Molecular Signatures Database are overrepresented in the set of synapse calcineurin genes (hypergeometric test P value: 0.002). Another possible explanation is the upregulation of RCAN1, which provides negative feedback to turn off NFAT—a transcription factor involved in the calcineurin pathway [57]. ITPKB2, which is upregulated, also deactivates NFAT through the activation of the AKT-PI3 kinase pathway and phosphorylation of NFAT by GSK3 [57]. Synapse genes could be downregulated in AD due to neuronal loss, but Hopp et al. [50] demonstrated downregulation in laser-captured HIPP neurons and also suggested the role of micro-RNAs. Future experiments that would either implicate CREB or possibly a micro-RNA as a key intermediate in AD pathology need to be conducted.
Genetic susceptibility also has a role in the calcium hypothesis. It has been previously studied in familial Alzheimer’s disease (FAD): for example, Tong et al. [58] has shown that familial Alzheimer’s disease PS1 mutations affect calcium release from endoplasmic reticulum through STIM1 (stromal interaction molecule 1). Here, we hypothesize that polymorphisms in the PTK2B and TOMM40 in sporadic AD have an effect on the calcium signaling pathway; our speculations are based on significant associations with the polymorphisms and downregulation of calcium signaling genes. PTK2B is a protein tyrosine kinase which is involved in calcium-induced regulation of ion channels; the SNP rs28834970 in the PTK2B locus was recently associated with altered splicing [59], potentially affecting its interaction with the glutamate receptor GRIN2A. TOMM40 interacts with the voltage-dependent anion channel, and both are located on the outer mitochondrial membrane, suggesting a potential role for TOMM40 in the calcium dysregulation of AD.
Another important consideration is the role of calcium signaling in other cell types. For example, in astrocytes, Ca2+-mediated CN/NFAT signaling leads to synapse dysfunction and neuroinflammation [60]. Norris suggests a positive-feedback cycle in which neurodegeneration leads to Ca2+ hyperactivity in astrocytes, which in turn promotes neuroinflammation [61]. Single-cell RNA sequencing of the aging and AD brain with the aid of in-silico modeling [62] promises to elucidate the details of this complex system in the future. Aging is another major factor in neurodegenerative disease, and a similar integrative approach with transcriptomics and genetics can be used to understand the interaction of aging and plaques on calcium in neurons and other cell types. A large control cohort with no pathologies or single pathology and a wide range of ages would be needed for such studies.
The hypothesis-based analyses of the calcium and the calcineurin pathways, which limit the number of comparisons, present new mechanisms without being penalized by harsh multiple-comparison adjustments that are required when performing unbiased searches on the whole genome. The combination of statistical analysis with visualization is innovative and produces interpretable results. However, there are limitations to these methods. Visualization using a bubble chart or force-directed graph involves the use of arbitrary cutoffs and has the potential to miss important genes that fall just below the chosen threshold. The size of the pathway and the number of dimensions that can be visualized are constrained. Neither compensatory mechanisms nor cross-talk between pathways are modeled. Furthermore, the force-directed graph displays the physical or pathway interactors of genes without the use of direction or statistics. In the differential gene expression analyses, the use of a composite clinical-neuropathological score is innovative and hereafter can be used to better characterize subjects with preclinical AD or to distinguish AD from other dementias. The differential expressional and eQTL analyses were conducted using the limma package [29], which builds a linear model and assumes a normal distribution. We normalized and transformed RNA-Seq count data using edgeR [27] and voom [28] to satisfy distributional assumptions, remove any skew or kurtosis to the extent possible, and account for any heteroscedasticity in the data. Preprocessing RNA-Seq data with voom allows us to use the same statistical test for both microarrays and RNA-Seq data.
As examples of the usefulness of pulling together data sets from multiple cohorts into a readily accessible format, we previously used part of this analytical framework to study angiogenesis stress signatures in mice [63] and calcineurin-activated genes in mice HIPP neurons [50]. Going forward, we imagine that this approach could be useful to many investigators, perhaps to study neuroinflammation, IFN signaling pathways, pathological aging, mitochondrial stress, auto-phagy, protein degradation, and microglial signatures and to test the various modules and genes implicated in the omics data sets developed by AMP-AD. We have made the methods and tools described in this article available for others to use. In future, we also plan to make all analyses and interactive visualizations of the AMP-AD data available through a DataLENS portal (alzdatalens.org). Researchers will be able to query and visualize the AMP-AD data using a list of genes of interest. DataLENS, which is complementary to the Agora platform, allows researchers to explore pathways of interest and to get a comprehensive view of all affected genes. We hope that the portal will serve as an useful resource for neuroscientists involved in the study of AD pathophysiology.
Supplementary Material
{kind=link}
RESEARCH IN CONTEXT.
Systematic Review: We did an extensive literature survey using PubMed to find integrative analyses of the AMP-AD omics data and computational methods used to analyze such studies.
Interpretation: Our analysis of the AMP-AD gene expression and genetics data and use of visualization methods for pathways exploration is novel. Through our findings in the molecular signaling pathways in late stages of AD, we elucidate a small piece of the complex role of calcium homeostasis in the disease.
Future Direction: We plan to deploy the data analytics and visualization through a portal for neuroscientists to study their genes and pathways of interest.
Acknowledgments
The project is funded by a grant from the Massachusetts Center for Alzheimer Therapeutics Science (MassCATS), a public-private partnership to discover new treatments for Alzheimer’s disease, organized through the Massachusetts Life Sciences.
The ROSMAP project was supported by funding from the National Institute on Aging (AG034504 and AG041232). Many data and biomaterials were collected from several sites funded by the National Institute on Aging (NIA) and National Alzheimer’s Coordinating Center (NACC, grant: #U01 AG016976). Amanda J. Myers, PhD (University of Miami, Department of Psychiatry) prepared the series. The directors, pathologist, and technicians involved are as follows: Rush University Medical Center, Rush Alzheimer’s Disease Center (NIH #AG10161): David A. Bennett, M.D. Julie A. Schneider, MD, MS, Karen Skish, MS, PA (ASCP)MT, Wayne T Longman. The Rush University portion of this study was supported by National Institutes of Health grants P30AG10161, R01AG15819, R01AG17917, R01AG36042, R01AG36836, U01AG46152, R01AG34374, R01NS78009, U18NS82140, R01AG42210, and R01AG39478 and the Illinois Department of Public Health. Quality control checks and preparation of the gene expression data were provided by the National Institute on Aging Alzheimer’s Disease Data Storage Site (NIAG-ADS, U24AG041689) at the University of Pennsylvania.
The MSBB data were generated from postmortem brain tissue collected through the Mount Sinai VA Medical Center Brain Bank and were provided by Dr. Eric Schadt from Mount Sinai School of Medicine.
Mayo study data were provided by the following sources: The Mayo Clinic Alzheimer’s Disease Genetic Studies, led by Dr. Nilufer Taner and Dr. Steven G. Younkin, Mayo Clinic, Jacksonville, FL, using samples from the Mayo Clinic Study of Aging, the Mayo Clinic Alzheimer’s Disease Research Center, and the Mayo Clinic Brain Bank. Data collection was supported through funding by NIA grants P50 AG016574, R01 AG032990, U01 AG046139, R01 AG018023, U01 AG006576, U01 AG006786, R01 AG025711, R01 AG017216, and R01 AG003949; NINDS grant R01 NS080820; CurePSP Foundation; and Mayo Foundation. Study data include samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05–901, and 1001 to the Arizona Parkinson’s Disease Consortium), and the Michael J. Fox Foundation for Parkinson’s Research.
Footnotes
Supplementary Data
Supplementary data related to this article can be found at https://doi.org/10.1016/joalz.2019.01.011.
References
- [1].Association As. 2018 Alzheimer’s disease facts and figures. Alzheimers Dement 2018;14:367–429. [Google Scholar]
- [2].Khachaturian AS, Hayden KM, Mielke MM, Tang Y, Lutz MW, Gustafson DR, et al. Future prospects and challenges for Alzheimer’s disease drug development in the era of the NIA-AA Research Frame-work. Alzheimers Dement 2018;14:532–4. [DOI] [PubMed] [Google Scholar]
- [3].Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB,et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018; 14:535–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hodes RJ, Buckholtz N. Accelerating Medicines Partnership: Alzheimer’s Disease (AMP-AD) Knowledge Portal Aids Alzheimer’s Drug Discovery through Open Data Sharing. Expert Opin Ther Targets 2016;20:389–91. [DOI] [PubMed] [Google Scholar]
- [5].Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013;153:707–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang M, Roussos P, McKenzie A, Zhou X, Kajiwara Y, Brennand KJ, et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med 2016;8:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J, et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline ofAlzheimer’s disease. Nat Neurosci 2018;21:811–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Readhead B, Haure-Mirande JV, Funk CC, Richards MA, Shannon P. Haroutunian V, et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Net-works by Human Herpesvirus. Neuron 2018;99:64–82.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gaiteri C, Mostafavi S, Honey CJ, De Jager PL, Bennett DA. Genetic variants in Alzheimer disease - molecular and brain network approaches. Nat Rev Neurol 2016;12:413–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mukherjee S, Russell JC, Carr DT, Burgess JD, Allen M, Serie DJ, et al. Systems biology approach to late-onset Alzheimer’s disease genome-wide association study identifies novel candidate genes validated using brain expression data and Caenorhabditis elegans experiments. Alzheimers Dement 2017;13:1133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 2013; 45:1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bennett DA, Yu L, Yang J, Srivastava GP, Aubin C, De Jager PL. Epi-genomics of Alzheimer’s disease. Transl Res 2015;165:200–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yu L, Chibnik LB, Srivastava GP, Pochet N, Yang J, Xu J, et al. Association of Brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol 2015;72:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014;82:756–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS. Overview and findings from the religious orders study. Curr Alzheimer Res 2012; 9:628–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the rush Memory and Aging Project. Curr Alzheimer Res 2012;9:646–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].SageBionetworks. AMP-AD Knowledge Portal: ROSMAP RNA-Seq. Available at: https://www.synapse.org/#!Synapse:syn3388564.
- [18].SageBionetworks. AMP-AD Knowledge Portal: ROSMAP Microarray. Available at: https://www.synapse.org/#!Synapse:syn3800853.
- [19].Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L, et al. A survey of genetic human cortical gene expression. Nat Genet 2007; 39:1494–9. [DOI] [PubMed] [Google Scholar]
- [20].Webster JA, Gibbs JR, Clarke J, Ray M, Zhang W, Holmans P, et al. Genetic control of human brain transcript expression in Alzheimer disease. Am J Hum Genet 2009;84:445–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chibnik LB, White CC, Mukherjee S, Raj T, Yu L, Larson EB, et al. Susceptibility to neurofibrillary tangles: role of the PTPRD locus and limited pleiotropy with other neuropathologies. Mol Psychiatry 2018;23:1521–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].SageBionetworks. AMP-AD Knowledge Portal: MSBB. Available at: https://www.synapse.org/#!Synapse:syn3159438.
- [23].Zou F, Chai HS, Younkin CS, Allen M, Crook J, Pankratz VS, et al. Brain expression genome-wide association study (eGWAS) identifies human disease-associated variants. Plos Genet 2012;8:e1002707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Carrasquillo MM, Zou F, Pankratz VS, Wilcox SL, Ma L, Walker LP, et al. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer’s disease. Nat Genet 2009;41:192–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Allen M, Carrasquillo MM, Funk C, Heavner BD, Zou F, Younkin CS, et al. Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci Data 2016; 3:160089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].SageBionetworks. AMP-AD Knowledge Portal: Mayo RNASeq. Available at: https://www.synapse.org/#!Synapse:syn3163039.
- [27].Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hochberg YBaY. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Statist SocSer B.57:289–300. [Google Scholar]
- [31].Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–59. [DOI] [PubMed] [Google Scholar]
- [32].Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991;41:479–86. [DOI] [PubMed] [Google Scholar]
- [33].Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Balsis S, Benge JF, Lowe DA, Geraci L, Doody RS. How Do Scores on the ADAS-Cog, MMSE, and CDR-SOB Correspond? Clin Neuropsy-chol 2015;29:1002–9. [DOI] [PubMed] [Google Scholar]
- [35].Huang H, Chanda P, Alonso A, Bader JS, Arking DE. Gene-based tests of association. Plos Genet 2011;7:e1002177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bostock M D3:Data-Driven Documents. Available at: https://d3js.org. [DOI] [PubMed]
- [37].Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science 1984;225:1168–70. [DOI] [PubMed] [Google Scholar]
- [38].Gomez-Isla T, Price JL, McKeel DW Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci 1996; 16:4491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kaufman LD, Pratt J, Levine B, Black SE. Antisaccades: a probe into the dorsolateral prefrontal cortex in Alzheimer’s disease. A critical review. J Alzheimers Dis 2010;19:781–93. [DOI] [PubMed] [Google Scholar]
- [40].Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016;44:D457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Alzheimer’s Association Calcium Hypothesis W. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement 2017;13:178–182.e17. [DOI] [PubMed] [Google Scholar]
- [43].Chen YG. Research Progress in the Pathogenesis of Alzheimer’s Disease. Chin Med J (Engl) 2018;131:1618–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 2010;38:W214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Stygelbout V, Leroy K, Pouillon V, Ando K, D’Amico E, Jia Y, et al. Inositol trisphosphate 3-kinase B is increased in human Alzheimer brain and exacerbates mouse Alzheimer pathology. Brain 2014; 137:537–52. [DOI] [PubMed] [Google Scholar]
- [46].Yu CE, Seltman H, Peskind ER, Galloway N, Zhou PX, Rosenthal E, et al. Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer’s disease: patterns of linkage disequilibrium and disease/marker association. Genomics 2007; 89:655–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ma XY, Yu JT, Wang W, Wang HF, Liu QY, Zhang W, et al. Association of TOMM40 polymorphisms with late-onset Alzheimer’s disease in a Northern Han Chinese population. Neuromolecular Med 2013; 15:279–87. [DOI] [PubMed] [Google Scholar]
- [48].Wan C, Borgeson B, Phanse S,Tu F, Drew K, Clark G, et al. Panorama of ancient metazoan macromolecular complexes. Nature 2015; 525:339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci 2010;30:2636–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hopp SC, Bihlmeyer NA, Corradi JP, Vanderburg C, Cacace AM, Das S, et al. Neuronal calcineurin transcriptional targets parallel changes observed in Alzheimer disease brain. J Neurochem 2018; 147:24–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Koffie RM, Hyman BT, Spires-Jones TL. Alzheimer’s disease: synapses gone cold. Mol Neurodegener 2011;6:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Irie M, Hata Y, Takeuchi M, Ichtchenko K, Toyoda A, Hirao K, et al. Binding of neuroligins to PSD-95. Science 1997;277:1511–5. [DOI] [PubMed] [Google Scholar]
- [53].Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008;59:214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Busche MA. In Vivo Two-Photon Calcium Imaging of Hippocampal Neurons in Alzheimer Mouse Models. Methods Mol Biol 2018; 1750:341–51. [DOI] [PubMed] [Google Scholar]
- [55].Wang X, Michaelis ML, Michaelis EK. Functional genomics of brain aging and Alzheimer’s disease: focus on selective neuronal vulnerability. Curr Genomics 2010;11:618–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang Y, Shi Y, Wei H. Calcium Dysregulation in Alzheimer’s Disease: ATarget for New Drug Development. J Alzheimers Dis Parkinsonism 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Crabtree GR, Schreiber SL. SnapShot: Ca2+-calcineurin-NFAT signaling. Cell 2009;138:210.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tong BC, Lee CS, Cheng WH, Lai KO, Foskett JK, Cheung KH. Familial Alzheimer’s disease-associated presenilin 1 mutants promote gamma-secretase cleavage of STIM1 to impair store-operated Ca21 entry. Sci Signal 2016;9:ra89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Raj T, Li YI, Wong G, Humphrey J, Wang M, Ramdhani S, et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat Genet 2018;50:1584–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sompol P, Norris CM. Ca(21), Astrocyte Activation and Calcineurin/ NFAT Signaling in Age-Related Neurodegenerative Diseases. Front Aging Neurosci 2018;10:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Norris CM. Calcineurin: directing the damage in Alzheimer disease: An Editorial for ‘Neuronal calcineurin transcriptional targets parallel changes observed in Alzheimer disease brain’ on page 24. J Neuro-chem 2018;147:8–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Khachaturian ZS, Lombardo J. In silico modeling system: a national research resource for simulation of complex brain disorders. Alzheimers Dement 2009;5:1–4. [DOI] [PubMed] [Google Scholar]
- [63].Bennett RE, Robbins AB, Hu M, Cao X, Betensky RA, Clark T, et al. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc Natl Acad Sci USA 2018;115:E1289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.