

Abstract
Recent reports indicate that inactivation of the RB, TP53 or PTEN tumour suppressor genes is detected in tumour stroma of oropharyngeal, breast and other human cancers. Mouse models have validated the tumour-promoting effects of deleting Rb, Pten or p53 in fibroblasts that converts them from normal fibroblasts to carcinoma associated fibroblasts (CAFs). The tumour-promoting activity of CAFs in these contexts was associated with increased paracrine signaling to tumour cells through production of specific growth factors, chemokines and MMPs by CAFs. The conversion of NOFs into CAFs through acquisition of specific mutations, such as loss of tumour suppressors, or deregulated expression of microRNAs or key epigenetic events, can clearly occur independently of genetic and epigenetic changes in tumour cells but an alternative source of CAFs that is being reconsidered is that CAFs derive from the tumour cells by EMT. Recent mouse models employing lineage-tracing techniques have suggested that this can take place in vivo and the extent to which this is relevant more broadly is discussed.
Keywords: carcinoma-associated fibroblasts, trophic support, EMT, lineage tracing, RB, Pten, p53, micro-RNAs, TGFβ
Introduction
The tumour microenvironment has emerged over the past 10–15 years as a key modulator of solid tumour initiation, progression, metastasis and therapeutic response, with evidence demonstrating that stromal cells both promote epithelial transformation by providing trophic support and respond and co-evolve with tumour cells in response to stresses [1]. The diversity of cell types in the tumour microenvironment also contributes to overall tumour heterogeneity. Given the dependence of tumour cells on stromal support as well as competition between tumour clones [2], changes in the tumour microenvironment are undoubtedly a major driving pressure in the tumour evolution that leads to drug resistance and malignant progression [3,4]. Thus, targeting the tumour microenvironment has emerged as a novel approach in cancer therapy. However, this approach relies on understanding the molecular changes differentiating malignant stroma from normal stroma. Reports in recent years have identified genetic mutations in tumour stroma in primary human cancers, including breast [5–8], colorectal [9], ovarian [10] and bladder cancers [11], as well as important epigenetic changes [12]. Further elegant studies in both genetically engineered [13,14] and xenograft mouse models [15,16] have also confirmed that genetic changes in the tumour microenvironment can promote tumourigenesis independent of initiating genetic events in tumour cells. Despite these important findings, it remains unclear to what extent genetic or indeed epigenetic changes in tumour stroma can be manipulated to modulate tumour growth in vivo. Given recent reports identifying molecular mechanisms of action for the RB, p53 and PTEN tumour suppressors in carcinoma-associated fibroblasts (CAFs) that make up a key component of the tumour stroma, this perspective focuses on how CAFs evolve and function. Specifically, we discuss the genetic basis of CAF induction, the cellular origin of CAFs and the significance of these new data for our understanding of tumour evolution and how to target the tumour microenvironment.
Effects of tumour suppressor gene inactivation on carcinoma-associated fibroblasts
Carcinoma-associated fibroblasts (CAFs) are a major component of the tumour microenvironment that can promote the transformation of normal epithelium as well as progression to carcinoma [15–20]. The CAF phenotype can be induced in normal fibroblasts by co-culture with carcinoma cells and this acquired CAF phenotype is stable even after tumour cells are removed [15,17,18]. This relationship suggests the induction of stable heritable changes in fibroblasts following growth in the presence of tumour cells, although the nature of these changes remains unclear. Certain characteristics of the CAF phenotype resemble changes seen in normal fibroblasts that become ‘activated’ in response to wounding, including the secretion of extracellular matrix (ECM), matrix-degrading enzymes/matrix metalloproteinases (MMPs) and signalling molecules, such as vascular endothelial growth factor (VEGF) and interleukin-6 (IL-6), that recruit endothelial cells and immune cells, respectively.
How ‘activated’ fibroblasts revert to normal or whether they are eliminated by cell death and phagocytosis after the completion of the wound-healing process remains unknown. Understanding how this ‘reversion’ is achieved seems likely to be relevant to the development of clinical approaches aimed at eliminating CAFs and reducing tumour burden. Interestingly, irradiated fibroblasts are able to promote the invasiveness of pancreatic tumour cells [21], suggesting that irradiation, although promoting a reactive phenotype in fibroblasts, also likely induces changes similar to those seen in CAFs. Senescent fibroblasts also behave in some ways like CAFs in their ability to induce tumour cell proliferation [22,23] and, conversely, CAFs mimic senescent fibroblasts through expression of the ‘senescent-associated secretory phenotype’ (SASP), which includes pro-inflammatory cytokines, chemokines and proteinases [20,24]. Some of these secreted molecules, such as SDF-1 and CCL5, have been shown to be key to the tumour-promoting activity of CAFs [19,25,26]. However, unlike CAFs, senescent fibroblasts cannot transform normal epithelia [22], suggesting that CAFs possess additional growth-inducing properties not present in senescent fibroblasts. Additional studies have also revealed heterogeneity amongst the fibroblast populations based on transcriptional profiles and positional memory [27,28]. Markers such as α-smooth muscle actin (α-SMA), vimentin, neural–glial antigen-2 (NG2) and fibroblast-specific protein-1 (FSP-1) have all been used to define different populations of fibroblasts in the tumour microenvironment [29]. Growing evidence indicates that FSP-1-positive fibroblasts represent a distinct subtype of CAF that possesses greater tumour-promoting activities [29–32] than the other populations, although the extent to which this population of fibroblasts is derived from other fibroblasts in the tumour by transdifferentiation, or how the FSP-1-positive population comes to be a more ‘activated’ type of CAF than the others, is not clear.
The critical role of CAFs in tumourigenesis highlights the importance of identifying the stable genetic, or more dynamic, epigenetic changes in CAFs that underlies their activity in carcinoma growth and progression. Clearly, epigenetic silencing and down-regulation of growth-suppressive genes, such as p16/INK4a/CDKN2A and p21/CDKN1A, could explain aspects of the CAF phenotype, particularly their enhanced proliferation, without the need to invoke genetic changes. Nevertheless, there is increasing evidence that genetic inactivation of tumour suppressors does indeed occur in tumour stroma. For example, TP53 and PTEN are somatically mutated in the stromal components of human breast cancer [6–8]. In addition, a specific role for p53 in prostate cancer stroma was identified by Hill et al [33], utilizing a GEMM model of prostate cancer in which the mutant TAg121 form of the SV40 large T antigen (which inactivates the pocket protein family of pRB, p107 and p130 but not p53) was expressed in a prostate-specific manner under the probasin promoter, resulting in prostate tumourigenesis. When these mice were crossed to p53-null mice, a massive increase in stromal involvement accompanied more rapid progression to carcinoma compared to p53 wild-type mice; the effect of p53 loss on tumour stroma was more obvious than the impact on tumour epithelia in this model. When the TAg121 transgenic mice were crossed to p53 heterozygous mice, the investigators also observed a more rapid and robust expansion of tumour stroma/mesenchyme compared to p53 wild-type mice, and laser capture analysis of tumour stroma revealed loss of heterozygosity for p53 in the stroma of these tumours, indicating a strong selective pressure for p53 loss, specifically in the tumour stroma. The authors concluded that loss of RB and proliferative control in prostate epithelial tumours induced a reaction in stromal fibroblasts, such as increased proliferation or cell survival, that was normally repressed by p53 [33].
These important in vivo studies revealing the role of p53 in tumour stroma have been followed by more targeted approaches in which relevant tumour suppressors were deleted specifically in CAFs. As previously mentioned, the PTEN tumour suppressor is somatically mutated in human breast cancer stroma [6]. Based on these findings, Trimboli and colleagues [34] targeted Pten loss to stromal fibroblasts in genetically engineered mice using the Fsp1-Cre deletor strain and crossed these mice to both the MMTV–Neu and MMTV–PyMT mouse models of mammary tumourigenesis. Fibroblast-specific deletion of Pten in this manner resulted in increased primary tumour growth in both models. Pten-deleted stroma deposited increased ECM and expressed increased levels of pro-inflammatory genes. Increased macrophage infiltration into tumours was also observed in these models. Mechanistically, the authors linked Pten loss in fibroblasts to increased Ras–Akt signalling, as well as increased activity of the Ets-2 transcription factor and associated increases in expression of MMP9, an Ets-2 target gene (Figure 1). Indeed, ablation of Ets-2 in Pten-deleted fibroblasts reduced tumour vasculature and resulted in reduced tumour burden. The authors also demonstrated that reduced expression of miR-320 as a result of Pten deletion promoted Ets-2 and MMP9 expression, as both genes are miR-320 targets [35]. Finally, the authors correlated reduced PTEN expression in primary human breast tumour stroma with increased levels of phospho(T72)-Ets-2 and phospho(S473)-Akt1 [34], providing important clinical support for their findings in mouse models.
Figure 1.

Loss of key tumour suppressor genes in CAFs promotes tumour cell progression to malignancy. Loss of RB in normal and carcinoma-associated fibroblasts promotes proliferation, inhibits differentiation and increases invasion through collagen of co-cultured keratinocytes and tumour epithelia [32,36]. The effect of RB loss in fibroblasts was dependent on production of keratinocyte growth factor (KGF) that acted on co-cultured epithelial cells to promote growth through the KGF receptor (FGFR2b) and signalling through AKT to Ets-2 and its target genes, such as MMPs. Similarly, loss of PTEN in CAFs promoted production of MMPs and chemokines through deregulation of Ets-2, achieved in part through phosphorylation of Ets-2 by Akt and also via loss of PTEN-induced repression of miR-320 that targets Ets-2.
More recent work from Pickard and colleagues [32,36] identified inactivation of the RB tumour suppressor in the stromal component of primary human breast and oropharyngeal cancers. High levels of the hyperphosphorylated inactive form of pRB were specifically detected in the FSP1-positive subset of CAFs in oropharyngeal cancers and, importantly, this was independent of HPV status [32]. The authors further examined the effect of RB loss on primary fibroblasts and demonstrated that RB-deficient human foreskin fibroblasts (HFFs) increased the proliferation and reduced the differentiation of co-cultured epithelial cells in three-dimensional (3D) culture [36]. This effect was not due to increased fibroblast proliferation and was phenocopied by over-expression of CDK6, a cyclin-dependent kinase that phosphorylates pRB, and conversely was inhibited by expression of phosphomutant pRB [36]. The authors showed that this non-cell autonomous effect of RB-deficient fibroblasts on co-cultured epithelial cells was due to increased expression of keratinocyte growth factor (KGF), which microarray analyses had previously shown to be up-regulated in Rb-null mouse fibroblasts compared to wild-type controls [37]. Knockdown of KGF or restoration of RB inhibited the effect of RB-deficient fibroblast co-culture on epithelial cells [36].
When the authors examined the effect of co-culture of RB-deficient fibroblasts on E6/E7 transformed keratinocytes, C33a cervical tumour cells, HCAT cervical tumour cells and MDA-MB-231 human breast cancer cells, they observed increased cell invasion through collagen [32]. Similar to results obtained previously with primary epithelial cells, over-expression of CDK6 in fibroblasts phenocopied RB depletion in promoting tumour cell invasion, whereas knockdown of KGF blocked this effect of RB-depleted fibroblasts on co-cultured tumour cells. Analogous to what was observed in Pten-deficient fibroblasts [34], signalling via Akt was required for the tumour-promoting activity of Rb-deficient HFFs, including KGF production and tumour cell invasion [38].
Intriguingly, Ets-2 appears to play a tumour-promoting role in both tumour cells [32] and in CAFs [34]. In response to fibroblast-secreted KGF, activation of the KGF receptor (FGFR2b) and downstream Akt signalling, E6/E7-transformed human keratinocytes expressed elevated levels of phosphorylated Ets-2 [32], similar to what was seen in Pten-deleted fibroblasts [34]. Expression of the Ets-2 oncogene in KGF-induced tumour cells promoted MMP1 expression and was required for tumour cell invasion. Furthermore, knockdown of the KGF receptor in tumour cells blocked the effect of exogenous KGF or co-culture with RB-deficient HFFs on tumour cell invasion and Ets-2 expression. Interestingly, the KGF receptor (FGFR2b) is amplified in some gastric and breast cancers and mutationally activated in a proportion of endometrial cancers [32]. These results suggest a model (Figure 1) whereby loss of RB in fibroblasts derepresses KGF expression, which acts in a paracrine manner on co-cultured/adjacent tumour cells through its receptor and Akt to promote Ets-2 expression and subsequent tumour cell invasion, in part as a result of increased production of MMPs and other known modulators of metastasis.
Understanding how CAFs emerge in the tumour microenvironment
The traditional model of tumour–stroma interactions posits that tumour cells recruit fibroblasts to the tumour, for example through production of TGFβ, which in turn modulates fibroblast activity, such as through effects on HGF production [18,20]. However, how the interaction between fibroblasts and tumour cells promotes conversion of the fibroblast into a CAF is not understood. Recent exploration of the role of the microenvironment in high-grade serous ovarian cancer (OvCa) revealed a role for microRNAs (miRNAs) in reprogramming normal fibroblasts into CAFs [26]. Mitra and colleagues [26] showed by miRNA profiling of primary OvCa-derived CAFs that although miR-155 is up-regulated, miR-31 and miR-214 are down-regulated in CAFs compared to normal fibroblasts (NOFs) from adjacent tissue. Furthermore, co-culture of NOFs with OvCa tumour cells induced conversion of NOFs to CAFs and was associated with increased expression of miR-155 and decreased expression of both miR-31 and miR-214. The authors went on to show that the cytokine CCL5 is a miR-214 target expressed by CAFs but not by NOFs. They showed that CCL5 induced a more metastatic phenotype in OvCa cells, similar to that seen with CAF co-culture, and that CCL5 was essential for the growth- and invasion-promoting properties of CAFs [26]. Nonetheless, the question remains as to how miR-155, miR-31 and miR-214 are deregulated during the NOF to CAF conversion – is their expression disrupted by mutation of key tumour suppressor genes? Both the RB and p53 tumour suppressor gene pathways are heavily implicated in human high-grade serous ovarian cancer [39] and it will be interesting to determine whether their role in HGSOvCa extends beyond their function in tumour cells themselves to the tumour microenvironment, as has been demonstrated in some of the other tumour types we have discussed above.
Beyond the traditional model of tumour cells recruiting normal fibroblasts (NOFs) via paracrine signalling and subsequently inducing the NOFs to become CAFs, other models have been proposed to explain the origins of CAFs [20,40,41]. These models include the possibility that CAFs are a kind of developmental precursor that is specifically recruited by tumour cells, whereas others suggest that CAFs are derived from bone marrow mesenchymal stem cells recruited to the tumour, where they undergo an alternative differentiation programme [41]. A particularly compelling alternative model postulates that CAFs are derived from tumour epithelial cells by epithelial-to-mesenchymal transition (EMT). Although the absence of a shared genetic mutation profile between tumour cells and CAFs has historically argued against EMT as an explanation for the origin of CAFs [20,41,42], more recent studies are challenging this view. For example, work illustrating a much higher degree of plasticity between the epithelial and mesenchymal components in tumours than previously appreciated comes from Rhim and colleagues [43], who used lineage tracing in the LSL-KRasG12D;p53fl/fl;Pdx1-Cre; (KPC) mouse model of pancreatic ductal adenocarcinoma (PDAC) to follow epithelial cells that had undergone Cre-mediated deletion. They showed that approximately 42% of lineage-marked YFP+ epithelial cells underwent EMT to E-cadherin-negative cells, but nevertheless retained their tumour-forming ability in transplanted mice. Inflammation has been shown to induce EMT and to suppress p16/INK4A/CDKN2A expression in vivo; this phenotype is detected early in KPC mice, as well as in human PDAC [44]. Both the E-cadherin-positive (epithelial) and E-cadherin-negative (mesenchymal) cells induced tumours in transplanted mice when derived from malignant PDAC but only the E-cadherin-negative cells exhibited tumour-forming ability when derived from early premalignant pancreatic intraepithelial neoplasias (PanINs). Furthermore, circulating YFP+ cells detected in mice prior to tumour formation exhibited both mesenchymal properties (E-cadherin-negative) and stem cell properties (CD24negCD44pos). This study therefore raises the provocative suggestion that the tumour stem cell, at least in this model, may actually originate in the stromal/mesenchymal compartment of tumours, and that therapeutic strategies need to be developed to specifically target this cell population [43]. Arguably, these and other studies highlighting the role of EMT in modulating the tumour microenvironment should also prompt a reconsideration of the role of TGFβ in tumour stroma. TGFβ is a well-defined inducer of EMT [45–47] and thus it is possible that what appear to be the effects of TGFβ in recruiting fibroblasts and regulating tumour cell–CAF interactions are in part the consequence of TGFβ-induced EMT.
As mentioned, the low rate of detection of tumour-specific mutations in the tumour stroma has been the major argument against EMT as the explanation for the origin of CAFs. However, several studies now report TP53 or PTEN mutations in the stroma with the same mutations detected in associated tumour epithelium [6,8,10]. Both p53 and RB have been shown to regulate EMT [48–52] and these key tumour suppressors may thus be having their significant impact on cancer incidence, because they occur in tumour stem cells, modulate the growth and function of both tumour epithelia and tumour stroma (and CAFs in particular) as discrete cell types, and also because they coordinate the interconversion of tumour epithelia and mesenchyme in a spatial and temporal manner. Intriguingly, p53 functions largely to repress EMT by inducing expression of miRNAs, such as miR200a/c and miR-130b, that suppress ZEB1/2, transcription factors required for EMT [53,54]. Together with work by Mitra and colleagues [26], described above, these data indicate that miRNAs may play a central role in programming the tumour microenvironment.
Nevertheless, reports of shared tumour and stromal genetic profiles remain uncommon in the literature [42,55]. One could postulate that key tumour suppressor mutations, such as those discussed above (p53, RB, PTEN ), are not always readily detectable in the stroma because not all cells in the tumour at any one time are derived from the tumour stem cell pool, and because each different population of cells is constantly in evolutionary/dynamic flux. Equally, epigenetic changes in CAFs modulating expression levels of key genes, such as p16 /INK4A/CDKN2A, and miRNAs likely also explain the absence of these mutations in some instances. In summary, a model explaining the origin of CAFs that takes account of all the data (Figure 2) is that the generation of CAFs through EMT from tumour epithelia operates in parallel with recruitment and conversion of NOFs by tumour cells into CAFs, as conventionally postulated. Moving forward with this enhanced understanding of the genetics and molecular mechanisms underlying the generation of CAFs should enable targeted approaches to eliminating these tumour-promoting cells. Such approaches may include the use of EMT inhibitors, neutralizing antibodies to key cytokines and chemokines produced by CAFs and, perhaps most innovative of all, the delivery of miRNA sponges through nanotechnology to inhibit CAF conversion.
Figure 2.
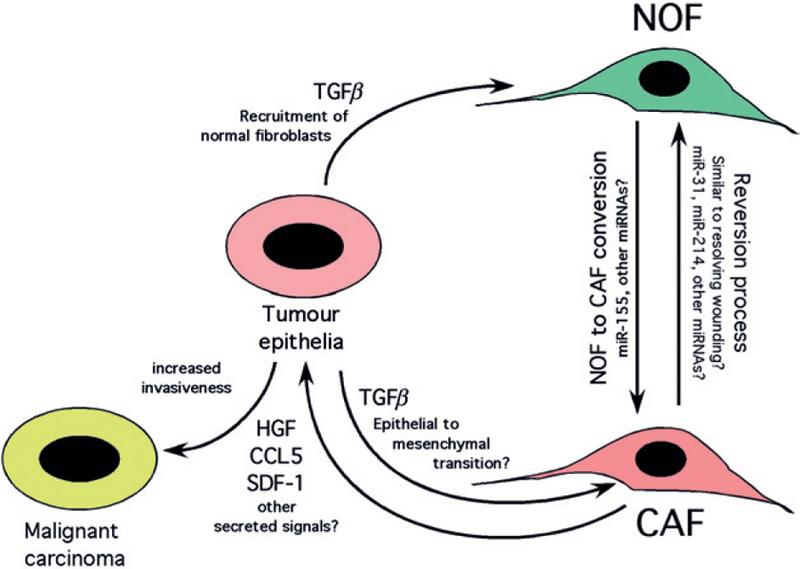
Dual mechanisms explaining the origin of CAFs and their tumour-promoting activity. Tumour cells recruit normal fibroblasts (NOFs) and promote their conversion to carcinoma-associated fibroblasts (CAFs) in part through deregulation of miRNAs, although how deregulation of these miRNAs is achieved is not known. CAFs also likely arise in some tumour types through EMT from tumour epithelia and thus harbour some of the same mutations, such as inactivation of p53, PTEN and RB, as the tumour cells from which they are derived. The extent to which CAFs are derived from NOFs as opposed to tumour epithelia via EMT is also unknown, and clearly may vary in real time as a function of selective pressures and prevalence of unique tumour clones and other cell types in the tumour microenvironment. CAFs promote tumour cell progression to invasiveness through secretion of key chemokines and MMPs, but may be susceptible to targeted therapies that promote their ‘reversion’ to NOFs, perhaps by sponging miRNAs that drive conversion, or by specific inhibitors of EMT.
Acknowledgements
The authors thank Marina Sharifi and Erin Mowers for critical reading of this manuscript. KFM is supported by a Program Project Development Grant (No. PPD/UC/01.12) from the Ovarian Cancer Research Foundation to study the tumour microenvironment.
Footnotes
No conflicts of interest were declared.
References
- 1.Coussens LM, Hanahan D. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012; 21: 309–322. [DOI] [PubMed] [Google Scholar]
- 2.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer 2008; 8: 56–61. [DOI] [PubMed] [Google Scholar]
- 3.Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet 2012; 13: 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer 2012; 12: 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moinfar F, Man YG, Arnould L, et al. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res 2000; 60: 2562–2566. [PubMed] [Google Scholar]
- 6.Kurose K, Gilley K, Matsumoto S, et al. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Gen 2002; 32: 355–357. [DOI] [PubMed] [Google Scholar]
- 7.Fukino K, Shen L, Matsumoto S, et al. Combined total genome loss of heterozygosity scan of breast cancer stroma and epithelium reveals multiplicity of stromal targets. Cancer Res 2004; 64: 7231–7236. [DOI] [PubMed] [Google Scholar]
- 8.Patocs A, Zhang L, Xu Y, et al. Breast-cancer stromal cells with TP53 mutations and nodal metastases. New Engl J Med 2007; 357: 2543–2551. [DOI] [PubMed] [Google Scholar]
- 9.Wernert N, Löcherbach C, Wellmann A, et al. Presence of genetic alterations in microdissected stroma of human colon and breast cancers. Anticancer Res 2001; 21: 2259–2264. [PubMed] [Google Scholar]
- 10.Tuhkanen H, Anttila M, Kosma VM, et al. Genetic alterations in the peritumoral stromal cells of malignant and borderline epithelial ovarian tumors as indicated by allelic imbalance on chromosome 3p. Int J Cancer 2004; 109: 247–245. [DOI] [PubMed] [Google Scholar]
- 11.Paterson RF, Ulbright TM, MacLennan GT, et al. Molecular genetic alterations in the laser-capture-microdissected stroma adjacent to bladder carcinoma. Cancer 2003; 98: 1830–1836. [DOI] [PubMed] [Google Scholar]
- 12.Hu M, Yao J, Cai L, et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Gen 2005; 37: 899–905. [DOI] [PubMed] [Google Scholar]
- 13.Bhowmick NA, Chytil A, Plieth D, et al. TGFβ signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004; 303: 848–851. [DOI] [PubMed] [Google Scholar]
- 14.DeNardo DG, Barreto JB, Andreu P, et al. CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009; 16: 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olumi AF, Grossfeld GD, Hayward SW, et al. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res 1999; 59: 5002–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuperwasser C, Chavarria T, Wu M, et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci USA 2004; 101: 4966–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayward SW, Wang Y, Cao M, et al. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res 2001; 61: 8135–8142. [PubMed] [Google Scholar]
- 18.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature 2004; 432: 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005; 121: 335–348. [DOI] [PubMed] [Google Scholar]
- 20.Kalluri R Fibroblasts in cancer. Nat Rev Cancer 2006; 6: 392–401. [DOI] [PubMed] [Google Scholar]
- 21.Ohuchida K, Mizumoto K, Murakami M, et al. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res 2004; 64: 3215–3222. [DOI] [PubMed] [Google Scholar]
- 22.Krtolica A, Parrinello S, Lockett S, et al. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA 2001; 98: 12072–12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capparelli C, Guido C, Whitaker-Menezes D, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle 2012; 11: 2285–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer 2009; 9: 81–94. [DOI] [PubMed] [Google Scholar]
- 25.Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007; 449: 557–563. [DOI] [PubMed] [Google Scholar]
- 26.Mitra AK, Zillhardt M, Hua Y, et al. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov 2012; 2: 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang HY, Chi JT, Dudoit S, et al. Diversity, topographic differentiation and positional memory in human fibroblasts. Proc Natl Acad Sci USA 2002; 99: 12877–12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rinn JL, Bondre C, Gladstone HB, et al. Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet 2006; 2: 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sugimoto H, Mundell TM, Kieran MW, et al. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther 2006; 5: 1640–1646. [DOI] [PubMed] [Google Scholar]
- 30.Orimo A, Weinberg RA. Heterogeneity of stromal fibroblasts in tumors. Cancer Biol Ther 2007; 6: e1. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J, Chen L, Xiao M, et al. FSP1+ fibroblasts promote skin carcinogenesis by maintaining MCP-1-mediated macrophage infiltration and chronic inflammation. Am J Path 2011; 178: 382–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pickard A, Cichon AC, Barry A, et al. Inactivation of Rb in stromal fibroblasts promotes epithelial cell invasion. EMBO J 2012; 31: 3092–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill R, Song Y, Cardiff RD, et al. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell 2005; 123: 1001–1011. [DOI] [PubMed] [Google Scholar]
- 34.Trimboli AJ, Cantemir-Stone CZ, Li F, et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009; 461: 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bronisz A, Godlewski J, Wallace JA, et al. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-321. Nat Cell Biol 2012; 14: 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pickard A, Cichon AC, Menges C, et al. Regulation of epithelial differentiation and proliferation by the stroma: a role for the retinoblastoma protein. J Invest Dermatol 2012; 132: 2691–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu H, Knabb JR, Spike BT, et al. Elevated Parp activity sensitizes Rb deficient cells to DNA damage-induced necrosis. Mol Cancer Res 2009; 7: 1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cichon AC, Pickard A, McDade SS, et al. AKT in stromal fibroblasts controls invasion of epithelial cells. Oncotarget 2013; 4: 1103–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.TCGA. Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474: 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Littlepage LE, Egeblad M, Werb Z. Coevolution of cancer and stromal cellular responses. Cancer Cell 2005; 7: 499–500. [DOI] [PubMed] [Google Scholar]
- 41.Orimo A, Weinberg RA. Stromal fibroblasts in cancer. Cell Cycle 2006; 5: 1597–1601. [DOI] [PubMed] [Google Scholar]
- 42.Hosein AN, Wu M, Arcand SL, et al. Breast carcinoma-associated fibroblasts rarely contain p53 mutations or chromosomal aberrations. Cancer Res 2010; 70: 5770–5777. [DOI] [PubMed] [Google Scholar]
- 43.Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012; 148: 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guerra C, Collado M, Navas C, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011; 19: 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiery JP, Acloque H, Huang RYJ, et al. Epithelial–mesenchymal transitions in development and disease. Cell 2009; 139: 871–890. [DOI] [PubMed] [Google Scholar]
- 46.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009; 9: 265–273. [DOI] [PubMed] [Google Scholar]
- 47.Ikushima H, Miyazono K. TGFβ signaling: a complex web in cancer progression. Nat Rev Cancer 2010; 10: 415–424. [DOI] [PubMed] [Google Scholar]
- 48.Chang CJ, Chao CH, Xia W, et al. p53 regulates epithelial–mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol 2011; 13: 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinho AV, Rooman I, Real FX. p53-dependent regulation of growth, epithelial–mesenchymal transition and stemness in normal pancreatic epithelial cells. Cell Cycle 2011; 10: 1312–1321. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y, Yan W, Chen X. Mutant p53 disrupts MCF-10A cell polarity in three-dimensional culture via epithelial-to-mesenchymal transitions. J Biol Chem 2011; 286: 16218–16228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arima Y, Inoue Y, Shibata T, et al. Rb depletion results in deregulation of E-cadherin and induction of cellular phenotypic changes that are characteristic of the epithelial-to-mesenchymal transition. Cancer Res 2008; 68: 5104–5112. [DOI] [PubMed] [Google Scholar]
- 52.Jiang Z, Deng T, Jones RE, et al. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J Clin Invest 2010; 120: 3296–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim T, Veronese A, Pichiorri F, et al. p53 regulates epithelial–mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med 2011; 208: 875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dong P, Karaayvaz M, Jia N, et al. Mutant p53 gain-of-function induces epithelial–mesenchymal transition through modulation of the miR–130b–ZEB1 axis. Oncogene 2013; 32: 3286–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qiu W, Hu M, Sridhar A, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Gen 2008; 40: 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]