

SUMMARY
The heterogeneity of small extracellular vesicles and presence of non-vesicular extracellular matter have led to debate about contents and functional properties of exosomes. Here, we employ high-resolution density gradient fractionation and direct immunoaffinity capture to precisely characterize the RNA, DNA, and protein constituents of exosomes and other non-vesicle material. Extracellular RNA, RNA-binding proteins and other cellular proteins are differentially expressed in exosomes and non-vesicle compartments. Argonaute 1–4, glycolytic enzymes and cytoskeletal proteins are absent from exosomes. We identify Annexin A1 as a specific marker for microvesicles that are shed directly from the plasma membrane. We further show that small extracellular vesicles are not vehicles of active DNA release. Instead, we propose a new model for active secretion of extracellular DNA through an autophagy- and multivesicular endosome-dependent, but exosome-independent mechanism. This study demonstrates the need for a reassessment of exosome composition and offers a framework for a clearer understanding of extracellular vesicle heterogeneity.
Keywords: exosomes, microvesicles, exomeres, extracellular vesicles, argonaute, extracellular RNA, extracellular DNA, annexin, autophagy, amphisomes
eTOC Blurb
A reassessment of exosome composition establishes the differential distribution of protein, RNA, and DNA between small extracellular vesicles and non-vesicular extracellular matter and establishes that small extracellular vesicles are not vehicles of active DNA release.
Graphical Abstract
INTRODUCTION
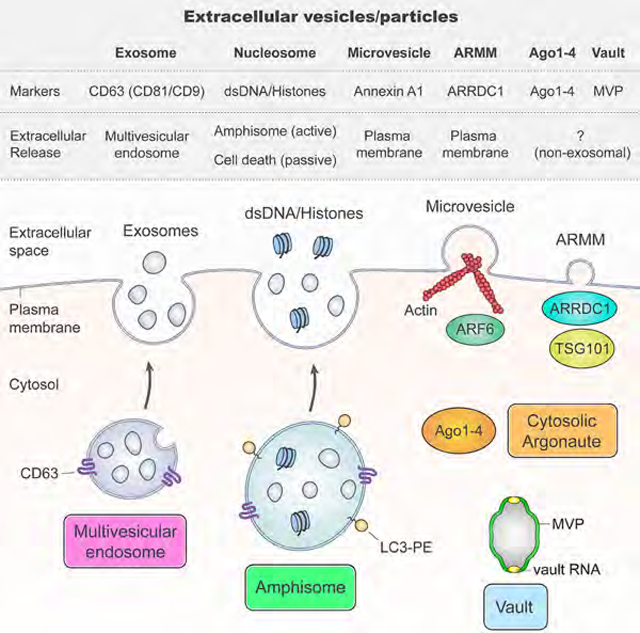
Cells release extracellular vesicles (EVs) of different sizes and intracellular origin. The heterogeneity of EVs and presence of non-vesicular extracellular nanoparticles pose major obstacles to our understanding of the composition and functional properties of distinct secreted components. Greater precision in assigning RNA, DNA and protein to their correct extracellular compartments and identifiying their mechanisms of secretion is crucial for identification of biomarkers and design of future drug interventions. Exosomes are 40–150 nm, endosome-derived, small extracellular vesicles (sEVs) secreted by most, if not all, cells. RNA (including mRNA, miRNA and other non-coding RNA), DNA and lipids are reported to be actively and selectively incorporated into intraluminal vesicles (ILVs), which reside within multivesicular endosomes (MVEs) and are the precursor of exosomes (van Niel et al., 2018). In addition to accounting for the presence of membrane proteins in exosomes, inward budding of endosomal membranes is thought to result in the engulfment of cytosolic proteins and other components to the lumen of ILVs (Mathieu et al., 2019; van Niel et al., 2018). Fusion of MVEs with the plasma membrane then releases ILVs into the extracellular space as exosomes. In contrast, microvesicles are 150–1000 nm large extracellular vesicles (lEVs) generated by shedding from the plasma membrane (Mathieu et al., 2019; van Niel et al., 2018). However, specific markers that distinguish microvesicles from exosomes are lacking.
Much of the recent interest in EVs was triggered by the discovery that exosomes function in the transport of secreted extracellular RNA (exRNA), including extracellular miRNA and mRNA transport (Skog et al., 2008; Valadi et al., 2007). Argonautes (Agos) are important miRNA-processing proteins, but the exosome-mediated secretion of human Ago proteins is an unsettled issue (Arroyo et al., 2011; Melo et al., 2014; Shurtleff et al., 2016). Other RNA-binding proteins (RBPs) have also been reported to be present in exosomes with possible roles for sorting of RNA (Mateescu et al., 2017; Shurtleff et al., 2016; Villarroya-Beltri et al., 2013). However, the heterogeneity of extracellular vesicles and nanoparticles, as well as differences in purification strategies, have confounded analyses.
Here, we employ high-resolution density gradient fractionation to separate sEVs from non-vesicular material, and direct immunoaffinity capture (DIC) to specifically isolate exosomes from other types of sEVs. DIC was performed without ultracentrifugation and using capture beads targeting classical exosomal tetraspanins. Comprehensive proteomic and nucleic acid analysis revealed that exRNA and proteins are differentially expressed between sEVs and non-vesicle compartments. Many RBPs linked to inclusion or loading of exRNA in exosomes, including Ago1–4, are not associated with classical exosomes displaying the exosomal markers CD63, CD81 and CD9 (Kowal et al., 2016; van Niel et al., 2018). Exosomes lack cytoskeletal elements and common glycolytic enzymes; the absence of these highly abundant cytosolic proteins suggests that exosome loading must be a highly regulated process. These studies were performed using human colon (DKO-1) and glioblastoma (Gli36) cancer cell lines. The major findings were validated in normal human kidney epithelial cells and human plasma. Exosomes are touted to be vehicles of extracellular DNA secretion, making them attractive targets as liquid biopsies for cancer patients. We provide evidence that double-stranded DNA (dsDNA) and DNA-binding histones are not carried by exosomes or any other type of sEV. Instead, we show that active secretion of extracellular dsDNA and histones can occur through an autophagy-and MVE-dependent, exosome-independent mechanism. Additionally, we identify Annexin A1 as a specific marker for classical microvesicles budding from the plasma membrane.
These findings clarify exosomal constituents and provide sounder footing for exploring their functional properties.
RESULTS
High-Resolution Density Gradient Fractionation Separates Small Extracellular Vesicles from Non-Vesicular Components
It is increasingly clear that “exosomal” samples contain a heterogeneous mixture of sEVs and non-vesicular compartments (operational definitions in Figure 1A and Table S1). Crude lEVs (P15) and crude sEVs (P120) were prepared from DKO-1 and Gli36 cells using conventional differential centrifugation (STAR Methods). These P120 preparations were highly enriched for the classical exosome markers CD63, CD81 and CD9 and devoid of gross contamination (Figure 1B). To further separate the membrane-enclosed sEVs from non-vesicular (NV) components, we employed high-resolution iodixanol gradients (STAR Methods). CD63 and CD81 were present in low-density fractions distinct from those containing the extracellular matrix protein Fibronectin and the ribosomal protein RPS3 (Figures 1C and S1A–C). Based on the presence of these exosomal markers, low-density fraction pools of purified sEVs and high-density pools of NV components were identified (Figures 1C and S1B–C). Subjecting the purified sEVs to a second round of density fractionation did not cause a reappearance of GAPDH and eEF1A1 in high-density fractions, indicating that components in the NV fractions are unlikely to arise de novo from damage to vesicles (Figure S1D). Only the low-density pool containing the gradient-purified sEVs was highly susceptible to detergent-mediated disruption (Figure 1D). The low-density pooled fractions contained cup-shaped vesicles of size and morphology consistent with sEVs/exosomes while these were absent from high-density fraction pools (Figures 1E and S1E–F). In summary, a high-resolution iodixanol density gradient separates small extracellular membrane vesicles bearing the hallmarks of sEVs/exosomes from non-vesicular components.
Figure 1. High-Resolution Density Gradient Fractionation Separates Small Extracellular Vesicles from Non-Vesicular Components.

(A) Nomenclature of extracellular vesicles and particles employed in this study.
(B) Immunoblots of DKO-1 and Gli36 whole cell lysates, large EVs (P15) and crude small EVs (P120) (STAR Methods). Equal quantities of protein were separated on SDS-PAGE gels, and membranes were blotted with indicated antibodies.
(C) Density gradient fractionation of DKO-1 and Gli36 crude small EVs (P120). After flotation of sample in high-resolution iodixanol gradients (STAR Methods), equal volumes of each fraction were loaded on SDS-PAGE gels, and membranes were blotted with indicated antibodies. NV, non-vesicular; sEV, small EV.
(D) Nanoparticle tracking analysis of pooled low (sEV) and high (non-vesicular) density fractions with or without pre-treatment with Triton X-100 detergent. For box plots the center lines mark the median; box limits indicate 25th and 75th percentiles; whiskers extend 1.5 times the interquartile range from 25th and 75th percentiles; n = 18 sample points; data from three independent experiments. Significant differences were assessed by one-way ANOVA and pairwise comparisons adjusted by the Holm-Bonferroni method; *p < 0.001; N.S., Not Significant.
(E) Negative stain transmission electron microscopy (TEM) of DKO-1 large EVs (P15), and pooled low (sEV) and high (NV) fractions obtained from high-resolution density gradients.
(F) Negative stain TEM of Gli36 pooled low (sEV) and high (NV) fractions. See also Figure S1 and Table S1.
Proteomic Profiling of Small Extracellular Vesicles and Non-Vesicular Fractions
To assess the protein composition of density gradient-purified sEVs and NVs, LC-MS/MS was performed (Figure S2A and Tables S2–4). Despite considerable overlap between the fractions in terms of identifiable proteins it became strikingly clear that gradient-purified sEVs are highly distinct from NVs (Figures 2A–C and S2B–C). Syntenin-1 (gene SDCBP) and ALIX (PDCD6IP) were among the most abundant proteins identified in the density gradient-purified sEV samples, and are two of the most highly expressed exosomal proteins found with more sophisticated purification methods (Kowal et al., 2016; Zhang et al., 2018). The most abundant proteins in the NV fractions were metabolic enzymes like GAPDH, PKM and ENO1, and cytosolic proteins including HSP90 and tubulins (Table S5). As expected, common exosomal markers were relatively overexpressed in gradient-purified sEVs (Figures 2D and S2D). Histones are frequently identified and among the most abundant proteins in proteomic analysis of sEVs/exosomes (Zhang et al., 2018). However, Histones H2A and H3 were highly enriched in NV fractions. LC-MS/MS results were validated, including the distinct association of histones with NV fractions (Figures 2E and S2E). Of the 25 most frequently reported proteins in the ExoCarta exosome database (Keerthikumar et al., 2016), many were more associated with NV fractions than purified sEV fractions, including GAPDH, PKM and HSP90 (Figures 2F and S2F). In summary, proteomic profiling reveals that after high-resolution iodixanol density gradient purification, some presumed exosomal proteins were more associated with NV fractions than with fractions containing sEVs/exosomes.
Figure 2. Differential Expression of Protein and RNA in Small Extracellular Vesicles and Non-Vesicular Fractions.

(A) Venn diagram representing the number of unique and overlapping proteins .
(B) Principal Component Analysis of the quantitative differences in spectral counts.
(C) Volcano plots of quantitative differences in proteins in sEV and NV fractions for DKO-1 samples. Black dots represent a four-fold or greater enrichment while orange dots represent less than four-fold enrichment. Dots above the dashed line represent proteins for which differences were significant (FDR < 0.05).
(D) Table of fold-change in spectral counts from proteomic profiling between sEV and NV pooled fractions for selected proteins chosen for validation by immunoblotting.
(E) Immunoblot validation of proteomic profiling. NV, non-vesicular; sEV, small EV.
(F) Heatmap of the 25 most commonly identified exosomal proteins from the ExoCarta exosome database form proteomic profiling of sEV and NV from DKO-1. Scale indicates intensity, defined as Δ(spectral counts – mean spectral counts)/standard deviation.
(G) Percentage of short RNA reads mapping to different types of small ncRNA for cellular and extracellular DKO-1 (left) and Gli36 (right) samples.
(H) Principal Component Analysis based on quantitative miRNA profiles.
(I) Heat map of the 50 most abundant miRNAs across all sample types. Scale indicates intensity, defined as Δ(read counts – mean read count)/standard deviation.
Differential Expression of RNA in Small Extracellular Vesicles and Non-Vesicular Fractions
Extracellular RNA (exRNA) was extracted from density gradient-purified sEV and NV pooled fractions. Distinct ribosomal RNA (rRNA) peaks (18S and 28S) were diminished in the extracellular lEV, sEV and NV samples, while small RNA species were enriched (Figure S2G). Short RNA sequencing (STAR Methods) revealed enrichment of rRNA fragments and other specific small RNAs or RNA fragments in the extracellular samples (Figure 2G). The overall pattern of miRNA expression was distinctly different not only between cellular and secreted miRNAs, but also between sEV and NV fractions (Figure 2H). Numerous miRNAs displayed significant differential distribution between purified sEV and NV fractions and between purified sEVs and their parental cells (Figure S2H and Tables S6–7). Many of the most abundant miRNAs were more associated with extracellular NV fractions than with either parental cells or sEV fractions (Figure 2I). Strong enrichment for extracellular transfer RNA (tRNA) fragments was also observed, particularly for lEV and sEV samples (Figure 2G). Y-box protein 1 (YBX1) is an RBP reported to be present in exosomes and responsible for sorting miRNA and tRNA into exosomes (Shurtleff et al., 2016; Shurtleff et al., 2017). However, YBX1 was not detected in either sEV or NV samples released from DKO-1 cells and was more abundant in Gli36 NV fractions than sEV fractions after gradient purification (Figure S2I). YBX1 is responsible for sorting miR-223 and miR-144 into HEK293T-derived exosomes (Shurtleff et al., 2016); however, both were detected with very low abundance in our short RNA-seq of DKO-1 and Gli36 cells and extracellular samples (Figure S2J). The 3’ termini of cellular miRNAs and purified sEV miRNAs were broadly similar, but the NV fraction miRNAs displayed marked trimming (Figure S2K). YRNAs and vault RNAs (VTRNA) were enriched in extracellular samples, with VTRNA in particular associated with NV fractions (Figure 2G). Long RNA sequencing revealed that the distribution of transcripts among lEVs, sEVs and NV fractions were similar, while protein-coding transcripts comprised a larger percentage of cellular samples compared to extracellular samples (Figure S2L). Strikingly, the overwhelming majority of cellular long RNA reads mapped to exonic regions, while the majority of extracellular reads mapped to intronic regions (Figure S2M). The abundance of lincRNA was greater for all extracellular samples compared to parental cells (Figure S2N) and the patterns of lincRNA expression were clearly distinct (Figure S2O). YRNA, and fragments or processed versions of YRNA, have repeatedly been reported to be present and enriched in crude samples of sEVs compared to cells (Chakrabortty et al., 2015; Nolte-’t Hoen et al., 2012). By short RNA-seq, we observed that YRNA was also enriched in lEVs and gradient-purified NV fractions (Figures 2G and S2P). However, when looking at primary full-length transcripts by long RNA-seq, extracellular enrichment was much less pronounced, but, once again, substantial amounts of full-length YRNA were associated with NV fractions (Figure S2Q). In summary, exRNAs are differentially distributed between purified sEV and extracellular non-vesicular fractions.
Secretion of Human Argonaute 1–4 is Independent of Exosomes
Exosome-mediated secretion of human Ago proteins and other components of the miRNA biogenesis machinery and RISC (Figure 3A) is an unsettled controversy (Arroyo et al., 2011; Melo et al., 2014; Shurtleff et al., 2016). Human Ago proteins are released from cells and can be recovered in the 120,000 × g pellet (P120) following ultracentrifugation (Figures 3B and S3A). None of the other common miRNA-associated proteins were detected in extracellular lEVs (P15) and crude sEVs (P120), including Drosha, DGCR8, Dicer, GW182 (gene TRNC6A) and TRBP (Figures 3B and S3A). Ago1 and Ago2 proteins can be found intracellularly in membrane-less cytoplasmic organelles processing bodies (P-bodies) and stress granules (Hubstenberger et al., 2017; Jain et al., 2016). The mRNA-decapping enzyme DCP1A is a canonical marker for P-bodies while G3BP1 and G3BP2 are canonical markers for stress granules (Hubstenberger et al., 2017). The existence of so-called “GW-bodies”, distinct from P-bodies and lacking DCP1A, has been suggested based on the localization of Ago2 with GW182 at MVEs (Gibbings et al., 2009). It is unlikely that extracellular Ago proteins arise from gross contamination of sEV samples with P-bodies, stress granules or GW bodies as DCP1A, GW182, Drosha and Dicer were all absent from crude sEVs (P120) (Figures 3B and S3A), and G3BP1 and G3BP2 were less abundant in extracellular samples compared to cells (Figure S3B). It is therefore unlikely that extracellular Ago proteins arise from gross contamination of sEV samples with P-bodies, stress granules or GW bodies. Detection of human Ago1–4 proteins was not due to contamination from bovine growth serum (Figure S3C). Ago2 was present in samples of crude sEVs isolated from freshly resected colorectal cancer tissue samples and adjacent grossly normal tissue (STAR Methods); however, the other miRNA-associated proteins DGCR8, Dicer and TRBP, were not (Figure 3C). In human plasma, Ago2 and HDL (ApoA1) were present in gradient-purified NV pool, but absent from the sEV pool (Figures 3D and S3D). The two ESCRT proteins TSG101 and ALIX were enriched in crude sEVs (P120) from cancer cell lines (Figure S3E) and gradient-purified sEVs (Figures 2E–F and 3E), but undetectable in plasma sEV (P120) samples (Figures S3E–F). All four human Ago proteins were strongly associated with NV fractions after high-resolution density gradient purification (Figures 3E and S3G), although the Gli36 cell line also had a minor population of Ago1–4 that was associated with purified sEV fractions (Figure 3E). The heat shock proteins HSP90 and HSC70, necessary for loading duplex miRNA into Ago proteins, were present in purified sEV and NV fractions (Figures 3E, 2F and S3D–F). To independently validate our high-resolution gradient density fractionation, we developed a DIC approach designed to specifically purify classical exosomes bearing the canonical tetraspanin markers CD63, CD81 and CD9 (Figure 4A). The term “direct” refers to two aspects of this approach: the use of magnetic beads directly conjugated to capture antibody, and the addition of the beads directly to sEV samples still in solution without any pre-concentration or ultracentrifugation steps to avoid aggregation/contamination of exosomes with other non-exosomal sEVs or NV components. CD81-, CD63-and CD9-positive classical exosomes did not contain Ago2, and CD81-positive exosomes did not contain Ago1, 3 or 4 (Figure 4B). Though we observed minor populations of Ago1–4 in Gli36 density gradient-purified sEV fractions, Ago proteins were absent in DIC-purified Gli36 exosomes, indicating that sEVs potentially carrying Ago1–4 must be distinct from exosomes (Figure 4C). Surprisingly, HSP90 was undetectable in classical exosomes (Figure 4B–C). In summary, cells secrete Ago1–4 independently of exosomes. No evidence could be uncovered that exosomes, or any other type of sEV, contain other major components of the miRNA biogenesis machinery.
Figure 3. Secretion of Human Argonaute and miRNA Biogenesis Machinery.

(A) Schematic of human miRNA biogenesis. Pri-miRNA is transcribed by RNA polymerase II and cropped by the Drosha/DGCR8 microprocessor complex to generate pre-miRNA. After export to the cytoplasm, Dicer cleaves the stem loop supported by TRBP. The miRNA/miRNA duplex is then loaded into Ago in an HSP90/HSC70-dependent manner. The miRNA duplex is unwound and the passenger strand is ejected. Ago and GW182 form the mature RISC complex necessary for gene silencing. Insert shows common proteins found in “GW bodies”, processing bodies and stress granules with * denoting proteins with increased abundance in conditions of cellular stress.
(B) Immunoblots of whole cell lysates, large EVs (P15) and crude small EVs (P120).
(C) Immunoblots of cell lysates from four colorectal cancers (CRC), adjacent normal and lymph node metastasis, and crude small EVs (P120) isolated from matched tissue/interstitial fluid (STAR Methods). N, normal; T, tumor; LM, lymph node metastasis.
(D) Immunoblots of high-resolution density gradient-purified sEV and NV samples isolated from plasma of three normal individuals (see Figure S3D).
(E) Immunoblots of high-resolution density gradient fractionation of crude small EVs (P120).
See also Figure S3.
Figure 4. Extracellular Release of RNA-Binding Protein and Vaults.

(A) Schematic illustration of the direct immunoaffinity capture (DIC) procedure. Magnetic beads directly conjugated to anti-CD63, anti-CD81, anti-CD9 antibodies or IgG were added directly to pre-cleared cell culture medium (STAR Methods) without prior ultracentrifugation or concentration. In parallel, conventional crude sEVs (P120) were prepared form the same pre-cleared cell culture medium.
(B-C) DIC of CD81-, CD63-and CD9-positive exosomes from (B) DKO-1 or (C) Gli36. Immunoblots of crude sEV pellet (P120) and bead-captured exosomes.
(D) Immunoblots of whole cell lysates, large EVs (P15) and crude small EVs (P120) obtained by ultracentrifugation.
(E) Immunoblots of high-resolution density gradient fractionation of crude small EVs (P120).
(F-H) DIC of CD81-, CD63-and CD9-positive exosomes. Immunoblot of crude sEV pellet (P120) and bead-captured exosomes from (F) DKO-1, (G) Gli36, and (H) cultured primary human renal epithelial cells. g.e, greater exposure.
(I) Structure and molecular composition of vaults.
(J) Non-fixed negative stain TEM of DKO-1 and Gli36 NV fractions. Red arrows indicate vault structures.
(K) Proteomic analysis of vault-associated proteins in purified sEV and NV generated by gradient density centrifugation. Data are mean ± SD. *p < 0.00001 for the quasiFDR.
(L) Short RNA-seq data for vault RNA in DKO-1 cells, large EVs (lEV), sEV and NV pooled fractions. RPM, reads per million.
(M) Long RNA-seq data for vault RNA in DKO-1 cells, large EVs (lEV), purified sEV and non-vesicular (NV) pooled fractions. FPKM, fragments per kilobase million.
See also Figure S4.
Extracellular Release of RNA-Binding Proteins and Vaults
Multiple RBPs have previously been linked to exosomes. We investigated hnRNPA2B1 (poly-A RNA-binding, miRNA), RPS3 and RPS8 (ribosome), EEF2 (translation), eEF1A1 (tRNA), PARK7/DJ1 (poly-A RNA-binding), GAPDH (unclear RNA-binding) and MVP (VTRNA), as these are among the top RBPs reported in exosomes in their respective functional category (Mateescu et al., 2017). Sorting of miRNA into exosomes has been attributed to sumolyation of the ribonucleoprotein hnRNPA2B1 (Villarroya-Beltri et al., 2013). Neither hnRNPA2B1, nor its approximately 10–12 kDa larger sumoylated form, could be detected in crude sEV (P120) preparations (Figure 4D). Ribosomal proteins are commonly found in exosomal preparations even when attempts have been made to improve sample purity beyond that obtainable by standard ultracentrifugation protocols (Kowal et al., 2016; Zhang et al., 2018). However, RPS3 and RPS8 were both associated with NV fractions (Figures 4E and S1B–C) and RPS3 and RPS8 were absent from exosomes bearing the classical tetraspanin markers (Figure 4F–G). GAPDH is widely reported to be present in exosomes and microvesicles (van Niel et al., 2018), but GAPDH was more associated with NV fractions than sEV fractions (Figures 1C, 2F, S1D and S2F). By DIC, GAPDH was not detectable in CD63-, CD81-and CD9-positive exosomes (Figure 4F–H). EEF2 was mostly associated with extracellular NV fractions, while some eEF1A1 was found to float at densities consistent with its presence in sEVs (Figures 4E, 2F, S1D and S2F); but neither protein was present in classical exosomes (Figure 4F–H). PARK7/DJ1 was distinctly associated with flotation in sEV fractions (Figure 4E), but, once again, not present in classical exosomes (Figure 4F–G). Ago2, GAPDH and EEF2 were not detected in CD81-positive exosomes derived from primary human renal epithelial cells (Figure 4H).
MVP is a proposed miRNA-binding protein responsible for sorting miRNA to exosomes (Teng et al., 2017). MVP was found in NV fractions (Figure 4E) and confirmed by DIC to not be associated with exosomes (Figure 4F–H). MVP is the major protein component (>70%) of vaults, large 41 nm by 72.5 nm ribonucleoprotein cytoplasmic particles (Figure 4I) (Rome and Kickhoefer, 2013). Humans express four non-coding cytoplasmic VTRNAs: VTRNA1–1 (98 bases), VTRNA1–2, VTRNA1–3 (88 bases) and VTRNA2–1 (102 bases) single exon transcripts. Some of the VTRNA also associate with Lupus La protein (La), although La is not present in highly purified vault samples (Rome and Kickhoefer, 2013). Analysis revealed intact vault structures in the NV pool (Figure 4J), while no such structures were observed in purified sEV pools (Figures 1E and S1E). The three vault proteins MVP, VPARP, and TEP1 were greatly enriched in the NV pool as was La protein (Figure 4K). Enrichment of VTRNA in exosomes has previously been reported (Nolte-’t Hoen et al., 2012). Short RNA-seq analysis confirmed enrichment of reads mapped to extracellular VTRNA with marked enrichment in the NV pool (Figure 4L). In contrast, long RNA-seq revealed marked cellular enrichment of VTRNA compared to extracellular samples (Figure 4M). Thus, extracellular VTRNA is predominantly either processed small VTRNA or fragments of VTRNA. In summary, the commonly reported exosomal RBPs investigated (Ago1–4, RPS3, RPS8, EEF2, eEF1A1, hnRNPA2B1, PARK7/DJ1, GAPDH and MVP) were absent from classical exosomes when advanced purification methods were employed.
Exosomes Exclude Common Glycolytic Enzymes and Lack a Cytoskeleton
As noted above, the glycolytic enzyme GAPDH was absent from classical exosomes (Figure 4F–H). Prompted by this finding, we examined Pyruvate Kinase M1/2 (PKM) and Enolase 1 (ENO1), two abundant cytosolic glycolytic enzymes that are among the 25 most commonly identified exosomal proteins. PKM was enriched in NV fractions, but ENO1 was enriched in sEV fractions after high-resolution density gradient fractionation (Figures 2F and S2F). However, both PKM and ENO1 were completely undetectable in classical exosomes when probed by DIC (Figure S4A). The widely expressed 14-3-3 proteins are involved in signal transduction and, as expected, enriched in sEV fractions (Figures 2F and S2F) but, surprisingly, absent from CD81-positive exosomes (Figure S4A). Actin, tubulin and keratins are highly abundant cellular cytoskeletal proteins observed in exosomal/sEV and microvesicle/lEV samples, even when more sophisticated purification techniques are employed (Kowal et al., 2016; Mathivanan et al., 2010; Zhang et al., 2018). Actins, tubulins and keratins were also present in our proteomic datasets for sEVs, including Keratin 8, 10, 18 and 19 (Tables S3 and S4). The presence of Keratin 8+18 and 19 in crude sEVs (P120) was confirmed (Figure S4B), while Keratin 10, a skin keratin notorious for being a contaminant in mass spectrometry analyses, was not detected by immunoblotting (data not shown). α-Actinin-4 is an actin-binding cytoskeleton protein reported to be enriched in lEVs over sEVs (Kowal et al., 2016); we observed a strong enrichment of α-Actinin-4 in lEVs (P15) (Figure S4B). Extracellular β-actin was present in one pool, consistent with its presence in sEVs, and one pool consistent with NV fractions; α-Actinin-4 was associated with the lightest fractions and α-tubulin with NV fractions (Figure S4C). We did not find evidence that classical CD63-, CD81-and CD9-positive exosomes possess the cytoskeletal constituents that make up actin filaments, microtubules or intermediate filaments (Figure S4D). Integrins on tumor exosomes have been reported to predict organ-specific metastasis (Hoshino et al., 2015). Integrin β1 was barely detectable on CD81-and CD63-positive exosomes while Integrin α2 was more abundant (Figure S4E–F), suggesting that Integrin β1 is more associated with non-exosomal sEVs. Considering that Integrin β1 is enriched in 300–900 nm microvesicles (Muralidharan-Chari et al., 2009), these vesicles may be sub-200 nm microvesicles. EGFR was present in classical exosomes, as previously reported (Higginbotham et al., 2016), but other sEVs must account for a significant part of extracellular EGFR (Figure S4E–F). This was also the case for the plasma membrane marker Na+/K+-ATPase (Figure S4E–F). Several members of the RAB GTPase family of proteins are implicated in sorting and trafficking of MVEs to the plasma membrane including RAB7, RAB11 and RAB27A (van Niel et al., 2018). However, these proteins do not appear to be informative markers for exosomes per se as relatively little of the extracellular RABs appear to be present in classical exosomes (Figure S4E–F). In summary, exosomes exclude highly abundant cytosolic enzymes, are vesicles without cytoskeletons, and proteins involved in exosome biogenesis cannot be assumed to be constituents of exosomes.
Annexin A1 is a Novel and Specific Marker of Microvesicles Distinct from Exosomes and ARMMs
Annexins are abundant membrane-associated proteins that have been identified as exosomal constituents (Kowal et al., 2016; van Niel et al., 2018; Zhang et al., 2018). Annexins were greatly enriched in DKO-1 and Gli36 purified sEV fractions (Figure 5A). Annexin A1, A2, A3 and V were enriched in lEVs (P15) compared to gradient-purified sEVs while the opposite was true for Annexin VII and XI (Figure 5A–B). The expression of Annexin A1 and A2 was shifted toward the lower densities compared to the exosomal marker CD9 (Figure 5C). Using a different formulation of the iodixanol gradient (Figure S5A, STAR Methods), designed to increase resolution at the lower flotation density range of sEVs, the buoyant density pattern of Annexin A1-and A2-positive sEVs was markedly shifted towards lower densities compared to TSG101 and ALIX (Figure 5D). Annexin A1 and A2 also floated at decidedly lower densities than the two lipid raft proteins, Flotillin-1 and Flotillin-2, while the epithelial marker EpCAM overlapped with both groups (Figure 5D). Both Annexin A1 and A2 were undetectable in CD81-, CD9-and CD63-positive exosomes (Figures 5E and S5B–D). Annexin V is a commonly observed exosomal protein (Figures 2F and S2F), but while the protein floats at densities consistent with EVs (Figure 5C), it was absent from CD63-, CD81-and CD9-positive exosomes (Figures 5E and S5B–D). Two other Annexins, VII and XI, both had flotation densities closer to that of CD9 than did Annexin A1 and A2 (Figure 5C). Annexin XI may be common to several types of sEV (Kowal et al., 2016), and we confirmed its presence in CD81-and CD9-positive exosomes (Figures S5B–C). The same pattern was observed for Annexin VII (Figure S5B–C). As Annexin A1 was absent from exosomes but present in samples of both small and large EVs, we investigated the possibility that Annexin A1 marks microvesicles. Structured illumination microscopy (SIM) revealed that Annexin A1 strongly stained the plasma membrane, and Annexin A1-positive vesicles could be observed to bud directly from the plasma membrane (Figures 5F and S5E). Annexin A1-positive vesicles had a size distribution of ~150 nm to 1 μm, consistent with classical microvesicles (Figure 5F and Table S1). The resolution limit of SIM did not allow vesicles smaller than ~150 nm to be conclusively resolved as budding vesicles. In addition to classical microvesicles, we also observed very large (~1–5 μm) plasma membrane blebbing (Figure 5G). The structure and size distribution of these protrusions were remarkably similar to that of large oncosomes released from prostate cancer cells (Minciacchi et al., 2017). ARF6 regulates abscission and shedding of microvesicles (Muralidharan-Chari et al., 2009) and possibly large oncosomes (Minciacchi et al., 2015). ARF6 was enriched in both lEVs and sEVs (Figure 5H), again suggesting the presence of potential <200 nm microvesicles. Arrestin-domain-containing protein 1 (ARRDC1)-mediated microvesicles (ARMMs) are a relatively recently described type of small (~40–100 nm) EV that bud directly from the plasma membrane, mediated by ARRDC1 and TSG101 (Nabhan et al., 2012; Wang and Lu, 2017). Both ARRDC1 and TSG101 were highly associated with sEVs, but much less so with lEVs (Figure 5H). ARRDC1 was largely absent from CD63-and CD81-positive DKO-1 vesicles (Figures 5I and S5B), while a faint signal for ARRDC1 was detected in CD9-positive DKO-1 vesicles (Figure 5I). In contrast, robust signals were observed for ARRDC1 in CD81-and CD9-positive Gli36 vesicles with expression levels similar (CD81-positive) or enriched (CD9-positive) compared with that of matched crude sEVs (P120) (Figure 5J). Expression of TSG101 was very weak in CD81-and CD63-positive vesicles, but stronger in CD9-positive vesicles (Figures 5I–J and S5B), suggesting TSG101 may be more associated with ARMMs than classical exosomes. In contrast, ALIX and Syntenin-1 were strongly associated with CD63-, CD81-and CD9-positive vesicles (Figures 4C, 4F–G, 5I–K and S4E–F). Annexin A1 expression spanned the range from small (~150 nm) to large (~1000 nm) EVs while ARMMs are small (~40–100 nm). Annexin A1 and A2 were absent from the tetraspanin-enriched vesicles, while ARRDC1 and TSG101 were present. Annexin A1 and A2 were enriched in lEVs, whereas ARRDC1 and TSG101 were enriched in sEVs. Based on the sum of the evidence, Annexin A1-and/or Annexin A2-positive EVs are distinct from both classical exosomes and ARMMs. Annexin A1-and A2-positive sEVs could be gradient-purified from human plasma (Figure S5F). DIC-purified CD81-positive plasma exosomes did not express Annexin A1 and A2, but did express the exosomal markers CD9, Syntenin-1 and Flotillin-1, as well as Annexin XI (Figure 5K). In summary, Annexin A1 and A2 are novel markers of population(s) of EVs distinct from classical exosomes and ARMMs. Biogenesis of Annexin A1-positive 150–1000 nm vesicles entails shedding from the plasma membrane, making Annexin A1 a specific molecular marker of classical microvesicles.
Figure 5. Annexin A1 is a Novel and Specific Marker of Microvesicles Distinct from Exosomes and ARMMs.

(A) Proteomic analysis of extracellular Annexins. (Left) Spectral counts for Annexins for cells, lEVs (P15) and gradient-purified sEV and NV samples for DKO-1, and (Right) for gradient-purified sEV and NV samples for Gli36. Data represent mean ± SD. n = 6.
(B) Immunoblot analysis of Annexin expression in cells, lEVs (P15), and crude sEVs (P120).
(C) High-resolution (12–36%) density fractionation of crude DKO-1 sEVs (P120).
(D) Gradient (6–30%) density fractionation of crude DKO-1 sEVs (P120).
(E) DIC of CD81-and CD9-positive exosomes from DKO-1 cells. Immunoblots of crude sEV pellet (P120) and bead-captured exosomes. g.e, greater exposure.
(F) Annexin A1-positive classical microvesicles. (Left) 3D Structured Illumination Microscopy (SIM) of DKO-1 cells stained for Annexin A1 and DAPI. Enlarged inserts displayed in greyscale at the bottom. (Right) Size distribution of Annexin A1-positive classical microvesicles imaged by 3D SIM of DKO-1 cells. Histogram of maximum width of vesicles shed at the plasma membrane. n = 221; data from four independent experiments.
(G) Annexin A1-positive large oncosomes. (Left) 3D SIM of DKO-1 cells stained for Annexin A1 and DAPI. Enlarged inserts displayed in greyscale at the bottom. (Right) Size distribution of Annexin A1-positive classical large oncosomes imaged by 3D SIM of DKO-1 cells. Histogram of maximum width of large oncosomes blebbing at the plasma membrane. n = 82; data from four independent experiments.
(H) Proteomic analysis of ARF6, ARRDC1 and TSG101 present in DKO-1 cells, lEV (P15), density gradient-purified sEV and NV fraction pools. Data are mean ± SD. n = 6.
(I-K) DIC of CD81-and CD9-positive exosomes from (I) DKO-1, (J) Gli36, and (K) human plasma. g.e, greater exposure.
See also Figure S5.
Release of Extracellular DNA and Histones from Human Cells is Independent of Exosomes and Small Extracellular Vesicles
It has previously been reported that dsDNA is present in cancer exosomes or sEVs (Montermini et al., 2015), and several studies identified dsDNA-binding histone proteins in exosomal preparations (Takahashi et al., 2017; Zhang et al., 2018). However, the observation that dsDNA-binding histones were associated with NV fractions rather than purified sEVs (Figures 2D–E and S2) prompted us to examine the release of DNA. Histones H2A, H3 and H4 were absent from the fractions dominated by exosomal/sEV marker proteins (Figures 6A and S6A). After DNA extraction from purified sEV and NV pools, it was apparent that dsDNA is present in high-density NV fractions, with post-extraction treatment of the purified nucleic acid with DNase and RNase confirming isolation of pure DNA (Figures 6B and S6B–C). To determine if extracellular dsDNA is protected from digestion, we pre-treated crude sEV (P120) samples with DNase I before proceeding with density gradient fractionation and DNA extraction. As before, no dsDNA was detected in sEV fractions, and the dsDNA present in NV fractions was highly susceptible to digestion by DNase I (Figures 6C and S6B). The majority of DNA was in the range of 1,000–10,000 base pairs with a peak around 6,000 base pairs (Figures 6D and S6D). Histones were not detected in association with CD81- or CD63-positive exosomes (Figure 6E). Likewise, it was not possible to detect any dsDNA associated with DIC-purified CD81-positive exosomes, confirming that exosomes do not natively contain dsDNA and do not associate with other particles/proteins that contain dsDNA (Figure 6F). The presence of histones and dsDNA in preparations of exosomes and other sEVs therefore appears to be an indicator of insufficient purification that largely disappears with improved methodology. In summary, extracellular dsDNA and histones are not associated with exosomes or any other type of sEV.
Figure 6. Release of Extracellular dsDNA and Histones from Human Cells is Independent of Exosomes and Small Extracellular Vesicles.

(A) Immunoblots of high-resolution density gradient fractionation of crude small EVs (P120).
(B) Quantification of DNA from gradient-fractionated sEV and NV pools extracted for DNA, and treated post-extraction with DNase I or RNase A/T1 to confirm identity as DNA. Data are mean ± SD. N.D., Not Detected; NV, non-vesicular; sEV, small EV.
(C) Quantification of DNA from samples pre-treated with DNase I (to eliminate unprotected DNA) before density gradient fractionation and extraction of DNA. Data are mean ± SD. N.D., Not Detected.
(D) Bioanalyzer electropherograms of size distribution of purified DNA in base pairs (bp) from DNase I pre-treated density gradient purified sEV and NV. DNA marker peaks at 35 bp and 10,380 bp. FU: fluorescence units.
(E) DIC of CD81-and CD63-positive exosomes.
(F) DIC of CD81-positive exosomes. DNA was extracted from bead-captured material and flowthrough material pelleted at 120,000 × g (P120). Data are mean ± SD.
See also Figure S6.
Active Secretion of DNA and Histones Through an Amphisome-Dependent Mechanism
It has been reported that dsDNA can be present inside MVEs (Takahashi et al., 2017) and cytoplasmic chromatin/DNA has recently been recognized as a driver of carcinogenesis and metastasis (Bakhoum et al., 2018; Dou et al., 2015). To investigate the presence of cytoplasmic DNA, we turned to multi-color SIM imaging. The presence of cytoplasmic DNA in the form of micronuclei was a consistently observable phenomenon (Figures 7A and S7A). Strikingly, CD63-positive intracellular compartments were readily observed among the micronuclei, while staining with CD9 was punctate (Figures 7A–B and S7A–B). The size and morphology of CD63-positive compartments were consistent with their identity as MVEs. Smaller MVEs frequently displayed a simpler and sometimes hollow appearance, whereas larger MVEs frequently displayed a multi-compartment appearance with internal staining (Figures 7C and S7C). Some CD63-positive compartments to co-localized with dsDNA, but peak staining intensities tended to be offset from each other, indicating that dsDNA does not co-localize with CD63-positive ILVs inside the larger compartment (Figure 7D). Histone H3 was also observed in some CD63-positive, MVE-like compartments (Figure 7E). Though the biochemical data clearly demonstrated that extracellular dsDNA and histones were not released in exosomes and sEVs (Figure 6), it was also apparent that cytoplasmic chromatin could be localized to CD63-positive compartments. In autophagy, the autophagosome fuses with the lysosome to generate an autolysosome for degradation of autophagosomal cargo. However, in a pre-lysosomal step, the autophagosome may undergo fusion with an MVE to form a hybrid organelle termed an amphisome (Eskelinen, 2005; Fader et al., 2008; Hessvik et al., 2016; Klionsky et al., 2014). Both the autophagosomal proteins p62 (Hessvik et al., 2016) and LC3 (Chen et al., 2017) co-localize with CD63-positive MVEs, indicative of amphisomes. Localization of p62 in CD63-positive compartments was observed, suggesting amphisomes (Figure 7F). For autophagosome formation to occur, cytosolic LC3 must be conjugated with phosphatidylethanolamine (PE) to generate the membrane-inserting LC3-PE form. Amphisomes were observed as hybrid organelles bearing both CD63 and LC3B (Figures 7G and S7D), suggesting some of the CD63-positive compartments previously observed to contain dsDNA and Histone H3 might represent amphisomes rather than MVEs. LC3B-PE interacts with Lamin B1 to mediate the autophagy-dependent degradation of cytoplasmic chromatin (Dou et al., 2015). We could localize dsDNA to CD63-and LC3B-positive amphisomes and some amphisomes were observed at the plasma membrane (Figure 7H). This indicates that rather than fusing with the lysosome the amphisomes may fuse with the plasma membrane. The presence of LC3-PE has been suggested to be a feature of autophagy-related exosomes (Chen et al., 2017; Hessvik et al., 2016). Both large and small EVs released from DKO-1 cells express LC3B-PE with particular enrichment in lEVs (P15) (Figure 7I), and LC3B-PE was detected in crude sEV preparations from CRC samples (Figures 7I and S7E). However, the flotation density pattern of LC3B-PE sEVs differed from that of the exosomal marker protein Syntenin-1 (Figure 7J), which suggested that LC3B-PE-expressing sEVs are not exosomes. DIC conclusively demonstrated that LC3B-PE and p62-expressing autophagic extracellular vesicles (AEVs) are distinct from CD63-, CD81-, and CD9-positive exosomes (Figure 7K). Since dsDNA and histones, while present in crude sEV samples, are exclusively released as non-vesicular entities, sEV-sized AEVs are not carriers of extracellular chromatin. Rather, the evidence supports a mechanism involving engulfment of cytoplasmic chromatin by an LC3B-PE-positive autophagosome, followed by merging with a CD63-positive MVE to form an amphisome that, upon fusion with the plasma membrane, releases dsDNA and histones independently of ILVs/exosomes (Figure 7L). This model of active secretion reconciles the major findings that 1) extracellular dsDNA and histones are not present in exosomes/sEVs, 2) cytoplasmic dsDNA and histones are localized to CD63-positive intracellular compartments, but not ILVs, 3) intracellular CD63-and LC3B-positive amphisomes localize with dsDNA, 4) cytoplasmic chromatin is cleared by an LC3B-dependent mechanism, and 5) CD63-and LC3B-positive amphisomes localize to the plasma membrane. In summary, dsDNA and histones co-localize with CD63-positive compartments, but dsDNA does not co-localize with ILVs. LC3B-and CD63-positive amphisomes contain dsDNA and amphisomes can be visualized at the plasma membrane. We therefore propose a model of active secretion of cytoplasmic DNA and histones through an autophagy-and MVE-dependent, but exosome-independent mechanism.
Figure 7. Active Secretion of DNA and Histones through an Amphisome-Dependent Mechanism.

(A) DKO-1 cells stained for DAPI, and endogenous CD63 and CD9, and imaged with 3D SIM. (Top) Z-stack projections. (Bottom) Enlarged inserts. See also Figure S7A.
(B) Z-slice image from (A), and greyscale enlarged insert.
(C) Size and structure of CD63-positive multivesicular endosomes (MVEs) by 3D SIM. (Top) Two types of CD63-positive MVE structure. Cropped images of DKO-1 cells stained for CD63. (Bottom) Data are mean ± SD. n = 57, data from five independent experiments.
(D) Cropped images of DKO-1 cells localized for CD63 and double-stranded DNA (dsDNA). (Left) Greyscale CD63 and dsDNA, and colorized merge. Yellow arrows in dsDNA channel indicate dsDNA peak intensity in line scans. Line scans were performed at yellow dotted lines in colorized merge. (Right) Line scans indicate drop of CD63 signal at dsDNA peak intensity. For line scan graphs, individual channels were normalized to display on graph. Scale bars, 250 nm.
(E) Cropped images of DKO-1 cells localized for CD63 and Histone H3 by 3D SIM. (Left) Greyscale CD63 and Histone H3, and colorized merge. Displayed are sequential image slices (125 nm) through the Z-stack. (Right) Enlarged image of the CD63-positive MVE with yellow line indicating longest axis. Scale bar, 200 nm.
(F) Cropped images of DKO-1 cells localized for CD63 and p62 by 3D SIM. (Left) Greyscale CD63 and p62, and colorized merge. Displayed are sequential image slices (125 nm) through the Z-stack. (Right) Enlarged image of the CD63-positive MVE with yellow line indicating longest axis. Scale bar, 200 nm.
(G) CD63-and LC3B-positive amphisomes in DKO-1 cells. Cells were stained for endogenous CD63 and LC3B. Greyscale CD63 and LC3B, colorized merge, and enlarged inserts. Yellow arrows in LC3B channel and enlarged inserts indicate LC3B localized to CD63-positive compartments. Scale bars for enlarged inserts, 200 nm.
(H) Localization of dsDNA in CD63-, LC3B-positive amphisomes in DKO-1 cells. (Left) Cropped images of cells localized for endogenous CD63, LC3B and dsDNA and imaged by three-color 3D SIM. X-Y axis “top-down view” of greyscale CD63, LC3B and dsDNA, and colorized merge. (Right) X-Z axis “side view”. Yellow arrow indicates CD63-positive compartment, and blue arrow indicates LC3B-positive compartments, both co-localized at the plasma membrane. Scale bars, 200 nm.
(I) Detection LC3B-PE positive EVs. (Left) Immunoblots of DKO-1 whole cell lysates, large EVs (P15) and crude small EVs (P120). (Right) Immunoblots of whole cell lysates and crude small EVs (P120) isolated from matched tissue/interstitial fluid of a colorectal cancer (CRC), adjacent normal and lymph node metastasis. N, normal; T, tumor; LM, lymph node metastasis.
(J) Gradient (6–30%) density fractionation of crude DKO-1 sEVs (P120).
(K) DIC of CD81-, CD63-and CD9-positive exosomes. Immunoblots of crude sEV pellet (P120) and bead-captured exosomes from DKO-1 (top) and Gli36 cells (bottom).
(L) Model of amphisome-dependent, exosome-independent secretion. For autophagy, cytosolic LC3 is lipidated by conjugation with phosphatidylethanolamine to form LC3-PE.
1) Nuclear membranes can bleb in a process dependent on LC3B and the nuclear lamina protein Lamin B1, causing the appearance of cytoplasmic chromatin fragments. 2) During autophagy, cytoplasmic components are sequestered as a phagophore begins to engulf material. 3) Continued expansion of autophagic membranes requires LC3-PE and results in formation of the double-membrane autophagosome. 4) As early endosomes develop to late endosomes, the pH decreases and continued invagination of limiting membranes generates intraluminal vesicles (ILVs). A fully developed CD63-positive multivesicular endosome (MVE) contains numerous ILVs. 5) Fusion of the autophagosome with a MVE causes degradation of the inner autophagosome membrane generating an amphisome, a single-membrane hybrid compartment. 6) The amphisome fuses with a lysosomal compartment to form the autolysosome followed by degradation of cargo, or alternatively, 7) the amphisome fuses with the plasma membrane causing extracellular release of dsDNA and histones, and separately, the ILVs as exosomes.
See also Figure S7.
DISCUSSION
Using two different yet highly complementary techniques, high-resolution density gradient fractionation and DIC, we demonstrate separation of sEVs from extracellular NV compartments, and the ability to include or exclude a molecular component to classical exosomes. Ultracentrifugation (≥100,000 × g) causes aggregation of vesicles (Jeppesen et al., 2014a; Linares et al., 2015) and to ensure that the DIC approach was not compromised, we used samples that had not been subjected to ultracentrifugation or concentration steps that might cause aggregation of vesicles. Surprisingly, many of the presumed components of exosomes (Kowal et al., 2016; van Niel et al., 2018; Zhang et al., 2018) were absent from classical exosomes expressing CD63, CD81 and CD9. This leaves open the question of hypothetical non-classical exosomes generated by the MVE biogenesis pathway, but lacking these three classical exosomal tetraspanin markers. There is evidence in support of ILV and MVE heterogeneity (Edgar et al., 2014; Stuffers et al., 2009; van Niel et al., 2011), and subpopulations of CD63/CD81/CD9-negative ILVs could conceivably be released as non-classical exosomes. Some proteins, including GAPDH, ENO1, 14-3-3, HSP90 and PARK7/DJ1, are present in sEVs, although they are absent from classical exosomes. Highly abundant cytosolic proteins, including GAPDH, PKM, ENO1, 14-3-3, HSP90, are absent from classical exosomes as are the common protein components of microfilaments, microtubules and intermediate filaments (Figures 4 and S4). The uptake of cytosolic constituents during classical exosome biogenesis is therefore not random - rather exosome loading must be a highly regulated process. In some ways, the NV fractions obtained with the high-resolution density gradient resemble the recently described non-membranous nanoparticles termed “exomeres” (Zhang et al., 2018) (Zhang et al., 2019). This includes lack of membrane-vesicular structure, depletion of membrane proteins, the presence of suggested exomere markers HSP90 and HSPA13 (Figures 3 and S3), as well as mitochondrial, cytoskeleton, metabolic, and Ago proteins (Zhang et al., 2019). We demonstrate here that Ago proteins are absent from classical exosomes and none of the other nuclear or cytosolic miRNA machinery, including Drosha, DGCR8, Dicer, TRBP and GW182, were detected extracellularly (Figures 3–4). Thus, exosomes do not carry the necessary molecular machinery to carry out cell-independent miRNA biogenesis as previously suggested (Melo et al., 2014). Given that many of the most abundant miRNAs are associated with extracellular NV fractions rather than with purified sEVs (Figure 2), detection of an extracellular miRNA is neither prima facie nor sufficient evidence that the secretion mechanism is due to exosome/EV release. None of the frequently reported exosomal RBPs (Hagiwara et al., 2015; Mateescu et al., 2017) that we investigated (Ago1–4, Annexin A2, RPS3, RPS8, EEF2, eEF1A1, MVP, PARK7/DJ1, hnRNPA2B1, GAPDH) were associated with classical CD63-, CD81- or CD9-positive exosomes. However, sEV fractions did display a significant presence of exRNA, and other investigators have shown that EV-associated exRNA can be protected from degradation by RNase (Shurtleff et al., 2017). Therefore, non-exosomal EVs may be significant carriers of exRNA and RBPs.
We have identified the membrane-associated proteins Annexin A1 and A2 as novel markers of non-exosomal small to large EVs and observed 150–1000 nm Annexin A1-positive microvesicles budding off the plasma membrane (Figure 5). Both proteins are absent from classical CD63-, CD81-and CD9-positive exosomes in vitro and in vivo (Figures 5 and S5). Annexin A1 thus appears to fit the criteria for a specific protein marker of classical shedding microvesicles, distinct from both exosomes and ARMMs. It is not yet clear if Annexin A1-and Annexin A2-positive EVs are identical. Phosphatidylserine is enriched in sucrose-gradient purified sEVs (Trajkovic et al., 2008); however, Annexin V is not present on classical exosomes (Figures 5 and S5). This likely indicates that phosphatidylserine is not exposed on the outer leaflet of exosomal membranes.
We demonstrate that dsDNA in the extracellular environment is not associated with exosomes or any other type of sEV (Figure 6), although it can co-purify with them using standard ultracentrifugation or sucrose density gradients (Takahashi et al., 2017). However, we find that dsDNA and histones are present intracellularly in CD63-positive compartments of a size consistent with MVEs, the site of ILV/exosome biogenesis, and we provide evidence that intracellular CD63-and LC3B-decorated compartments may traffic their dsDNA cargo to the plasma membrane (Figure 7). We propose a model of autophagy-and MVE-dependent, but exosome-independent, active secretion of dsDNA and histones (Figure 7L). The model rests on a number of recent discoveries: 1) presence of cytoplasmic chromatin in cancer, and the autophagy (LC3B)-mediated nuclear-to-cytoplasmic transport and degradation of chromatin fragments (Bakhoum et al., 2018; Dou et al., 2015); 2) existence of amphisomes (Berg et al., 1998; Klionsky et al., 2014) (Figure 7); 3) presence of dsDNA within MVEs/amphisomes independent of ILVs (Figure 7); 4) presence of amphisomes at the plasma membrane and absence of dsDNA and histones in exosomes (Figures 6–7). Engulfment of cytoplasmic components and subsequent amphisome-dependent extracellular cargo release could conceivably be a more general mechanism of secretion for diverse non-membranous molecular component assemblies such as NV fractions and exomeres.
In summary, we have demonstrated that improved methodology for exosome isolation allows more precise determination of the molecular composition of classical exosomes. Specifically, the classical tetraspanin-enriched exosomes contain a more limited repertoire of the diverse molecules present in the extracellular milieu than has previously been assumed (summarized in Figure S7F). The conceptual and practical framework provided here is a step toward a better understanding of the heterogeneity of extracellular vesicles and particles (summarized in Table S1). It will be of utmost importance for the future therapeutic potential and design of treatment interventions to correctly identify the compartment and mechanisms by which specific DNA, RNA and proteins are secreted in human disease.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Robert J. Coffey (robert.coffey@vumc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture
Human colon cancer cell lines DKO-1 (male), DiFi (female) and SW620 (male), human Gli36 glioblastoma cells and human MDA-MB-231 (female) breast cancer cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 100 μg ml−1 penicillin-streptomycin (Gibco Invitrogen, Carlsbad, CA, USA) at 37°C in a 5% CO2 humidified incubator. Cells were maintained by passage every 3–4 days at 70–80% confluence, and were routinely tested for mycoplasma contamination (Universal Mycoplasma Detection Kit, ATCC, Manassas, VA, USA). Primary cultures of human renal proximal tubule epithelial cells were generated from transplant discards purchased from Innovative BioTherapies (Ann Arbor, MI, USA). Primary cultures for production of extracellular vesicles was initiated at passage 2, and cells were maintained in DMEM supplemented with 2 μg ml−1 Normocin, Insulin-Transferrin-Selenium (ITS), epidermal growth factor (EGF), hydrocortisone and T3 thyroid hormone (complete formulation available upon request).
Human samples
Fresh peripheral blood from healthy individuals, and resected patient colorectal cancer tumor and adjacent normal tissue was processed immediately without any freezing of samples as described in detail below. All procedures on peripheral blood specimens and human tissue were approved and performed in accordance with the Vanderbilt University Medical Center Institutional Review Board.
METHOD DETAILS
Extracellular vesicle isolation from cultured cells grown in dishes
Cell-conditioned medium was collected from approximately 90% confluent MDA-MB-231 cells grown for 48h in 150 mm cell culture dishes with DMEM containing FBS depleted of bovine serum extracellular vesicles (EVs) by 24h ultracentrifugation at 120,000 × g. Cell-conditioned medium was collected from approximately 90% confluent DiFi and SW620 cells grown for 48h in 150 mm cell culture dishes with DMEM without FBS. Cell viability was assessed using trypan blue exclusion and only medium from cultures with >95% viability was used for isolation of EVs. Cell-conditioned medium was collected from approximately 95% confluent primary human kidney epithelial cells grown for 96h in cell culture flasks with DMEM without FBS. The collected media was first subjected to a centrifugation step of 400 × g for 10 min at RT to pellet and remove cells. All following centrifugation steps were performed at 4°C. Next, the supernatant was spun at 2,000 × g for 20 min to remove debris and apoptotic bodies. Then, to pellet and collect large EVs (lEVs), the supernatant was centrifuged at 15,000 × g for 40 min. The resulting lEV pellet (P15) was resuspended in a large volume of phosphate-buffered saline (PBS) followed by ultracentrifugation at 15,000 × g for 40 min to wash the sample. To remove any remaining any lEVs, the media supernatant from the first 15,000 × g step was passed through a 0.22 μm pore PES filter (Millipore). This supernatant (pre-cleared medium) was next subjected to ultracentrifugation at 120,000 × g for 4h in a SW 32 Ti Rotor Swinging Bucket rotor (k factor of 204, Beckman Coulter, Fullerton, CA) to sediment small EVs (sEVs). The crude sEV pellet (P120) was resuspended in a large volume of PBS followed by ultracentrifugation at 120,000 × g for 4h to wash the sample. At no time during the process were samples subjected to temperatures below 4°C.
Extracellular vesicle isolation from cultured cells grown in bioreactors
DKO-1 and Gli36 cells were maintained in CELLine Adhere 1000 (CLAD1000) bioreactors (INTEGRA Biosciences AG, Zizers, Switzerland) at 37°C in a 5% CO2 humidified incubator, essentially as previously described (Jeppesen et al., 2014b; Mitchell et al., 2008). Cell-conditioned medium was harvested from bioreactors every 48h, starting from one week after inoculation of the bioreactor and continuing for a period of 4 weeks. Pellets of lEVs (P15) and sEVs (P120) were generated as described. At no time during the process were samples subjected to temperatures below 4°C.
Extracellular vesicle isolation from human plasma samples
Blood was drawn into BD Vacutainer Blood Collection Tubes (BD Bioscience) containing either Acid Citrate Dextrose Solution A or Buffered Sodium Citrate as anticoagulantsThe first tube drawn was discarded. Further processing of samples was initiated within 20 min of blood draw. Plasma was generated by first centrifugation of the blood at 3000 × g for 15 min and then a second round of centrifugation of the supernatant at 3000 × g for 15 min tube to ensure that no platelets remained (see also Figure S3D). The resulting plasma samples were immediately diluted ~1:20 in ice cold PBS and spun at 15,000 × g for 40 min to pellet and remove lEVs and microparticles. The supernatant was filtered through a 0.22 μm pore PES filter (Millipore). Clarified supernatants were subjected to ultracentrifugation at 120,000 × g for 4h in a SW 32 Ti Swinging Bucket rotor (k factor of 204, Beckman Coulter, Fullerton, CA) to sediment sEVs. Pellets of crude sEVs (P120) were resuspended in ice-cold PBS, tubes filled with PBS, and then subjected to ultracentrifugation at 120,000 × g for 4h. The washed pellet was resuspended again and subjected to a second wash step, again at 120,000 × g for 4h. The washed pellet was resuspended in ice-cold PBS. Crude plasma sEV samples were further purified by high-resolution iodixanol density gradients fractionation (see below and Figure S3D). At no time during the process were plasma or plasma sEVs subjected to temperatures below 4°C.
Extracellular vesicle isolation from human tissue samples
Crude sEVs were isolated from the interstitial space/tumor microenvironment of human tissue as previously established (Asai et al., 2015; Vella et al., 2017), with some modifications. Briefly, fresh tumor or fresh adjacent normal tissue was immediately after resection dissected on ice. The pieces of tissue was then incubated at 37°C in 250 units/ml Collagenase type I (Worthington, Lake Wood, NJ, USA) for 30 min after which the tissue was returned to ice and diluted with PBS containing Complete Mini Protease Inhibitor Cocktail and PhosSTOP Phosphatase Inhibitor Cocktail (both from Roche). The solution of tissue was gently disassociated by pipetting (10 ml pipette). After dissociation, the sample was applied to first a 60 μm then a 40 μm mesh filter (BD Biosciences, San Jose, CA, USA) to remove any remaining larger particles from the single-cell suspension. The supernatant containing disassociated cells and extracellular vesicles was spun at 400 × g for 10 min to pellet cells. The cell pellet was washed once in PBS by resuspension and repelleting, and cellular proteins extracted as described below. Crude sEVs (P120) were then purified from the supernatant in the same manner as described above for cell culture sEVs. At no time during the process were samples subjected to temperatures below 4°C.
Protein extraction from cells, large EVs and small EVs
To extract cellular proteins, cultured cells and tumor cell from solid colorectal tumors were collected, washed twice with ice-cold PBS, and solubilized in cell lysis buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, 60 mM octyl β-D-glucopyranoside) to which complete Mini Protease Inhibitor Cocktail and PhosSTOP phosphatase inhibitor cocktail (both from Roche), and 2.0 mM Pefabloc (Sigma-Aldrich, St Louis, MO, USA) was added immediately before use. Lysed samples were incubated on ice for 30 min. Cell lysates were then centrifuged at 10,000 × g at 4 °C to remove cellular debris. Protein content of cell lysates was quantified by a Direct Detect Infrared Spectrometer (Millipore). After the final wash step of ultracentrifugation in PBS, lEV (P15) and sEV (P120) samples were lysed, and proteins extracted as described above for cell samples.
Immunoblot analysis
Samples of lysed cells, lEVs and sEVs/exosomes were prepared in lithium dodecyl sulfate (LDS) sample buffer, heated to 70°C for 10 min, or incubated at RT for 30 min, before being loaded on gels. The samples were separated on 4–12% or 12% SDS-PAGE Bis-Tris gels (Life Technologies) under either reducing or non-reducing conditions, depending on the subsequent use of primary antibody, before being transferred to Immobilon-FL PVDF Transfer Membranes (EMD-Millipore). Membranes were blocked for 1h in 5% non-fat dry milk, 5% bovine serum albumin, or Odyssey TBS Blocking Buffer (LI-COR Biosciences, Lincoln, NE, USA), depending on the primary antibody subsequently used. Primary antibodies used for immunoblotting are described in Supplementary Table X. For chemiluminescence detection of proteins, HRP-conjugated anti-rabbit IgG (Cell Signaling Technology), anti-rat IgG H&L (Abcam) and anti-mouse IgG (Cell Signaling Technology) secondary antibodies, and Western Lightning Plus-ECL Substrate (PerkinElmer) were used. For fluorescence detection of proteins, IRDye 680RD anti-mouse IgG (H+L), Highly Cross Adsorbed, IRDye 800CW anti-rabbit IgG (H+L), Highly Cross Adsorbed and IRDye 800CW anti-rat IgG (H+L), Highly Cross Adsorbed (LI-COR) secondary antibodies was used. Detection was with an Odyssey Fc Imaging System (LI-COR). Relative protein levels were determined using Image Studio software (LI-COR).
High-resolution (12–36%) iodixanol density gradient fractionation
Iodixanol (OptiPrep) density media (Sigma-Aldrich, St Louis, MO, USA) were prepared in ice-cold PBS immediately before use to generate discontinuous step (12–36%) gradients. Crude pellets of sEVs (P120) were resuspended in ice-cold PBS and mixed with ice-cold iodixanol/PBS for a final 36% iodixanol solution. The suspension was added to the bottom of a centrifugation tube and solutions of descending concentrations of iodixanol in PBS were carefully layered on top yielding the complete gradient. Identical, gradients without sample were generated in the same manner for later determination of fraction densities (Figure S1A). The bottom-loaded 12–36% gradients were subjected to ultracentrifugation at 120,000 × g for 15h at 4°C using a SW41 TI Swinging Bucket rotor (k factor of 124, Beckman Coulter). Twelve individual fractions of 1 ml were collected from the top of the gradient. From the duplicate gradient, fraction densities were measured using a refractometer (ATAGO, Tokyo, Japan). For Nanoparticle Tracking Analysis (NTA), fractions were pooled and diluted in particle-free PBS. For immunoblotting, each individual 1 ml fraction was transferred to new ultracentrifugation tubes, diluted 12-fold in PBS and subjected to ultracentrifugation at 120,000 × g for 4h at 4°C using a SW41 TI swinging bucket rotor. The resulting pellets were lysed in cell lysis buffer (see below) for 30 min on ice. For RNA and DNA extraction, fractions were pooled, transferred to new ultracentrifugation tubes, diluted approximately 6-fold in PBS and subjected to ultracentrifugation at 120,000 × g for 4h at 4°C using a SW 32 Ti Swinging Bucket rot or.
6–30% iodixanol density gradient fractionation
Crude pellets of sEVs (P120) were resuspended in ice-cold PBS and mixed with ice-cold iodixanol/PBS for a final 30% iodixanol solution. The suspension was added to the bottom of a centrifugation tube and solutions of descending concentration of iodixanol in PBS were carefully layered on top yielding the complete gradient. Identical, gradients without sample were generated in the same manner for later determination of fraction densities (Figure S5A). The bottom-loaded 6–30% gradients were subjected to ultracentrifugation at 120,000 × g for 15h at 4°C using a SW41 TI Swinging Bucket rotor (k factor of 124, Beckman Coulter). Ten individual fractions of 1 ml were collected from the top of the gradient.
Particle size and concentration measurement by Nanoparticle Tracking Analysis (NTA)
Samples in solution were analyzed by nanoparticle tracking using a NanoSight LM10 system (NanoSight Ltd, Amesbury, UK) configured with a 405 nm laser and a high sensitivity sCMOS camera (OrcaFlash2.8, Hamamatsu C11440, NanoSight Ltd). Prior to analysis, samples were either treated with 0.1% (v/v) Triton X-100 (Sigma) (Osteikoetxea et al., 2015) for 10 min at RT, or mock treated (control). Analysis using the NTA-software (version 2.3, build 0006 beta 2) was essentially as previously described (Jeppesen et al., 2014a; Jeppesen et al., 2014b). In brief, each sample of particles was diluted in particle-free PBS, and introduced manually. After optimization, settings were kept constant between measurements for that session. Ambient temperature was recorded manually and did not exceed 25°C. Approximately 20–40 particles were in the field of view and the typical concentration was approximately 2 × 108 to 8 × 108 particles/ml for each measurement. Six videos of 60 seconds duration, with a 10-second delay between measurements, were recorded for each independent replicates (n = 3), generating data for each sample type that is the average of 18 individual measurements.
Negative stain transmission electron microscopy
With fixation
Samples were first fixed with 2.5% gluteraldehyde in 0.1M sodium cacodylate buffer at RT for 30 min. Samples were then deposited on formvar carbon-coated grids (Electron Microscopy Sciences, Hatfield, PA) for 1 min followed by negative staining with 2% uranyl acetate for 30s at RT. Imaging was performed on a Philips/FEI T-12 Transmission Electron Microscope. Micrographs were captured with a 2k × 2k CCD camera (Advanced Microscopy Techniques, Woburn, MA).TEM was performed in part through the use of the Vanderbilt Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, DK59637 and EY08126).
Without fixation (for imaging of vault structures)
Partially-purified and highly purified exosome fractions (usually 0.2–0.5 mg/ml total protein concentration) were prepared for transmission electron microscopy (TEM) by absorption of samples onto 1–2 nm thick carbon film mounted on carbon-coated holey-film grids for 5 min at 4°C. This was accomplished by floating the grid on 25 μl of sample. Following sample adsorption, grids were quickly and gently blotted on filter paper and immediately floated for 5 min on 1 mL of 1% uranyl acetate at 4°C and dried on filter paper. Imaging was performed on a JEM 1200EX microscope. Micrographs were captured with a BioScan 600W digital camera (Gatan Inc., Pleasanton, CA) using Gatan’s DigitalMicrograph software.
In all cases, TEM was performed on fresh sample of EVs that had not been subjected to freezing temperatures at any step in purification or processing.
Mass spectrometry and data analysis
Samples were re-suspended in 100 μL of trifluoroethanol (TFE) and 100 μL of 100 mM ammonium bicarbonate, pH 8. Samples were sonicated three times for 20s with 30s incubation on ice in between. The resulting homogenate was heated with shaking at 1000 rpm for 1h at 60°C followed by a second series of s onication steps. Protein content of samples was measured and two aliquots of 100 μg (two process replicates per sample) was reduced with 50 mM dithiothreitol (DTT) at 60°C for 30 min followed by alkylation with 100mM iodoacetamide (IAM) in the dark at room temperature for 20 min. The lysate was diluted with the appropriate volume of 50 mM ammonium bicarbonate, pH 8.0, to reduce the TFE concentration to 10%. Trypsin was added at a ratio of 1:50 (w:w) and digestion allowed to proceed overnight at 37°C. The digested mixture was frozen at −80°C and lyophilized to dryness. The lyophilized samples were re-suspended in 350 μL of HPLC-grade water and vortexed vigorously for 1 min followed by desalting using an Oasis HLB 96-well Elution plate (30 μm, 5 mg, Waters Corp., Milford, MA) pre-washed with 500 μL of acetonitrile and equilibrated with 750 μL of HPLC-grade water. Flow-through was discarded, plates washed with 500 μL of HPLC-grade water, and peptides eluted with 80% acetonitrile followed by vacuum drying and reconstitution using 2% acetonitrile/0.1% formic acid.
Peptides from each replicate were analyzed in three technical replicates on a Q Exactive mass spectrometer equipped with an Easy nLC-1000 (Thermo Scientific). A 2 μL injection volume of peptides was separated on a PicoFrit (New Objective, Woburn, MA) column (75 μm ID × 120 mm, 10 μm ID tip) packed with ReproSil-Pur C18-AQ resin (3 μm particle size and 120 Å pore size). Peptides were eluted using a flow rate of 300 nL/min, and the mobile phase solvents consisted of water containing 0.1% formic acid (solvent A) and acetonitrile containing 0.1% formic acid (solvent B). A 155 min gradient was performed, consisting of the following: 0−10 min, increase to 5% B; 10−40 min, 5−10% B; 40–140 min, 35% B, 140–145 min, 90% B and held at 90% B for 5 min before returning to the initial conditions of 2% B. Mass spectra were acquired over the scan range of m/z 300–1800 at a resolution of 70,000 (AGC target 3 ×106 and 64 ms maximum injection time). Data-dependent scans of the top 20 most abundant ions were selected for fragmentation with HCD using an isolation width of 2 m/z, 27% normalized collision energy and a resolution of 35,000 (AGC target 2 ×105 and 100 ms maximum injection time). Dynamic exclusion was set to 60 sec.
Tandem spectra were searched against the human RefSeq database (v. 3.56) using the Myrimatch algorithm (v 1.6.75) and MS GF+. The database incorporated both the forward and reversed sequences to allow for determination of false discovery rate (FDR). The searches were performed allowing a static modification of +57 on cysteine (for carboxyamidomethylation from iodoacetamide) and dynamic modifications of +16 on methionine (oxidation). Semi-tryptic peptides were considered in the search parameters. Peptide and fragment ion tolerances were set to ±0.01 and 0.5 Da, respectively. The data were filtered and assembled with the IDPicker algorithm (v. 2.6.165) using a 5% FDR for all peptides with parsimony applied (minimum protein reporting) and requiring at least two peptides (minimum peptide length of seven amino acids). Spectral count differences between cell lines were analyzed using QuasiTel of using a quasiFDR value of < 0.05 and requiring at least a 4-fold change in expression.
RNA extraction from cells, large EVs and small EVs, and RNA analysis
RNA was extracted using miRCURY RNA Isolation Cell and Plant Kit (Exiqon, 300110) according to the manufacturer’s protocol with elution in a volume of 50 μl. RNA was quantified and 260/280 ratio assessed using a NanoDrop 2000 (Thermo Scientific), and Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, California, CA, USA. Profiles of RNA species present in samples were generated using Agilent RNA Nano 6000 and Agilent RNA Pico 6000 kits on a 2100 Bioanalyzer instrument (Agilent Technologies).
Short RNA library preparation and sequencing
Short RNA libraries were prepared using NEBNext Small RNA Library Prep Set for Illumina (New England BioLabs Inc., Ipswich, MA, USA). Briefly, 3′ adapters were ligated to total input RNA followed by hybridization of multiplex single read (SR) reverse transcription (RT) primers and ligation of multiplex 5′ SR adapters. RT was performed using ProtoScript II RT for 1h at 50°C. Immediately after RT reactions, amplification was performed for 15 PCR cycles using LongAmp Taq 2× master mix with Illumina-indexed primers to uniquely barcode each sample. The purified material was size selected for 146–148 bp range using 3% dye-free agarose gel cassettes on a Pippin Prep instrument (Sage Science Inc., Beverly, MA, USA). Post-PCR material was purified using QIAquick PCR purification kits (Qiagen Inc.) and the yield and concentration of the prepared libraries were assessed using a Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, California, CA, USA) and DNA 1000 chips on a Agilent 2100 Bioanalyzer (Applied Biosystems, Carlsbad, CA, USA), respectively. For sequencing, library quantification was performed using qPCR-based KAPA Biosystems Library Quantification kits (Kapa Biosystems, Inc., Woburn, MA, USA). Each library was diluted to a final concentration of 1.25 nM and pooled in equimolar ratios prior to clustering. Cluster generation was carried out on a cBot v8.0 using an Illumina Truseq Single Read Cluster Kit v3.0. Single-end sequencing was performed to generate at least 15 million reads per sample on an Illumina HiSeq2000 using a 50-cycleTruSeq SBSHSv3 reagent kit (50 bp single-end reads). Clustered flow cells were sequenced for 56 cycles, consisting of a 50-cycle read, followed by a 6-cycle index read. Image analysis and base calling were performed using the standard Illumina pipeline consisting of Real Time Analysis version v1.17 and demultiplexed using bcl2fastq converter with default settings.
Long RNA library preparation and sequencing
For long RNA library preparation a Ribo-zero Magnetic Gold rRNA removal kit (Epicenter, IIlumina Inc.) was used to remove ribosomal RNA from the total RNA. First strand synthesis was performed using NEBNext RNA first strand synthesis module (New England BioLabs Inc., Ipswich, MA, USA). Immediately thereafter, directional second strand synthesis was performed using NEBNExt Ultra Directional second strand synthesis kit. Following this, cDNAs were used for standard library preparation protocol using NEBNext DNA Library Prep Master Mix Set for Illumina. End-repair was performed followed by polyA addition and custom adapter ligation. After ligation, material was individually barcoded with unique in-house genomics service lab primers. Library quality and concentration were assessed by a Qubit 2.0 Fluorometer and DNA 1000 chips on an Agilent 2100 Bioanalyzer. Accurate quantification for sequencing applications was determined using the qPCR-based KAPA Biosystems Library Quantification kit (Kapa Biosystems, Inc., Woburn, MA). Each library was diluted to a final concentration of 12.5 nM and pooled equimolar prior to clustering. Paired-end sequencing was performed on all samples (100 bp paired-end directional reads). Raw reads were de-multiplexed using a bcl2fastq conversion software v1.8.3 (Illumina, Inc.) with default settings.
Direct Immunoaffinity Capture (DIC) of exosomes
Cell-conditioned medium was immediately after collection subjected to differential centrifugation at 400 × g, 2,000 × g and 15,000 × g at 4°C. The supernatant was filtered through a 0.22 μm pore PES filter (Millipore) to generate pre-cleared medium. All following steps were also performed at 4°C. Pre-cleared medium was split three ways, one portion were incubated with magnetic beads directly conjugated to anti-mouse antibodies directed at CD63, CD81 or CD9 (ThermoFisher Scientific), one portion incubated with magnetic beads conjugated to mouse IgG, and incubation with allow to proceed with nutation and rotation for 16h (Figure 4A). The third portion was subjected to ultracentrifugation and washing to generate a crude sEV pellet as described above (P120). After incubation, the beads were washed four times in ice-cold 0.1% BSA-PBS (pH 7.4, filtered through a 0.22 μm membrane), and finally, washed one time in PBS pH 7.4 (pH 7.4, filtered through a 0.22 μm membrane). Immediately following the last wash, the exosome-loaded beads were resuspended first in cell lysis buffer and LDS sample buffer for 30 min on ice, and then resuspended in LDS sample buffer followed by heating to 70°C for 10 min to release proteins. The beads were removed from the suspension by using a magnet and the clarified lysate used for immunoblot analysis. In some cases, immediately following the last wash step, the exosome-loaded beads were lysed in for DNA extraction (Figure S6E and see below). DIC of exosomes directly from human plasma was performed as described above except that the supernatant in these cases was raw, undiluted and unfiltered, plasma (generated by two rounds of 3000 × g centrifugation for 15 min).
DNA extraction and analysis
Small EV samples were generated by iodixanol density gradient fractionation as described above and pooled fractions were resuspended in PBS for DNA extraction. For some experiments, samples were either pre-treated for 30 min with DNase I (New England Biolabs) at 50U/ml in reaction buffer (10 mM Tris-HCl, 2.5mM MgCl2, 0.5 mM CaCl2), or mock treated in reaction buffer, followed by DNase I deactivation. Extraction of DNA was performed using a DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer’s instructions for cultured cells. Elution of DNA was in 100 μl of elution buffer. For some experiments extracted DNA was for some experiments treated with either RNase A/T1 (Thermo Fisher Scientific) at 100 μg/ml RNase A and 250U/ml RNase T1, or DNase I (New England Biolabs) at 200U/ml in reaction buffer (10 mM Tris-HCl, 2.5mM MgCl2, 0.5 mM CaCl2). DNA concentrations were measured by a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific), and a Qubit 3.0 fluorometer using Qubit dsDNA HS and dsDNA BR kits (Thermo Fisher Scientific). Presence of contaminating RNA was assessed using the Qubit fluorometer with a Qubit RNA HS kit. The size of purified DNA was evaluated with microfluidic High Sensitivity DNA kits on a Bioanalyzer 2100 instrument (Agilent, Santa Clara, CA, USA).
For magnetic bead-capture experiments for DNA extraction, conditioned-media from DKO-1 and Gli36 cells in culture were subjected first to pre-clearing of larger vesicles and debris as described above, and then incubated with anti-CD81-beads or IgG overnight at 4°C under rotation and tilting. Beads were collected and washed as described above. The flowthrough from capture experiments was subjected to 120,000 g ultracentrifugation, resuspended and washed in PBS with another round of 120,000 g ultracentrifugation to pellet sEVs. Both beads and flowthrough sEV pellets were lysed and DNA extracted as described above.
Structured Illumination Microscopy (SIM)
3D SIM imaging and processing was performed on a Nikon N-SIM structured illumination platform equipped with an Andor DU-897 EMCCD camera and a SR Apo TIRF 100X (1.49 NA, WD 0.12) oil immersion objective. Samples were imaged in PBS at RT. For calibration, 100 nm fluorescent (360/430 nm, 505/515 nm, 560/580 nm and 660/680 nm) beads (TetraSpeck™ Microspheres, Thermo Fisher Scientific, Waltham, MA, USA) were fixed and imaged. Images were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).SIM was performed in part through the use of the Vanderbilt University Nikon Center of Excellence.
Immunofluorescence staining
Cells prepared for SIM were cultured on 35-mm culture dishes with 1.5 coverslip and 14 mm glass diameter (P35G-0.170–14-C, MatTek Corporation, Ashland, MA, USA) to approximately 50% confluence. DKO-1 cells were fixed with 4% paraformaldehyde in PBS at RT for 20 min and then extracted for 5 min with 1% Triton X-100 in 4% paraformaldehyde in PBS as previously described (Fenix et al., 2016). Cells were washed three times in PBS. Cells were blocked in 10% bovine serum albumin (BSA) in PBS. Primary antibodies were diluted in 10% BSA and incubation was overnight at 4°C. Cells were washed three times. The secondary Alexa Flour antibodies were prepared in blocking buffer and centrifuged at 13,000 rpm for 10 min before incubation on cells for 1h at RT.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using the SPSS Statistical Analysis System (version 22.0; SPSS Inc, Chicago, IL) and R (The R foundation). Normal distribution of data sets was assessed by Shapiro-Wilks test, skewness and kurtosis, and visual inspection of Q-Q plots. Equality of variance was tested by Levene’s test. Sphericity was assessed where appropriate by Mauchly’s test. All data are presented as mean ± SD unless otherwise stated. To assess differences between two groups, two-tailed unpaired Student’s t-test. When three or more groups were compared one-way ANOVA was performed for an overall difference. If significant, this was followed by multiple comparisons with significance between groups corrected by the Holm-Bonferroni procedure. Differences were considered to be significant for values of p < 0.05. For RNA-seq and mass spectrometry data the false discovery rate for multiple comparisons were controlled with the Benjamini–Hochberg procedure. For mass spectrometry, the spectral count difference between samples was considered to be significant for quasiFDR values of p < 0.05 (see above).
DATA AND SOFTWARE AVAILABILITY
Datasets for RNA-seq have been deposited in GEO: GSE125905
Supplementary Material
Figure S1. High-Resolution Density Gradient Fractionation Separates Small Extracellular Vesicles from Non-Vesicular Components, Related to Figure 1. (A) Density profile of fractions obtained after high resolution iodixanol density gradient. An empty gradient (tube with iodixanol gradient but containing no sEV sample) was used run opposite an identical sample gradient and density of fractions was measured by refractometry.
(B-C) High-resolution density fractionation of (B) DiFi and (C) primary human renal epithelial cell-derived crude sEVs (P120). After flotation of sample in an iodixanol gradient equal volumes of each fraction were loaded on SDS-PAGE gels, and membranes were blotted with indicated antibodies. NV, non-vesicular; sEV, small EV.
(D) Two-round high-resolution density fractionation of DKO-1 cell-derived crude sEVs (P120). After a first round flotation of sample in an iodixanol gradient, purified sEVs (from fractions 1–6) were subjected to a second round of flotation in an identical iodixanol gradient.
(E) Negative stain EM of pooled Low (sEV) density fractions obtained by floatation of crude DKO-1 and Gli36 sEV pellets (P120) in iodixanol density gradients. Sample preparation was without a fixation step. Imaging was performed on a JEM 1200EX microscope.
(F) Size distribution of gradient-purified sEVs imaged by EM as in (C). Histograms of the diameter of measured vesicles. n = 504 for DKO-1 and n = 400 for Gli36.
Table S3. Proteomic analysis of DKO-1 Cell, large EV, and high-resolution density gradient-purified sEV and non-vesicular (NV) samples, Related to Figure 2.
Table S4. Proteomic analysis of Gli36 high-resolution density gradient-purified sEV and non-vesicular (NV) samples, Related to Figure 2.
Figure S2. Differential Expression of Protein and RNA in Small Extracellular Vesicles and Non-Vesicular Fractions, Related to Figure 2. (A) Schematic of experimental setup for proteomics analysis. After flotation on density gradients, separate pools of low (sEV) and high (NV) density fraction pools were subjected to LC-MS/MS.
(B) Venn diagram representing the number of proteins unique and overlapping between sample types.
(C) Volcano plots of quantitative differences in proteins in sEV and NV fractions. Black dots represent a four-fold or greater enrichment while orange dots represent less than four-fold enrichment. Dots above the dashed line represent proteins for which differences were significant (FDR < 0.05).
(D) Table of the fold-change in spectral counts from proteomic profiling between sEV and NV fractions for selected proteins chosen for validation by immunoblotting.
(E) Immunoblot validation of proteomic profiling.
(F) Heatmap of the 25 most commonly identified exosomal proteins from the ExoCarta exosome database form proteomic profiling of sEV and NV from DKO-1. Scale indicates intensity, defined as Δ(spectral counts – mean spectral counts)/standard deviation.
(G) Bioanalyzer electropherograms of the size distribution (in nucleotides) of RNA extracted from cells, lEV (P15) and gradient-purified sEV and NV samples, as measured by RNA Pico (DKO-1) or RNA Nano (Gli36) chips. lEV, large EV; sEV, small EV; NV, non-vesicular.
(H) Volcano plots of miRNA expression patterns in sEV and NV fractions for DKO-1 (left) and Gli36 (right).
(I) Spectral counts for YBX1 in cells, lEVs, purified sEV and NV fractions generated by high-resolution density gradient centrifugation for DKO-1 (left) and Gli36 (right). Data represent mean ± SD. N.D., Not Detected.
(J) Expression level of miR-223 and miR-144 for DKO-1 and Gli36 samples. Data represent mean ± SD. RPM, reads per million bases.
(K) 3’ trimming and tailing of miRNA. The changes in either 3’ nucleotide additions (tailing) or 3’ resection (trimming) compared to full length miRNA sequences (intact) from cells, lEVs (P15), and density gradient-purified sEV (low density) and NV (high density) fractions. n = 4 biological replicates for each sample type, representative examples displayed.
(L) Percentage of long RNA reads mapping to different types of transcripts for cellular and extracellular DKO-1 samples.
(M) Percentage of long RNA reads mapping to exonic and intronic gene regions for cellular and extracellular DKO-1 samples.
(N) Principal Component Analysis based on the quantitative lincRNA profiles of cellular and extracellular DKO-1 samples generated by long RNA-seq.
(O) Violin plot of lincRNA expression for cellular and extracellular DKO-1 samples, based on GENCODE annotation. The shapes indicate kernel density estimation; the thick black lines indicate the middle two quadrants in the distribution; and the white dots indicates the median.
(P) Short RNA-seq data for YRNA in DKO-1 cells, lEV, sEV and NV fraction pools. RPM, reads per million.
(Q) Long RNA-seq data for YRNA in DKO-1 cells, lEV, sEV and NV fraction pools. FPKM, fragments per kilobase per million reads.
Figure S3. Argonaute Protein But Not Other miRNA Processing Proteins Are Secreted From Human Cells, Related to Figure 3. (A) Immunoblot analysis of DKO-1, Gli36 and MDA-MB-231 whole cell lysate, large EVs (P15) and small EVs (P120).
(B) Proteomic analysis of P-body and stress granule proteins. Spectral counts for DCP1A, G3BP1, G3BP2 and TIA-1 present in DKO-1 cells, lEV (P15), density gradient-purified sEV and NV fraction pools. Data represent mean ± SD. N.D., Not Detected.
(C) Immunoblot analysis of crude sEV (P120) from bovine growth serum, exosome-depleted serum-supplemented DMEM and DKO-1 cells. BGS, bovine growth serum; EDM, exosome-depleted media.
(D) Schematic of the generation of purified plasma sEV and NV fraction pools by high-resolution iodixanol density gradients. RBCs, red blood cells; WBCs, white blood cells.
(E) Immunoblot analysis of crude DKO-1, Gli36 and plasma sEVs (P120).
(F) TSG101 immunoblot analysis of crude DKO-1 and plasma sEV samples (P120).
(G) Crude DKO-1, Gli36, DiFi and primary renal epithelial sEVs obtained by ultracentrifugation at 120,000 × g (P120) were fractionated by floatation on high-resolution iodixanol gradients, and twelve fractions of equal volume were collected for analysis by immunoblotting.
Figure S4. Exosomes Exclude Common Glycolytic Enzymes and Lack a Cytoskeleton, Related to Figure 4. (A) Direct immunoaffinity capture of CD81-, CD63-, and CD9-positive exosomes. Immunoblots of crude sEV pellet (P120) and bead-captured exosomes from DKO-1 and Gli36 cells.
(B) Immunoblots of DKO-1 whole cell lysates, large EVs (P15) and crude small EVs (P120) obtained by ultracentrifugation.
(C) High-resolution density fractionation of crude sEVs (P120). NV, non-vesicular; sEV, small EV.
(D) Direct immunoaffinity capture of CD81-, CD63-, and CD9-positive exosomes from DKO-1, Gli36 and primary renal epithelial cells. Immunoblots of crude sEV pellet (P120) and bead-captured exosomes. g.e, greater exposure.
(E-F) Direct immunoaffinity capture of CD81-, CD63-, and CD9-positive exosomes from (E) DKO-1 and (F) Gli36 cells. Immunoblots of crude sEV pellet (P120) and bead-captured exosomes.
Figure S5. Annexin A1 is a Novel and Specific Marker of Microvesicles Distinct from Exosomes and ARMMs, Related to Figure 5. (A) Density profile of fractions obtained after 6–30% iodixanol density gradient fractionation. An empty gradient (tube with iodixanol gradient but containing no sEV sample) was used run opposite an identical sample gradient and density of fractions was measured by refractometry.
(B-D) Direct immunoaffinity-capture of CD63-, CD81-, and CD9-positive exosomes from (B) DKO-1, (C) Gli36, and (D) primary renal epithelial cells. Immunoblots of crude sEV pellet (P120) and bead-captured exosomes. g.e, greater exposure.
(E) Annexin A1-positive classical microvesicles. (top) Structured Illumination Microscopy (SIM) of DKO-1 cells stained for Annexin A1. Enlarged inserts displayed to the right. (Bottom) Size of Annexin A1-positive classical microvesicles measured at the maximum width.
(F) High-resolution (12–36%) density gradient sEV and NV fraction pools purified from plasma samples (see Figure S3D). For immunoblotting, equal volumes (representing equal portions of the total) were separated on SDS-PAGE gels, and membranes were blotted with indicated antibodies.
Figure S6. Release of Extracellular dsDNA and Histones from Human Cells is Independent of Exosomes and Small Extracellular Vesicles, Related to Figure 6. (A) High-resolution iodixanol gradient fractionation of DiFi cell-derived (top) and primary human renal epithelial cell-derived (bottom) crude sEVs (P120) analyzed by immunoblotting. NV, non-vesicular; sEV, small extracellular vesicle.
(B) Schematic of experimental design for Figure 6B–C. P120 sample was resupended and either loaded directly at the bottom of the iodixanol gradient (Figure 6B) or treated with DNase I to digest unprotected DNA (Figure 6C) before loading. After 15h of centrifugation, fractions from the now approximately linear gradient were collected from the top to generate sEV and NV pools. Material was washed and pelleted at 120,000 × g followed by DNA extraction. For Figure 6B, the extracted nucleic acid was treated with DNase I or RNase A/T1 before analysis.
(C) Quantification of DNA from gradient-fractionated sEV and NV pools obtained from primary human cultures of renal epithelial cells. Data represent mean ± SD of three technical measurements of one biological replicate. N.D., Not Detected.
(D) Bioanalyzer electropherograms of the size distribution of purified DNA in base pairs (bp) from DNase I pre-treated density gradient-fractionated, pooled, exosomal DKO-1 samples, as measured with the DNA High Sensitvity chip. DNA marker peaks at 35 bp and 10,380 bp. FU: fluorescence units.
(E) Schematic of experimental design for Figure 6E–F. Pre-cleared medium was obtained after differential centrifugation steps of 400–15,000 × g (see STAR Methods).
Figure S7. Active Secretion of DNA and Histones Through an Amphisome-Dependent Mechanism, Related to Figure 7. (A) DKO-1 cells stained for DAPI, and endogenous CD63 and CD9, and imaged with 3D SIM. Greyscale images of Z-stack projection in Figure 7A. MVE, multivesicular endosome.
(B) Cropped images of DKO-1 cells localized for CD63 and DNA (DAPI) by 3D SIM. (Left) Greyscale CD63 and DNA, and colorized merge. Displayed are sequential image slices (125 nm) through the Z-stack. Blue arrow indicates the cell nucleus; yellow arrow indicates a micronuclei. (Right) Enlarged image of the CD63-positive MVE with yellow line indicating the longest axis. Scale bar, 200 nm.
(C) DKO-1 cells were after permeabilization stained for CD63 and wheat germ agglutinin (WGA), and imaged with 3D SIM. (Left) Low magnification image of standard Y-X axis “Top-Down View”. (Middle) Enlarged yellow inserts. Scale bar, 200 nm. (Right) Z-X axis “Side View” of blue insert. Blue arrows indicate CD63-positive MVE. Yellow asterisks indicate CD63-positive MVE at the plasma membrane.
(D) CD63-and LC3B-positive amphisomes in DKO-1 cells. Cells were stained for endogenous CD63 and LC3B. Greyscale CD63 and LC3B, and colorized merge. White arrows indicate LC3B localized to CD63-positive compartments.
(E) Immunoblots of whole cell lysate and crude small EVs (P120) isolated from matched tissue/interstitial fluid of colorectal cancers (CRC) and adjacent normal. N, normal; T, tumor.
(F) Reassessment of exosome composition. Schematic illustration of revised composition of CD63/CD81/CD9-positive classical exosomes based on the data generated in this study. Proteins not examined here but associated with exosomes based on other studies are not included. The principal fractions containing the CD63/CD81/CD9 proteins after high-resolution density gradient centrifugation indicates that the majority of classical exosomes have buoyant densities in iodixanol-PBS from 1.08–1.13 g/ml, but can be found ranging from 1.05–1.15 g/ml. In contrast, small Annexin A1-positive classical microvesicles were principally observed at densities of 1.05–1.10 g/ml, but can be found ranging from 1.03–1.10 g/ml. ARMM, Arrestin-domain-containing protein 1 (ARRDC1)-mediated microvesicle; EV, extracellular vesicle; MVE, multivesicular endosome.
Highlights.
Human cells release Argonaute 1–4 and major vault protein independently of exosomes
Annexin A1 is a specific marker of microvesicles shed from the plasma membrane
Small extracellular vesicles do not contain DNA
Active secretion of cytosolic DNA occurs through an amphisome-dependent mechanism
ACKNOWLEDEGMENTS
This publication is part of the NIH Extracellular RNA Communication Consortium paper package and was supported by the NIH Common Fund’s exRNA Communication Program. We thank Emily J. Poulin for editing the manuscript. The work was funded by NIH grants F31 HL136081 to A.M.F., R35 GM125028 to D.T.B., U01-EB021214 to W.H.F., and R35 CA197570 and U19 CA179514 to R.J.C. The authors acknowledge the support Vanderbilt Digestive Diseases Research Center P30 DK058404 and Vanderbilt-Ingram Cancer Center P30 CA068485.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova-Agadjanyan EL, Stirewalt DL, et al. (2011). Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A 108, 5003–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kugler S, and Ikezu T (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18, 1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg TO, Fengsrud M, Stromhaug PE, Berg T, and Seglen PO (1998). Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem 273, 21883–21892. [DOI] [PubMed] [Google Scholar]
- Chakrabortty SK, Prakash A, Nechooshtan G, Hearn S, and Gingeras TR (2015). Extracellular vesicle-mediated transfer of processed and functional RNY5 RNA. RNA 21, 1966–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YD, Fang YT, Cheng YL, Lin CF, Hsu LJ, Wang SY, Anderson R, Chang CP, and Lin YS (2017). Exophagy of annexin A2 via RAB11, RAB8A and RAB27A in IFN-gamma-stimulated lung epithelial cells. Sci Rep 7, 5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crescitelli R, Lasser C, Szabo TG, Kittel A, Eldh M, Dianzani I, Buzas EI, and Lotvall J (2013). Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, et al. (2015). Autophagy mediates degradation of nuclear lamina. Nature 527, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JR, Eden ER, and Futter CE (2014). Hrs-and CD63-dependent competing mechanisms make different sized endosomal intraluminal vesicles. Traffic 15, 197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen EL (2005). Maturation of autophagic vacuoles in Mammalian cells. Autophagy 1, 1–10. [DOI] [PubMed] [Google Scholar]
- Fader CM, Sanchez D, Furlan M, and Colombo MI (2008). Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 9, 230–250. [DOI] [PubMed] [Google Scholar]
- Fenix AM, Taneja N, Buttler CA, Lewis J, Van Engelenburg SB, Ohi R, and Burnette DT (2016). Expansion and concatenation of non-muscle myosin IIA filaments drive cellular contractile system formation during interphase and mitosis. Mol Biol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbings DJ, Ciaudo C, Erhardt M, and Voinnet O (2009). Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol 11, 1143–1149. [DOI] [PubMed] [Google Scholar]
- Hagiwara K, Katsuda T, Gailhouste L, Kosaka N, and Ochiya T (2015). Commitment of Annexin A2 in recruitment of microRNAs into extracellular vesicles. FEBS Lett 589, 4071–4078. [DOI] [PubMed] [Google Scholar]
- Hessvik NP, Overbye A, Brech A, Torgersen ML, Jakobsen IS, Sandvig K, and Llorente A (2016). PIKfyve inhibition increases exosome release and induces secretory autophagy. Cell Mol Life Sci 73, 4717–4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham JN, Zhang Q, Jeppesen DK, Scott AM, Manning HC, Ochieng J, Franklin JL, and Coffey RJ (2016). Identification and characterization of EGF receptor in individual exosomes by fluorescence-activated vesicle sorting. J Extracell Vesicles 5, 29254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, et al. (2015). Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristov M, Erl W, Linder S, and Weber PC (2004). Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood 104, 2761–2766. [DOI] [PubMed] [Google Scholar]
- Hubstenberger A, Courel M, Benard M, Souquere S, Ernoult-Lange M, Chouaib R, Yi Z, Morlot JB, Munier A, Fradet M, et al. (2017). P-Body Purification Reveals the Condensation of Repressed mRNA Regulons. Mol Cell 68, 144–157 e145. [DOI] [PubMed] [Google Scholar]
- Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, and Parker R (2016). ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 164, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen DK, Hvam ML, Primdahl-Bengtson B, Boysen AT, Whitehead B, Dyrskjot L, Orntoft TF, Howard KA, and Ostenfeld MS (2014a). Comparative analysis of discrete exosome fractions obtained by differential centrifugation. J Extracell Vesicles 3, 25011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen DK, Nawrocki A, Jensen SG, Thorsen K, Whitehead B, Howard KA, Dyrskjot L, Orntoft TF, Larsen MR, and Ostenfeld MS (2014b). Quantitative proteomics of fractionated membrane and lumen exosome proteins from isogenic metastatic and nonmetastatic bladder cancer cells reveal differential expression of EMT factors. Proteomics 14, 699–712. [DOI] [PubMed] [Google Scholar]
- Keerthikumar S, Chisanga D, Ariyaratne D, Al Saffar H, Anand S, Zhao K, Samuel M, Pathan M, Jois M, Chilamkurti N, et al. (2016). ExoCarta: A Web-Based Compendium of Exosomal Cargo. J Mol Biol 428, 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Eskelinen EL, and Deretic V (2014). Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes… wait, I’m confused. Autophagy 10, 549–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, and Thery C (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A 113, E968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares R, Tan S, Gounou C, Arraud N, and Brisson AR (2015). High-speed centrifugation induces aggregation of extracellular vesicles. J Extracell Vesicles 4, 29509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateescu B, Kowal EJ, van Balkom BW, Bartel S, Bhattacharyya SN, Buzas EI, Buck AH, de Candia P, Chow FW, Das S, et al. (2017). Obstacles and opportunities in the functional analysis of extracellular vesicle RNA - an ISEV position paper. J Extracell Vesicles 6, 1286095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu M, Martin-Jaular L, Lavieu G, and Thery C (2019). Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol 21, 9–17. [DOI] [PubMed] [Google Scholar]
- Mathivanan S, Lim JW, Tauro BJ, Ji H, Moritz RL, and Simpson RJ (2010). Proteomics analysis of A33 immunoaffinity-purified exosomes released from the human colon tumor cell line LIM1215 reveals a tissue-specific protein signature. Mol Cell Proteomics 9, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo SA, Sugimoto H, O’Connell JT, Kato N, Villanueva A, Vidal A, Qiu L, Vitkin E, Perelman LT, Melo CA, et al. (2014). Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 26, 707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minciacchi VR, Freeman MR, and Di Vizio D (2015). Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol 40, 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minciacchi VR, Spinelli C, Reis-Sobreiro M, Cavallini L, You S, Zandian M, Li X, Mishra R, Chiarugi P, Adam RM, et al. (2017). MYC Mediates Large Oncosome-Induced Fibroblast Reprogramming in Prostate Cancer. Cancer Res 77, 2306–2317. [DOI] [PubMed] [Google Scholar]
- Mitchell JP, Court J, Mason MD, Tabi Z, and Clayton A (2008). Increased exosome production from tumour cell cultures using the Integra CELLine Culture System. J Immunol Methods 335, 98–105. [DOI] [PubMed] [Google Scholar]
- Montermini L, Meehan B, Garnier D, Lee WJ, Lee TH, Guha A, Al-Nedawi K, and Rak J (2015). Inhibition of oncogenic epidermal growth factor receptor kinase triggers release of exosome-like extracellular vesicles and impacts their phosphoprotein and DNA content. J Biol Chem 290, 24534–24546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, and D’Souza-Schorey C (2009). ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol 19, 1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabhan JF, Hu R, Oh RS, Cohen SN, and Lu Q (2012). Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc Natl Acad Sci U S A 109, 4146–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte-’t Hoen EN, Buermans HP, Waasdorp M, Stoorvogel W, Wauben MH, and t Hoen PA (2012). Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic Acids Res 40, 9272–9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteikoetxea X, Sodar B, Nemeth A, Szabo-Taylor K, Paloczi K, Vukman KV, Tamasi V, Balogh A, Kittel A, Pallinger E, et al. (2015). Differential detergent sensitivity of extracellular vesicle subpopulations. Org Biomol Chem 13, 9775–9782. [DOI] [PubMed] [Google Scholar]
- Rome LH, and Kickhoefer VA (2013). Development of the vault particle as a platform technology. ACS Nano 7, 889–902. [DOI] [PubMed] [Google Scholar]
- Schiller M, Bekeredjian-Ding I, Heyder P, Blank N, Ho AD, and Lorenz HM (2008). Autoantigens are translocated into small apoptotic bodies during early stages of apoptosis. Cell Death Differ 15, 183–191. [DOI] [PubMed] [Google Scholar]
- Shurtleff MJ, Temoche-Diaz MM, Karfilis KV, Ri S, and Schekman R (2016). Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurtleff MJ, Yao J, Qin Y, Nottingham RM, Temoche-Diaz MM, Schekman R, and Lambowitz AM (2017). Broad role for YBX1 in defining the small noncoding RNA composition of exosomes. Proc Natl Acad Sci U S A 114, E8987–E8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr., Carter BS, Krichevsky AM, and Breakefield XO (2008). Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 10, 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuffers S, Sem Wegner C, Stenmark H, and Brech A (2009). Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 10, 925–937. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Okada R, Nagao K, Kawamata Y, Hanyu A, Yoshimoto S, Takasugi M, Watanabe S, Kanemaki MT, Obuse C, et al. (2017). Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun 8, 15287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Ren Y, Hu X, Mu J, Samykutty A, Zhuang X, Deng Z, Kumar A, Zhang L, Merchant ML, et al. (2017). MVP-mediated exosomal sorting of miR-193a promotes colon cancer progression. Nat Commun 8, 14448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, and Simons M (2008). Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319, 1244–1247. [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, and Lotvall JO (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9, 654–659. [DOI] [PubMed] [Google Scholar]
- van Niel G, Charrin S, Simoes S, Romao M, Rochin L, Saftig P, Marks MS, Rubinstein E, and Raposo G (2011). The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev Cell 21, 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niel G, D’Angelo G, and Raposo G (2018). Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. [DOI] [PubMed] [Google Scholar]
- Vella LJ, Scicluna BJ, Cheng L, Bawden EG, Masters CL, Ang CS, Willamson N, McLean C, Barnham KJ, and Hill AF (2017). A rigorous method to enrich for exosomes from brain tissue. J Extracell Vesicles 6, 1348885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, Perez-Hernandez D, Vazquez J, Martin-Cofreces N, Martinez-Herrera DJ, Pascual-Montano A, Mittelbrunn M, and Sanchez-Madrid F (2013). Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun 4, 2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, and Lu Q (2017). Plasma membrane-derived extracellular microvesicles mediate non-canonical intercellular NOTCH signaling. Nat Commun 8, 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Freitas D, Kim HS, Fabijanic K, Li Z, Chen H, Mark MT, Molina H, Martin AB, Bojmar L, et al. (2018). Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat Cell Biol 20, 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. High-Resolution Density Gradient Fractionation Separates Small Extracellular Vesicles from Non-Vesicular Components, Related to Figure 1. (A) Density profile of fractions obtained after high resolution iodixanol density gradient. An empty gradient (tube with iodixanol gradient but containing no sEV sample) was used run opposite an identical sample gradient and density of fractions was measured by refractometry.
(B-C) High-resolution density fractionation of (B) DiFi and (C) primary human renal epithelial cell-derived crude sEVs (P120). After flotation of sample in an iodixanol gradient equal volumes of each fraction were loaded on SDS-PAGE gels, and membranes were blotted with indicated antibodies. NV, non-vesicular; sEV, small EV.
(D) Two-round high-resolution density fractionation of DKO-1 cell-derived crude sEVs (P120). After a first round flotation of sample in an iodixanol gradient, purified sEVs (from fractions 1–6) were subjected to a second round of flotation in an identical iodixanol gradient.
(E) Negative stain EM of pooled Low (sEV) density fractions obtained by floatation of crude DKO-1 and Gli36 sEV pellets (P120) in iodixanol density gradients. Sample preparation was without a fixation step. Imaging was performed on a JEM 1200EX microscope.
(F) Size distribution of gradient-purified sEVs imaged by EM as in (C). Histograms of the diameter of measured vesicles. n = 504 for DKO-1 and n = 400 for Gli36.
Table S3. Proteomic analysis of DKO-1 Cell, large EV, and high-resolution density gradient-purified sEV and non-vesicular (NV) samples, Related to Figure 2.
Table S4. Proteomic analysis of Gli36 high-resolution density gradient-purified sEV and non-vesicular (NV) samples, Related to Figure 2.
Figure S2. Differential Expression of Protein and RNA in Small Extracellular Vesicles and Non-Vesicular Fractions, Related to Figure 2. (A) Schematic of experimental setup for proteomics analysis. After flotation on density gradients, separate pools of low (sEV) and high (NV) density fraction pools were subjected to LC-MS/MS.
(B) Venn diagram representing the number of proteins unique and overlapping between sample types.
(C) Volcano plots of quantitative differences in proteins in sEV and NV fractions. Black dots represent a four-fold or greater enrichment while orange dots represent less than four-fold enrichment. Dots above the dashed line represent proteins for which differences were significant (FDR < 0.05).
(D) Table of the fold-change in spectral counts from proteomic profiling between sEV and NV fractions for selected proteins chosen for validation by immunoblotting.
(E) Immunoblot validation of proteomic profiling.
(F) Heatmap of the 25 most commonly identified exosomal proteins from the ExoCarta exosome database form proteomic profiling of sEV and NV from DKO-1. Scale indicates intensity, defined as Δ(spectral counts – mean spectral counts)/standard deviation.
(G) Bioanalyzer electropherograms of the size distribution (in nucleotides) of RNA extracted from cells, lEV (P15) and gradient-purified sEV and NV samples, as measured by RNA Pico (DKO-1) or RNA Nano (Gli36) chips. lEV, large EV; sEV, small EV; NV, non-vesicular.
(H) Volcano plots of miRNA expression patterns in sEV and NV fractions for DKO-1 (left) and Gli36 (right).
(I) Spectral counts for YBX1 in cells, lEVs, purified sEV and NV fractions generated by high-resolution density gradient centrifugation for DKO-1 (left) and Gli36 (right). Data represent mean ± SD. N.D., Not Detected.
(J) Expression level of miR-223 and miR-144 for DKO-1 and Gli36 samples. Data represent mean ± SD. RPM, reads per million bases.
(K) 3’ trimming and tailing of miRNA. The changes in either 3’ nucleotide additions (tailing) or 3’ resection (trimming) compared to full length miRNA sequences (intact) from cells, lEVs (P15), and density gradient-purified sEV (low density) and NV (high density) fractions. n = 4 biological replicates for each sample type, representative examples displayed.
(L) Percentage of long RNA reads mapping to different types of transcripts for cellular and extracellular DKO-1 samples.
(M) Percentage of long RNA reads mapping to exonic and intronic gene regions for cellular and extracellular DKO-1 samples.
(N) Principal Component Analysis based on the quantitative lincRNA profiles of cellular and extracellular DKO-1 samples generated by long RNA-seq.
(O) Violin plot of lincRNA expression for cellular and extracellular DKO-1 samples, based on GENCODE annotation. The shapes indicate kernel density estimation; the thick black lines indicate the middle two quadrants in the distribution; and the white dots indicates the median.
(P) Short RNA-seq data for YRNA in DKO-1 cells, lEV, sEV and NV fraction pools. RPM, reads per million.
(Q) Long RNA-seq data for YRNA in DKO-1 cells, lEV, sEV and NV fraction pools. FPKM, fragments per kilobase per million reads.
Figure S3. Argonaute Protein But Not Other miRNA Processing Proteins Are Secreted From Human Cells, Related to Figure 3. (A) Immunoblot analysis of DKO-1, Gli36 and MDA-MB-231 whole cell lysate, large EVs (P15) and small EVs (P120).
(B) Proteomic analysis of P-body and stress granule proteins. Spectral counts for DCP1A, G3BP1, G3BP2 and TIA-1 present in DKO-1 cells, lEV (P15), density gradient-purified sEV and NV fraction pools. Data represent mean ± SD. N.D., Not Detected.
(C) Immunoblot analysis of crude sEV (P120) from bovine growth serum, exosome-depleted serum-supplemented DMEM and DKO-1 cells. BGS, bovine growth serum; EDM, exosome-depleted media.
(D) Schematic of the generation of purified plasma sEV and NV fraction pools by high-resolution iodixanol density gradients. RBCs, red blood cells; WBCs, white blood cells.
(E) Immunoblot analysis of crude DKO-1, Gli36 and plasma sEVs (P120).
(F) TSG101 immunoblot analysis of crude DKO-1 and plasma sEV samples (P120).
(G) Crude DKO-1, Gli36, DiFi and primary renal epithelial sEVs obtained by ultracentrifugation at 120,000 × g (P120) were fractionated by floatation on high-resolution iodixanol gradients, and twelve fractions of equal volume were collected for analysis by immunoblotting.
Figure S4. Exosomes Exclude Common Glycolytic Enzymes and Lack a Cytoskeleton, Related to Figure 4. (A) Direct immunoaffinity capture of CD81-, CD63-, and CD9-positive exosomes. Immunoblots of crude sEV pellet (P120) and bead-captured exosomes from DKO-1 and Gli36 cells.
(B) Immunoblots of DKO-1 whole cell lysates, large EVs (P15) and crude small EVs (P120) obtained by ultracentrifugation.
(C) High-resolution density fractionation of crude sEVs (P120). NV, non-vesicular; sEV, small EV.
(D) Direct immunoaffinity capture of CD81-, CD63-, and CD9-positive exosomes from DKO-1, Gli36 and primary renal epithelial cells. Immunoblots of crude sEV pellet (P120) and bead-captured exosomes. g.e, greater exposure.
(E-F) Direct immunoaffinity capture of CD81-, CD63-, and CD9-positive exosomes from (E) DKO-1 and (F) Gli36 cells. Immunoblots of crude sEV pellet (P120) and bead-captured exosomes.
Figure S5. Annexin A1 is a Novel and Specific Marker of Microvesicles Distinct from Exosomes and ARMMs, Related to Figure 5. (A) Density profile of fractions obtained after 6–30% iodixanol density gradient fractionation. An empty gradient (tube with iodixanol gradient but containing no sEV sample) was used run opposite an identical sample gradient and density of fractions was measured by refractometry.
(B-D) Direct immunoaffinity-capture of CD63-, CD81-, and CD9-positive exosomes from (B) DKO-1, (C) Gli36, and (D) primary renal epithelial cells. Immunoblots of crude sEV pellet (P120) and bead-captured exosomes. g.e, greater exposure.
(E) Annexin A1-positive classical microvesicles. (top) Structured Illumination Microscopy (SIM) of DKO-1 cells stained for Annexin A1. Enlarged inserts displayed to the right. (Bottom) Size of Annexin A1-positive classical microvesicles measured at the maximum width.
(F) High-resolution (12–36%) density gradient sEV and NV fraction pools purified from plasma samples (see Figure S3D). For immunoblotting, equal volumes (representing equal portions of the total) were separated on SDS-PAGE gels, and membranes were blotted with indicated antibodies.
Figure S6. Release of Extracellular dsDNA and Histones from Human Cells is Independent of Exosomes and Small Extracellular Vesicles, Related to Figure 6. (A) High-resolution iodixanol gradient fractionation of DiFi cell-derived (top) and primary human renal epithelial cell-derived (bottom) crude sEVs (P120) analyzed by immunoblotting. NV, non-vesicular; sEV, small extracellular vesicle.
(B) Schematic of experimental design for Figure 6B–C. P120 sample was resupended and either loaded directly at the bottom of the iodixanol gradient (Figure 6B) or treated with DNase I to digest unprotected DNA (Figure 6C) before loading. After 15h of centrifugation, fractions from the now approximately linear gradient were collected from the top to generate sEV and NV pools. Material was washed and pelleted at 120,000 × g followed by DNA extraction. For Figure 6B, the extracted nucleic acid was treated with DNase I or RNase A/T1 before analysis.
(C) Quantification of DNA from gradient-fractionated sEV and NV pools obtained from primary human cultures of renal epithelial cells. Data represent mean ± SD of three technical measurements of one biological replicate. N.D., Not Detected.
(D) Bioanalyzer electropherograms of the size distribution of purified DNA in base pairs (bp) from DNase I pre-treated density gradient-fractionated, pooled, exosomal DKO-1 samples, as measured with the DNA High Sensitvity chip. DNA marker peaks at 35 bp and 10,380 bp. FU: fluorescence units.
(E) Schematic of experimental design for Figure 6E–F. Pre-cleared medium was obtained after differential centrifugation steps of 400–15,000 × g (see STAR Methods).
Figure S7. Active Secretion of DNA and Histones Through an Amphisome-Dependent Mechanism, Related to Figure 7. (A) DKO-1 cells stained for DAPI, and endogenous CD63 and CD9, and imaged with 3D SIM. Greyscale images of Z-stack projection in Figure 7A. MVE, multivesicular endosome.
(B) Cropped images of DKO-1 cells localized for CD63 and DNA (DAPI) by 3D SIM. (Left) Greyscale CD63 and DNA, and colorized merge. Displayed are sequential image slices (125 nm) through the Z-stack. Blue arrow indicates the cell nucleus; yellow arrow indicates a micronuclei. (Right) Enlarged image of the CD63-positive MVE with yellow line indicating the longest axis. Scale bar, 200 nm.
(C) DKO-1 cells were after permeabilization stained for CD63 and wheat germ agglutinin (WGA), and imaged with 3D SIM. (Left) Low magnification image of standard Y-X axis “Top-Down View”. (Middle) Enlarged yellow inserts. Scale bar, 200 nm. (Right) Z-X axis “Side View” of blue insert. Blue arrows indicate CD63-positive MVE. Yellow asterisks indicate CD63-positive MVE at the plasma membrane.
(D) CD63-and LC3B-positive amphisomes in DKO-1 cells. Cells were stained for endogenous CD63 and LC3B. Greyscale CD63 and LC3B, and colorized merge. White arrows indicate LC3B localized to CD63-positive compartments.
(E) Immunoblots of whole cell lysate and crude small EVs (P120) isolated from matched tissue/interstitial fluid of colorectal cancers (CRC) and adjacent normal. N, normal; T, tumor.
(F) Reassessment of exosome composition. Schematic illustration of revised composition of CD63/CD81/CD9-positive classical exosomes based on the data generated in this study. Proteins not examined here but associated with exosomes based on other studies are not included. The principal fractions containing the CD63/CD81/CD9 proteins after high-resolution density gradient centrifugation indicates that the majority of classical exosomes have buoyant densities in iodixanol-PBS from 1.08–1.13 g/ml, but can be found ranging from 1.05–1.15 g/ml. In contrast, small Annexin A1-positive classical microvesicles were principally observed at densities of 1.05–1.10 g/ml, but can be found ranging from 1.03–1.10 g/ml. ARMM, Arrestin-domain-containing protein 1 (ARRDC1)-mediated microvesicle; EV, extracellular vesicle; MVE, multivesicular endosome.
Data Availability Statement
Datasets for RNA-seq have been deposited in GEO: GSE125905