

Abstract
Background
Heavy metal ATPases (HMAs) are responsible for Cd translocation and play a primary role in Cd detoxification in various plant species. However, the characteristics of HMAs and the regulatory mechanisms between HMAs and microRNAs in wheat (Triticum aestivum L) remain unknown.
Results
By comparative microRNA and transcriptome analysis, a total three known and 19 novel differentially expressed microRNAs (DEMs) and 1561 differentially expressed genes (DEGs) were found in L17 after Cd treatment. In H17, by contrast, 12 known and 57 novel DEMs, and only 297 Cd-induced DEGs were found. Functional enrichments of DEMs and DEGs indicate how genotype-specific biological processes responded to Cd stress. Processes found to be involved in microRNAs-associated Cd response include: ubiquitin mediated proteolysis, tyrosine metabolism, and carbon fixation pathways and thiamine metabolism. For the mRNA response, categories including terpenoid backbone biosynthesis and phenylalanine metabolism, and photosynthesis - antenna proteins and ABC transporters were enriched. Moreover, we identified 32 TaHMA genes in wheat. Phylogenetic trees, chromosomal locations, conserved motifs and expression levels in different tissues and roots under Cd stress are presented. Finally, we infer a microRNA-TaHMAs expression network, indicating that miRNAs can regulate TaHMAs.
Conclusion
Our findings suggest that microRNAs play important role in wheat under Cd stress through regulation of targets such as TaHMA2;1. Identification of these targets will be useful for screening and breeding low-Cd accumulation wheat lines.
Electronic supplementary material
The online version of this article (10.1186/s12864-019-5939-z) contains supplementary material, which is available to authorized users.
Keywords: Wheat, RNA sequencing, microRNA, HMA
Background
Cadmium (Cd) contamination of soils is rapidly increasing from both human activities and other environmental causes [1]. Deleterious effects of Cd are dramatic and varied in both humans and plants. In humans, Cd ingestion leads to osteoporosis and emphysema, among other diseases [2, 3]. In plants, high Cd results in a wide range of biochemical and physiological disorders, inhibiting yield and quality [4]. Unfortunately, even moderate levels of Cd in soils can result in dramatic Cd accumulation in leaves or grains [5, 6]. Contamination in the food chain with Cd thus poses a major widespread hazard to human health [7–9]. Wheat, one of the most important worldwide crops, is grown on nearly 20% of cultivated land and serves as a staple food source for 30% of humanity [10, 11]. Previous studies indicated that compared with other cereals, wheat accumulates Cd primarily via roots, eventually accumulating Cd in edible grains [12, 13]. Therefore, it is very important to understand wheat responses to Cd in order to improve wheat growth, grain quality, and human welfare.
MicroRNAs (miRNAs) are typically 21 nucleotide endogenous non-coding RNAs that play key roles in regulating gene expression post-transcriptionally, usually via cleavage or translational repression of target mRNAs [14, 15]. In plants, miRNAs are involved in various physiological processes and play important roles in growth and development [16, 17], and numerous miRNAs participate in responses to biotic and abiotic stresses [18–21]. Deep-sequencing of developing wheat grains revealed 605 miRNAs [22] and 57 conserved miRNA families have been identified in wheat hybrid necrosis [23]. Hundreds of miRNAs were differentially expressed in leaf and roots tissues in wheat under drought stress, respectively [24]. Individual analysis of miRNAs or mRNAs have been widely reported under various stressors. However, the interplay of Cd, regulatory miRNAs, and the transcriptome is not at all understood, so there is still an obvious gap in our understanding of their interplay in a major crop.
P1B-ATPases, also known as heavy metal ATPases (HMAs), are proteins that use ATP to pump a wide range of cations across membranes against electrochemical gradients [25]. AtHMA2 and AtHMA4 act as pumps to efflux Zn/Cd out of cells, playing critical role in root-shoot Zn/Cd translocation through xylem loading [26]. The HMA3 proteins, such as AtHMA3, TcHMA3 and OsHMA3, are typically located in tonoplasts and are not regulated through Zn or Cd; these proteins play a role in pumping Zn and/or Cd into vacuoles [27–29]. Much available evidence indicates that HMAs play an important role in heavy metal transmembrane transport. However, there is little understanding of HMAs in the wheat genome and their roles in wheat under Cd stress.
Therefore, to gain an understanding of the global transcriptional response and its interplay with miRNAs and HMAs in wheat during Cd stress, we used next generation transcriptome and miRNA sequencing to profile the responses of low- and high- cadmium-accumulating wheat cultivars in response to Cd. Additionally, we paid special attention to regulatory dynamics of the HMA gene family. We further verified by qRT-PCR significantly differentially expressed of miRNAs, mRNAs and TaHMAs. Finally, we performed for gene ontology and pathway analysis, as well as depicted general functional landscapes of miRNAs-TaHMAs expression. We expect that these findings will provide a meaningful resource for the understanding the global transcriptional response to Cd and how these are modulated by miRNAs.
Results and discussion
Differences in cd accumulation in grains among wheat germplasms
We first screened 185 wheat germplasms for grain Cd levels using ICP-MS (Additional file 1: Table S1). By group, the average concentrations of Cd in grain decreased in the order of CMC > Chuanyu > CIMMYT, with significant differences between group averages (Fig. 1a). The tolerance limit of Cd in food is 0.1 mg/kg set by Ministry of Health of PRC (People’s Republic of China). Thus, there were 15 (28.85%) cultivars that exceeded the tolerance limit in Chuanyu group, 1 (1.79%) in the CIMMYT group, and 56 (76.71%) in the CMC group (Fig. 1b). Our results revealed that the average grain Cd concentration of four durum wheat genotypes was 0.224 mg/kg, and ranged from 0.191 to 0.371 mg/kg. These four durum cultivars contained higher grain Cd concentrations compared with bread wheat (Additional file 2: Figure S1), in agreement with previous studies [30–32]. Previous work showed that durum wheat cultivars grown in a variety test in South Dakota contained similar average grain Cd concentrations (0.22 mg/kg, ranged from 0.13 to 0.25 mg/kg) [33], indicating that our results are good representatives of world-wide sampling.
Fig. 1.

a Grain Cd concentration among three major wheat groups. The Chuanyu group included 52 wheat cultivars, the CIMMYT group included 56 wheat cultivars, and the CMC group included 73 wheat cultivars. b Percentage of low Cd concentration (≤ 0.0579 mg/kg), moderate Cd concentrations (< 0.1 mg/kg) and exceeding Cd concentration standard (≥ 0.1 mg/kg) wheat in each of the three groups
Differentially expressed miRNAs and mRNAs between L17Cd and L17CK, H17Cd and H17CK
To illuminate the underlying molecular mechanism, we chose cultivars strongly contrasting for Cd content: a low-Cd-accumulating cultivar (Chuanyu 17, hereafter L17) and a high-Cd-accumulating cultivar (a Chuanyu 17 sib-line, hereafter H17) to determine differentially expressed miRNAs and mRNAs in roots. We generated 12 small RNA sequencing libraries from three biological replicates each for L17 and H17 roots under control and CdCl2 treatment and sequenced them on an Illumina HiSeq TM 2500. The major characteristics of these libraries are summarized in Table 1. Between 18.8 and 32.4 million total raw reads per library were obtained. After quality filtration, between 12.6 and 20.4 million clean reads were obtained per replicate.
Table 1.
Read statistics in small RNA libraries by genotype
| Type | L17CK | L17Cd | H17CK | H17Cd |
|---|---|---|---|---|
| Raw reads | 29,634,338 ± 12,235,540 | 23,765,742 ± 5,243,896 | 18,786,733 ± 2,145,550 | 32,365,435 ± 7,244,882 |
| Clean reads | 20,430,195 ± 8,138,193 | 14,922,483 ± 7,059,238 | 12,556,082 ± 1,740,984 | 19,285,729 ± 5,444,137 |
| Unique clean reads | 3,232,322 ± 981,148 | 2,426,636 ± 1,239,154 | 2,313,643 ± 314,127 | 2,580,979 ± 647,449 |
| Wheat genome-matched read (redundant) | 17,858,724 ± 7,263,840 | 12,629,611 ± 5,018,126 | 8,991,791 ± 1,289,534 | 14,299,057 ± 3,343,128 |
| Wheat genome-matched read (unique) | 2,180,300 ± 664,560 | 1,604,078 ± 683,985 | 1,313,826 ± 142,562 | 1,481,392 ± 264,618 |
| Wheat genome-matched read (redundant %) | 87.14 ± 1.14 | 86.43 ± 5.95 | 71.61 ± 2.78 | 74.81 ± 3.80 |
| Wheat genome-matched read (unique %) | 67.45 ± 0.56 | 67.82 ± 5.18 | 56.93 ± 1.82 | 58.12 ± 4.71 |
In each wheat samples, data is shown as the mean of the three libraries generated ± standard deviation
After comparing the distributions of miRNA expression values in the samples after normalization and correlation analysis of miRNA expression levels between samples and samples (Additional file 2: Figure S2), we next performed miRNA expression analysis and found many clear contrasts in miRNA expression between genotypes and groups. Two miRNAs were upregulated in L17Cd (treated with 100 μM CdCl2 for 24 h Cd) and one was downregulated compared with L17CK (CdCl2-free). In the high-Cd-accumulating genotype (H17), 12 miRNAs in H17Cd (treated with 100 μM CdCl2 for 24 h Cd) were downregulated compared with H17CK (CdCl2-free) (Table 2). Regarding novel miRNAs in L17, 10 miRNAs were upregulated and nine were downregulated in L17Cd compared with L17CK, while in H17, 49 miRNAs were upregulated and eight were downregulated in H17Cd compared with H17CK (Table 3). In addition, one uniquely expressed miRNA between L17Cd and L17CK, one uniquely expressed miRNA between H17Cd and H17CK, and two expressed miRNAs were commonly expressed. These results indicated that tae-miR9664-3p and tae-miR159a were upregulated in L17Cd group compared with that in L17CK group, while they were downregulated in H17Cd group compared with that in H17CK group. Moreover, according to our RNA sequencing data, the putative target mRNAs (such as Traes_7BL_66C9AAA60, Traes_1BS_1E568BC28, Traes_5DL_BDE25A669, Traes_2DS_47DDC83DC and Traes_5BS_C3B48D8CC) for upregulated tae-miR9664-3p and the putative target mRNAs (such as Traes_3AL_F09C7CA19, Traes_7AS_2831C6473, Traes_7DS_0FFF2FB02 and Traes_4AL_03723C140) for upregulated tae-miR159a were downregulated in L17Cd group compared with that in L17CK group. Previous work demonstrated that tae-miR-9664 was expressed in the flag leaf and developing seed of wheat (Triticum aestivum L.) [34]. Tae-miR159a played positive roles in wheat response to Puccinia striiformis f.sp.tritici through the regulation of TaMyb3 expression [35]. Our results suggested that contrasting Cd-accumulating ability of L17 and H17 might associated with the up-regulation or down-regulation between tae-miR9663-3p, tae-miR159a and their target genes. We also found that tae-miR9774 was uniquely down-regulated between L17Cd and L17CK, while tae-miR398 was specifically down-regulated between H17Cd and H17CK. A previous study demonstrated that tae-miR-9774 was expressed in wheat leaves, stems, roots and young spikes [36]. miR398 was induced by UVB light and heat stress in Arabidopsis but was inhibited by ABA, salinity, cold, and oxidative stress [37, 38]. miR398 can regulate copper superoxide dismutase (CSD) 1 and 2 that were both induced by salinity treatment [37, 39]. These results suggested that miRNAs could participate in the growth and development of wheat and also function in wheat response to both biotic and abiotic stress. To our knowledge, this is the first study to identify differentially expressed miRNAs in response to Cd accumulation in multiple wheat genotypes contrasting for Cd accumulation.
Table 2.
Known miRNAs identified in the 12 wheat sRNA libraries
| Comparison | miRNA family | miRNA_id | log2FoldChange | P | Down |
|---|---|---|---|---|---|
| L17Cd/L17CK | miR159a | tae-miR159a | 0.87 | 3.347E-05 | Up |
| miR9664 | tae-miR9664-3p | 0.53 | 0.0427885 | Up | |
| miR9774 | tae-miR9774 | −2.29 | 0.0279111 | Down | |
| H17Cd/H17CK | miR159a | tae-miR159a | −1.04 | 5.42E-11 | Down |
| miR167 | tae-miR167a | −1.47 | 0.0379267 | Down | |
| mi395 | tae-miR395b | −2.40 | 0.0038538 | Down | |
| miR397 | tae-miR397-5p | −1.50 | 0.019139 | Down | |
| miR398 | tae-miR398 | −0.99 | 0.0250389 | Down | |
| miR5048 | tae-miR5048-5p | −0.91 | 0.0247939 | Down | |
| miR7757 | tae-miR7757-5p | −1.37 | 0.0001656 | Down | |
| miR9662 | tae-miR9662a-3p | −1.10 | 7.92E-06 | Down | |
| miR9664 | tae-miR9664-3p | −0.68 | 0.0203362 | Down | |
| miR9669 | tae-miR9669-5p | −1.50 | 0.0258714 | Down | |
| miR9674 | tae-miR9674b-5p | −0.92 | 0.0109898 | Down | |
| miR9778 | tae-miR9778 | −1.30 | 1.11E-07 | Down |
Table 3.
Summary of newly identified novel miRNAs in the 12 wheat sRNA libraries
| Comparison | miRNA_id | log2 (Fold Change) | P | Down | Mature sequence(5'-3') | Length |
|---|---|---|---|---|---|---|
| L17Cd/L17CK | 2B_20696a | 1.69 | 0.0270291 | Up | CUCGCCGGUCGCGCGUCCUCC | 21 |
| 2B_21940a | 2.33 | 0.0462198 | Up | AUGACACGGGGAGGUAGC | 18 | |
| 2B_39534a | 6.11 | 0.0176417 | Up | AGGCUGGAGGAACGUAGG | 18 | |
| 2B_40590a | 2.00 | 0.0383352 | Up | GUGUGGAGCAAAAGGGUG | 18 | |
| 4B_10717a | 4.18 | 0.0021475 | Up | CCCCGGGUGCGAGCUCCU | 18 | |
| 4B_12465a | 5.45 | 0.0221443 | Up | CCUCCUGGUGAGCCUGCG | 18 | |
| 4B_4791a | 1.96 | 0.0091409 | Up | UGGGGUUGUGGCAAUGGCC | 19 | |
| 4B_519a | 2.40 | 0.0050086 | Up | AAUCUGGUUGUCGCCUCC | 18 | |
| 4B_5711a | 4.20 | 0.0024414 | Up | GUGAAGAGACAUGGAGGU | 18 | |
| 4B_9306a | 1.13 | 0.0050015 | Up | UAGAAUGGCUGGUGCUAUGGA | 21 | |
| 2B_19887a | -4.18 | 0.0352225 | Down | ACGGACCGCGCUACUACUAUAAGU | 24 | |
| 2B_22021a | -1.39 | 0.016954 | Down | AUGGAAGACGUGGUAGCC | 18 | |
| 2B_28100a | -1.27 | 0.0412731 | Down | AGGUAGAGAGGAAGGUGGG | 19 | |
| 2B_33543a | -3.40 | 0.0480069 | Down | CUCGCCGGAGGAGCGUGC | 18 | |
| 2B_38598a | -2.15 | 0.0214449 | Down | CUGCUGGUGCUGUAGCCCU | 19 | |
| 4B_17875 | -4.36 | 0.0138807 | Down | UUCCAAUUUACUCGUCGUGGU | 21 | |
| 4B_4344a | -2.68 | 0.0327648 | Down | GGUGUGGACCAGGUUUCC | 18 | |
| 4B_87a | -4.35 | 0.0106819 | Down | CGCGUGCAGGAUGAAGCC | 18 | |
| 4B_8797 | -2.08 | 0.0458935 | Down | ACUCACUCUGUAAACUAAUAUAAG | 24 | |
| H17Cd/H17CK | 2B_19349a | 4.00 | 1.83E-06 | Up | AAACAGUCUGUAAAGCCCC | 19 |
| 2B_20999a | 2.42 | 1.06E-14 | Up | CUACGGGGGAAAGCAGGG | 18 | |
| 2B_21614a | 2.02 | 0.0023063 | Up | UGUUGAAGUAGCUGGAAC | 18 | |
| 2B_21995a | 1.41 | 0.002425 | Up | CGGAGAGGGGAGUGAAGU | 18 | |
| 2B_22021a | 2.93 | 9.43E-27 | Up | AUGGAAGACGUGGUAGCC | 18 | |
| 2B_22879a | 3.19 | 0.0120242 | Up | CGGCCAUCGUGAGGUUAGUGC | 21 | |
| 2B_25173a | 2.36 | 0.0000977 | Up | GACUUUGAUCCAGAGAUCA | 19 | |
| 2B_27180a | 1.86 | 0.0252563 | Up | GUUGGUCGGGAGUUCGAUCCU | 21 | |
| 2B_28100a | 1.76 | 4.01E-07 | Up | AGGUAGAGAGGAAGGUGGG | 19 | |
| 2B_28714a | 1.46 | 0.0001773 | Up | AUGAGAGGACGGACAAGG | 18 | |
| 2B_29589a | 1.98 | 0.0040311 | Up | GAGUUUGGGAGUCUGUGUGU | 20 | |
| 2B_32148 | 1.66 | 0.0364154 | Up | UCAGUGCAAUCCCUCUGGAAU | 21 | |
| 2B_33858a | 1.58 | 0.0226808 | Up | CGUGCCAGCAGCCGCGGA | 18 | |
| 2B_37122a | 1.33 | 0.0002491 | Up | UCUGGUGGAUGCCUUGGG | 18 | |
| 2B_37173a | 1.15 | 0.0031618 | Up | AGAGGGUGGUGGAGUUUCU | 19 | |
| 2B_38197a | 5.03 | 1.57E-10 | Up | UUGUUGGAUAUGUUGGUU | 18 | |
| 2B_40139a | 2.06 | 0.0018813 | Up | GUGGAUGCCUUGGCGAUC | 18 | |
| 2B_40352a | 2.74 | 0.0074044 | Up | CAAUGGUAGAGCAGAGGC | 18 | |
| 2B_42257a | 1.93 | 0.0031222 | Up | GGCGGCGGACGGGUGAGC | 18 | |
| 2B_42905a | 0.91 | 0.0142497 | Up | GAGGUGCUGCAUGGCUGG | 18 | |
| 2B_43522a | 1.91 | 0.0003365 | Up | UGAGGUUUGAGAGGGGCU | 18 | |
| 4B_10203a | 1.29 | 0.0034214 | Up | GAGGCGAUGAAGGACGGG | 18 | |
| 4B_10621a | 1.88 | 0.0391309 | Up | GGGUGAAGCUGUGGCGCG | 18 | |
| 4B_10870a | 2.94 | 6.11E-06 | Up | UGGUUCUGUAUGGAAGUG | 18 | |
| 4B_12067a | 1.14 | 0.0151284 | Up | AUGAACGCUAGCGGGAGG | 18 | |
| 4B_12312a | 3.38 | 0.0022606 | Up | GAGUGGUCGAGCGGGACGG | 19 | |
| 4B_13500a | 2.41 | 0.0044361 | Up | GGUUCUGUAUGGAAGUGG | 18 | |
| 4B_13640a | 1.37 | 0.0041213 | Up | AGGUCUGGGUUCGAGUCC | 18 | |
| 4B_1373a | 2.42 | 0.0134111 | Up | GCUUGUCGCUGGUUUGAG | 18 | |
| 4B_15373a | 0.92 | 0.0420051 | Up | GUGGCUCCACGUGGGCGGGC | 20 | |
| 4B_15496a | 1.63 | 0.0036066 | Up | UGCGGGAUGGAGCAGUCG | 18 | |
| 4B_15927a | 2.37 | 2.51E-08 | Up | AUGCUAGUCGAGCGGAUA | 18 | |
| 4B_16269a | 3.22 | 3.8E-11 | Up | AGGUCGACGGUUCGAAUCC | 19 | |
| 4B_16562a | 1.34 | 0.0445484 | Up | CGGAGGUCGCGGGUUCGA | 18 | |
| 4B_16625a | 1.94 | 0.0000161 | Up | UCCUGGGGUUGGAGAAGG | 18 | |
| 4B_18394a | 1.24 | 0.007484 | Up | GCGGCGAGAGCGGGUCGCU | 19 | |
| 4B_1943a | 1.85 | 0.0000179 | Up | ACAGUGGAUGCCUUGGCA | 18 | |
| 4B_2104a | 1.61 | 0.0136994 | Up | AAGCCUCUGGCGAAUGGG | 18 | |
| 4B_2324a | 3.28 | 0.0050367 | Up | GAUUUAUCGGGGAAGGAU | 18 | |
| 4B_2847a | 3.91 | 0.0001901 | Up | GAGGAAGGUGGAUGACGG | 18 | |
| 4B_4039a | 3.73 | 0.0200414 | Up | GAACGGAAGUUGGGGGCG | 18 | |
| 4B_4609a | 3.46 | 0.0000312 | Up | GAUCUUGAUGUGGUGUGCA | 19 | |
| 4B_4703a | 2.74 | 1.23E-07 | Up | GUGAUUCAGCGGCGGACG | 18 | |
| 4B_5897a | 1.35 | 0.0138282 | Up | AGGAGGGUGGGGAUGAGGU | 19 | |
| 4B_5947a | 2.75 | 0.0124835 | Up | GAGUGGUCGAGCGGGACGG | 19 | |
| 4B_6530a | 2.05 | 3.8E-10 | Up | GCUCGUGUCGUGAGCUGG | 18 | |
| 4B_841a | 1.64 | 0.0087137 | Up | GAAGGUCGUGGGUUCGUG | 18 | |
| 4B_88a | 3.20 | 0.0471916 | Up | AGACGUUUCCGUCGACUACGAAA | 23 | |
| 4B_974a | 1.79 | 0.0009387 | Up | UAUCGUGGGUUCGAAUCC | 18 | |
| 2B_19630a | -1.52 | 0.0134799 | Down | CAGCCUGCCAACCCUGGG | 18 | |
| 2B_23520a | -5.38 | 3.24E-07 | Down | GCUUCGGCGGUAUCCUCAU | 19 | |
| 2B_27285a | -1.72 | 0.0294342 | Down | GAUGGGUCUCUGAGGGAU | 18 | |
| 2B_40624a | -1.93 | 0.0212515 | Down | UGGCGAUGAGGAACGGAA | 18 | |
| 2B_40888a | -2.07 | 0.0151122 | Down | GGCGGAGGCUGCGGGUUU | 18 | |
| 4B_11590a | -2.33 | 0.0093351 | Down | GAGGGGUGUUGAUUUUCCGG | 20 | |
| 4B_7298a | -1.77 | 0.0132316 | Down | GAGGGGUGUUGAUUUUCCGG | 20 | |
| 4B_7309a | -2.87 | 0.0008129 | Down | GAACGGAAGUUGGGGGCG | 18 |
aHighly conserved miRNAs
After checking the distributions of expression values after normalization and correlation analysis of expression levels between samples (Additional file 2: Figure S3), we next performed differential expression analysis of mRNA transcripts. In the low-Cd-accumulating genotype (L17), a total of 1561 mRNAs were detected to be differentially expressed with fold change > 2.0, P < 0.05 (volcano plot filtering) and FDR < 0.05 (Additional file 2: Figure S4, Fig. 2a). Among them, 1208 and 353 mRNAs were upregulated and downregulated (fold change > 2.0, P < 0.05 and FDR < 0.05) in L17 roots compared with controls, respectively. In the high-Cd-accumulating genotype (H17), a total of 297 mRNAs were found to be differentially expressed with a fold change > 2.0, P < 0.05 (volcano plot filtering) and FDR < 0.05 (Additional file 2: Figure S4, Fig. 2b). Among them, 204 and 93 mRNAs were upregulated and downregulated (fold change > 2.0, P < 0.05 and FDR < 0.05) in H17 roots compared with controls, respectively.
Fig. 2.

Differentially expressed mRNAs. a Heat map of differentially expressed mRNA with fold changes ≥2.0, P < 0.05 and false discovery rate (FDR) < 0.05 between L17Cd and L17CK. b Heat map of differentially expressed mRNA with fold changes ≥2.0, P < 0.05 and FDR < 0.05 between H17Cd and H17CK
Verification of differentially expressed miRNAs and mRNAs
To confirm the results of the differential expression analyses, eight Cd-induced miRNAs and six Cd-induced mRNAs were selected for qRT-PCR expression analysis according to their fold change and GO analysis. For microRNAs, compared with L17CK group, tae-miR159a, tae-miR9446-3P, microRNA-2B_39534* were increased in L17Cd group, while tae-miR9774, microRNA-2B_28100* were decreased (Fig. 3a). Compared with H17CK group, microRNA-2B_28100* was increased in H17Cd group, while tae-miR159a, tae-miR395b, tae-miR398, tae-miR7757-5P and tae-miR9664-3P microRNA-2B_28100* (Fig. 3a). For mRNAs, compared with L17CK group, Traes_4AS_8944253DC was increased in L17Cd group, while Traes_2AL_E5172BD9D was decreased (Fig. 3b). Compared with H17CK group, Traes_4AS_8944253DC, Traes_5AL_B4E8A3115 and Traes_2BS_1484A7516 were increased in H17Cd group, while Traes_6DS_C59B6322F and Traes_2DS_01A0E5F7A (Fig. 3b). Hence, the qRT-PCR data verified the small RNA and RNA sequencing results.
Fig. 3.

qRT-PCR verification of differential expression analysis. a Relative expression levels of five miRNAs shown comparing L17Cd and L17CK (left) and six miRNAs are shown comparing H17Cd and H17CK (right). The heights of the columns stand for the fold changes (log2 transformed) computed from both qRT-PCR and small RNA sequencing data. b Relative expression levels of two mRNAs, comparing L17Cd and L17CK (left) and five mRNAs, shown comparing H17Cd and H17CK (right). The heights of the columns stand for the fold changes (log2 transformed) computed from the qRT-PCR and RNA sequencing data. A good correlation of qRT-PCR results and RNA sequencing data are shown through comparing such two results in both cases
GO and KEGG enrichment analysis
It is well known that miRNAs play essential roles in plant growth and development and also control plant responses to biotic and abiotic stresses by regulating targeted gene expression [23, 40]. Therefore, Gene Ontology (GO) analysis of targeted mRNAs of miRNAs can real the role of differentially expressed miRNAs. Enriched GO categories between L17Cd and L17CK groups, H17Cd and H17CK groups, according to the gene number enriched in GOs, the top 10 biological process (BP), molecular function (MF) and cellular component (CC) for downregulated miRNAs and upregulated miRNAs were shown in (Additional file 2: Figure S5). Overlaying gene sets (Additional file 2: Figure S6) revealed that down-regulated miRNAs in L17 consisted of genes involved in ubiquitin-dependent protein catabolic processes and cell wall organization, whereas up-regulated miRNAs in L17 consist of those involved in protein glycosylation and oxidoreductase activity. Down-regulated miRNAs in H17 consist of those involved in vesicle-mediated transport, and response to heat, whereas up-regulated miRNAs in H17 consist of those involved in photosynthetic electron transport chain and iron ion binding.
For mRNAs, GOs between L17Cd and L17CK groups, H17Cd and H17CK groups, according to the gene number enriched in GOs, the top 10 BP, MF and CC for upregulated mRNAs and downregulated mRNAs were shown in (Additional file 2: Figure S7). Based on the results of Venn diagrams, we found that up-regulated mRNAs in L17 prefer to cell wall macromolecule catabolic process and response to stress, whereas down-regulated mRNAs in L17 prefer to cell wall organization and DNA replication; up-regulated mRNAs in H17 prefer to photosynthesis and nicotianamine biosynthetic process, whereas down-regulated mRNAs in H17 prefer to defense response and metal ion transport (Additional file 2: Figure S6).
Analysis of pathway enrichment for miRNAs between L17Cd and L17CK groups revealed a diverse set of processes responsive to Cd (Fig. 4a, b). Among these pathways, starch and sucrose metabolism was the most prominent pathway targeted by downregulated miNAs (Fig. 4a), whereas glycolysis / gluconeogenesis was the top enriched pathway for up-regulated miRNAs (Fig. 4b). In the H17Cd and H17CK contrasts we found that carbon metabolism was the most prominent pathway targeted by downregulated miNAs (Fig. 4c), whereas photosynthesis was the most prominent pathway targeted by upregulated miNAs (Fig. 4d). For mRNAs differentially regulated between L17Cd and L17CK groups, the top 10 pathways associated with up- or down-regulated mRNAs are given in Fig. 5. Among these pathways, phenylpropanoid biosynthesis was the top pathway in upregulated mRNAs (Fig. 5a), whereas starch and sucrose metabolism was the top one in downregulated mRNAs (Fig. 5b). Pathways enriched in the H17Cd and H17CK groups are given in Fig. 5c, where glutathione metabolism was most prominent. On the other hand, phenylpropanoid biosynthesis was the most prominent in down-regulated mRNAs (Fig. 5d).
Fig. 4.

KEGG enrichment analysis of differentially-expressed miRNA-targeted genes. The top 10 pathways enriched in targeted genes are given in response to miRNAs: a down-regulated between L17Cd and L17CK; b up-regulated between L17Cd and L17CK; c down-regulated between H17Cd and H17CK; and d up-regulated between H17Cd and H17CK. The x-axis represent the number of genes enriched
Fig. 5.

KEGG pathway analysis for differentially-expressed mRNAs. The top 10 pathways enriched in differentially expressed mRNAs are given for the contrasts: a up-regulated between L17Cd and L17CK; b down-regulated between L17Cd and L17CK; c up-regulated between H17Cd and H17CK; and d down-regulated between H17Cd and H17CK. The x-axis represent the gene number enriched in pathways
Our results are complement previous work in other systems. Feng et al. [41] found enrichment of genes differentially-expressed in response to Cd in two sweet sorghum genotypes. Enriched in the high Cd-accumulating sweet sorghum genotype were genes that Sb04g033320, Sb04g033330, and Sb01g012440. Enriched in the low Cd-accumulating sweet sorghum genotypes were genes that Sb02g006960, Sb01g043400, and Sb10g022390. Other studies have indicated that transporters for essential elements such as Fe2+, Zn2+ and Ca2+ are involved in Cd uptake and transport [42]. Numerous heavy metal transporters, such as ATP-binding cassette transporters (ABC), natural resistance-associated macrophage proteins (Nramp) and heavy metal ATPases (HMA) participate in the acquisition, distribution, and homeostasis of Cd in plants [27, 43, 44]. The MAPK signaling pathway is also part of the miRNA-target gene network. MAPK signaling cascades are one of the most conserved pathways in plants and are known to modulate plant response to abiotic stress [45]. Taken together, these studies and ours indicate that the interaction effects of multiple miRNAs are involved in the regulation of diverse physiological systems to relieve the toxicity induced by Cd.
Genes related to transport
Between L17Cd and L17CK groups 223 up-regulated genes and 183 down-regulated genes are classified as transporters based on functional annotation. Between H17Cd and H17CK, 162 up-regulated genes and 134 down-regulated genes were transporter genes according to the functional annotation of the differentially expressed genes (Additional file 3). Of these, Traes_2AL_9B175F3DA, Traes_2AS_95611CAD2, Traes_2DL_D5F2272A9, Traes_2BS_3212EB7DF, Traes_7BL_39745BF3F, Traes_7DL_44DA42073, Traes_2DL_2A1D45D02, Traes_3DL_4EE343C68, Traes_5BL_4BCE2DBEF, Traes_2BS_3212EB7DF, Traes_3AS_CBB5F7EB6, Traes_4DL_560FD0832, Traes_7AL_3E508AA9C, Trae_7DL_A5269C73F, Trae_7AL_8304348B7 and Trae_7BL_A4BE6BD10 are related to metal ion.
Genome-wide identification and phylogenetic analysis of HMA gene in wheat
Through the availability of the wheat genome sequence, it is now possible to identify all the HMA family members in wheat. Here we identify a total of 32 HMA members in the wheat genome (Table 4) based on the genome sequence. We further performed a BLASTN search against the wheat expressed sequence tag (EST) using these HMAs queries to verify the existence of these wheat HMAs. Results showed that most of the TaHMAs’ we found in the genome sequence were supported by EST hits. Among the supported TaHMA genes, TaHMA1;4 has the largest hits of ESTs, with 61, followed by TaHMA6;3 with 53. The lengths of these predicted TaHMA proteins varied from 419 aa to 1702 aa with the putative molecular weights (Mw) ranging from 44.07 to 185.21 kDa and theoretical isoelectric points (PI) ranging from 5.23 to 8.21. Eight HMA proteins are known in A.thaliana and nine in O.sativa, which range from 542 aa to 1171aa and from 792 aa to 1060 aa, respectively [46]. Using full-length HMA protein sequences from Triticum aestivum, A.thaliana and O.sativa, we constructed a phylogentic tree to determine the phylogenetic relationship between HMA proteins from Triticum aestivum and those from other species. The HMA gene families could be divided into three groups: the Cu/Ca/Zn/Cd/Co-ATPases group, the Zn/Cd/Pb/Co-ATPase group, and the Cu-ATPase group [47]. TaHMA1;1-TaHMA1;5 belong to the Cu/Ca/Zn/Cd/Co-ATPases group, TaHMA2;1-TaHMA2;6, TaHMA3;1-TaHMA3;2 belong to the Zn/Cd/Pb/Co-ATPase group, TaHMA4;1-TaHMA4;3, TaHMA5;1-TaHMA5;3, TaHMA6;1-TaHMA6;3, TaHMA7;1-TaHMA7;3, TaHMA8;1-TaHMA8;3, TaHMA9;1-TaHMA9;3 belong to the Cu-ATPase group (Fig. 6). To our knowledge, it is first genome-wide analysis of the HMA family in wheat.
Table 4.
Identified putative wheat HMA genes
| HMAs | Subfamily gene name | Ensemble Wheat Gene ID | Chromosome location | Length (bp) | Exons | Introns | Amino acid length (aa) | PI | MW (kDA) | EST count |
|---|---|---|---|---|---|---|---|---|---|---|
| TaHMA1;1 | TaHMA1 | Trae_5AL_C89EEBE50 | TGACv1_scaffold_375043_5AL:69226–81,875 | 12,650 | 16 | 15 | 790 | 6.31 | 84.44 | 2 |
| TaHMA1;2 | Trae_5BL_F83C809F0 | TGACv1_scaffold_405147_5BL:88295–97,375 | 9081 | 13 | 12 | 480 | 8.21 | 51.96 | 2 | |
| TaHMA1;3 | nd | TGACv1_scaffold_556453_7AL:106656–112,341 | 5686 | 8 | 7 | 419 | 6.56 | 44.07 | 20 | |
| TaHMA1;4 | Trae_7BL_041308E74 | TGACv1_scaffold_578186_7BL:25106–34,662 | 9557 | 14 | 13 | 832 | 6.90 | 88.37 | 61 | |
| TaHMA1;5 | Trae_7DL_FBCC75BBC | TGACv1_scaffold_603768_7DL:23146–31,623 | 8478 | 12 | 11 | 748 | 6.98 | 78.96 | 39 | |
| TaHMA2;1 | TaHMA2 | Trae_7DL_A5269C73F | TGACv1_scaffold_602651_7DL:105511–113,285 | 7742 | 10 | 9 | 1003 | 6.53 | 108.33 | 34 |
| TaHMA2;2 | Trae_7AL_8304348B7 | TGACv1_scaffold_556712_7AL:90458–108,176 | 17,719 | 9 | 8 | 985 | 6.29 | 106.46 | 21 | |
| TaHMA2;3 | Trae_7BL_A4BE6BD10 | TGACv1_scaffold_579416_7BL:14127–24,069 | 9282 | 10 | 9 | 1023 | 6.51 | 110.32 | 37 | |
| TaHMA2;4 | Trae_7BL_0CF58CF4E | TGACv1_scaffold_577252_7BL:118443–123,897 | 5455 | 9 | 8 | 766 | 7.83 | 81.09 | 3 | |
| TaHMA2;5 | Trae_7AL_6AE850114 | TGACv1_scaffold_557470_7AL:61198–66,965 | 5768 | 10 | 9 | 939 | 7.11 | 99.55 | 6 | |
| TaHMA2;6 | Trae_7DL_EED4BED66 | TGACv1_scaffold_603863_7DL:43200–49,689 | 5572 | 8 | 7 | 800 | 6.44 | 84.37 | 6 | |
| TaHMA3;1 | TaHMA3 | Trae_5BL_D6C3DC326 | TGACv1_scaffold_404346_5BL:222670–226,813 | 4108 | 6 | 5 | 829 | 6.05 | 86.82 | 3 |
| TaHMA3;2 | nd | TGACv1_scaffold_375473_5AL:29654–33,170 | 3517 | 6 | 5 | 816 | 6.29 | 85.66 | 2 | |
| TaHMA3;3 | nd | TGACv1_scaffold_435190_5DL:5098–8725 | 3628 | 6 | 5 | 853 | 6.12 | 89.09 | 3 | |
| TaHMA4;1 | TaHMA4 | Trae_6DS_26C5A0A44 | TGACv1_scaffold_543118_6DS:56997–63,045 | 6049 | 10 | 9 | 687 | 6.01 | 73.46 | 30 |
| TaHMA4;2 | Trae_6BS_A8B960E60 | TGACv1_scaffold_515718_6BS:7391–13,506 | 6116 | 9 | 8 | 980 | 5.44 | 105.63 | 29 | |
| TaHMA4;3 | Trae_6AS_6F306F27E | TGACv1_scaffold_485988_6AS:25642–29,385 | 3744 | 7 | 6 | 687 | 6.01 | 73.47 | 25 | |
| TaHMA5;1 | TaHMA5 | Trae_2DL_51FF05F66 | TGACv1_scaffold_157956_2DL:22600–32,475 | 9876 | 6 | 5 | 1000 | 5.65 | 108.18 | 9 |
| TaHMA5;2 | Trae_2AL_D0EABF355 | TGACv1_scaffold_099043_2AL:1541–7440 | 5900 | 6 | 5 | 1011 | 5.66 | 109.11 | 9 | |
| TaHMA5;3 | Trae_2BL_19B3E60AA | TGACv1_scaffold_130949_2BL:4724–10,512 | 5789 | 6 | 5 | 994 | 5.88 | 107.49 | 9 | |
| TaHMA6;1 | TaHMA6 | Trae_6BS_9A12C2A1D | TGACv1_scaffold_513141_6BS:286354–292,595 | 6242 | 9 | 8 | 974 | 5.73 | 104.50 | 50 |
| TaHMA6;2 | Trae_6DS_9FA053DF8 | TGACv1_scaffold_542481_6DS:33526–39,252 | 5727 | 9 | 8 | 1042 | 5.53 | 111.76 | 50 | |
| TaHMA6;3 | Trae_6AS_9321C1C5B | TGACv1_scaffold_486033_6AS:36396–41,875 | 5480 | 9 | 8 | 997 | 5.23 | 106.85 | 53 | |
| TaHMA7;1 | TaHMA7 | Trae_7AS_766146E70 | TGACv1_scaffold_569059_7AS:131450–140,663 | 9214 | 17 | 16 | 947 | 7.26 | 99.55 | 22 |
| TaHMA7;2 | Trae_7BS_8EC4B41E4 | TGACv1_scaffold_592600_7BS:48974–58,635 | 9662 | 17 | 16 | 952 | 7.23 | 100.02 | 26 | |
| TaHMA7;3 | Trae_7DS_04F16455B | TGACv1_scaffold_622783_7DS:19856–29,376 | 9521 | 17 | 16 | 952 | 6.98 | 99.94 | 26 | |
| TaHMA8;1 | TaHMA8 | Trae_4BL_89775421A | TGACv1_scaffold_640736_U:185896–195,006 | 9111 | 17 | 16 | 903 | 6.07 | 94.48 | 37 |
| TaHMA8;2 | Trae_4AS_622EEFE10 | TGACv1_scaffold_306691_4AS:30689–37,419 | 6731 | 17 | 16 | 900 | 5.85 | 94.16 | 37 | |
| TaHMA8;3 | Trae_4DL_385639507 | TGACv1_scaffold_344605_4DL:1306–16,767 | 15,462 | 20 | 19 | 1702 | 6.93 | 185.21 | 30 | |
| TaHMA9;1 | TaHMA9 | Trae_7AL_7A2639A1B | TGACv1_scaffold_558946_7AL:1236–7780 | 6545 | 9 | 8 | 1001 | 5.29 | 106.89 | 17 |
| TaHMA9;2 | Trae_7DL_DF97DD324 | TGACv1_scaffold_604439_7DL:28926–35,180 | 6255 | 9 | 8 | 1001 | 5.40 | 106.78 | 18 | |
| TaHMA9;3 | Trae_7BL_EFF0E2E31 | TGACv1_scaffold_579154_7BL:35573–41,675 | 6103 | 9 | 8 | 1001 | 5.28 | 106.77 | 16 |
Fig. 6.
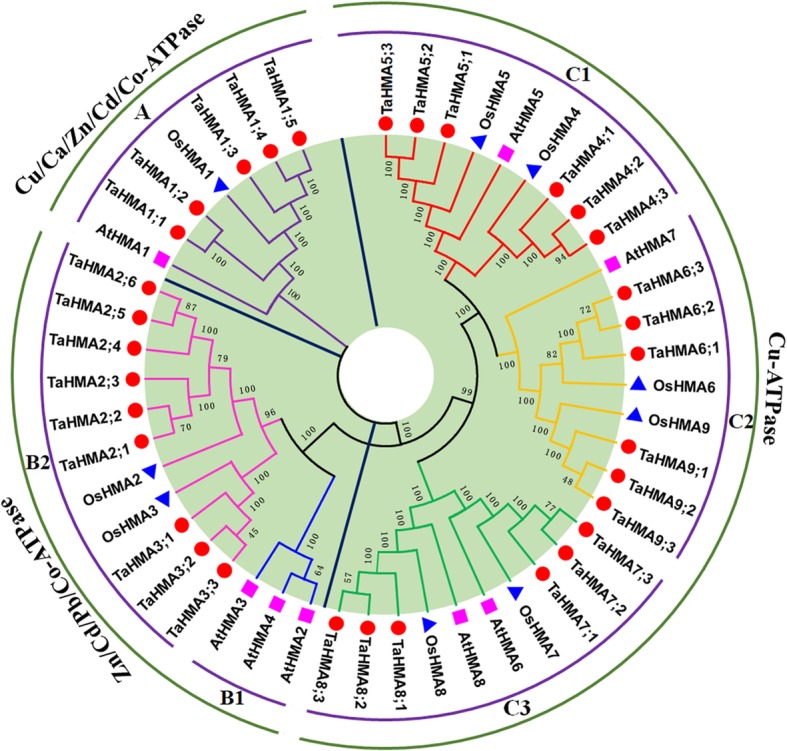
Neighbour-Joining phylogenetic tree of HMA proteins from wheat (Ta), Aradidopsis (At), and rice (Os). HMA proteins were used to establish the phylogenetic tree with MEGA6.0. TaHMA proteins are marked in red. An unrooted Neighbour-Joining analysis was performed with pairwise deletion and Poisson correction
Chromosomal location, gene structure and conserved motif analysis of TaHMAs
Chromosome localization analysis revealed that 31 of the TaHMA genes were distributed on 2A, 2B, 2D, 4A, 4D, 5A, 5B, 5D, 6A, 6B, 6D, 7A, 7B and 7D, of which 7A, 7B, 7D contained the greatest number of HMA genes, while one TaHMA did not have a corresponding chromosome (Table 4). To further understand the features of the genes of the HMA family, the exon/intron structures of TaHMA genes were analyzed. As shown in Table 1, the number of introns varied from 6 to 20. TaHMA8;3 has the largest number of introns (19), with TaHMA3;1, TaHMA3;2, TaHMA3;3, TaHMA5;1, TaHMA5;2 and TaHMA5;3 having the fewest introns (5). Although the exons varied among TaHMA genes, the most closely related members have similar gene structures. To predict and verify domains in the TaHMAs proteins, we used the Multiple EM for Motif Elicitation (MEME) motif search tool. Ten corresponding consensus motifs were detected (Additional file 2: Figure S8, Table 5). The number of motifs varied among the TaHMA proteins. Motif 2, motif 6, motif 8, motif 9 and motif 10 were observed in all TaHMA proteins. Motif 1 and motif 3 were observed in all TaHMA proteins except TaHMA1;3.
Table 5.
Motif sequences for HMAs identified by MEME analysis
| Motif | Width | Multilevel consensus sequence |
|---|---|---|
| 1 | 50 | LNLBGYJHVRATKVGSNSALAKIVRLVEEAQMSKAPVQRLADKVAKYFVP |
| 2 | 41 | TPTAVMVATGVGARRGVLIKGGDVLESLANIKAIAFDKTGT |
| 3 | 50 | VGDVIKVLPGEKVPVDGVVVDGRSHVBESMLTGESAPVAKZVGSEVIGGT |
| 4 | 50 | AIGSGTAVAIEAADVVLMSNBLEDVPTAIDLSRKTFRTIRQNYVWAVAYN |
| 5 | 41 | KFFEESGMJVFFFLLGKYLEVLAKGKASDAMSKLMELAPET |
| 6 | 29 | ELQKRGGPVAMVGDGINDAPALAAADVGM |
| 7 | 50 | IVGIPVAAGALFPFTGFRLPPWLAGACMAFSSVSVVCSSLLLRLYKKPRH |
| 8 | 26 | SMGIKSVMLTGDNWAAAQAVAKZVGI |
| 9 | 29 | ELLYLVASAESNSEHPLAKAIVEYAQSFS |
| 10 | 29 | EEVADFEILPGEGVYAEIDGKKVLVGNKR |
Motif numbers corresponded to the motifs in Additional file 2: Figure S8
Tissue-specific expression patterns of TaHMA genes
Using available RNA-seq data (the wheat expression database, http://wheat.pw.usda.gov/WheatExp/) for five different tissues, the tissue specificity of the TaHMA genes was investigated to focus on the temporal and spatial expression patterns and putative functions of HMA genes in the growth and development. According to FPKM values, we found that the expression levels of the TaHMAs varied significantly in different tissues (Fig. 7). Most HMAs were found to be expressed in five detected organs. TaHMA1;1, TaHMA1;2, TaHMA2;5 and TaHMA4;3 showed weak expression in all tissues, while TaHMA6;1, TaHMA9;1, TaHMA9;2 and TaHMA9;3 had strong expression. TaHMA1;4, TaHMA2;1, TaHMA2;2, TaHMA2;4, TaHMA2;5, TaHMA7;1, TaHMA7;2, TaHMA7;3, TaHMA8;1, TaHMA8;2 and TaHMA8;3 were differentially expressed in 5 organs.
Fig. 7.

Heat map of the expression profiles of 29 TaHMA genes in five different tissues (grain, leaf, root, spike and stem). Log2 transformed FPKM values are represented. The red or blue colors indicate higher or lower relative abundance, respectively. Z represent Zadoks scale, a decimal code for the growth stages of cereals. P-value< 0.05 were regarded as statistically significant
Expression pattern of TaHMA genes under cd stress
To characterize potential roles of TaHMA genes in response to Cd stresses, expression of all TaHMA genes in response to Cd stress were investigated using our RNA sequencing data. Overall, the 32 wheat HMA genes exhibited diverse expression patterns under Cd challenge. In L17, the expression levels of TaHMA1;2, TaHMA1;4, TaHMA1;5, TaHMA2;2, TaHMA5;1, TaHMA6;1, TaHMA6;3, TaHMA7;1, TaHMA7;2, TaHMA7;3, TaHMA8;1, TaHMA8;2 and TaHMA9;2 were down-regulated under Cd stresses, while, the expression levels of TaHMA2;1, TaHMA2;3, TaHMA2;5, TaHMA2;6, TaHMA4;1, TaHMA4;2, TaHMA4;3, TaHMA5;2, TaHMA5;3, TaHMA6;2, TaHMA8;3 and TaHMA9;1, TaHMA9;3 were up-regulated (Fig. 8a). In H17, the expression levels of TaHMA1;5, TaHMA5;1, TaHMA6;1, TaHMA6;2, TaHMA6;3, TaHMA7;2, TaHMA7;3, TaHMA9;1, TaHMA9;2 and TaHMA9;3 were down-regulated under Cd stresses, while, the expression levels of TaHMA1;2, TaHMA1;4, TaHMA2;1, TaHMA2;2, TaHMA2;3, TaHMA2;5, TaHMA2;6, TaHMA4;1, TaHMA4;2, TaHMA4;3 TaHMA5;2, TaHMA5;3, TaHMA7;1 TaHMA8;1, TaHMA8;2 and TaHMA8;3 were both up-regulated (Fig. 8b).
Fig. 8.

Heat map of the expression profiles of TaHMA genes under Cd treatment. a Expression profiles of TaHMA genes in L17Cd and L17CK groups. b Expression profiles of TaHMA genes in H17Cd and H17CK groups. Log2 transformed FPKM values were used to create the heat map. Red or blue indicates higher or lower relative abundance, respectively
Integration analysis miRNAs and TaHMAs
To explore the regulatory association between miRNAs and TaHMAs, microRNA-TaHMAs we performed a expression network analysis using targetfinder in plant [48] which was shown using Cytoscape v3.6 (http://www.cytoscape.org/). Results showed that microRNA-2B_36279* can regulate TaHMA2;1, TaHMA2;2, TaHMA2;4, TaHMA2;5 and TaHMA2;6, while microRNA-4B_11876*, microRNA-4B_3407*, microRNA-4B_16562*, microRNA-4B_13629* and microRNA-2B_40139* can regulate TaHMA3;1 (Fig. 9), and microRNA-2B_28883* can regulate TaHMA1;4. To our knowledge, it is the first time to perform microRNA-TaHMA regulatory network in wheat.
Fig. 9.

Relationships between miRNAs and TaHMAs given by co-expression network analysis
Response of selected TaHMA genes to exogenous cd stress
HMAs have been implicated in the transmembrane transport of metals, such as Zn, Cd, Pb and Cu in different plant species [46, 49–51]. Therefore, we were interested to determine the response of select HMA genes under Cd stress. We thus selected eight TaHMA genes (TaHMA1;4-TaHMA3;1) belonging to the Cu/Ca/Zn/Cd/Co-ATPase group and the Zn/Cd/Pb/Co-ATPase group to determine their relative expression levels in roots under Cd stress using qRT-PCR. Results showed that relative expression levels of TaHMA1;4, TaHMA1;5, TaHMA2;1 and TaHMA3;1 were significantly down-regulated under Cd treatment compared with those under control conditions in high-accumulating wheat genotype (H17), while relative expression levels of TaHMA1;4, TaHMA1;5, TaHMA2;1 andTaHMA2;2 were significantly up-regulated under Cd treatment compared with those under control conditions in low-accumulating wheat genotype (L17) (Fig. 10). Relative expression of TaHMA1;5 in H17, TaHMA2;1 in L17 was consistent with our transcriptome sequencing data. Interestingly, expression data from other TaHMA genes in H17 and L17 treated with Cd were not consistent with sequencing data. A possible explanation for this inconsistence is the number of samples used on this experiment. Previous work indicated that AtHMA1 is responsible for Cu to the stroma, exporting Zn2+ form the chloroplast, or as a Ca2+/heavy metal transporter to the intracellular organelle [52–54]. AtHMA2 and AtHMA4 in A.thaliana are involved in xylem loading of Zn as well as Cd [55–59]. Compared with wild-type plants, plants overexpressing AtHMA3 exhibited a 2–3-fold increase in Cd accumulation [28]. AtHMA5 is responsible for the Cu translocation from roots to shoots or Cu detoxification of roots [60, 61]. AtHMA6 (also known as PAA1) is involved in transporting Cu over the chloroplast envelope, whereas AtHMA8 (PAA2) most likely transports Cu into the thylakoid lumen to supply plastocyanin [62, 63]. AtHMA7 has been proposed to delivery Cu to ethylene receptors and Cu homeostasis in the seedlings [64, 65]. Among rice HMA characterized, OsHMA1 transports Zn and Cd [66], OsHMA2 is responsible for Cd accumulation [67], OsHMA5 is involved in loading Cu to the xylem of the roots and other organs [68], and OsHMA9 is responsible for transporting Zn, Cu, Pb and Cd [69]. In wheat, TaHMA2 is a plasma membrane-localized Zn/Cd transporter that pumps Zn/Cd into apoplast [26]. Our results indicated that Cd stress induced the up-regulation of TaHMA2;1 in L17.
Fig. 10.

Relative expression of TaHMAs in high- (H17) and low- (L17) cadmium-accumulating wheat cultivars under Cd stress. Relative expression levels of TaHM1-TaHMA3 were analyzed using qRT-PCR. H17 and L17 wheat samples were treated with 100 μM Cd. H17 and L17 wheat samples growing under Cd-free conditions were used as controls
Conclusions
We present here a broad range of genome-wide data surrounding Cd response in contrasting wheat lines. A total three known and 19 novel differentially expressed microRNAs (DEMs), and 1561 differentially expressed genes (DEGs) were found in L17 after Cd treatment. In H17 we found 12 known and 57 novel DEMs, 297 Cd-induced DEGs. Moreover, we present the identification of 32 TaHMA genes in wheat. Finally, we provide microRNA-TaHMA expression networks that suggest that miRNAs can regulate TaHMAs which need to be validated experimentally in future. Taken together, our results indicate that microRNAs may play important roles in wheat under Cd stress through regulating target genes such as TaHMA2;1. This information will be useful for screening and breeding low-Cd accumulation wheat genotypes.
Methods
Study site and wheat germplasms
The Shifang heavy industry and Cd-polluted area was selected as our study site in the northeast of Sichuan province. Low grain Cd accumulative wheat cultivars were selected from the Cd-polluted area as suitable for planting in Cd pollution farmland. A total of 185 wheat germplasms (provided by Chengdu Institute of Biology, Chinese Academy of Sciences) were chosen to include wide genetic diversity. Detailed information of wheat cultivars were shown in Additional file 1: Table S1. The 185 wheat germplasms could be divided into two groups, including 181 bread wheat (Triticum aestivum L.) and 4 durum wheat (Triticum turgidum L. var. durum) groups. The 181 bread wheats were further divided into three groups, including 52 Chuanyu wheat cultivars (Chuanyu group), 56 International Maize and Wheat Improvement Center (CIMMYT) materials (CIMMYT group) and 73 Chinese modern cultivars (CMC group).
Plant growth conditions and cd treatment
A low-Cd-accumulating cultivar (Chuanyu 17, namely L17) and its sib-line H17 (a high-Cd-accumulating cultivar) from Chuanyu group were used in the study. Seeds were sterilized by soaking in 2% H2O2 for 10 min and fully rinsed with deionized water. After sterilizing, the seeds were soaked in deionized water at room temperature for 6 h, and then planted hydroponically in Hoagland solution in growth chambers at 24 ± 2 °C and 50% relative humidity with a photoperiod of 16 h light/8 h dark. One-week-old seedlings were treated with 0 (CK) and 100 μM CdCl2 for 24 h (Cd). Roots from the plants of similar size were harvested separately and washed three times with deionized water. Roots from three biological replicates were frozen in liquid nitrogen immediately and stored at − 80 °C for small RNA and transcriptome sequencing.
Determination of grain and root cd concentrations
For wheat grain, whole grain was ground into flour (< 1 mm). Flour was dried at 80 °C for 12 h and then digested by HNO3 and H2O2 in a microwave digester. Roots from three plants as replicates for each of CK and Cd treatments were dried at 70 °C to a constant weight and then also digested by HNO3 and H2O2 in a microwave digester. All grain samples were processed together with quality controls. Cd concentration was measured using inductively couple plasma-mass spectrometry (ICP-MS, NexIONTM 300, USA) following the manufacturer’s instructions [70]. A Certified Reference material (CRM; GBW10020, provided by the National Research Center for CRM, China) was applied to assess the precision of the analytical procedures for plant material.
RNA extraction
One week-old L17 and H17 seedlings were treated with 0 (CK) and 100 μM CdCl2 (Cd) for 24 h. Then, roots of 12 seedlings (each treatment for three biological replicates) were collected for RNA extraction. Total RNA were extracted using the TRIzol Reagent (Invitrogen, USA) following the manufacturer’s protocol. RNA integrity was evaluated using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), and each sample had an RNA integrity number (RIN) > 8.0.
Small RNA sequencing and analysis
cDNA libraries were built using SuperScript II Reverse Transcriptase basing on the manufacturer’s instructions. Resultant libraries were then sequenced on the Illumina HiSeqTM 2500 sequencing platform (OEbiotech Company, Shanghai, China). Raw reads were converted into sequence data by base calling. After reads with low-quality, 5′ primer contaminants and ploy (A), without 3’adapter and insert tag, shorter than 15 nt and longer than 41 nt from the raw reads were filtered, the clean reads were obtained and mapped against the wheat genome. Clean reads were annotated with miRBase (version 21.0, http://www.mirbase.org/) to identify know miRNAs using Bowtie software. To identify novel miRNAs, the remaining unannotated clean reads that could be aligned to the genome were analyzed by miRDeep2 and RNAfold. The criteria for designation as a novel miRNA were as follows: length of 18-24 nt, precursors with a signature hairpin structure and the formation of maturation achieved by Dicer, minimum free energy of precursors of less than 18 kcal/mol, a minimum support number for the maturity sequence of precursors of at least 5, and potential miRNA sequence with less than 3 nt mispairing in the sequence of the mature and perfectly matched middle sequence. Differentially expressed miRNAs (DEMs) were identified with the threshold of p value < 0.05. While the p value was calculated with the DEG algorithm in the R package. The targets of DEMs were predicted by using targetfinder [48].
Transcriptome sequencing
cDNA libraries were constructed using the TruSeq Stranded mRNA LTSample prep kit (Illumina, Sam Diego, CA, USA) according to the manufacturer’s instructions. Resultant libraries were sequenced on the Illumina HiSeqTM 2500 sequencing platform (OEbiotech Company, Shanghai, China). Raw reads were processed using NGS QC toolkit. The reads containing poly-N and the low quality reads were removed to obtain the clean reads. Clean reads were then mapped to wheat reference genome (ftp://ftp.ensemblgenomes.org/pub/plants/release-28/fasta/triticum_aestivum/dna/Triticum_aestivum.IWGSC1.0+popseq.28.dna.toplevel.fa.gz) [71] using hisat2. Fragments per kilobase of transcript sequence per million base pairs sequenced (FPKM) value of each gene was calculated using cufflinks, and the read counts of each gene were obtained by htseq-count. Differentially expressed genes (DEGs) were identified using the DESeq. R package functions estimateSizeFactors and nbinomTest. P value < 0.05, fold Change > 2 and false discovery rate (FDR) < 0.05 were set as the threshold for significantly differential expression. Hierarchical cluster analysis of DEGs was performed to explore genes expression pattern.
Gene ontology (GO) and KEGG pathway analysis
For miRNAs, GO enrichment and KEGG pathway enrichment analysis of different expressed miRNA-target-Gene were performed using R based on the hypergeometric distribution. For mRNAs, GO enrichment and KEGG pathway enrichment analysis of DEGs were performed using R based on the hypergeometric distribution [72].
Identification of HMA gene family in wheat
The HMA gene family was identified following the method as described by Li et al. with some modifications [73]. First, to construct a local protein database, all the wheat (T.aestivum L.) protein sequences were downloaded from the Ensemble database (http://plants.ensembl.org/index.html). Then, the database were searched with 17 known HMA gene sequences collected from A.thaliana (8) and O.sativa (9) using the local BLASTP program with an e-value of le-5 and identity of 50% as the threshold. Moreover, a self-blast of these sequences was performed to remove redundant sequences. The physical localizations of all candidate HMA genes were checked and redundant sequences with the same chromosome location were rejected. To analyze whether there were domains belonging to the HMA gene family, the online tool InterProScan 5 (http://www.ebi.ac.uk/interpro/search/sequence search) [74] was used. Finally, to verify the existence of all the obtained sequences, BLASTN similarity search against the wheat ESTs deposited in the NCBI database were performed. The theorectical pI (isoelectric point) and Mw (molecular weight) of the putative HMA from T.aestivum L were calculated using compute pI/Mw tool online (http://web.expasy.org/compute_pi/), respectively.
Gene structure construction, protein domain and motif analysis
Gene structure information was obtained from the Ensemble plants database (http://plants.ensembl.org/index.html). All full-length amino acid sequences of the TaHMAs were also used to identify conserved domain motifs by the Multiple Em for Motif Elicitation (MEME) tool [75]. The parameters were set as follows: maximum numbers of different motifs, 10; minimum motif width, 6; maximum motif width, 50.
Phylogenetic analysis
The HMA protein sequences from A.thaliana, O.sativa and T.aestivum L. were performed for multiple alignments by CLUSTALW and the results of alignment were used to construct phylogenetic tree using the NJ method in MEGA (version 6.0) [76]. Bootstrap test method was emplyed and the replicate was set to 1000.
The TaHMA gene expression analysis by RNA-seq data
To study the expression of TaHMA genes in different organs, the wheat expression database (http://wheat.pw.usda.gov/WheatExp/) was used to investigate the differential expression of TaHMAs. The FPKM (fragments per kilobase of transcript per million fragments mapped) value was calculated for each HMA gene, the log2 transformed FPKM value of the TaHMA genes were used for heat map generation. And p-value < 0.05 were taken as statistically significant threshold [77].
Quantitative real time polymerase chain reaction (qRT-PCR) analysis
Eight miRNAs and six genes with different expression patterns and eight TaHMA genes were selected for validation by quantitative real-time RT-PCR (qRT-PCR). Total RNA was extracted from roots of L17 and H17 seedlings treated with 0 (CK) or 100 μM CdCl2 (Cd) for 24 h. For mRNA, first strand cDNA was synthesized using HiScript II Q RT SuperMix (Vazyme, R223–1). For miRNA, the first strand cDNA was synthesized using miScript II Reverse Transcription Kit (Qiagen, 218,161). The qRT-PCR was carried out using QuantiFast® SYBR® Green PCR kit (Qiagen, 204,054) according to the manufacturer’s instructions. The primers used in the qRT-PCR analyses were listed in (Additional file 4). GAPDH and 5S were used as internal genes for mRNAs and miRNAs, respectively. Three technical replicates were performed for each gene. The expression levels were calculated from the 2-ΔΔCt value.
Statistical analysis
All data were represented as mean ± SD and analyzed with SPSS20.0 software. For multiple group comparison, ANOVA with least significant difference was applied. P < 0.05 was considered statistically significant.
Additional files
Grain Cd concentration among wheat germplasms. (XLSX 16 kb)
Figure S1. Grain Cd concentration between bread wheat and durum wheat. Figure S2. A Box plot of miRNA. B Correlation analysis of expression levels of miRNAs between samples and samples. Figure S3. A Box plot of mRNA. B Correlation analysis of expression levels of mRNAs between samples and samples. Figure S4. A The volcano plot illustrates the distribution of mRNA expression fold changes vs P values between L17Cd and L17CK. B The volcano plot illustrates the distribution of the data in mRNA profiles between H17Cd and HL17CK. Red, green and black points in the volcano plot represents significantly upregulated mRNAs, significantly downregulated mRNAs and not differential expressed mRNAs, respectively. Figure S5. GO enrichment analysis of differentially-expressed miRNA-targeted genes. The top 10 GOs enriched in targeted genes are given in response to miRNAs: (A) down-regulated and (B) up-regulated between L17Cd and L17CK; (C) down-regulated (D) up-regulated between H17Cd and H17CK. The y-axis represent the number of genes enriched. Figure S6. Venn diagrams of mRNA Gene Ontology (GO) enrichment results. Overlap of GO results of differentially expressed microRNAs (DEMs) between L17Cd and L17CK, H17Cd and H17CK. B Venn diagram for GO of DEGs between L17Cd and L17CK, H17Cd and H17CK. Figure S7. GO analysis for differentially-expressed mRNAs. The top 10 GOs enriched in differentially expressed mRNAs are given for the contrasts: (A) up-regulated and (B) down-regulated between L17Cd and L17CK; (C) up-regulated and (D) down-regulated between H17Cd and H17CK. The y-axis represent the gene number enriched in GOs. Figure S8. Ten wheat HMAs motifs were identified by MEME tools and indicated by different color. Motif location and combined p-value were represented. (ZIP 1392 kb)
Functional annotation of transport genes. (XLSX 27 kb)
Primers for genes. (XLSX 10 kb)
Acknowledgements
We thank Xian Fu and Pengfei Xiang for technical support.
Abbreviations
- Cd
Cadmium
- Chuanyu
Chuanyu wheat cultivars
- CIMMYT
International Maize and Wheat Improvement Center
- CMC
Chinese modern cultivars
- DEGs
Differentially expressed genes
- DEMs
Differentially expressed microRNAs
- FDR
False discovery rate
- FPKM
Fragments per kilobase of transcript per million fragments mapped
- GO
Gene ontology
- HMAs
Heavy metal ATPases
- MEME
Multiple Em for Motif Elicitation
- qRT-PCR
Quantitative real time quantitative-polymerase chain reaction
Authors’ contributions
MZ and YW contributed to the experimental design and manuscript drafting. MZ, SGZ and RL performed the research. MZ performed bioinformatic analysis. MZ, LL and CHZ contributed reagents/materials/analysis tools. LY supported the revision of the manuscript and drafted the manuscript revision with MZ and YW. All authors read and approved the final manuscript.
Funding
This work was supported by the “13th Five-year Plan” for National Key Research and Development (Grant No. 2016YFD0102000). This work was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme [grant number ERC-StG 679056 HOTSPOT], via a grant to L.Y. The funding body had no role in the design of the study, collection, analysis, or interpretation of data, or in writing the manuscript.
Availability of data and materials
The dataset and materials presented in the investigation is available by request from the corresponding author.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Min Zhou, Email: cherry62088@163.com.
Shigang Zheng, Email: zhsg528@163.com.
Rong Liu, Email: LiuR0512@163.com.
Lu Lu, Email: 761173084@qq.com.
Chihong Zhang, Email: 664881538@qq.com.
Lei Zhang, Email: zhanglei_7810@163.com.
Levi Yant, Email: Levi.Yant@nottingham.ac.uk.
Yu Wu, Phone: 86-28-82890317, Email: wuyugood@126.com.
References
- 1.Khan MA, Khan S, Khan A, Alam M. Soil contamination with cadmium, consequences and remediation using organic amendments. Sci Total Environ. 2017;601-602:1591–1605. doi: 10.1016/j.scitotenv.2017.06.030. [DOI] [PubMed] [Google Scholar]
- 2.Kazantzis G. Cadmium, osteoporosis and calcium metabolism. Biometals. 2004;17:493–498. doi: 10.1023/B:BIOM.0000045727.76054.f3. [DOI] [PubMed] [Google Scholar]
- 3.Straif K, Benbrahim-Tallaa L, Baan R, Grosse Y, Secretan B, El GF, et al. A review of human carcinogens--Part C: metals, arsenic, dusts, and fibres. Lancet Oncol. 2009;10:453. doi: 10.1016/S1470-2045(09)70134-2. [DOI] [PubMed] [Google Scholar]
- 4.Das P, Samantaray S, Rout GR. Studies on cadmium toxicity in plants: a review. Environ Pollut. 1997;98:29–36. doi: 10.1016/S0269-7491(97)00110-3. [DOI] [PubMed] [Google Scholar]
- 5.Zorrig W, Rouached A, Shahzad Z, Abdelly C, Davidian JC, Berthomieu P. Identification of three relationships linking cadmium accumulation to cadmium tolerance and zinc and citrate accumulation in lettuce. J Plant Physiol. 2010;167:1239–1247. doi: 10.1016/j.jplph.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 6.Uraguchi S, Kamiya T, Sakamoto T, Kasai K, Sato Y, Nagamura Y, et al. Low-affinity cation transporter (OsLCT1) regulates cadmium transport into rice grains. Proc Natl Acad Sci U S A. 2011;108:20959–20964. doi: 10.1073/pnas.1116531109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang M, Zhou S, Sun B, Zhao Q. Heavy metals in wheat grain: Assessment of potential health risk for inhabitants in Kunshan, China. Sci Total Environ. 2008;405:54. doi: 10.1016/j.scitotenv.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Dai XP, Feng L, Ma XW, Zhang YM. Concentration Level of Heavy Metals in Wheat Grains and the Health Risk Assessment to Local Inhabitants from Baiyin, Gansu, China. Adv Mater Res. 2012;518-523:951–956. doi: 10.4028/www.scientific.net/AMR.518-523.951. [DOI] [Google Scholar]
- 9.Satarug S, Baker JR, Urbenjapol S, Haswellelkins M, Reilly PE, Williams DJ, Moore MR. A global perspective on cadmium pollution and toxicity in non-occupationally exposed population. Toxicol Lett. 2003;137:65–83. doi: 10.1016/S0378-4274(02)00381-8. [DOI] [PubMed] [Google Scholar]
- 10.Gill BS, Appels R, Botha-Oberholster AM, Buell CR, Bennetzen JL, Chalhoub B, et al. A workshop report on wheat genome sequencing: International Genome Research on Wheat Consortium. Genetics. 2004;168:1087–1096. doi: 10.1534/genetics.104.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayer KFX, Rogers J, Doležel J, Pozniak C, Eversole K, Feuillet C, et al. A chromsome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;345:1251788. doi: 10.1126/science.1251788. [DOI] [PubMed] [Google Scholar]
- 12.Jafarnejadi AR, Homaee M, Sayyad G, Bybordi M. Large scale spatial variability of accumulated cadmium in the wheat farm grains. Soil Sediment Contam Int J. 2011;20:98–113. doi: 10.1080/15320383.2011.528472. [DOI] [Google Scholar]
- 13.Greger M, Lofstedt M. Comparison of uptake and distribution of cadmium in different cultivars of bread and durum wheat. Crop Sci. 2004;44:111–113. doi: 10.2135/cropsci2004.5010. [DOI] [Google Scholar]
- 14.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 15.Voinnet O. Origin, biogenesis, and activity of plant microRNAs. Cell. 2009;136:669–687. doi: 10.1016/j.cell.2009.01.046. [DOI] [PubMed] [Google Scholar]
- 16.Zhang B, Pan X, Cobb GP, Anderson TA. Plant microRNA: A small regulatory molecule with big impact. Dev Biol. 2006;289:3–16. doi: 10.1016/j.ydbio.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 17.Sunkar R, Chinnusamy V, Zhu J, Zhu JK. Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 2007;12:301–309. doi: 10.1016/j.tplants.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Sunkar R, Kapoor A, Zhu JK. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell. 2006;18:2051–2065. doi: 10.1105/tpc.106.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdel-Ghany SE, Pilon M. MicroRNA-mediated systemic down-regulation of copper protein expression in response to low copper availability in Arabidopsis. J Biol Chem. 2008;283:15932–15945. doi: 10.1074/jbc.M801406200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujii H, Chiou TJ, Lin SI, Aung K, Zhu JK. A miRNA involved in phosphate-starvation response in Arabidopsis. Curr Biol. 2005;15:2038. doi: 10.1016/j.cub.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 21.Liu HH, Tian X, Li YJ, Wu CA, Zheng CC. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA. 2008;14:836–843. doi: 10.1261/rna.895308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng F, Liu H, Wang K, Liu L, Wang S, Zhao Y, Yin J, Li Y. Development-associated microRNAs in grains of wheat (Triticum aestivum L.) BMC Plant Biol. 2013;13:140. doi: 10.1186/1471-2229-13-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou J, Cheng Y, Yin M, Yang E, Gong W, Liu C, et al. Identification of novel miRNAs and miRNA expression profiling in wheat hybrid necrosis. PLoS One. 2015;10:e0117507. doi: 10.1371/journal.pone.0117507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akdogan G, Tufekci ED, Uranbey S, Unver T. miRNA-based drought regulation in wheat. Funct Integr Genomics. 2016;16:221–233. doi: 10.1007/s10142-015-0452-1. [DOI] [PubMed] [Google Scholar]
- 25.Møller JV, Juul B, le Maire M. Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim Biophys Acta. 1996;1286:1–51. doi: 10.1016/0304-4157(95)00017-8. [DOI] [PubMed] [Google Scholar]
- 26.Tan J, Wang J, Chai T, Zhang Y, Feng S, Li Y, Zhao H, Liu H, Chai X. Functional analyses of TaHMA2, a P(1B)-type ATPase in wheat. Plant Biotechnol J. 2013;11:420–431. doi: 10.1111/pbi.12027. [DOI] [PubMed] [Google Scholar]
- 27.Miyadate H, Adachi S, Hiraizumi A, Tezuka K, Nakazawa N, Kawamoto T, et al. OsHMA3, a P1B-type of ATPase affects root-to-shoot cadmium translocation in rice by mediating efflux into vacuoles. New Phytol. 2011;189:190–199. doi: 10.1111/j.1469-8137.2010.03459.x. [DOI] [PubMed] [Google Scholar]
- 28.Morel M, Crouzet J, Gravot A, Auroy P, Leonhardt N, Vavasseur A, Richaud P. AtHMA3, a P1B-ATPase allowing Cd/Zn/Co/Pb vacuolar storage in Arabidopsis. Plant Physiol. 2009;149:894–904. doi: 10.1104/pp.108.130294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ueno D, Milner MJ, Yamaji N, Yokosho K, Koyama E, Clemencia Zambrano M, et al. Elevated expression of TcHMA3 plays a key role in the extreme Cd tolerance in a Cd-hyperaccumulating ecotype of Thlaspi caerulescens. Plant J. 2011;66:852–862. doi: 10.1111/j.1365-313X.2011.04548.x. [DOI] [PubMed] [Google Scholar]
- 30.Hart JJ, Welch RM, Norvell WA, Sullivan LA, Kochian LV. Characterization of cadmium binding, uptake, and translocation in intact seedlings of bread and durum wheat cultivars. Plant Physiol. 1998;116:1413–1420. doi: 10.1104/pp.116.4.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hart JJ, Welch RM, Norvell WA, Kochian LV. Transport interactions between cadmium and zinc in roots of bread and durum wheat seedlings. Physiol Plant. 2002;116:73–78. doi: 10.1034/j.1399-3054.2002.1160109.x. [DOI] [PubMed] [Google Scholar]
- 32.Greger M, Lofstedt M. Comparison of uptake and distribution of cadmium in different cultivars of bread and durum wheat. Crop Sci. 2004;44:501–507. doi: 10.2135/cropsci2004.5010. [DOI] [Google Scholar]
- 33.Erdman JA, Moul RC. Mineral composition of small-grain cultivars from a uniform test plot in South Dakota. J Agric Food Chem. 1982;30:169–174. doi: 10.1021/jf00109a037. [DOI] [Google Scholar]
- 34.Han R, Jian C, Lv J, Yan Y, Chi Q, Li Z, et al. Identification and characterization of microRNAs in the flag leaf and developing seed of wheat (Triticum aestivum L.) BMC Genomics. 2014;15:289. doi: 10.1186/1471-2164-15-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng H, Zhang Q, Li H, Wang X, Wang X, Duan X, Wang B, Kang Z. vsiRNAs derived from the miRNA-generating sites of pri-tae-miR159a based on the BSMV system play positive roles in the wheat response to Puccinia striiformis f. sp. tritici through the regulation of taMyb3 expression. Plant Physiol Biochem. 2013;68:90–95. doi: 10.1016/j.plaphy.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 36.Wei B, Cai T, Zhang R, Li A, Huo N, Li S, et al. Novel microRNAs uncovered by deep sequencing of small RNA transcriptomes in bread wheat (Triticum aestivum L.) and Brachypodium distachyon (L.) Beauv. Funct Integr Genomics. 2009;9:499–511. doi: 10.1007/s10142-009-0128-9. [DOI] [PubMed] [Google Scholar]
- 37.Jia X, Wang WX, Ren L, Chen QJ, Mendu V, Willcut B, Dinkins R, Tang X, Tang G. Differential and dynamic regulation of miR398 in response to ABA and salt stress in Populus tremula and Arabidopsis thaliana. Plant Mol Biol. 2009;71:51–59. doi: 10.1007/s11103-009-9508-8. [DOI] [PubMed] [Google Scholar]
- 38.Guan Q, Lu X, Zeng H, Zhang Y, Zhu J. Heat stress induction of miR398 triggers a regulatory loop that is critical for thermotolerance in Arabidopsis. Plant J. 2013;74:840–851. doi: 10.1111/tpj.12169. [DOI] [PubMed] [Google Scholar]
- 39.Naya L, Paul S, Valdes-Lopez O, Mendoza-Soto AB, Nova-Franco B, Sosa-Valencia G, Reyes JL, Hernandez G. Regulation of copper homeostasis and biotic interactions by microRNA 398b in common bean. PLoS One. 2014;9:e84416. doi: 10.1371/journal.pone.0084416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khraiwesh B, Zhu JK, Zhu J. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim Biophys Acta. 2012;1819:137. doi: 10.1016/j.bbagrm.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng J, Jia W, Lv S, Bao H, Miao F, Zhang X, et al. Comparative transcriptome combined with morpho-physiological analyses revealed key factors for differential cadmium accumulation in two contrasting sweet sorghum genotypes. Plant Biotechnol J. 2018;16:558–571. doi: 10.1111/pbi.12795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lux A, Martinka M, Vaculik M, White PJ. Root responses to cadmium in the rhizosphere: a review. J Exp Bot. 2011;62:21–37. doi: 10.1093/jxb/erq281. [DOI] [PubMed] [Google Scholar]
- 43.Verrier PJ, Bird D, Burla B, Dassa E, Forestier C, Geisler M, et al. Plant ABC proteins--a unified nomenclature and updated inventory. Trends Plant Sci. 2008;13:151–159. doi: 10.1016/j.tplants.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 44.Thomine S, Wang R, Ward JM, Crawford NM, Schroeder JI. Cadmium and iron transport by members of a plant metal transporter family in Arabidopsis with homology to Nramp genes. Proc Natl Acad Sci U S A. 2000;97:4991–4996. doi: 10.1073/pnas.97.9.4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raghuram B, Sheikh AH, Sinha AK. Regulation of MAP kinase signaling cascade by microRNAs in Oryza sativa. Plant Signal Behav. 2014;9:e972130. doi: 10.4161/psb.29804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams LE, Mills RF. P1B-ATPases–an ancient family of transition metal pumps with diverse functions in plants. Trends Plant Sci. 2005;10:491–502. doi: 10.1016/j.tplants.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 47.Hermand V, Julio E, de Borne FD, Punshon T, Ricachenevsky FK, Bellec A, Gosti F, Berthomieu P. Inactivation of two newly identified tobacco heavy metal ATPases leads to reduced Zn and Cd accumulation in shoots and reduced pollen germination. Metallomics. 2014;6:1427–1440. doi: 10.1039/C4MT00071D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fahlgren N, Carrington JC. miRNA Target Prediction in Plants. Methods Mol Biol. 2010;592:51–57. doi: 10.1007/978-1-60327-005-2_4. [DOI] [PubMed] [Google Scholar]
- 49.Grotz N, Guerinot ML. Molecular aspects of Cu, Fe and Zn homeostasis in plants. Biochimica et Biophysica Acta (BBA)-Molecular. Cell Res. 2006;1763:595–608. doi: 10.1016/j.bbamcr.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 50.Argüello JM, Eren E, González-Guerrero M. The structure and function of heavy metal transport P 1B-ATPases. Biometals. 2007;20:233. doi: 10.1007/s10534-006-9055-6. [DOI] [PubMed] [Google Scholar]
- 51.Burkhead JL, Reynolds KAG, Abdel-Ghany SE, Cohu CM, Pilon M. Copper homeostasis. New Phytol. 2009;182:799–816. doi: 10.1111/j.1469-8137.2009.02846.x. [DOI] [PubMed] [Google Scholar]
- 52.Seigneurin-Berny D, Gravot A, Auroy P, Mazard C, Kraut A, Finazzi G, et al. HMA1, a new Cu-atpase of the chloro plast envelope, is essential for growth under adverse light conditions. J Biol Chem. 2006;281:2882–2892. doi: 10.1074/jbc.M508333200. [DOI] [PubMed] [Google Scholar]
- 53.Moreno I, Norambuena L, Maturana D, Toro M, Vergara C, Orellana A, Zurita-Silva A, Ordenes VR. AtHMA1 is a thapsigargin-sensitive Ca2+/heavy metal pump. J Biol Chem. 2008;283:9633–9641. doi: 10.1074/jbc.M800736200. [DOI] [PubMed] [Google Scholar]
- 54.Kim YY, Choi H, Segami S, Cho HT, Martinoia E, Maeshima M, Lee Y. AtHMA1 contributes to the detoxification of excess Zn (II) in Arabidopsis. Plant J. 2009;58:737–753. doi: 10.1111/j.1365-313X.2009.03818.x. [DOI] [PubMed] [Google Scholar]
- 55.Hussain D, Haydon MJ, Wang Y, Wong E, Sherson SM, Young J, Camakaris J, Harper JF, Cobbett CS. P-type ATPase heavy metal transporters with roles in essential zinc homeostasis in Arabidopsis. Plant Cell. 2004;16:1327–1339. doi: 10.1105/tpc.020487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Verret F, Gravot A, Auroy P, Leonhardt N, David P, Nussaume L, Vavasseur A, Richaud P. Overexpression of AtHMA4 enhances root-to-shoot translocation of zinc and cadmium and plant metal tolerance. FEBS Lett. 2004;576:306–312. doi: 10.1016/j.febslet.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 57.Wong CKE, Cobbett CS. HMA P-type ATPases are the major mechanism for root-to-shoot Cd translocation in Arabidopsis thaliana. New Phytol. 2009;181:71–78. doi: 10.1111/j.1469-8137.2008.02638.x. [DOI] [PubMed] [Google Scholar]
- 58.Wong CKE, Jarvis RS, Sherson SM, Cobbett CS. Functional analysis of the heavy metal binding domains of the Zn/Cd-transporting ATPase, HMA2, in Arabidopsis thaliana. New Phytol. 2009;181:79–88. doi: 10.1111/j.1469-8137.2008.02637.x. [DOI] [PubMed] [Google Scholar]
- 59.Yamaji N, Xia XJ, Mitani-Ueno N, Yokosho K, Ma JF. Preferential delivery of Zn to developing tissues in rice is mediated by a P-type ATPases, OsHMA2. Plant Physiol. 2013:113.216564. [DOI] [PMC free article] [PubMed]
- 60.Andrés-Colás N, Sancenón V, Rodríguez-Navarro S, Mayo S, Thiele DJ, Ecker JR, Puig S, Peñarrubia L. The Arabidopsis heavy metal P-type ATPase HMA5 interacts with metallochaperones and functions in copper detoxification of roots. Plant J. 2006;45:225–236. doi: 10.1111/j.1365-313X.2005.02601.x. [DOI] [PubMed] [Google Scholar]
- 61.Kobayashi Y, Kuroda K, Kimura K, Southron-Francis JL, Furuzawa A, Kimura K, et al. Amino acid polymorphisms in strictly conserved domains of a P-type ATPase HMA5 are involved in the mechanism of copper tolerance variation in Arabidopsis. Plant Physiol. 2008;148:969–980. doi: 10.1104/pp.108.119933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shikanai T, Müller-Moulé P, Munekage Y, Niyogi KK, Pilon M. PAA1, a P-type ATPase of Arabidopsis, functions in copper transport in chloroplasts. Plant Cell. 2003;15:1333–1346. doi: 10.1105/tpc.011817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abdel-Ghany SE, Müller-Moulé P, Niyogi KK, Pilon M, Shikanai T. Two P-type ATPases are required for copper delivery in Arabidopsis thaliana chloroplasts. Plant Cell. 2005;17:1233–1251. doi: 10.1105/tpc.104.030452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Woeste KE, Kieber JJ. A strong loss-of-function mutation in RAN1 results in constitutive activation of the ethylene response pathway as well as a rosette-lethal phenotype. Plant Cell. 2000;12:443–455. doi: 10.1105/tpc.12.3.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Binder Brad M., Rodríguez Fernando I., Bleecker Anthony B. The Copper Transporter RAN1 Is Essential for Biogenesis of Ethylene Receptors inArabidopsis. Journal of Biological Chemistry. 2010;285(48):37263–37270. doi: 10.1074/jbc.M110.170027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takahashi R, Ishimaru Y, Shimo H, Ogo Y, Senoura T, Nishizawa NK, Nakanishi H. The OsHMA2 transporter is involved in root-to-shoot translocation of Zn and Cd in rice. Plant Cell Environ. 2012;35:1948–1957. doi: 10.1111/j.1365-3040.2012.02527.x. [DOI] [PubMed] [Google Scholar]
- 67.Ueno D, Yamaji N, Kono I, Huang CF, Ando T, Yano M, Ma JF. Gene limiting cadmium accumulation in rice. Proc Natl Acad Sci U S A. 2010;107:16500–16505. doi: 10.1073/pnas.1005396107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deng F, Yamaji N, Xia J, Ma JF. A member of heavy metal P-type ATPase OsHMA5 is involved in xylem loading of copper in rice. Plant Physiol. 2013:113.226225. [DOI] [PMC free article] [PubMed]
- 69.Lee S, Kim Y-Y, Lee Y, An G. Rice P1B-type heavy-metal ATPase, OsHMA9, is a metal efflux protein. Plant Physiol. 2007;145:831–842. doi: 10.1104/pp.107.102236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arnold BJ, Lahner B, DaCosta JM, Weisman CM, Hollister JD, Salt DE, Bomblies K, Yant L. Borrowed alleles and convergence in serpentine adaptation. Proc Natl Acad Sci U S A. 2016;113:8320–8325. doi: 10.1073/pnas.1600405113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feng N, Song G, Guan J, Chen K, Jia M, Huang D, et al. Transcriptome profiling of wheat inflorescence development from spikelet initiation to floral patterning identified stage-specific regulatory genes. Plant Physiol. 2017;174:1779–1794. doi: 10.1104/pp.17.00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li N, Xiao H, Sun J, Wang S, Wang J, Chang P, et al. Genome-wide analysis and expression profiling of the HMA gene family in Brassica napus under cd stress. Plant Soil. 2018;426:365–381. doi: 10.1007/s11104-018-3637-2. [DOI] [Google Scholar]
- 74.Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34:W369–WW73. doi: 10.1093/nar/gkl198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pearce S, Vazquez-Gross H, Herin SY, Hane D, Wang Y, Gu YQ, Dubcovsky J. WheatExp: an RNA-seq expression database for polyploid wheat. BMC Plant Biol. 2015;15:299. doi: 10.1186/s12870-015-0692-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Grain Cd concentration among wheat germplasms. (XLSX 16 kb)
Figure S1. Grain Cd concentration between bread wheat and durum wheat. Figure S2. A Box plot of miRNA. B Correlation analysis of expression levels of miRNAs between samples and samples. Figure S3. A Box plot of mRNA. B Correlation analysis of expression levels of mRNAs between samples and samples. Figure S4. A The volcano plot illustrates the distribution of mRNA expression fold changes vs P values between L17Cd and L17CK. B The volcano plot illustrates the distribution of the data in mRNA profiles between H17Cd and HL17CK. Red, green and black points in the volcano plot represents significantly upregulated mRNAs, significantly downregulated mRNAs and not differential expressed mRNAs, respectively. Figure S5. GO enrichment analysis of differentially-expressed miRNA-targeted genes. The top 10 GOs enriched in targeted genes are given in response to miRNAs: (A) down-regulated and (B) up-regulated between L17Cd and L17CK; (C) down-regulated (D) up-regulated between H17Cd and H17CK. The y-axis represent the number of genes enriched. Figure S6. Venn diagrams of mRNA Gene Ontology (GO) enrichment results. Overlap of GO results of differentially expressed microRNAs (DEMs) between L17Cd and L17CK, H17Cd and H17CK. B Venn diagram for GO of DEGs between L17Cd and L17CK, H17Cd and H17CK. Figure S7. GO analysis for differentially-expressed mRNAs. The top 10 GOs enriched in differentially expressed mRNAs are given for the contrasts: (A) up-regulated and (B) down-regulated between L17Cd and L17CK; (C) up-regulated and (D) down-regulated between H17Cd and H17CK. The y-axis represent the gene number enriched in GOs. Figure S8. Ten wheat HMAs motifs were identified by MEME tools and indicated by different color. Motif location and combined p-value were represented. (ZIP 1392 kb)
Functional annotation of transport genes. (XLSX 27 kb)
Primers for genes. (XLSX 10 kb)
Data Availability Statement
The dataset and materials presented in the investigation is available by request from the corresponding author.