

Abstract
In this chapter, we discuss a method to determine the affinity and specificity of nearly all single-point mutants for a full-length protein binder. This method combines deep sequencing, comprehensive mutagenesis, yeast surface display, and fluorescence-activated cell sorting. This approach has been used to study sequence-function relationships for protein-protein interactions. The data can be used to determine the fine conformational epitope on the protein binder.
Keywords: Deep sequencing, Yeast surface display, Nicking mutagenesis, FACS, Conformational epitope mapping
1. Introduction
Delineating the sequence determinants of stability, affinity, and specificity of protein-protein interactions (PPI) has been a major research goal for decades. The classical approach to study PPI sequence-function relationships has been “alanine-scanning” mutagenesis in which residues are individually mutated to alanine and assayed by assorted biophysical techniques [1, 2]. The change of binding affinity upon mutation gives a reasonable measure of the importance of the perturbed residue. However, such classical mutagenesis and screening studies are extremely labor-intensive.
Recently, transformative methods utilizing large-scale mutagenesis, surface display, and next-generation sequencing (NGS) have been developed to obtain relative binding contributions of individual residues by testing thousands of PPI mutants in a single experiment [3–8]. All methods share a similar framework: a population containing mutants of one PPI partner is prepared and cloned into a surface-display vector. The population is selected and/or screened for positive or negative binding to the other partner, and then the selected and unselected populations are deep sequenced and analyzed. Finally, the change in frequency for each library member is calculated and converted to a relative binding score [8]. In the method developed by our lab [7], we utilize yeast surface display (YSD) [9, 10] as it affords quantitative screening via fluorescence-activated cell sorting (FACS) [5, 6, 11]. Compared with competing methods using YSD [5, 6, 12], our approach is faster and less expensive albeit with a limited dynamic range of approximately tenfold change in binding affinity centered about the wild-type sequence. Accordingly, our method is suitable for fine maturation of PPI affinity and specificity or to determine fine conformational epitopes.
In this chapter, we provide a detailed protocol to determine relative binding affinities and conformational epitope maps for PPIs (overview in Fig. 1). We cover creation of single-site saturation mutagenesis (SSM) libraries using nicking mutagenesis [13], transformation of libraries into yeast by the method of Gietz and Woods [14], screening of the SSM YSD library using FACS, DNA preparation for sequencing on an Illumina platform, and data analysis to determine a relative binding score and conformational epitope map. Relative binding calculations and estimated errors are carried out according to methods described in Kowalsky et al. [7]. Note: we assume the end user has (1) one PPI partner successfully induced and displayed in a YSD format with the other partner biotinylated and with (2) reproducible measurement of the apparent dissociation constant using protocols as described in Chao et al. [9].
Fig. 1.
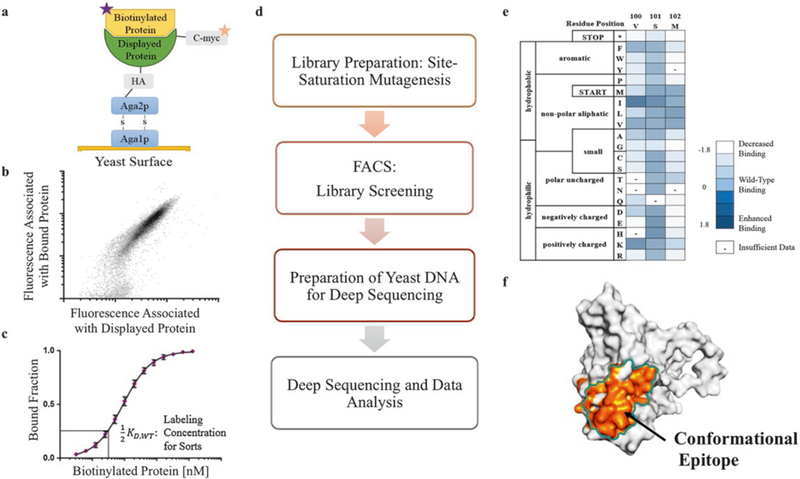
A streamlined process required for PPI characterization using deep sequencing and mutagenesis analysis. (a-c) Requirements for the pipeline: the binding activity of two proteins is measured using yeast surface display coupled to flow cytometry, and the relative dissociation constant is determined using yeast clonal titrations. The top panel is adapted from Chao et al. [9]. (d) The workflow covered in this chapter to characterize protein-protein interactions. (e, f) Deep sequencing results can be visualized as a heatmap and used to determine the conformational epitope of one member of the interaction
2. Materials
2.1. Yeast and Bacteria Strains and Plasmid
Yeast strain: Saccharomyces cerevisiae strain EBY100 is available at American Type Culture Collection and prepared to be chemically competent according to Gietz and Woods [14] (see a shortened protocol in Subheading 3.1.4).
Bacterial strain: Escherichia coli strain XL1-Blue high-efficiency electrocompetent cells are available through Agilent Technologies. Other competent cells with at least 1 × 109 transformants per μg of plasmid can be used.
Yeast display vectors: The YSD vector used, pETconNK, is freely available on Addgene (plasmid #81169) [15]. The gene of interest is inserted between NdeI and XhoI restriction sites.
2.2. Nicking Site Saturation Mutagenesis (SSM) Library Preparation
All enzymes and buffers for SSM library preparation with nicking mutagenesis are from New England Biolabs Inc. (NEB) unless noted otherwise.
2.2.1. Reagents, Media, and plates
pETconNK plasmid containing gene of interest (freshly pre-pared from a dam+ bacterial strain).
Nuclease-free water (NFH2O, Integrated DNA Technologies).
Custom mutagenic primers (see Subheading 3.1.1).
SEC_Rev primer: 5′-CAAGTCCTCTTCAGAAATAAGCTT TTGTTC – 3′.
T4 polynucleotide kinase buffer.
10× CutSmart Buffer.
5× Phusion HF Buffer.
10 mM ATP.
50 mM DTT.
50 mM NAD+.
10 mM dNTPs.
50% v/v sterile glycerol solution using deionized H2O.
TB media: 4.76% w/v of TB powder (premixed) and 0.8% v/v of glycerol. Sterilize by autoclaving.
-
LB agar plates: 2.5% w/v of LB powder (premixed) and 1.5% w/v of agar. Sterilize by autoclaving.
*Add kanamycin to a final concentration of 30 pg/mL when preparing the small plates to calculate the transformation efficiency and the large bioassay dishes for SSM libraries (see Subheading 3.2.1).
2.2.2. Enzymes
10 U/μL T4 polynucleotide kinase.
10 U/μL Nt.BbvCI.
10 U/μL Nb.BbvCI.
100 U/μL exonuclease III.
20 U/μL exonuclease I.
2 U/μL Phusion High-Fidelity DNA Polymerase.
40 U/μL Taq DNA ligase.
-
20 U/pL DpnI.
*Diluent for all enzymes required for Subheading 3.2.1 is 1× NEB CutSmart Buffer.
2.2.3. Equipment and Materials
Zymo Clean and Concentrator-5 kit (Zymo Research).
Corning square bioassay dishes, 245 mm × 245 mm × 25 mm (Sigma-Aldrich).
Chemically Competent Library Yeast Transformation
2.3.1. Yeast Solutions and Plates
Growth media: Synthetic dextrose medium supplemented with casamino acids (SDCAA): 2% w/v dextrose (D-glucose), 67% w/v yeast nitrogen base without amino acids (Sigma-Aldrich), 0.5% w/v Bacto casamino acids technical (BD Biosciences), 0.54% w/v Na2HPO4, and 0.856% w/v Na2HPO4-H2O. Filter sterilize. Add 1% v/v of 10,000 U/mL penicillin-streptomycin immediately prior to growth to prevent bacterial contamination.
Induction media: Synthetic galactose medium supplemented with casamino acids (SGCAA): prepare like SDCAA but with 2% w/v of galactose instead of dextrose.
SDCAA agar plate: 0.54% w/v Na2HPO4, 0.856% w/v Na2HPO4-H2O, 18.2% w/v sorbitol, and 1.5% w/v agar. Sterilize by autoclaving. 2% w/v dextrose (D-glucose), 0.67% w/v yeast nitrogen base without amino acids, 0.5% w/v Bacto casamino acids technical. Sterilize by filtrating. Add the filter-sterilized solution into the cool autoclaved mix (approximately below 50 °C) at 1:10 ratio. Store for up to 6 months at 4 °C.
Yeast storage buffer: 20% w/v glycerol, 20 mM HEPES, and 150 mM NaCl pH 7.5. Filter sterilize.
2.3.2. Reagents
10 mg/mL salmon sperm DNA (Invitrogen).
50% w/v polyethylene glycol (PEG), filter sterilize.
1 M lithium acetate, LiOAc.
2.4. Library Screening
2.4.1. Buffers and Reagents
Phosphate buffered saline (PBS) at pH 7.4: 0.8 w/v NaCl, 0.02% w/v KCl, 0.144% w/v Na2HPO4, and 0.024% w/v KH2PO4. Sterilize by filtrating.
Phosphate buffered saline with bovine serum albumin (PBS-BSA) at pH 7.4: prepare as PBS and supplemented with 0.1% w/v bovine serum albumin (BSA). Sterilize by filtrating.
Anti-c-myc-FITC antibody, FITC (Miltenyi Biotec).
Streptavidin, R-phycoerythrin conjugate, SAPE (Thermo Fisher).
Biotinylated PPI partner protein (see Note 1).
2.5. Deep Sequencing Preparation of Yeast DNA
2.5.1. Buffers and Reagents
TE media: 10 mM Tris-HCl at pH 8.0 and 0.1 mM EDTA.
5 U/μL Zymolyase (Zymo Research).
10× lambda nuclease buffer (NEB).
SYBR Gold Nucleic Acid Gel Stain (Thermo Fisher).
Agencourt AMPure XP (Beckman Coulter).
Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies).
70% v/v ethanol.
2.5.2. Enzymes
2.5.3. Equipment
5000 U/mL lambda nuclease (NEB).
Zymo Research Yeast Plasmid Miniprep II kit.
Qiagen mini-prep kit.
96-well magnetic plate.
3. Methods
3.1. Library Preparation: Site Saturation Mutagenesis (SSM)
Because a protein of250 amino acids is encoded by a 750 bp gene, separate SSM libraries are prepared for the gene of interest (Fig. 2a) to allow compatibility with 250 bp paired end (PE) Illumina MiSeq sequencing reads (see Note 2 for considerations for library preparation).
Fig. 2.
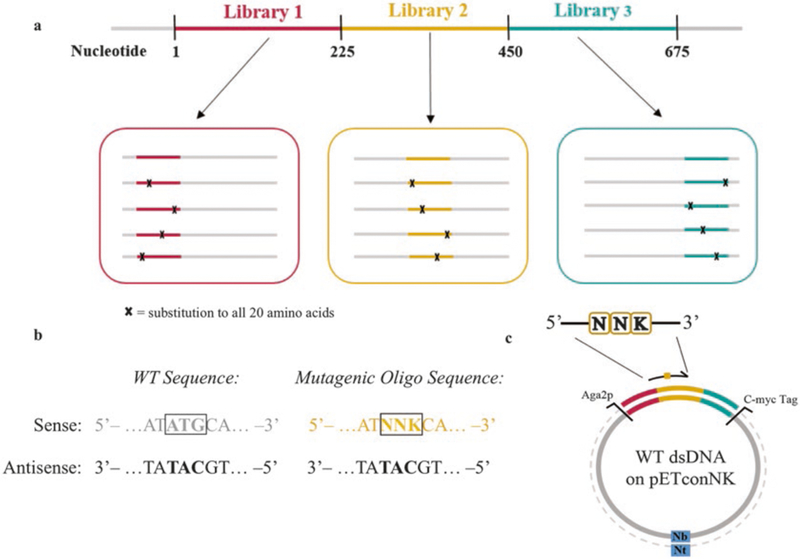
Essential considerations needed for preparing site saturation mutagenesis (SSM) libraries. (a) The gene of interest is segmented in multiple libraries containing contiguous sections of 200–250 bp. Here, sections of 225 bp are shown for compatibility with 250 bp PE Illumina MiSeq sequencing. (b) Each mutagenic oligo contains an “NNK” codon to cover all possible 20 amino acids. (c) An Nt.BbvCI restriction enzyme (Nt) is used to create a nick on the sense strand. Mutagenic oligos are designed to be complementary to the antisense ssDNA template
3.1.1. Design of Mutagenic Oligonucleotides
SSM libraries are created using degenerative oligonucleotides containing a “NNK” codon to cover all possible point mutations, where N represents any of the A/T/G/C, and K represents T/G. Mutagenic oligos are designed to be complementary to the wild-type template sequence as determined by the orientation of the BbvCI restriction site on the pETconNK vector (Fig. 2b, c; see Note 3).
Design your mutagenic oligos using QuikChange Primer Design Program (www.agilent.com). Use a degenerate “NNK” codon to cover all possible 20 amino acids at each codon position.
Order the mutagenesis oligos on a 500 pmol DNA Plate Oligo from Integrated DNA Technologies and resuspend to 10 μM in TE, pH 8.
3.1.2. Preparation of SSM Libraries by Nicking Mutagenesis
This protocol is exactly as described in Wrenbeck et al. [16]. All reactions should be prepared on ice unless otherwise stated.
To phosphorylate the oligos, make a mixture comprising 5 μL of each NNK mutagenic primer.
Into a PCR tube, add 20 μL of the 10 μM mutagenic primer mixture, 2.4 μL of T4 polynucleotide kinase buffer, 1 μL of 10 mM ATP, and 1 μL of T4 polynucleotide kinase. Incubate the reaction mixture at 37 °C for 1 h.
At the same time and in a separate PCR tube, add 18 μL of NFH2O, 3 μL of T4 polynucleotide kinase buffer, 7 μL of 100 μM SEC_Rev primer, 1 μL of 10 mM ATP, and 1 μL of T4 polynucleotide kinase. Incubate the reaction mixture at 37 °C for 1 h.
Store phosphorylated oligos at −20 °C.
The day of mutagenesis, dilute phosphorylated mutagenic primers 1:1000 and SEC_Rev primer 1:20 using NFH2O (see Note 4).
For the preparation of ssDNA template strand, in a PCR tube, add 0.76 pmol of dsDNA plasmid (approximately 2–3 μg), 2 μL of 10× CutSmart Buffer, 1 μL of 1:10 diluted exonuclease III (final concentration of 10 U/μL), 1 μL of Nt.BbvCI, 1 μL of exonuclease I, and NFH2O to 20 μL final volume.
Place the tube in a preheated (37 °C) thermal cycle with the following program: 60 min at 37 °C, 20 min at 80 °C, and hold at 4 °C.
To proceed with the comprehensive codon mutagenesis on the first strand, in each PCR tube, add 26.7 μL NFH2O, 20 μL of 5× Phusion HF Buffer, 4.3 μL 1:1000 diluted phosphorylated mutagenic oligos, 20 μL of 50 mM DTT, 1 μL of 50 mM NAD+, 2 μL of 10 mM dNTPs, 5 μL of Taq DNA ligase, and 1 μL of Phusion High-Fidelity DNA Polymerase. Mix the tube content briefly.
Place the tube into a preheated (98 °C) thermal cycler with the following program: 2 min at 98 °C, 15× cycles of 30 s at 98 °C, 45 s at 55 °C and 7 min at 72 °C, followed by a final incubation at 45 °C for 20 min, and hold at 4 °C. Add additional 4.3 μL oligo at the beginning of cycles 6 and 11.
Purify each reaction using a Zymo Clean and Concentrate kit to a final volume of 15 μL using NFH2O according to the manufacturer’s instructions.
To degrade the template strand, transfer 14 μL of the purified DNA product to a new PCR tube, and add 2 μL of 10× CutSmart Buffer, 2 μL of 1:50 diluted exonuclease III (final concentration of 2 U/μL), 1 μL of 1:10 Nb.BbvCI (final concentration of 1 U/μL), and 1 μL of exonuclease I.
Place the reaction tube in a preheated (37 °C) thermal cycle with the following program: 60 min at 37 °C, 20 min at 80 °C, and hold at 4 °C.
To synthesize the second mutagenic strand, add 27.7 μL NFH2O, 20 μL of 5× Phusion HF Buffer, 3.3 μL of 1:20 diluted phosphorylated SEC_REV primer, 20 μL of 50 mM DTT, 1 μL of 50 mM NAD+, 2 μL of 10 mM dNTPs, 5 μL of Taq DNA ligase, and 1 μL of Phusion High-Fidelity DNA Polymerase to the same reaction mixture. Mix the tube content briefly.
Place the tube in a preheated (98 °C) thermal cycler with the following program: 30 s at 98 °C, 45 s at 55 °C, 10 min at 72 °C, 20 min at 45 °C, and hold at 4 °C.
Add 2 μL of DpnI into each reaction tube, and incubate the reaction for 60 min at 37 °C to degrade methylated and hemi-methylated wild-type DNA.
Purify each reaction using a Zymo Clean and Concentrate kit to a final volume of 6 μL using NFH2O according to the manufacturer’s instructions.
Transform the entire 6 μL reaction product into E. coli XL1-Blue following standard electrocompetent transformation protocol [17].
After recovery, bring the final volume of the transformation to 2–2.5 mL with additional sterile media (TB media).
Prepare six tenfold serial dilutions and plate 10 μL of each. To calculate the transformation efficiency, the next day count the section that contains between 10 and 100 colonies. It is important to obtain at least 99.9% coverage of the theoretical diversity of the library (see Note 5).
Spread the remaining cells onto the prepared large bioassay dishes.
Place in a 37 °C humidity-controlled incubator overnight when bioassay dishes have dried.
3.1.3. Extraction of dsDNA SSM Library Plasmid
On the next day, scrape the large plates using between 5 and 10 mL TB media, and collect the cells in a 50 mL centrifuge tube.
Vortex the cell suspension, and extract the library plasmid DNA of a 1 mL aliquot of the cell suspension using a Qiagen mini-prep kit. Additional mini-preps can be done if large amounts of library DNA are required.
Store the rest of the cells at −80 °C by resuspending the pellet in 3 mL of 50% v/v glycerol.
3.1.4. Chemically Competent Library Yeast Transformation
Competent yeast can be prepared up to 6 months ahead of time.
Grow the EBY100 cells in 500 mL YPD to an OD600 of 1.2 and then harvest at 4000 × g for 5 min.
Resuspend the pelleted cells in 250 mL sterile H2O and repellet.
Resuspend the pelleted cells in 10 mL of 100 mM LiOAc and repellet.
Resuspend in 3.5 mL of 100 mM LiOAc, and then add 1.5 mL of 50% v/v glycerol and the mixture vortexed.
Prepare aliquots of 210 μL of cells to a tube, and store at −80 °C. Do not snap-freeze cells.
Boil 30 pL of salmon sperm DNA at 97 °C for 10 min.
Add 720 μL of 50% PEG, 108 μL of 1 M LiOAc, and 30 μL of boiled salmon sperm DNA to 210 μL of chemically competent EBY100 cells.
Vortex hard until there is a uniform mixture.
Add 5 μg of library plasmid to the mixture and vortex briefly.
Incubate the mixture at 30 °C for 30 min.
Heat shock the cells by incubating at 42 °C for 20 min.
Pellet the cells by spinning at 18,000 × g for 30 s.
Resuspend the cells pellet in 1 mL of SDCAA media, and let stand for 5 min.
Prepare six tenfold serial dilutions from the suspension, and plate on SDCAA plates using 10 μL of each. Incubate for 2–3 days at 30 °C to calculate transformation efficiency (see Note 5).
Add the remaining culture into 100 mL of SDCAA media. Grow for 30 h at 30 °C and 250 rpm.
On the next day, resuspend the cell culture at OD600 = 1 in 50 mL of SDCAA media.
Grow overnight at 30 °C and 250 rpm.
Prepare multiple cells stocks by pelleting, resuspending in yeast storage buffer to an OD600 = 1, and storing in 1 mL aliquots (approximately 1 × 107 cells) at −80 °C. Do not snap-freeze cells (see Note 6).
3.2. Library Screening
3.2.1. Preparation of Labeling Reactions
For each PPI partner to analyze, thaw a 1 mL aliquot as prepared on previous section, spin down at 2500 × g for 3 min, and remove the supernatant.
Resuspend the pellet in 1 mL SDCAA media, and grow for 4–6 h at 30 °C and 250 rpm.
Spin down the cells at 2500 × g for 3 min, and reinoculate at OD600 = 1.0 in 1 mL of SGCAA media. Induce overnight using the predetermined induction conditions (see Note 7).
Spin down the cells at 2500 × g for 3 min, wash with 1 mL of ice-cold PBS-BSA, and spin down again.
Resuspend the cells in ice-cold PBS-BSA at an OD600 = 2.0.
-
In PBS-BSA, label 1 mL (2 × 107) cells with the biotinylated protein at half of the apparent dissociation constant, and incubate at room temperature for 30 min using a tabletop mixer. Vary the total reaction volume to ensure that the number of biotinylated protein is at least tenfold higher than the PPI partner that is displayed on the yeast cell surface. For example, assuming a 10:1 partner/displayed protein ratio at a typical PPI apparent dissociation constant of 10 nM, 2 × 107 cells (1 mL) should be labeled with 5 nM biotinylated partner protein (half of the apparent dissociation constant). The total reaction volume is calculated following Eq. 1. Thus, label 1 mL of cells 2305 μL of PBS-BSA with 16.6 μL of 1 μM partner stock solution in
(1) Spin down at 2500 × g for 5 min, wash the pellet with 5 mL of PBS-BSA and spin down, and remove supernatant again. In this and subsequent steps, PBS-BSA should be ice-cold, the tabletop centrifuge should be refrigerated, and all tubes should be kept on ice and protected from light.
Label cells with 60 μL of FITC, 50 μL of SAPE, and 1.89 mL of PBS-BSA, vortex briefly, and incubate the labeled cells on ice for 10 min.
Repeat step 7.
Leave the cell pellet on ice until ready to sort.
3.2.2. Sorting Conditions Set-Up
Set Gate1, Gate2, and Gate3 on your cell sorter as shown in Fig. 3.
Add 4 mL of ice-cold PBS-BSA to the cell pellet, mix by vor-texing, and transfer to a FACS-compatible tube.
Obtain the reference population by sorting 240,000 cells (see Note 8) using Gate1+ (Fig. 3a).
Obtain the displayed population by sorting 240,000 cells using Gate1+/Gate2+ (Fig. 3b).
Obtain the bound population for each PPI by sorting 240,000 cells using Gate1+/Gate2+/Gate3+ (Fig. 3c).
Recover the collected cells in 5 mL of SDCAA media for approximately 30 h at 30 °C and 250 rpm.
Prepare cells stocks by storing 1 mL of OD600 = 4 cell stocks in yeast storage buffer and at −80 °C.
Fig. 3.

Sorting gates used for library screening. Yeast SSM libraries are labeled with biotinylated complementary protein at half of the apparent dissociation constant. Next, SSM libraries are sorted using three different gates as shown: (a) Gatel set with the light scatter parameters for yeast, forward scatter/side scatter; (b) Gate2 set on the forward scatter and the fluorescence channel for displayed protein (FITC); and (c) Gate3 set on the fluorescence channel for displayed protein and fluorescence channel for bound protein. Gate3 is configured to collect the top 5–10% of the bound population
3.3. Deep Sequencing Preparation of Yeast DNA
3.3.1. Primer Design and Library Amplification Test
Yeast DNA is prepared for deep sequencing using a two-step PCR amplification: the first step is with a gene-specific primer set (“inner” primers), while the second step uses an invariant set of “outer” primers (Fig. 4). Inner primers are designed to be complementary to adjacent 5′ and 3′ ends of each library followed by an Illumina universal primer sequence (Fig. 4a). The following rules needs to be considered to determine these regions:
Fig. 4.
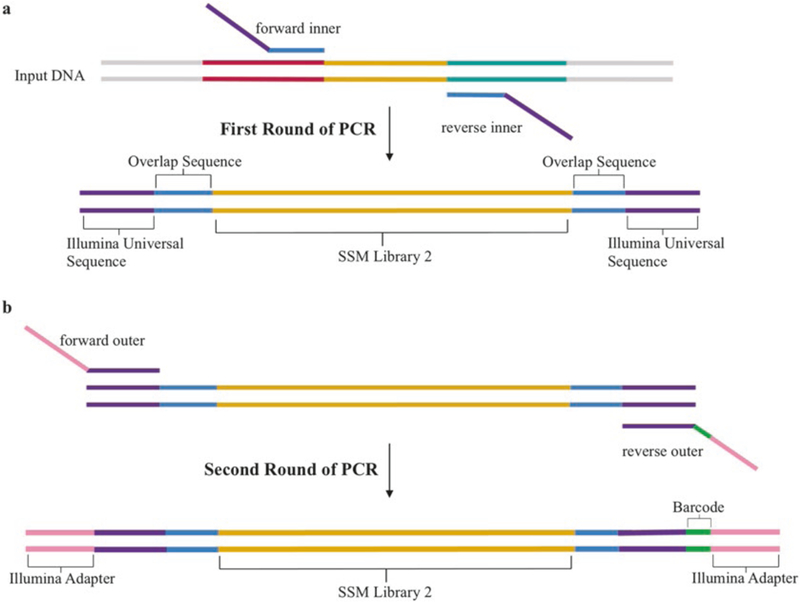
PCR steps performed for deep sequencing preparation of SSM libraries. Sequential PCR reactions to amplify the genes of interest and attach the Illumina adapters are shown for SSM library 2 (gold). (a) After extracting the plasmid DNA from yeast cells, SSM libraries are amplified by PCR using a set of inner primers containing a segment that overlaps with the gene of interest (light blue) and the Illumina universal sequence (purple). (b) A second round of PCR is performed to attach the Illumina adapter sequence using a set of outer primers which contain an overlapping region to the Illumina universal sequence (purple), a unique barcode on the reverse primer (green), and Illumina adapter sequences (yellow)
The length of the segment section plus the library should not be longer than 250 base pairs.
Design the segment region to have a melting temperature of 53–56 °C using the NEB Phusion melting point calculator using Phusion High-Fidelity Polymerase.
Once the gene-specific sequence is designed, append the conserved primer sequence as shown in Table 1.
-
Upon receiving the inner primers, we recommend performing a PCR verification with wild-type plasmid as a template to confirm a single band of the expected size.
Further steps for yeast DNA deep sequencing preparation require the addition of universal primers to add the Illumina adapters and barcodes. Universal primers are designed using the TruSeq small RNA oligo sequences. The forward primer is the same for all preparations, while the reverse primer contains an indexing bar-code that allows multiplexing of samples on an Illumina lane (Fig. 4b; full sequences shown in Table 1).
Table 1.
Gene amplification and Illumina adapter primers to prepare samples for deep sequencing
| Inner primers for library amplification | |
|---|---|
| Primer name | Sequence |
| Inner_FWD | 5′-gttcagagttctacagtccgacgatc < segment that overlaps to sense strand > – 3′ |
| Inner_REV | 5′-ccttggcacccgagaattcca < segment that overlaps to antisense strand > – 3′ |
| Outer primers to add the Illumina adapters and barcodes | |
| Illumina_FWD | 5′-aatgatacggcgaccaccgagatctacacgttcagagttctacagtccgacgatc - 3’ |
| Illumina_REV | 5′-caagcagaagacggcatacgagatnnnnnngtgactggagttccttggcacccgagaattcca - 3′ |
Bold, Illumina adapter; nnnnnn, indexing barcode (see Kowalsky et al. [18] for complete set); and italic, Illumina universal sequence
3.3.2. Yeast Plasmid DNA Preparation for Deep Sequencing
Thaw an aliquot of the stored yeast library by hand, spin down at 2500 × g for 3 min, and remove the supernatant.
Resuspend the pellet cells in 200 μL of Solution 1 and add 5 μL of 5 U/μL of Zymolyase.
Incubate at 37 °C for 4 h, and mix once per hour.
Perform one freeze-thaw cycle in dry ice/EtOH bath and 42 °C incubation.
Add 200 μL of Solution 2, mix briefly, and incubate for 3–5 min at room temperature.
Add 400 μL of Solution 3, mix well, and centrifuge at 17,000 × g for 5 min.
Transfer the supernatant to a Qiagen mini-prep column, and spin down for 1 min at 17,000 × g.
Add 700 μL of PB buffer, and spin down for 30 s at 17,000 × g.
Add 700 μL of PE buffer, and spin down for 30 s at 17,000 × g.
Repeat step 9.
Take out supernatant, and spin down again at 17,000 × g for 1 min to dry the column.
Transfer the column to a new clean 1.5 mL microfuge tube, add 30 μL of elution buffer, and spin down for 1 min at 17,000 × g.
Reload the column with the eluate, and spin down again. Store 15 μL of eluate, and proceed with the remaining 15 μL.
For the purification of plasmid from the yeast preparation, in a PCR tube, add 15 μL of dsDNA plasmid, 2 μL of exonuclease I, 1 μL of lambda nuclease, and 2 μL of 10× lambda buffer.
Place the mixture in a preheated (30 °C) thermocycler with the following cycle: 90 min at 30 °C, 20 min at 80 °C, and hold at 4 °C.
Clean the PCR product following the standard procedure from Qiagen mini-prep PCR cleanup, and elute in 30 μL of TE buffer.
Store 15 μL of eluate, and proceed with the remaining 15 μL.
For the gene library amplification, in a PCR tube, add 10 μL of 5× Phusion HF Buffer, 18.5 μL of NFH2O, 1 μL of 10 mM dNTPs, 2.5 μL of 10 μM of forward inner primer, 2.5 μL of 10 μM of reverse inner primer, 15 μL of dsDNA template, and 0.5 μL of Phusion High-Fidelity Polymerase.
Place the tube in a preheated (98 °C) thermocycler with the following cycle: 30 s at 98 °C, 14× cycles of 5 s at 98 °C, 15 s at 53 °C, and 15 s at 72 °C, follow by a final incubation for 10 min at 72 °C, and a hold at 4 °C.
Add 1.87 μL of 1:10 diluted exonuclease I.
Place the tube back in the thermocycler with the following cycle: 30 min at 37 °C, 5 min at 95 °C, and a hold at 4 °C.
In a new PCR tube, add 10 μL of 5× Phusion HF Buffer, 32.5 μL of NFH2O, 1 μL of 10 mM dNTPs, 2.5 μL of 10 μM of forward outer primer, 2.5 μL of 10 μM of reverse outer primer, 1 μL of dsDNA template from previous PCR amplification, and 0.5 μL of Phusion High-Fidelity Polymerase.
Repeat the same PCR cycle used for the inner primers.
Run 5 μL of PCR product on 2% agarose gel, and visualize with SYBR Gold. It is important to verify that you have single clear band before proceeding (see Note 9).
Purify and clean the PCR product using Agencourt AMPure XP following the manufacturer’s instructions for the 96-well format procedure.
Measure the concentration of each sample.
Stored the purify product at −20 °C.
3.3.3. dsDNA Quantification Using Quant-iT PicoGreen
At this point, samples are ready for deep sequencing. Follow the instructions for the Illumina MiSeq 2×250 bp submission from your sequencing facility. Usually, each Illumina MiSeq sequencing holds between 10 and 15 million reads per lane. Based on the read depth and library size, calculate the amount of reads necessary for each sample—our group uses approximately 500,000 reads per sample and multiplexes 20–30 samples per lane. Individual samples are quantified and mixed together in a single vial. The following procedure was adopted from the Invitrogen MP 07581 manual. The final yield should be about 1–4 ng in 40 μL.
Allow the Quant-iT reagent to warm to room temperature while covered in foil.
Prepare a 200-fold dilute solution of Quant-iT into TE buffer using a foil covered culture tube. (Example: 25 μL of PicoGreen reagent into 4.975 mL of TE). This solution should be prepared and used the day of the experiment.
Beginning with a 50 ng/mL stock of a kit-supplied lambda DNA standard, prepare a blank and a 1:2 standard curve (0, 1.56, 3.12,…, 25 ng/mL) using the first column of a 96-well black plate.
In a black 96-well plate, add 2.5 μL of each sample to 97.5 μL of TE in wells.
Carry out extra dilutions as necessary if the concentration is too high.
Add 100 μL of diluted PicoGreen solution to DNA samples and standard samples, mix briefly, and incubate for 5 min at room temperature covered with foil to protect from light.
Measure the fluorescence of the samples (excitation ~480 nm, emission ~520 nm).
Subtract the fluorescence value of the reagent blank from that of each of the samples.
Use the corrected data to generate a standard curve of fluorescence versus DNA standard concentrations, and calculate the concentrations of each sample. In our hands, the final concentration is between 5 and 40 ng/μL.
Mix equivalent mass amounts of samples in a single 1.5 mL Eppendorf tube, and send to your sequencing facility.
3.4. Data Analysis
Custom scripts used in the data analysis are available at GitHub (user: JKlesmith). Sample command lines and instructions are provided at the same source.
Use the modified version of Enrich 0.2 software as describe in Kowalsky et al. [18] to compute the enrichment ratios of individual mutants for the DNA sequencing results from Illumina MiSeq run (Fig. 5 see Note 10). Enrich 0.2 [8] documentation is available at http://depts.washington.edu/sfields/soft-ware/enrich/docs/0.2/enrich.html. The output from Enrich 0.2 is required as input for the remaining steps. The wild-type protein sequence is also required as input for the following steps.
The relative binding of each variant on the displayed and bound population is calculated using a custom Python script called QuickNormalize.py (see Note 11). The output from this script is a .csv file that can be read by multiple programs. In our lab we use Microsoft Excel to visualize the data as heatmaps and to carry out the data analysis (see Note 12).
Calculate the Shannon entropy for each variant on the displayed and bound population using a custom script called FACSEntropy.py -the output file is a .csv. The entropy values are used to discriminate those residues that participate in the protein-protein interaction and to determine the conformational epitope following the cut-off analysis flowchart as shown in Fig. 6 (also see Note 13).
Calculate the reportable statistics using QuickStat.py script. Statistics will report the reads passing through enrich; the percentage of possible codon substitutions observed; the percent of reads with none, one, and multiple nonsynonymous mutations; and the coverage of possible single nonsynonymous mutations.
Fig. 5.
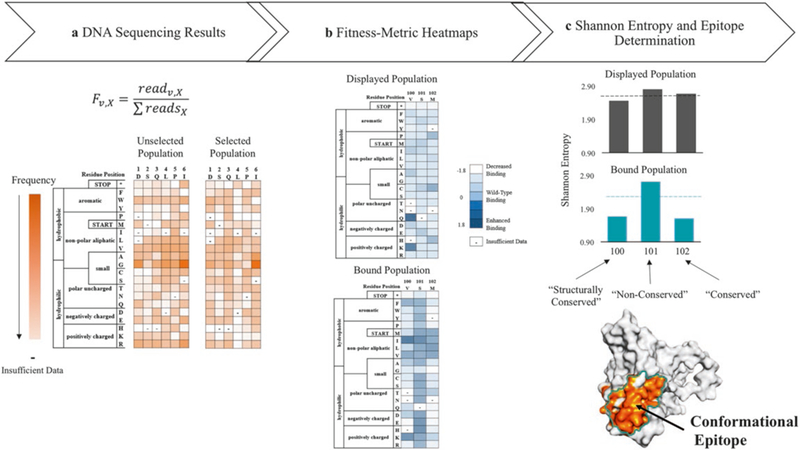
Deep sequencing results and data analysis used to determine the conformational epitope, (a) DNA sequencing results are processed using Enrich 0.2 software [19] to calculate the frequency, Fv,x, of each point mutant, v, for each position, x, in the primary sequence, (b) The frequency data of each variant from different populations is transformed into heatmaps comparing the relative fluorescence of each variant in the displayed population (top) and the bound population (bottom) against the unselected population, (c) Heatmaps are used to calculate the Shannon entropy for each residue on the displayed (black) and bound populations (turquoise). Next, the entropy is used to determine the conserved and non-conserved positions, which allows for identification of the conformational epitope
Fig. 6.

Flowchart of analysis used to determine the conformational epitope
4. Notes
The PPI partner protein is chemically biotinylated following the instructions for EZ-Link NHS-Biotin Reagents (ThermoFisher). We prefer chemical biotinylation to genetically encoded biotinylation (e.g., AviTag) as the former has a higher fluorescence signal. If proteins are small, covalent labeling with multiple biotins may disrupt the structure; in such a case, we recommend genetically fusing the PPI partner to a carrier-like maltose-binding protein or an IgG Fc. Anecdotally, we have noticed cleaner results with PPI partners with monovalent interactions and for that reason recommend creating a Fab if the PPI partner is a mAb.
The following rules apply for preparing separate mutagenesis libraries: (1) the length of each library should be divisible by three to avoid splitting a codon; (2) the gene should be segmented into libraries with a maximum length of225 base pairs for Illumina 250 bp paired-end sequencing (273 base pairs for 300 bp paired-end sequencing); and (3) libraries should be similar in length (±three nucleotides).
In some cases, the gene sequence of interest also contains a BbvCI restriction site. If the site is in the same orientation as the site on the pETconNK plasmid, continue the protocol as usual. If the BbvCI site is in the opposite direction as the site on the pETconNK plasmid, use the YSD plasmid pETCON (Addgene # 41522) as this plasmid does not contain a nicking site. The orientation of the BbvCI may not be in the same way as exists on the pETconNK plasmid. For example, if the nicking site is in the opposite direction from Fig. 2b, Nb.BbvCI (not Nt.BbvCI) should be used first to create the ssDNA wildtype template; otherwise follow the protocol as described in Subheading 3.1.2.
We recommend preparing phosphorylated oligos no earlier than the day before the nicking mutagenesis procedure. Avoid repeated freeze-thaw cycles.
For a library with NNK SSM at 75 amino acids, the theoretical library size is 2400 nucleotide variants. The percentage theoretical coverage is described by the following equation: %coverage = . In the above case, 16,500 transformants will give 99.9% coverage.
At this point, cells could be inoculated in fresh SGCAA media for Library Screening Preparation (Subheading 3.2) or frozen aliquots can be prepared for long-term storage. We often reinoculate the cells in SGCAA media to an OD600 = 1 and induce at 22 °C to confirm that the mutagenesis libraries displays on the yeast surface and binds the PPI partner. We prepare between 20 and 48 aliquots for long-term storage.
Induction temperature should be the same as used to prepare the PPI partner in a YSD format according to Chao et al. [9]. For each new YSD protein, our lab tests induction of surface display at 18, 20, 22, and 30 °C.
It is important that the number of collected cells should be at least 100-fold higher than the theoretical library size to avoid complexity bottlenecks. For example, at least 240,000 cells should be collected for each sorted population for a library with NNK SSM at 75 amino acids with a theoretical library size of 2400 nucleotide variants.
If the correct band size was not obtained from the second PCR product, we recommend troubleshooting by running 5 μL of the first PCR product on a 2% agarose gel and visualized on SYBR-Gold to identify which PCR amplification did not work. We recommend staining the gel on SYBR Gold for at least 1 h to resolve low-intensity bands. Fewer or more cycles of each PCR could be used to improve the product.
Our group routinely analyzes the quality of the Illumina sequencing data using FastQC available online at http://www.bioinformatics.babraham.ac.uk/projects/fastqc. Poor quality reads can hinder the data analysis using Enrich 0.2. The quality of the Illumina sequencing data is highest for the forward read and the first 150 bp. For issues where quality is poor on the reverse read, perform Enrich only for the forward read. We have also performed Enrich for short segments of the reads where the quality is highest.
-
The relative binding (ζi) for variant i is defined as
(2) where is the mean fluorescence of variant i and is the mean fluorescence of wild type. There are a number of assumptions used to calculate relative binding—see Kowalsky et al. [7] for further details.
Positions with insufficient data at more than ten substitutions should be excluded from analysis.
In the current experimental set-up, discriminating mutations that disrupt the interface and maintain the overall fold between those that destabilize the structure is difficult to determine, as unfolded mutants still predominantly display on the yeast surface [4, 20]. However, a recent study confirms that destabilizing mutations display with fewer copies on the yeast surface than stabilizing mutations, at least for proteins with >200 residues [ 15 ]—for small proteins destabilizing mutants appear to display at the same rate as stable mutants (T.A.W. and A.M.C., unpublished data). To further identify mutations that stabilize larger proteins, a FACS protocol is used with a sort gate set to collect the top 5% of the displaying population. For library screening, 2 × 106 yeast cells per mL, in PBS-BSA, are labeled with 1 μL of anti-c-myc-FITC per 2 × 105 yeast cells. The population is sorted using a gate that collects the top 5% of the displaying population. Shannon entropy obtained from this study is used to identify structurally conserved positions.
Acknowledgments
This work was supported by NSF CAREER (Award #1254238) to T.A.W. and a NIH T32 Biotechnology Training Grant (Award # T32-GM110523) to A.M.C.
References
- 1.Weiss GA, Watanabe CK, Zhong A et al. (2000) Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc Natl Acad Sci U S A 97:8950–8954. 10.1073/pnas.160252097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chao G, Cochran JR, Dane Wittrup K (2004) Fine epitope mapping of anti-epidermal growth factor receptor antibodies through random mutagenesis and yeast surface display. J Mol Biol 342:539–550. 10.1016/j.jmb.2004.07.053 [DOI] [PubMed] [Google Scholar]
- 3.Fowler DM, Fields S (2014) Deep mutational scanning: a new style of protein science. Nat Methods 11:801–807. 10.1038/nmeth.3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitehead TA, Chevalier A, Song Y et al. (2012) Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol 30:543–548. 10.1038/nbt.2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Blarcom T, Rossi A, Foletti D et al. (2015) Precise and efficient antibody epitope determination through library design, yeast display and next-generation sequencing. J Mol Biol 427:1513–1534. 10.1016/j.jmb.2014.09.020 [DOI] [PubMed] [Google Scholar]
- 6.Doolan KM, Colby DW (2015) Conformation-dependent epitopes recognized by prion protein antibodies probed using mutational scanning and deep sequencing. J Mol Biol 427:328–340. 10.1016/j.jmb.2014.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kowalsky CA, Faber MS, Nath A et al. (2015) Rapid fine conformational epitope mapping using comprehensive mutagenesis and deep sequencing. J Biol Chem 290:26457–26470. 10.1074/jbc.M115.676635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fowler DM, Araya CL, Fleishman SJ et al. (2010) High-resolution mapping of protein sequence-function relationships. Nat Methods 7:741–746. 10.1038/nMeth.1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chao G, Lau WL, Hackel BJ et al. (2006) Isolating and engineering human antibodies using yeast surface display. Nat Protoc 1:755–768. 10.1038/nprot.2006.94 [DOI] [PubMed] [Google Scholar]
- 10.Van DJA, Wittrup KD (2014) Yeast surface display for antibody isolation: library construction, library screening, and affinity maturation. Methods Mol Biol 1131:151–181. 10.1007/978-1-62703-992-5_10 [DOI] [PubMed] [Google Scholar]
- 11.Mata-Fink J, Kriegsman B, Yu HX et al. (2013) Rapid conformational epitope mapping of anti-gp120 antibodies with a designed mutant panel displayed on yeast. J Mol Biol 425:444–456. 10.1016/j.jmb.2012.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adams RM, Mora T, Walczak AM et al. (2016) Measuring the sequence-affinity landscape of antibodies with massively parallel titration curves. Elife 5:5980–5985. 10.7554/eLife.23156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wrenbeck EE, Klesmith JR, Stapleton JA et al. (2016) Plasmid-based one-pot saturation mutagenesis. Nat Methods 13:928–930. 10.1038/nmeth.4029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gietz RD, Schiestl RH (2007) High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2:31–34. 10.1038/nprot.2007.13 [DOI] [PubMed] [Google Scholar]
- 15.Klesmith JR, Bacik J-P, Wrenbeck EE et al. (2017) Trade-offs between enzyme fitness and solubility illuminated by deep mutational scanning. Proc Natl Acad Sci U S A 114:2265–2270. 10.1073/pnas.1614437114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wrenbeck E, Klesmith J, Stapleton J, Whitehead T (2016) Nicking mutagenesis: comprehensive single-site saturation mutagenesis. Protoc Exch. 10.1038/protex.2016.061 [DOI] [Google Scholar]
- 17.Sambrook J, Russell DW (2006) Transformation of E. coli by electroporation. CSH Protoc 2006:pdb.prot3933. 10.1101/pdb.prot3933 [DOI] [PubMed] [Google Scholar]
- 18.Kowalsky CA, Klesmith JR, Stapleton JA et al. (2015) High-resolution sequence-function mapping of full-length proteins. PLoS One 10:e0118193. 10.1371/journal.pone.0118193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fowler DM, Araya CL, Gerard W, Fields S (2010) Enrich: software for analysis of protein function by enrichment and depletion of variants. Bioinformatics 27:3430–3431. 10.1093/bioinformatics/btr577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kowalsky CA, Whitehead TA (2016) Determination of binding affinity upon mutation for type I dockerin-cohesin complexes from Clostridium thermocellum and Clostridium cellulolyticum using deep sequencing. Proteins 84:1914–1928. 10.1002/prot.25175 [DOI] [PubMed] [Google Scholar]