

Introduction
A method allowing for efficient and quantitative coexpression of multiple heterologous proteins in neurons in vivo would be highly valuable for many applications in neuroscience. To date, different approaches, such as internal ribosomal entry site (IRES) elements (Douin et al., 2004), bidirectional or double promoters (Baron et al., 1995), or coinfection with multiple viral vectors (Mastakov et al., 2002), are commonly used. These strategies, however, mostly suffer from the fundamental problem that coexpression of the heterologous proteins is unreliable and far from quantitative. In the case of IRES-mediated coexpression, for example, the upstream elements in these bicistronic mRNA open reading frames (ORFs) are more strongly transcribed than the IRES-controlled downstream protein (Hennecke et al., 2001).
An alternative and more promising approach involves the use of self-processing viral peptide bridges. So-called 2A or 2A-like peptide sequences separate different protein coding sequences in a single ORF transcription unit of Picornaviridae (Ryan et al., 1991). The 2A peptide sequences from different members of the picornavirus family share a highly conserved motif of only 18 aa, mediating the cleavage between the C-terminal glycine and the N-terminal proline of the downstream 2B sequence (Ryan et al., 1991). Initially, the 2A peptide cleavage was thought to be mediated by an autoproteolytic event (Ryan et al., 1991), and 2A peptides were called “self-cleaving peptides.” Ultimately, a ribosomal-skip mechanism was proposed, and 2A and 2A-like sequences are now referred to as CHYSELs (cis-acting hydrolase elements) rather than self-cleaving peptides (Donnelly et al., 2001). Linking proteins with 2A or 2A-like peptide sequences results in cellular expression of multiple, discrete proteins (in essentially equimolar quantities) derived from a single ORF (de Felipe et al., 2006).
The 2A peptide-mediated cleavage has previously been shown to result in coexpression of functional heterologous proteins in cell lines (Lengler et al., 2005; de Felipe et al., 2006), embryonic stem cells (Hasegawa et al., 2007), and neurons (Furler et al., 2001). In some cases, however, C-terminal 2A peptide sequences resulted in impaired protein function (Lengler et al., 2005; Hasegawa et al., 2007). Surprisingly, the potential of this system has not been exploited in cellular and systems neuroscience. It has not been addressed whether 2A peptide-mediated coexpression of heterologous proteins is quantitative in neurons and expression levels of fluorescent reporters would be high enough for in vivo electrophysiological and imaging applications. Another important issue is to ensure that 2A peptide transcripts leave neuronal viability and function intact. Furthermore, it was unclear whether, in neurons, this technology could be extended to coexpress more than two proteins, including secreted and membrane proteins, which are critical players for understanding neural circuit function.
To address these questions, we analyzed the efficiency of coexpression of various proteins in neurons in vitro and in vivo, using a slightly modified Thosea asigna virus 2A peptide bridge (de Felipe et al., 2006) containing a short C-terminal linker sequence composed of four amino acid residues. With this approach, we demonstrate efficient and reliable functional coexpression of multiple cytosolic, secreted and membrane proteins in neurons in vivo. We show that viral coexpression of the codon-improved Cre recombinase (iCre) (Shimshek et al., 2002) or an improved tetracycline-controlled transactivator (itTA) (Krestel et al., 2004) with fluorescent proteins (FPs) is compatible with in vitro and in vivo electrophysiological and imaging approaches, and can be combined with inducible genetic mouse models to enable targeted manipulation of faithfully labeled neuronal subgroups. Furthermore, by using two 2A sequences, three proteins can be simultaneously expressed under the control of a single promoter. In this last approach, we show that coexpression of a fluorescent reporter and two light-activated membrane proteins Channelrhodopsin2 (ChR) (Gradinaru et al., 2007) and Halorhodopsin (NpHR) (Gradinaru et al., 2008) may greatly simplify gene delivery for reliable bidirectional optical control of neuronal activity.
Materials and Methods
Plasmid constructs
The Thosea asigna virus 2A peptide coding sequence together with N-terminal CTC and C-terminal GCACCGGGATCCACC flanking sequences was synthesized and subcloned between BamHI and AgeI sites of plasmid pAAV-6P-SEWB (Shevtsova et al., 2005). The iCre gene sequence (Shimshek et al., 2002) was cloned between the synapsin promoter and the 2A peptide sequence to generate pAAV-Syn-iCre2A-EGFP. The pAAV-CMV-iCre2A-EGFP was constructed by substituting of the CMV promoter for the synapsin promoter in construct pAAV-Syn-iCre2A-EGFP. The Venus sequence was inserted as PCR product by 5′-blunt end 3′-BsrGI ligation into pAAV-Syn-iCre2A-EGFP vector in place of EGFP. The itTA (Krestel et al., 2004) and Kusabira Orange (KO) (Karasawa et al., 2004) gene sequences were generated by PCR and were used to replace iCre and Venus sequences in pAAV-Syn-iCre2A-Venus, respectively. The iCre sequence was replaced by human Noggin PCR fragment to generate pAAV-Syn-Noggin2A-Venus. The ChR (H134R) (Chlamydomonas reinhardtii Channelrhodopsin2) (Gradinaru et al., 2007) and tDimer sequences (Campbell et al., 2002) were generated by PCR to replace iCre and Venus, respectively, in pAAV-Syn-iCre2A-Venus to construct plasmid pAAV-Syn-ChR2A-tDimer. For construction of pAAV-Syn-NpHR2A-Venus, the eNpHR (enhanced Natronomonas pharaonis Halorhodopsin) (Gradinaru et al., 2008) sequence was generated by PCR and replaced iCre in pAAV-Syn-iCre2A-Venus. The triple expression vector pAAV-Syn-ChR2A-NpHR2A-Venus was constructed by generation of NpHR and 2A-Venus sequences by PCR and successive insertion into pAAV-Syn-ChR2A-tDimer. All ChR and NpHR containing expression cassettes were shuttled into pAAV-CAG-shuttle (Applied Viromics). For construction of plasmid pHD-iCre2A-Venus, the iCre2A-Venus fragment was isolated from plasmid pAAV-Syn-iCre2A-Venus with EcoRI and BsrGI and subcloned between BglII and XhoI in plasmid pHD-iCre (Shimshek et al., 2002) by 5′- and 3′- blunt ligation to replace iCre sequence. All plasmids were sequenced after cloning.
Transfection of cultured cells and staining assays
Primary hippocampal neuron culture transfection was done with Lipofectamine 2000 (Invitrogen) at day 5 in vitro (DIV 5) as described previously (Dalby et al., 2004). HEK293 cells were transfected (Shimshek et al., 2002) with pAAV-CMV-iCre2A-EGFP. Other plasmids (pHD-iCre2A-Venus and pHD-iCre) were separately cotransfected with control plasmid (600 ng/well) pHD-AP into CV1/lacZ indicator cells in a 24-well plate (Shimshek et al., 2002). Activities of β-galactosidase and alkaline phosphatase were detected by X-Gal and Fast Red (Roche) stainings 48 h after transfection. The Cre-activity was evaluated by the ratio of blue to red cells, which reflects the fraction of cells with Cre-activity among the total number of transfected cells.
Preparation and delivery of recombinant adeno-associated viruses
Recombinant adeno-associated viruses (rAAVs) serotype 1 and 2 were generated and purified by iodixanol gradients as described before (Zolotukhin et al., 1999; Zhu et al., 2007). Since all viral constructs express FPs, titers were determined by infection of hippocampal primary neuron cultures. For this dissociated hippocampal neurons (Brewer et al., 1993) of E18 rats were plated at a density of 5 × 104 cells per well in 24-well plates coated with poly-l-lysine (Sigma). Neurons were infected with rAAVs at DIV 4 with 0.1 μl of virus. Two weeks later, fluorescent cells were counted to calculate the infectious titer, and rAAV preparations showing a titer of 107–108 infectious particles/ml were stored at −70°C and used for further experiments. For the amygdala infection, purified rAAVs (serotype 7) were obtained from Penn Vector Core (titers: rAAV-Syn-ChR-2A-tDimer, 5.36 × 1012 genome copies/ml; rAAV-Syn-ChR-2A-NpHR-2A-Venus, 2.61 × 1012 genome copies/ml). In the dissociated primary hippocampal neuron cultures, the expression of rAAV-transduced proteins was evaluated 10 d postinfection (DPI 10, with 0.5 × 104–105 infectious particles per well of a 24-well plate) by Western blots or immunohistochemistry. For in vivo expression, viruses were delivered into the brains of deeply anesthetized (ketamine/xylazine mix or isoflurane) 5- to 6-week-old mice by stereotactic injections as described in detail (Cetin et al., 2006). Stereotactic coordinates relative to bregma were as follows: anteroposterior (AP) −2.1 mm, lateral (Lat) ±1.6 mm, depth 0.3–1.6 mm, for hippocampus and cortex, and AP −1.7 mm, Lat ± 3.3 mm, depth 4.4 mm for amygdala. During each injection, 0.3 μl of purified rAAVs (1.0 × 107–108 infectious particles/ml for hippocampus and cortex infection; 2–5 × 1012 genome copies/ml for amygdala infection) was delivered by stereotactic injection. All procedures were performed according to the guidelines of the local animal use and care committees.
Protein extraction and Western blot analysis
Transfected HEK293 cells and cultured primary hippocampal neurons were harvested with lysis buffer (50 mm Tris-HCl, pH 7.6; 5 mm MgCl2; 130 mm NaCl; 10 mm KCl; 1% Triton X-100; 5% glycerin) 48 and 24 h after transfection, respectively, while infected primary hippocampal neurons were harvested 10 d after virus application. Brain tissue (cortex and hippocampus) was collected 2 weeks after virus infection and homogenized in ice-cold buffer (25 mm HEPES, pH 7.4) containing a protease inhibitor cocktail (Complete; Roche). From rAAV-infected neuron cultures, 10 μg of total cell lysate and 20 μl of culture medium were separated by SDS-PAGE (10% separating and 4% stacking gels) and transferred to nitrocellulose membranes. Western blots were probed with polyclonal rabbit anti-Cre (1:8000, Covance), monoclonal mouse anti-GFP (1:10,000, Clontech), polyclonal rabbit anti-tTA (1:10,000, provided by Prof. Dr. Wolfgang Hillen, Institute of Biology, Friedrich-Alexander University, Erlangen, Germany), monoclonal rat anti-Noggin (1:1000, R&D Systems), and polyclonal rabbit anti-DsRed (1:1000, Clontech) antibodies. Horseradish peroxidase-linked anti-rabbit, anti-mouse, or anti-rat was used as secondary antibody (1:15,000, Vector Laboratories), and blots were visualized by enhanced chemiluminescence (ECL kit, GE Healthcare). Quantification of Western blots was done using ImageJ (NIH).
Immunohistochemistry
Cultured primary hippocampal neurons at DPI 10 or brains from virus-injected mice at DPI 14 were fixed with 4% paraformaldehyde/PBS, and 70 μm coronal sections were obtained from the brains. Immunostaining was performed with polyclonal rabbit anti-Cre primary antibody (1:3000, Covance) and Cy3-coupled anti-rabbit secondary antibody (1:200, Jackson ImmunoResearch). Activity of β-gal in brain slices of Rosa26R mice (Soriano, 1999) was visualized by X-Gal staining (Shimshek et al., 2002).
Slice preparation and transfection of slice cultures
Standard procedures were used to prepare acute, 300-μm-thick brain slices (Bissière et al., 2003). Hippocampal slices were cut from 6- to 7-week-old mice infected with rAAVs for 2–3 weeks, and amygdala slices were prepared from 8- to 10-week-old mice infected with rAAVs for 4–6 weeks. Hippocampal slice cultures were prepared from 5- to 7-d-old rat pups as previously described (Stoppini et al., 1991) and maintained in culture for 1–2 weeks. Plasmid DNA was prepared and biolistic transfection using a Gene Gun (Bio-Rad) was performed as described previously (Ehrlich and Malinow, 2004). Expression was allowed for 7–10 d before recording.
Whole-cell recordings
Neurons were visualized using infrared and fluorescence microscopy. The ACSF contained 124 mm NaCl, 2.7 mm CaCl2, 1.3 mm MgCl2, 2.7 mm KCl, 26 mm NaHCO3, and 0.4 mm NaH2PO4. Whole-cell recordings were obtained from neighboring control and infected or transfected pyramidal neurons in hippocampus or principal neurons in the basolateral amygdala. Patch electrodes (5–6 MΩ) were pulled from borosilicate glass and filled with solution containing 130 mm K-gluconate, 5 mm KCl, 10 mm HEPES, 0.6 mm EGTA, 4 mm Mg-ATP, 0.4 mm Na-GTP, and 10 mm Na2-phosphocreatine (290 mOsm, pH 7.2). For AMPA/NMDA ratios, K-gluconate and KCl were replaced by 115 mm Cs-methylsulfonate and 20 mm CsCl. Data were acquired using a Multiclamp 700B and pClamp software (Molecular Devices), and analyzed offline using Igor Pro (WaveMetrics). Resting membrane potential was measured after break-in, and input resistance was calculated from the slope of a linear fit to the current-voltage relation or steady state voltage steps. Excitatory synaptic inputs from Schaffer collaterals were evoked and measured as previously described (Ehrlich and Malinow, 2004). Paired-pulse ratios were obtained at a 50 ms interval. Light-evoked responses were measured using K-gluconate internal solution. For some experiments, a light-emitting diode (470 nm) (Cairn Research) was used to stimulate ChR, in other experiments, a metal-halide lamp was used in combination with a blue excitation filter (WIBA2, Olympus) for ChR and a yellow excitation filter (FF01–593/40-25, AHF) for NpHR and a fast shutter (Uniblitz). Data are shown as mean ± SEM; n is the number of cells or cell pairs. For statistical comparisons, paired or unpaired Student's t tests were used, as applicable.
In vivo two-photon imaging
Two-photon imaging was performed 3 weeks after virus injection into somatosensory cortex using a custom-built two-photon microscope (Hasan et al., 2004). Mice were anesthetized with urethane (1.5 mg/g body weight), and body temperature was maintained at 37°C. A custom-built head plate with an imaging window (4 × 3 mm) was glued to the top of the skull using cyanoacrylate (Uhu) and attached to a fixed metal bar. The microscope objective was positioned so that the optical axis was perpendicular to the surface of the cortex. Two-photon image sequences were collected in regions of interest with a MicroMax, 512 × 512 back-illuminated CCD camera (Roper Scientific).
Results
Efficient 2A peptide cleavage and coexpression in primary neurons
To examine 2A-mediated coexpression in neurons, we generated a modified 2A peptide bridge from Thosea asigna virus (de Felipe et al., 2006) by adding 4 aa to the C-terminal proline (Fig. 1A). Because C-terminal 2A peptide sequences had previously been reported to interfere with protein function (Lengler et al., 2005; Hasegawa et al., 2007), we reasoned that the addition of 4 aa may favor reproducible processing efficiency at the cleavage site by creating a constant environment.
Figure 1.

Reliable coexpression of heterologous proteins using 2A peptide bridges in cultured neurons. A, Top, Amino acid residues of 2A peptide from Thosea asigna virus. The cleavage site is indicated by the arrow. Amino acid residues in red were added to keep the C-terminal environment constant between different constructs. Bottom, Diagram of rAAV vector constructs. B, 2A-mediated self-processing in primary hippocampal neuron cultures. Western blots of protein extracts from rAAV-infected hippocampal neurons at DPI 10. The 40.8 kDa iCre2A fusion protein and the 27.3 kDa Venus were detected, and to a very small extent, the 68.1 kDa precursor full-length protein. I, Neurons infected with rAAV-Syn-iCre2A-Venus. II+III, Neurons coinfected with the activator virus rAAV-Syn-tTA and rAAV-Ptetbi-iCre-Venus. Ctrl, Noninfected neurons. C, Confocal images of primary hippocampal neurons infected with rAAV-Syn-iCre2A-Venus at DPI 10. Neurons coexpress cleaved and freely diffusible Venus (green) and nuclear restricted iCre (red, visualized with Cy3-coupled antibody), indicating that the 2A fusion construct was cleaved successfully in situ. Scale bars, 10 μm.
First, we infected rat primary hippocampal neurons with rAAV encoding a 2A fusion protein between the nuclear-localized iCre (Shimshek et al., 2002) and the FP Venus (Nagai et al., 2002) under the control of the human synapsin promoter (Fig. 1A). Ten days after infection, we found in Western blots of cell lysates high levels of released iCre and Venus together with low levels of the full-length protein, indicating efficient 2A self-processing (Fig. 1B). The molecular weight (MW) of the released iCre2A was slightly bigger than “wild-type” iCre (39 kDa) due to the short, remaining 2A tail of 17 amino acid residues (40.8 kDa). When compared with a similar construct expressed from a cytomegalovirus (CMV) promoter in heterologous HEK293 cells (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material), the balance between newly synthesized full-length iCre2A-FP and the released iCre2A and FPs appeared much more in favor of cleaved products in neurons (Fig. 1B), indicating that the 2A strategy may be particularly suitable for coexpression in nondividing cells such as neurons or in the brain. We confirmed that the enzymatic activity of the released iCre2A fusion was completely intact by assaying Cre-mediated activation of lacZ expression in the Cre-reporter cell line CV1 (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). The 2A self-processing was also supported by immunostaining of rAAV-infected primary hippocampal neurons, which detected iCre exclusively in the nucleus and Venus mainly in the cytosol. Even without enhancement, Venus expression was high enough to visualize fine cellular processes (Fig. 1C).
To directly compare the 2A approach with conventional, dual-promoter-mediated coexpression, we used a bidirectional Ptet-bi promoter construct (Baron et al., 1995) (Fig. 1B; supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Using this bidirectional promoter, iCre levels were much lower than those obtained from the iCre2A-Venus fusion construct when normalized to the levels of Venus (data not shown). Assuming that iCre and Venus are released approximately in a one-to-one ratio from the iCre2A-Venus fusion construct, the amount of iCre was approximately sixfold higher with the 2A strategy than with the bidirectional promoter, demonstrating that expression levels at the left and right side of this bidirectional promoter were very different. In summary, using our modified 2A peptide sequence results in effective processing of the expressed fusion protein and allows for the reliable and quantitative coexpression of proteins in heterologous cells and particularly in neurons.
Functional coexpression in intact brain and applications for electrophysiology and imaging ex vivo and in vivo
To examine 2A peptide-mediated coexpression in the brain in vivo, we injected rAAV-Syn-iCre2A-Venus into cortex and hippocampus of adult Cre-reporter Rosa26R mice (Soriano, 1999). Two weeks later, the expression of the Cre-dependent lacZ reporter gene indicated successful delivery of functional iCre2A (Fig. 2A). At the injection sites, cytosolic Venus fluorescence and nuclear iCre staining were colocalized in the same neurons, confirming efficient self-processing of iCre2A and Venus in vivo (Fig. 2B,C). In Western blots, we detected exclusively the released iCre2A and Venus, but not the full-length iCre2A-Venus fusion protein (Fig. 2D).
Figure 2.
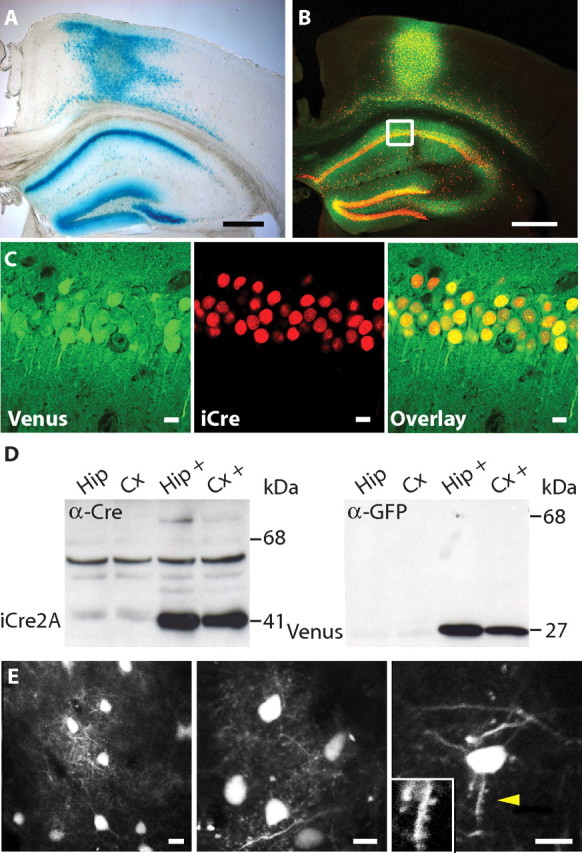
Functional and reliable coexpression using 2A peptide bridges in vivo and its application in life imaging. A, LacZ staining of a brain section from a Rosa26R mouse 2 weeks after stereotactic injection of rAAV-Syn-iCre2A-Venus, indicating that the released iCre2A protein was functional in vivo. Only infected brain regions showed blue LacZ signal. Scale bars, 500 μm. B, Confocal image of a brain section from a wild-type mouse injected with rAAV-Syn-iCre2A-Venus at DPI 14. Scale bars, 500 μm. C, Higher magnification of the boxed CA1 region. Coexpression of Venus (green) and iCre (red, visualized with Cy3-coupled antibody) was seen in virtually all infected neurons. Scale bars, 10 μm. D, Western blots of extracts from infected brain regions demonstrate near complete 2A peptide self-processing in vivo. Tissues were dissected from brains of noninjected and rAAV-Syn-iCre2A-Venus injected C57BL/6 mice at DPI 14. The 40.8 kDa iCre2A (left) and the 27.3 kDa Venus (right) were clearly detected, the 68.1 kDa full-length fusion protein was hardly visible. Hip, Hippocampus; Cx, cortex; +, infected with rAAV. E, In vivo two-photon images of a mouse injected with purified rAAV-Syn-iCre2A-Venus in somatosensory cortex at DPI 21. Cell bodies of neurons in superficial layers as well as their fine processes were imaged at a depth of 150–200 μm. Spines are indicated by arrowhead and zoom-in image is shown. Scale bars, 10 μm.
Next, we examined whether expression levels of FPs would be sufficient to enable high-resolution imaging of neurons and their processes in the intact brain in vivo. Indeed, 3 weeks after infection with rAAV-Syn-iCre2A-Venus, fluorescence was strong enough to allow for imaging of cortical neurons in anesthetized mice by two-photon microscopy. The labeled neurons and their processes, including spines, were readily visible in the superficial cortical layers (Fig. 2E). These experiments not only demonstrate that expression levels were high enough for subcellular imaging, but also that our rAAVs were purified well enough to avoid immune responses, cell damage, or necrosis.
To confirm at a more subtle level that rAAV-infected neurons expressing 2A-constructs displayed the same physiological parameters as uninfected counterparts, we recorded from Venus-expressing cells in acute brain slices ex vivo 3 weeks after infection. Hippocampal pyramidal neurons infected with rAAV-Syn-iCre2A-Venus were viable and easy to identify by their green fluorescence (Fig. 3A). The postsynaptic properties (measured by AMPA to NMDA ratio) and presynaptic function (measured by paired-pulse ratio) were indistinguishable from neighboring nonfluorescent cells (Fig. 3B–E). We also found no difference in the passive electrical properties (supplemental Table 1, available at www.jneurosci.org as supplemental material), intrinsic excitability, and spiking behavior between infected and control neurons (Fig. 3F,G). Hence, rAAV-driven coexpression of 2A self-processing proteins does not influence neuronal properties and induce cell toxicity. We conclude that our 2A peptide strategy has the powerful potential to combine genetic manipulations with the expression of FPs in subgroups of neurons, a prerequisite for functional studies using optical and electrophysiological high-resolution analysis in vivo, in particular for long-term investigations.
Figure 3.

Unaltered electrophysiological properties of neurons expressing 2A constructs. Viral expression of 2A constructs had no effect on viability, spiking behavior, and synaptic transmission. A, Sparsely infected CA1 region in an acute hippocampal slice of rAAV-Syn-iCre2A-Venus at DPI 14–21, where Venus-expressing cells are clearly seen. B, AMPA-EPSCs recorded from a pair of CA1 neurons in response to paired pulses at −70 mV; control (black) and infected cell (green). Calibration: 20 pA, 40 ms. C, No difference in amplitudes of AMPA-EPSCs, which were −54.4 ± 9.2 pA and −57.4 ± 9.7 pA in control and infected neurons, respectively (n = 11 pairs, p = 0.68). D, No alterations in paired-pulse ratios (PPRs) of AMPA-EPSCs, which were 1.91 ± 0.09 and 1.79 ± 0.09 in control and infected neurons, respectively (n = 11 pairs, p = 0.56). E, AMPA- and NMDA-mediated EPSCs recorded from a pair of CA1 neurons at −70 mV and +40 mV; control (black) and infected cell (green). Calibration: 20 pA, 50 ms. There was no change in AMPA/NMDA ratios, which were 3.80 ± 0.63 (n = 6) and 4.09 ± 0.84 (n = 5) in control and infected neurons, respectively (p = 0.74). F, Indistinguishable electrical properties of control and infected CA1 neurons. Responses of a control (black) and an infected (green) neuron to current injections of −100, 0, +100, and +200 pA. Calibration: 20 mV, 100 ms. G, No difference in input–output curves (control, n = 8, infected, n = 7; p > 0.72).
Functional coexpression of itTA or secreted proteins with fluorescent proteins
To demonstrate that the self-processing activity of the modified 2A peptide was independent of the fused protein domains, we extended our strategy to other activators used for conditional gene expression, and other FPs. We generated rAAVs expressing a fusion protein between the itTA (Krestel et al., 2004) and the red FP KO (Karasawa et al., 2004) (Fig. 4A). When expressed in cultured primary hippocampal neurons, itTA2A was released (Fig. 4B). To provide proof of principle that itTA2A and KO were coexpressed and active in vivo, we coinjected rAAV-Syn-itTA2A-KO and a tTA responder virus rAAV-Ptetbi-iCre-Venus (Zhu et al., 2007) into the cortex and hippocampus of wild-type mice. We observed activation of the tTA responder genes in infected neurons, as assessed by Venus fluorescence. The FPs, Venus and KO, colocalized in most neurons at the injection site, suggesting a high rate of neurons coinfected with both viruses (Fig. 4C,D). Even when we consider the constraints of coinfection and promoter leakiness of the responder virus (Zhu et al., 2007), our data provide strong evidence that 2A self-processing with our modified sequence is independent of the fused proteins and can be used universally.
Figure 4.

Use of 2A strategy to express other activators, fluorescent proteins, and secreted proteins. A, Diagram of the rAAVs for dual virus activator-responder approach to test for itTA and KO coexpression by 2A self-processing. B, Western blots of protein extracts from rAAV-Syn-itTA2A-KO-infected dissociated hippocampal neuron in culture at DPI 10. The 30.5 kDa itTA2A fusion protein was detected and 55.4 kDa full-length protein was faintly visible. I+II, Coinfected neurons. Ctrl, Noninfected neurons. C, Confocal images of a brain section from a mouse coinjected with rAAV-Syn-itTA2A-KO and the responder virus rAAV-Ptetbi-iCre-Venus 14 d after injection. Cells expressing itTA are labeled with the coexpressed red fluorescent protein KO. The itTA-driven Venus expression from the responder virus was detected by green fluorescence. Scale bars, 200 μm. D, At higher magnification, many neurons were fluorescently labeled in the infected cortical area, and most of them showed coexpression of KO and Venus as expected. Scale bars, 50 μm. E, Left, Dissociated primary hippocampal neurons at DPI 10 with rAAV-Syn-Noggin2A-Venus. Right, Western blots from rAAV-Syn-Noggin2A-Venus-infected primary hippocampal cultures at DPI 10. Three different forms of Noggin were detected in hippocampal neuron lysate, and only the 32 kDa secreted form was detected in the culture medium. Ctrl, Noninfected neurons.
To examine whether our 2A approach is also effective for fusion proteins synthesized at the rough endoplasmic reticulum (ER) that undergo cotranslational secretion, we introduced the secreted protein Noggin with a signal peptide at its N terminus as a 2A fusion with Venus (Fig. 4E). We infected primary hippocampal neuron cultures with rAAV-Syn-Noggin2A-Venus, and when Venus fluorescence could be detected 10 d later, we analyzed cell lysates and the culture medium by Western blotting. In cell lysates, three proteins with MWs of 32, 30, and 26 kDa could be detected. The 26 kDa form represents the unmodified Noggin2A, the 30 kDa protein the glycosylated Noggin2A, and the 32 kDa protein is the mature Noggin2A, which is glycosylated and contains a cystine knot motif that is resistant to regular reduction procedures (Groppe et al., 2002). Importantly, in the medium, we only detected the 32 kDa isoform, suggesting successful secretion of Noggin2A after its release from the Noggin2A-Venus fusion (Fig. 4E). As in previous experiments, the full-length Noggin2A-Venus fusion protein was not detected in cell lysates or in the medium. We conclude that 2A self-processing also occurs at the rough ER, and can be successfully applied to reliably coexpress secreted proteins and fluorescent markers in individual neurons.
Coexpression of multiple membrane proteins and fluorescent reporters
Another highly desirable application is to faithfully coexpress untagged membrane proteins, such as ion channels or receptors, together with FPs. Quantitative coexpression of several membrane proteins together with FPs is particularly useful for studying function of circuits and synapses. Here, we tested this possibility using one or multiple 2A peptide bridges. We generated fusion constructs between the light-gated ion channel ChR or the ion pump NpHR and FPs, and a triple construct coexpressing ChR, NpHR, and an FP using two 2A elements (Fig. 5A). The function of these light-activated proteins is easily assessed by electrophysiology and permits bidirectional light-gated control of neuronal activity (Gradinaru et al., 2007). We first transfected all pAAV constructs biolistically into hippocampal slice cultures before producing the respective viruses. Transfected cells were viable and exhibited normal basic electrical properties when compared with neighboring control neurons (supplemental Table 1, available at www.jneurosci.org as supplemental material). Importantly, FPs were always localized in the cytosol and in the nucleus, indicating that they were freely diffusible, and therefore, were cleaved products of the 2A fusion proteins (Fig. 5A). Neurons transfected with ChR2A-tDimer, identified by red fluorescence, showed large inward currents (in voltage-clamp mode) or depolarizing responses (in current-clamp mode) when exposed to blue light (Fig. 5B). Train stimulation reliably induced action potentials, even at high frequencies (Fig. 5C). When NpHR2A-Venus-transfected cells, identified by green fluorescence, were exposed to yellow light, outward light-induced currents (in voltage-clamp mode) and hyperpolarizing responses (in current-clamp mode) were recorded (Fig. 5E). Light-evoked hyperpolarizations significantly reduced action potential firing (Fig. 5F). For bidirectional optogenetic control, we expressed a construct encoding both light-activated proteins together with a soluble FP (pAAV-Syn-ChR2A-NpHR2A-Venus) (Fig. 5A). All transfected neurons responded to both blue and yellow light (Fig. 5H). Light-activated currents were of similar or even larger amplitude when compared with cells transfected with only one of the light-gated proteins, demonstrating efficient self-processing and equal function when expressed from the dual versus triple construct (Fig. 5D,G,J). Moreover, efficient, bidirectional control of action potential firing was achieved in the same neurons (Fig. 5H,I). To test suitability of this method for in vivo applications, we generated rAAVs expressing ChR2A-tDimer and ChR2A-NpHR2A-Venus. Amygdala neurons were infected in vivo, and light-gated activity was subsequently assayed ex vivo in acute brain slices. For both constructs, infected neurons showed no signs of cellular toxicity, and had stable resting membrane potentials and electrical properties (data not shown). All neurons infected with rAAV-Syn-ChR2A-tDimer displayed light-evoked currents, and in 69% (n = 20/29) expression levels were high enough to induce light-evoked spiking with high fidelity (Fig. 5K). For rAAV-Syn-ChR2A-NpHR2A-Venus, all neurons with Venus fluorescence showed light-evoked currents. Effective bidirectional optic control of neural activity (spiking or attenuation of spiking) was achieved in 77% (n = 14/18) of recorded cells (Fig. 5L). Although all recorded cells showed light-evoked currents (Fig. 5M), in some cells they were not large enough to control action potential firing. This could be explained by heterogeneous expression levels caused by differences in infection rates or in the number of infecting virus particles per cell.
Figure 5.

Functional coexpression of multiple membrane proteins and a fluorescent reporter. A, Left, Diagram of pAAV plasmids. Right, Infrared and fluorescence images of a plasmid-transfected pyramidal neuron in an organotypic slice expressing ChR2A-NpHR2A-Venus. Scale bar, 10 μm. B, Light responses in a transfected pyramidal neuron expressing ChR2A-tDimer in organotypic slices. Blue light (2 ms) elicited an inward current in voltage clamp (VC) and a spike in current-clamp mode (CC). Calibration: VC, 2000 pA, 100 ms; CC, 40 mV, 100 ms. C, Light-induced spiking at different stimulation frequencies, and summary of spike probability as a function of frequency in transfected cells (blue symbols; n = 13). No spikes were seen in control cells (black symbols; n = 6). D, Blue light-evoked ChR-currents were −2009 ± 304 pA (n = 9) versus −3 ± 1 pA (n = 6) in transfected versus control cells (p < 0.01). E, Light responses in a transfected pyramidal neuron expressing NpHR2A-Venus. Yellow light (50 ms) elicited an outward current in VC, and a hyperpolarization in CC mode. Calibration: VC, 40 pA, 50 ms; CC, 10 mV, 50 ms. F, Attenuation of spiking in pAAV-Syn-NpHR2A-Venus-transfected cells. Spikes evoked by current steps of increasing size in absence and presence of yellow light. The average spike ratio (number of spikes with/without light) was reduced for several step sizes in transfected (yellow symbols; n = 12) versus control cells (black symbols; n = 12, p < 0.02). G, Yellow light-evoked NpHR currents were 55 ± 7 pA (n = 12) versus 4 ± 1 pA (n = 12) in transfected versus control cells (p < 0.01). H, Light responses in a pyramidal neuron expressing ChR2A-NpHR2A-Venus. Blue light (2 ms) elicited a spike, and yellow light (50 ms) elicited a hyperpolarization. Calibration: Top, 20 mV, 100 ms; bottom, 2 mV, 100 ms. High fidelity spiking in transfected cells (blue symbols; n = 7), but not in control cells (black symbols; n = 9). I, In the same cells, NpHR activation attenuated neural firing evoked by short current steps. The average spike ratio was significantly reduced for the first two step sizes in transfected (yellow symbols; n = 8) versus control cells (black symbols; n = 6, p < 0.05). J, Blue light-evoked ChR-currents were −1803 ± 342 pA (n = 7) versus −10 ± 3 pA (n = 9) in transfected versus control cells (p < 0.01). Yellow light-evoked NpHR-currents were 111 ± 15 pA (n = 7) versus 4 ± 1 pA (n = 9) in transfected versus control cells (p < 0.01). K–M, Light responses of amygdala neurons in acute slices after rAAV infection in vivo. K, Blue light (2 ms) induced an inward current or spiking in a neuron infected with rAAV Syn-ChR2A-tDimer. Calibration: VC, 100 pA, 100 ms; CC, 20 mV, 100 ms. Spike probability of infected neurons at different stimulation frequencies (n = 18). L, Blue (2 ms) or yellow (50 ms) light evoked a spike or hyperpolarization, respectively, in a neuron infected with rAAV-Syn-ChR2A-NpHR2A-Venus. Calibration: Top, 20 mV, 100 ms; bottom, 0.5 mV, 100 ms. Blue light stimulation induced spiking with high fidelity (n = 8). Yellow light pulses effectively reduced spiking for the first two current steps compared with no light stimulation (n = 14, p < 0.01). M, Light-evoked currents in cells infected with either rAAV-Syn-ChR-2A-tDimer (double) or rAAV-Syn-ChR2A-NpHR2A-Venus (triple). Blue light-evoked currents were −365 ± 52 pA (double) and −243 ± 43 pA (triple). Yellow light-evoked currents were 11 ± 2 pA (triple).
In addition to our functional data suggesting efficient self-processing of the ChR and NpHR constructs, we sought to confirm this biochemically. We used plasmid transfection or virus infection to express rAAV-Syn-ChR2A-tDimer and rAAV-Syn-ChR2A-NpHR2A-Venus in primary hippocampal neuron cultures. Western blot analysis of these constructs revealed undetectable levels of the full-length proteins (89.9 kDa and 98.8 kDa, respectively) and released tDimer or Venus (supplemental Fig. 2B,C, available at www.jneurosci.org as supplemental material). The Venus signal intensity from the rAAV triple construct was comparable to that from rAAV-Syn-iCre2A-Venus-infected neurons (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material), indicating that for the triple construct, FP expression levels were not severely compromised. Together, our data strongly argue that the 2A strategy can be used to quantitatively coexpress up to two functional membrane proteins together with fluorescent markers in slice cultures and in vivo for labeling as well as faithful bidirectional optogenetic control of individual neurons.
Discussion
Considering the difficulties to achieve reliable coexpression of heterologous proteins using current approaches, such as IRES (Douin et al., 2004) elements, bidirectional or dual promoters (Baron et al., 1995), cotransfection of several plasmids, or coinfection with multiple viruses (Mastakov et al., 2002), it is surprising that the use of 2A peptide bridges has not received more attention (Furler et al., 2001; de Felipe et al., 2006). One particular advantage of the 2A system is the use of only a single promoter that can be cell-type specific. A second one is its small size, and therefore the potential to design constructs with several 2A elements for expression of multiple proteins, even in vector systems with stringent size restrictions. And thirdly, in view of the well justified assumption that the cleaved proteins from a 2A fusion protein are translated and released in equimolar ratios (de Felipe et al., 2006), the 2A system would allow for quantification of the coexpressed proteins by measuring the level of the fluorescent reporter within and across experiments.
In the present study, we demonstrate that the 2A peptide approach can be used in neurons and in the brain in vivo for reliable and quantitative coexpression. When compared with heterologous cells, 2A cleavage was always more efficient in neurons and in the brain with undetectable levels of unprocessed 2A fusion proteins. Most importantly, all 2A fusion proteins retained their biological activities, without detectable consequences for neuronal viability and function.
In contrast to our findings, previous studies reported that reporters such as DsRed and the humanized Renilla GFP with the FMDV 2A peptide at their C terminus, were functionally inactive (Lengler et al., 2005; Hasegawa et al., 2007), or mislocalization of the second gene products occurred (de Felipe and Ryan, 2004). A careful design of the 2A fusion constructs, and perhaps use of the additional amino acid residues to retain a stable environment for self-processing at the cleavage sites may be a key feature that made our approach successful and broadly applicable. However, we cannot exclude that other proteins with critical C-terminal elements such as glutamate receptor subunits (Kessels and Malinow, 2009) or neomycin phosphotransferase II (Reiss et al., 1984) may not be able to function when expressed as 2A fusion proteins.
In all our experiments, the levels of FPs were high enough for both imaging in vivo and targeting of cells for electrophysiology, but a direct comparison with single expression of FP is difficult. In virus-mediated gene transfer, expression levels always depend on a number of factors, such as the gene itself, the viral titer, and the mode of application in vivo. We expressed fluorescent reporters as C-terminal partners in the 2A fusion proteins. As the translation efficiency of the 2A fusion proteins is determined by translation initiation at the start codon of the variable N-terminal encoded proteins, additional studies with N-terminal FPs would be needed to clarify whether the sensitivity of the FPs suffers in the 2A approach. However, for most applications a C-terminal located FP should be considered, since most likely the activity of the FPs reflects the expression efficiency of the N-terminal proteins as well. A limiting factor for interpreting quantitative, equimolar expression of the N- and C-terminal proteins in the 2A fusion constructs is the potential of different half lives of the released proteins. Stabilizing and nonstabilizing motifs in the individual proteins could lead to unbalanced protein turnover and thus different levels, even when they are expressed from a single ORF.
In our study, we emphasize that efficient coexpression of potent genetic regulators, such as iCre and tTA, in 2A fusions with different FPs permitted the reliable visualization of conditionally regulated neurons in fixed tissue and in vivo. Furthermore, we provide clear evidence that the 2A peptide bridge approach can be applied to secreted and membrane proteins, and up to three functionally intact proteins can be expressed linked by several 2A peptides. This allows, for instance, to reliably control the activity of the defined neurons in a bidirectional manner by quantitative coexpression of ChR and NpHR.
Overall, we conclude that the 2A peptide bridge is a very versatile tool for coexpression of multiple heterologous proteins in neurons in vitro and in vivo, which will greatly facilitate molecular and functional analysis in cellular and systems neuroscience.
Footnotes
Editor's Note: Toolboxes are intended to briefly highlight a new method or a resource of general use in neuroscience or to critically analyze existing approaches or methods. For more information, see http://www.jneurosci.org/misc/itoa.shtml.
This work was supported by the Volkswagen-Foundation (AZ:I/80-704), the Swiss National Science Foundation, the Austrian Science Fund, the German Research Foundation (SFB636/A4 and SP602/2-1 to R.S.), and the Novartis Research Foundation. Dox treatment and rAAV injection were performed under the license of Regierungspräsidium Karlsruhe 35-9815.82-A49/06 and 35-9815.81-A56/05. We thank Prof. Peter H. Seeburg for support and discussion. We thank Annette Herold, Sabine Grünwald, and Dr. Günter Giese for technical help. We also thank Daniela Gerosa-Erni and Dr. Thomas Oertner for providing hippocampal slice cultures, Prof. Wolfgang Hillen for providing tTA antibody, and Dr. Karl Deisseroth for providing Channelrhodopsin and Halorhodopsin plasmids.
References
- Baron et al., 1995.Baron U, Freundlieb S, Gossen M, Bujard H. Co-regulation of two gene activities by tetracycline via a bidirectional promoter. Nucleic Acids Res. 1995;23:3605–3606. doi: 10.1093/nar/23.17.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissière et al., 2003.Bissière S, Humeau Y, Lüthi A. Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat Neurosci. 2003;6:587–592. doi: 10.1038/nn1058. [DOI] [PubMed] [Google Scholar]
- Brewer et al., 1993.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Campbell et al., 2002.Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci U S A. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetin et al., 2006.Cetin A, Komai S, Eliava M, Seeburg PH, Osten P. Stereotaxic gene delivery in the rodent brain. Nat Protoc. 2006;1:3166–3173. doi: 10.1038/nprot.2006.450. [DOI] [PubMed] [Google Scholar]
- Dalby et al., 2004.Dalby B, Cates S, Harris A, Ohki EC, Tilkins ML, Price PJ, Ciccarone VC. Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods. 2004;33:95–103. doi: 10.1016/j.ymeth.2003.11.023. [DOI] [PubMed] [Google Scholar]
- de Felipe and Ryan, 2004.de Felipe P, Ryan MD. Targeting of proteins derived from self-processing polyproteins containing multiple signal sequences. Traffic. 2004;5:616–626. doi: 10.1111/j.1398-9219.2004.00205.x. [DOI] [PubMed] [Google Scholar]
- de Felipe et al., 2006.de Felipe P, Luke GA, Hughes LE, Gani D, Halpin C, Ryan MD. E unum pluribus: multiple proteins from a self-processing polyprotein. Trends Biotechnol. 2006;24:68–75. doi: 10.1016/j.tibtech.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Donnelly et al., 2001.Donnelly ML, Luke G, Mehrotra A, Li X, Hughes LE, Gani D, Ryan MD. Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage’ mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal ‘skip’. J Gen Virol. 2001;82:1013–1025. doi: 10.1099/0022-1317-82-5-1013. [DOI] [PubMed] [Google Scholar]
- Douin et al., 2004.Douin V, Bornes S, Creancier L, Rochaix P, Favre G, Prats AC, Couderc B. Use and comparison of different internal ribosomal entry sites (IRES) in tricistronic retroviral vectors. BMC Biotechnol. 2004;4:16. doi: 10.1186/1472-6750-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich and Malinow, 2004.Ehrlich I, Malinow R. Postsynaptic density controls AMPA-receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furler et al., 2001.Furler S, Paterna JC, Weibel M, Büeler H. Recombinant AAV vectors containing the foot and mouth disease virus 2A sequence confer efficient bicistronic gene expression in cultured cells and rat substantia nigra neurons. Gene Ther. 2001;8:864–873. doi: 10.1038/sj.gt.3301469. [DOI] [PubMed] [Google Scholar]
- Gradinaru et al., 2007.Gradinaru V, Thompson KR, Zhang F, Mogri M, Kay K, Schneider MB, Deisseroth K. Targeting and readout strategies for fast optical neural control in vitro and in vivo. J Neurosci. 2007;27:14231–14238. doi: 10.1523/JNEUROSCI.3578-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru et al., 2008.Gradinaru V, Thompson KR, Deisseroth K. eNpHR: a Natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biol. 2008;36:129–139. doi: 10.1007/s11068-008-9027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groppe et al., 2002.Groppe J, Greenwald J, Wiater E, Rodriguez-Leon J, Economides AN, Kwiatkowski W, Affolter M, Vale WW, Belmonte JC, Choe S. Structureal basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature. 2002;420:636–642. doi: 10.1038/nature01245. [DOI] [PubMed] [Google Scholar]
- Hasan et al., 2004.Hasan MT, Friedrich RW, Euler T, Larkum ME, Giese G, Both M, Duebel J, Waters J, Bujard H, Griesbeck O, Tsien RY, Nagai T, Miyawaki A, Denk W. Functional fluorescent Ca2+ indicator proteins in transgenic mice under TET control. PloS Biol. 2004;2:e163. doi: 10.1371/journal.pbio.0020163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa et al., 2007.Hasegawa K, Cowan AB, Nakatsuji N, Suemori H. Efficient multicistronic expression of a transgene in human embryonic stem cells. Stem Cells. 2007;25:1707–1712. doi: 10.1634/stemcells.2006-0813. [DOI] [PubMed] [Google Scholar]
- Hennecke et al., 2001.Hennecke M, Kwissa M, Metzger K, Oumard A, Kröger A, Schirmbeck R, Reimann J, Hauser H. Composition and arrangement of genes define the strength of IRES-driven translation in bicistronic mRNAs. Nucleic Acids Res. 2001;29:3327–3334. doi: 10.1093/nar/29.16.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasawa et al., 2004.Karasawa S, Araki T, Nagai T, Mizuno H, Miyawaki A. Cyan-emitting and orange-emitting fluorescent proteins as a donor/acceptor pair for fluorescence resonance energy transfer. Biochem J. 2004;381:307–312. doi: 10.1042/BJ20040321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels and Malinow, 2009.Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krestel et al., 2004.Krestel HE, Shimshek DR, Jensen V, Nevian T, Kim J, Geng Y, Bast T, Depaulis A, Schonig K, Schwenk F, Bujard H, Hvalby Ø, Sprengel R, Seeburg PH. A genetic switch for epilepsy in adult mice. J Neurosci. 2004;24:10568–10578. doi: 10.1523/JNEUROSCI.4579-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengler et al., 2005.Lengler J, Holzmüller H, Salmons B, Günzburg WH, Renner M. FMDV-2A sequence and protein arrangement contribute to functionality of CYP2B1-reporter fusion protein. Anal Biochem. 2005;343:116–124. doi: 10.1016/j.ab.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Mastakov et al., 2002.Mastakov MY, Baer K, Symes CW, Leichtlein CB, Kotin RM, During MJ. Immunological aspects of recombinant adeno-associated virus delivery to the mammalian brain. J Virol. 2002;76:8446–8454. doi: 10.1128/JVI.76.16.8446-8454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai et al., 2002.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Reiss et al., 1984.Reiss B, Sprengel R, Schaller H. Protein fusion with the kanamycin resistance gene from transposon Tn5. EMBO J. 1984;3:3317–3322. doi: 10.1002/j.1460-2075.1984.tb02297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan et al., 1991.Ryan MD, King AM, Thomas GP. Cleavage of foot-and-mouth disease virus polyprotein is mediated by residues located within a 19 amino acid sequence. J Gen Virol. 1991;72:2727–2732. doi: 10.1099/0022-1317-72-11-2727. [DOI] [PubMed] [Google Scholar]
- Shevtsova et al., 2005.Shevtsova Z, Malik JM, Michel U, Bähr M, Kügler S. Promoters and serotypes: targeting of adeno-associated virus vectors for gene transfer in the rat central nervous system in vitro and in vivo. Exp Physiol. 2005;90:53–59. doi: 10.1113/expphysiol.2004.028159. [DOI] [PubMed] [Google Scholar]
- Shimshek et al., 2002.Shimshek DR, Kim J, Hübner MR, Spergel DJ, Buchholz F, Casanova E, Stewart AF, Seeburg PH, Sprengel R. Codon-improved Cre recombinase (iCre) expression in the mouse. Genesis. 2002;32:19–26. doi: 10.1002/gene.10023. [DOI] [PubMed] [Google Scholar]
- Soriano, 1999.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Stoppini et al., 1991.Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Zhu et al., 2007.Zhu P, Aller MI, Baron U, Cambridge S, Bausen M, Herb J, Sawinski J, Cetin A, Osten P, Nelson ML, Kügler S, Seeburg PH, Sprengel R, Hasan MT. Silencing and un-silencing of tetracycline-controlled genes in neurons. PLoS ONE. 2007;2:e533. doi: 10.1371/journal.pone.0000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotukhin et al., 1999.Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]