

Abstract
In vivo experience induces changes in synaptic NMDA receptor (NMDAR) subunit components, which are correlated with subsequent modifications of synaptic plasticity. However, little is known about how these subunit changes regulate the induction threshold of subsequent plasticity. At hippocampal Schaffer collateral–CA1 synapses, we first examined whether a recent history of neuronal activity could affect subsequent synaptic plasticity through its actions on NMDAR subunit components. We found that prior activity history produced by priming stimulations (PSs) across a wide range of frequencies (1–100 Hz) could induce bidirectional changes in the NR2A/NR2B ratio, which governs the threshold for subsequent long-term potentiation/long-term depression (LTP/LTD). Manipulating the NR2A/NR2B ratio through partial NR2 subunit blockade mimicked the PS regulation of the LTP/LTD threshold. Our results demonstrate that activity-dependent changes in the NR2A/NR2B ratio can be critical factors in metaplastic regulation of the LTP/LTD threshold.
Introduction
The Bienenstock–Cooper–Munro (BCM) theory, proposed in the early 1980s, was considered as a complement to Hebb's theory by proposing that active synapses could grow stronger or weaker depending on whether their postsynaptic activity fell above or below a “modification threshold” called θm (Bienenstock et al., 1982; Deisseroth et al., 1995; Abraham and Bear, 1996; Abraham and Tate, 1997; Bear, 2003; Abraham, 2008), also called the long-term potentiation/long-term depression (LTP/LTD) crossover point (θLTP/LTD). The properties of synaptic plasticity could primarily be affected by the recent history of synaptic or cellular activity. This property could keep the network of modifiable synapses within a useful dynamic range and contribute to homeostasis (Bear, 2003; Abraham, 2008).
NMDA receptors (NMDARs) consist of the obligatory NR1 subunit in combination with NR2A–D and NR3A–B subunits (Sheng et al., 1994; Perez-Otano et al., 2001). NR2A and NR2B subunits have longer intracellular C-tails than the NR1 subunit, which endow them the capability to interact with neighboring postsynaptic density proteins (Osten et al., 2000). Therefore, NR2A and NR2B mainly determine the properties of the NMDAR. Many forms of synaptic plasticity depend on NMDAR activation and subsequent increase of intracellular Ca2+ (Morris et al., 1986; Cline et al., 1987; Kleinschmidt et al., 1987). Modulation of NMDAR activation and the biochemical downstream cascade after Ca2+ entry through the NMDAR are favorable targets for metaplasticity expression (Abraham and Tate, 1997).
A series of elegant investigations by Bear's laboratory in layer 3 of the visual cortex demonstrated that the “sliding threshold” is an attractive mechanism based on experience-dependent alterations in NMDARs (Kirkwood et al., 1996; Quinlan et al., 1999a,b; Philpot et al., 2001, 2003, 2007; Shouval et al., 2002; Bear, 2003; Iny et al., 2006). The coincident alterations in the NR2A/NR2B ratio and in the θLTP/LTD inspired the assumption that changes in NR2A/NR2B lead to sliding of θLTP/LTD (Bear, 1995; Quinlan et al., 1999a,b; Philpot et al., 2007; Yashiro and Philpot, 2008). They put forward a hypothesis that activity-dependent changes in NR2A/NR2B are involved in metaplastic regulation of the LTP/LTD threshold (Morishita et al., 2007; Yashiro and Philpot, 2008). Although a recent study using genetic ablation of NR2A suggests an obligatory role of NR2A for metaplasticity in the visual cortex, and thus adds new support to this hypothesis (Philpot et al., 2007), there is no direct examination whether and how activity-dependent changes in the NR2A/NR2B ratio take an active role in bidirectional metaplastic regulation of the LTP/LTD threshold. Resolving this question will be very helpful to understand in depth the role of NR2 subunits in the regulation of synaptic plasticity.
In the present study, we first studied the effects of priming stimulations (PSs) at different frequencies on the properties of NMDAR-mediated synaptic responses and θLTP/LTD in CA1 pyramidal neurons. We then demonstrated that the changes in NR2A/NR2B and θLTP/LTD are highly correlated. Finally, through manipulation of the NR2A/NR2B ratio using partial NR2A or NR2B blockade, we provided direct evidence for the active role of changes in NR2A/NR2B in bidirectional metaplastic regulation of θLTP/LTD.
Materials and Methods
Hippocampal slice preparation
Male Sprague Dawley rats, 2–3 weeks old, were anesthetized with ethyl ether and decapitated. The entire hippocampus was removed from the brain. Coronal brain slices (350 μm thickness) were cut using a vibrating blade microtome in ice-cold artificial CSF (ACSF) containing (in mm) 126 NaCl, 2.5 KCl, 1 MgCl2, 1 CaCl2, 1.25 KH2PO4, 26 NaHCO3, and 20 glucose. ACSF was bubbled continuously with carbogen (95% O2/5% CO2) to adjust the pH to 7.4. Fresh slices were incubated in a chamber with carbogenated ACSF and recovered at 34°C for at least 1.5 h, before they were transferred to a recording chamber.
Electrophysiological studies
PSs across a range of frequencies (1, 5, 10, 20, 50, and 100 Hz) were applied to Schaffer fibers through a constant current pulse delivered by a bipolar tungsten electrode. To avoid PSs that cause persistent changes in EPSPs, we used a dual-recording technique [one pipette for the field EPSP (fEPSP) recording and another one for the whole-cell EPSC recording (Malinow et al., 1989)] in the preliminary experiments and found that, at the intensity producing one-fifth maximal fEPSPs, PSs across different frequencies consistently failed to induce persistent changes under these two recording configurations. In our subsequent formal experiments, we used only a single electrode. Each time before we delivered PSs, we first recorded fEPSPs to set the intensity to the one-fifth maximal level. Immediately after we finished the PSs, we rapidly switched from the pipette containing saline solution (resistance, 2–5 MΩ) for recording fEPSPs to a pipette for whole-cell EPSC recordings and only started the 5 min baseline EPSC recording 20 min later. So LTP/LTD induction with the whole-cell patch configuration was conducted 25 min after the PSs were finished. The slices were perfused with bicuculline immediately after PSs were delivered; therefore, we can achieve an equilibrium concentration and make sure the baseline data are real EPSCs in before induction of plasticity. Conventional whole-cell recordings were made with patch pipettes containing (in mm) 17.5 CsCl, 2 MgCl2, 0.5 EGTA, 10 HEPES, 4 ATP, and 5 QX-314, with the pH adjusted to 7.2 by CsOH. Hippocampal slices were perfused with 34°C ACSF that was bubbled continuously with carbogen (95% O2/5% CO2) to adjust the pH to 7.4. Evoked synaptic responses were evoked at 0.05 Hz, except during the induction of LTP or LTD. LTP was induced with 200 synaptic stimuli at 2 Hz during a 2.5 min depolarization to 0 mV (Chen et al., 1999; Zhao et al., 2008). LTD was induced with 300 synaptic stimuli at 1 Hz during depolarization to −45 mV (Zhu et al., 2002; Liu et al., 2004; Toyoda et al., 2005). The threshold response was elicited with stimulation of 300 pulses of 1 Hz during depolarization to −30 mV (Artola et al., 1990; Toyoda et al., 2005), a protocol that we refer to as the “threshold protocol” here. This stimulating protocol might induce a response at the crossover point (θLTP/LTD), whereas the PSs allow it to now elicit either LTP or LTD. The current intensity of test stimuli (25–50 μA) was set to produce half-maximal EPSCs. EPSCs were recorded in ACSF perfusion medium containing bicuculline methiodide (BMI; 10 μm) to block GABAA receptor-mediated inhibitory synaptic currents. CA1 neurons were viewed under upright microscopy (ECLIPSE E600-FN, Nomasky; Nikon) and recorded with an Axopatch-200B amplifier (Molecular Devices). Changes in LTP/LTD production were examined during the last 5 min of recording. Data were low-pass filtered at 2 kHz and acquired at 5–10 kHz. The series resistance (R) was always monitored during recording for fear of resealing the ruptured membrane, which will cause changes in both the kinetics and amplitude of the EPSCs. Cells in which the R or capacitance deviated by >20% from initial values were excluded from the analysis. Also, cells with R > 20 MΩ at any time during the recording were excluded from the analysis. Data were collected with pClamp 9.2 software and analyzed using Clampfit 9.2 (Molecular Devices).
The NR2A/NR2B ratio was measured using a specific NR2B antagonist, ifenprodil (3 μm), to distinguish between the NR2A and NR2B components. The area under the evoked NMDAR-mediated EPSC curves before and after ifenprodil application (i.e., the ifenprodil-sensitive component) was considered as current mediated by the NR2B subunit. The remaining current component was considered mediated by the NR2A subunit. The data were averaged and analyzed using Minianalysis software (Synaptosoft).
Drugs.
Blockers, including AP-5, NBQX (1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide hydrate), and ifen-prodil were purchased from Sigma-Aldrich. BMI was purchased from Tocris. NR2A antagonist NVP-AAM077 was kindly provided by Novartis.
Data analysis.
Within-group comparisons were performed using two-tailed t tests, and differences between groups were compared using ANOVA post hoc comparisons. An ANOVA post hoc least significant difference (LSD) test was used when equal variances were assumed. Differences were considered significant when p < 0.05, and significance for the homogeneity of variance test was set at 0.1. Data are shown as the mean ± SEM.
Results
PSs at different frequencies exert differential influences on subsequent LTP/LTD induction
We first examined whether and how PSs at different frequencies could change the subsequent LTP induction. PSs across a range of frequencies (1, 5, 10, 20, 50, and 100 Hz) was delivered to the Schaffer's fibers of hippocampal slices. The pulse number for the stimulation at each frequency was fixed at 600 (Wagner et al., 1998; Wang and Wagner, 1999; Mockett et al., 2002). The intensity of the PSs was set to produce one-fifth maximal fEPSPs. PSs at this intensity failed to produce any persistent changes in either fEPSPs or EPSCs (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Immediately after we finished the PSs, we rapidly switched from the pipette containing saline solution (resistance, 2–5 M) for recording fEPSPs to the pipette for whole-cell EPSC recordings. It was reported that the effect of prior activity history on subsequent synaptic activity could last at least 30 min but decay in <1 h (Huang et al., 1992; Abraham and Bear, 1996). To avoid the decay of the effect exerted by prior activity history and make our results between different groups comparable, we started to collect baseline EPSCs only ∼20 min after the PSs were finished, using a whole-cell patch-clamp recording technique on hippocampal CA1 neurons. Therefore, recording of whole-cell EPSCs only started 20 min later. After 5 min of stable baseline recording of evoked EPSCs with a perfusate containing GABAA receptor antagonist BMI (10 μm), LTP was induced in CA1 pyramidal neurons with 200 synaptic stimuli at 2 Hz during a 2.5 min depolarization to 0 mV (Chen et al., 1999; Zhao et al., 2008). This pairing protocol elicited a marked potentiation of EPSC amplitude (150.5 ± 3.7% of baseline; n = 6; p < 0.001) (Fig. 1A). Interestingly, we found that low-frequency PSs (LPS; 1 or 5 Hz) significantly elevated the magnitude of subsequent potentiation (1 Hz: 201.3 ± 11.3% of baseline compared with control LTP, p < 0.001, n = 7; 5 Hz: 175.8 ± 5.0% of baseline compared with control LTP, p < 0.01, n = 7; ANOVA post hoc LSD test) (Fig. 1A,B,G). In contrast, high-frequency PSs (HPS; 50 or 100 Hz) significantly decreased the magnitude of subsequent potentiation (50 Hz: 106.4 ± 3.3% compared with control LTP, p < 0.001, n = 8; 100 Hz: 109.5 ± 4.1% compared with control LTP, p < 0.001, n = 7) (Fig. 1E–G). PSs at 10 and 20 Hz did not lead to marked changes in the potentiation level (10 Hz: 154.7 ± 5.9% compared with control LTP, p > 0.05, n = 7; 20 Hz: 146.2 ± 5.6% of baseline compared with control LTP, p > 0.05, n = 7) (Fig. 1C,D,G). These data suggest that PSs at different frequencies exert differential effects on subsequent LTP induction.
Figure 1.
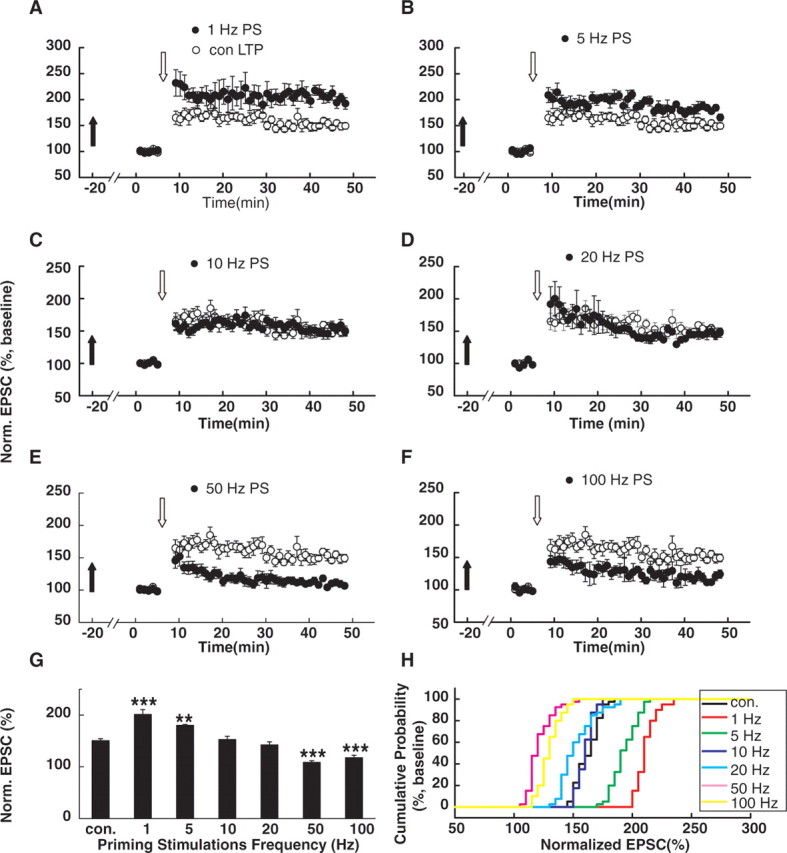
PSs at different frequencies exert differential influences on subsequent LTP induction. A, B, Summary graphs comparing LTP in CA1 cells delivered with 600 pulses of 1 Hz (A; n = 7) or 5 Hz (B; n = 7) LPS with LTP in interleaved control cells without PSs. PSs were applied ∼20 min before the baseline recording. LTP was induced in CA1 neurons with 200 synaptic stimuli on the Schaffer fibers at 2 Hz during a 2.5 min depolarization to 0 mV in the presence of the GABAA antagonist BMI (10 μm). Obviously, 1 and 5 Hz LPSs facilitated subsequent LTP induction by increasing the potentiation magnitude. Filled arrows refer to the time points that the PSs were delivered. Open arrows refer to the time points when the LTP-producing pairing protocols were applied. C, D, Summary graphs comparing LTP in cells primed with 600 pulses of 10 Hz (C; n = 7) or 20 Hz (D; n = 7) PSs with LTP in interleaved control cells without PSs. The 10 and 20 Hz PSs failed to exert any significant effect on subsequent LTP induction. E, F, Summary graphs comparing LTP in CA1 cells primed with 600 pulses of 50 Hz (E; n = 8) or 100 Hz (F; n = 7) HPS with LTP in interleaved control cells without PSs. The 50 and 100 Hz HPS suppressed subsequent LTP induction by decreasing the potentiation magnitude. G, Summary of data comparing LTP in cells primed with stimulations at different frequencies. **p < 0.01; ***p < 0.001; ANOVA post hoc LSD test versus control. H, Cumulative distributions of normalized EPSCs in cells primed with different stimulations and in interleaved control cells without PSs. Norm., Normalized; con., control.
We next examined the effects of PSs at different frequencies on the subsequent LTD induction. LTD was induced in CA1 pyramidal neurons with 300 synaptic stimuli at 1 Hz during depolarization to −45 mV (Zhu et al., 2002; Liu et al., 2004; Toyoda et al., 2005). This pairing protocol elicited a marked depression of the EPSC amplitude (65.0 ± 1.9% of baseline; p < 0.001; n = 7) (Fig. 2A). We found that LPS (1 or 5 Hz) significantly suppressed subsequent LTD by decreasing the magnitude of depression (1 Hz: 85.7 ± 1.7% of baseline compared with control LTD, p < 0.001, n = 7; ANOVA post hoc LSD test; 5 Hz: 79.6 ± 2.8% of baseline, compared with control LTD, p < 0.001, n = 8) (Fig. 2A,B,G). In contrast, HPSs (50 or 100 Hz) significantly facilitated subsequent LTD by increasing the magnitude of depression (50 Hz: 49.1 ± 1.8% of baseline compared with control LTD, p < 0.001, n = 7; 100 Hz: 45.4 ± 3.2% of baseline, p < 0.001, n = 8) (Fig. 2E–G). PSs at 10 and 20 Hz did not cause significant changes in depression level (10 Hz: 67.5 ± 1.9% of baseline compared with control LTD, p > 0.05, n = 7; 20 Hz: 66.9 ± 2.4% of baseline compared with control LTD, p > 0.05, n = 7) (Fig. 2C,D,G). These data suggest that PSs at different frequencies exert differential effects on subsequent LTD induction.
Figure 2.

PSs with different frequencies exert differential influences on subsequent LTD induction. A, B, Summary graphs comparing LTD in CA1 cells primed with 600 pulses of 1 Hz (A; n = 7) or 5 Hz (B; n = 8) LPS with LTD in interleaved control cells without PSs. PSs were applied ∼20 min before the baseline recording. LTD was induced in CA1 neurons with 300 synaptic stimuli of the Schaffer fibers at 1 Hz during depolarization to −45 mV in the presence of the GABAA antagonist BMI (10 μm). Obviously, 1 or 5 Hz LPS suppressed subsequent LTD induction by decreasing the depression magnitude. Filled arrows refer to the time points when the PSs were delivered. Open arrows refer to the time points when the LTD-producing pairing protocols were applied. C, D, Summary graphs comparing LTD in cells primed with 600 pulses of 10 Hz (C; n = 7) and 20 Hz (D; n = 7) PSs with LTD in interleaved control cells without PSs. The 10 and 20 Hz PSs failed to exert any significant effect on the subsequent LTD induction. E, F, Summary graphs comparing LTP in CA1 cells primed with 600 pulses of 50 Hz (E; n = 7) or 100 Hz (F; n = 8) HPS with LTD in interleaved control cells without PSs. The 50 and 100 Hz HPS facilitated subsequent LTD induction by increasing the depression magnitude. G, Summary of data comparing LTD in cells primed with PSs at different frequencies. ***p < 0.001, ANOVA post hoc LSD test versus control. H, Cumulative distributions of normalized EPSCs in cells priming with different stimulations and in interleaved control cells without PSs. Norm., Normalized; con., control.
PSs at different frequencies induce sliding of θLTP/LTD
Previous studies have suggested that the mechanisms for regulating the threshold for inducing synaptic plasticity and the magnitude of the expressed plasticity could be different (Lu et al., 2001; Philpot et al., 2007); thus, we also investigated whether the above observed changes in LTP/LTD magnitude after PSs could be attributable to a shifting θLTP/LTD, defined as the LTP/LTD crossover point. A stimulating protocol that is above the LTD induction threshold but below the LTP induction threshold and induces neither LTP nor LTD might induce a response at the crossover point, whereas PSs allow this protocol to now elicit either LTP or LTD. Previous studies have examined the persistent changes in synaptic responses using various pairing protocols with different depolarization levels. Those studies showed that one protocol (stimulation with 300 pulses at 1 Hz depolarization to −30 mV) that was at a depolarization level set between inducing LTP and LTD was very reliable and consistently failed to elicit a threshold response (Artola et al., 1990; Toyoda et al., 2005). Indeed, we made similar observations that no significant persistent changes in EPSC amplitude were detected with this protocol (99.6 ± 1.7% of baseline; n = 6; p > 0.05). Therefore, we called this induction protocol the threshold protocol.
Then, we investigated whether PSs at different frequencies could affect subsequent threshold responses induced by this threshold protocol. Interestingly, after 600-pulse LPS (1 and 5 Hz), LTP was produced with the threshold protocol (1 Hz: 128.9 ± 2.3% of baseline compared with control, p < 0.05, n = 6; 5 Hz: 124.3 ± 6.0% of baseline, p < 0.01, n = 6; ANOVA post hoc LSD test) (Fig. 3A,B,E,F). In contrast, HPS at 50 or 100 Hz led to the production of LTD (50 Hz: 70.9 ± 6.3% of baseline compared with control, p < 0.05, n = 7; 100 Hz: 65.7 ± 7.9% of baseline, p < 0.05, n = 6) (Fig. 3C,D,G,H). These data suggest bidirectional regulation of θLTP/LTD by prior activity history. LPS led to left-shifting of θLTP/LTD and facilitation of subsequent LTP induction, whereas HPS led to right-shifting of θLTP/LTD and facilitation of subsequent LTD production.
Figure 3.
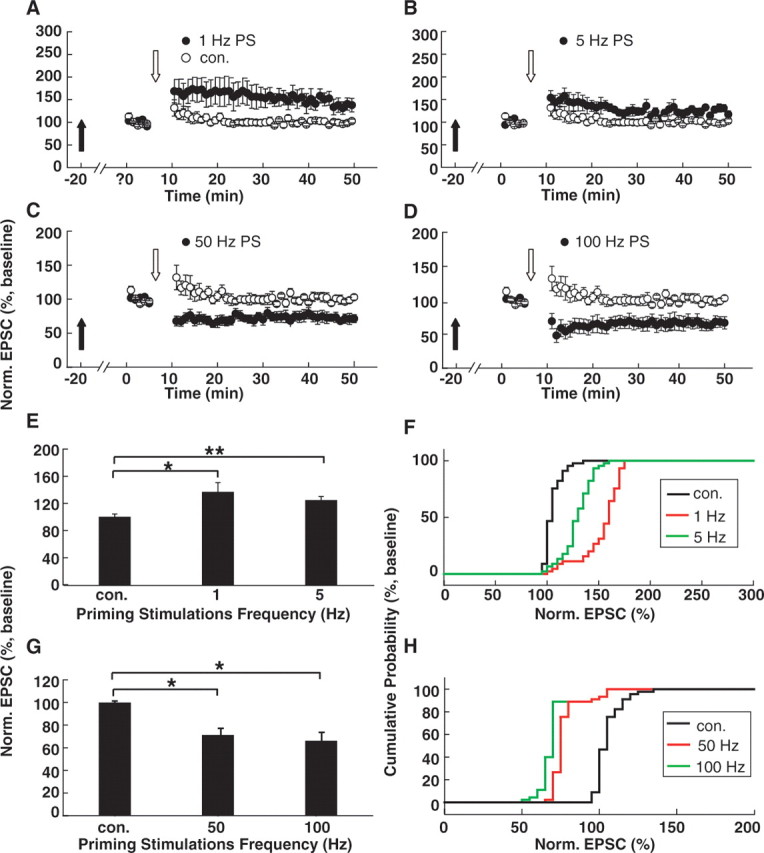
PS-induced shifting of the θLTP/LTD. A, B, LPS led to a leftward shift in the θLTP/LTD. Simulation with 300 pulses at 1 Hz during depolarization to −30 mV, which we termed as the threshold protocol, failed to induce any persistent changes in the evoked EPSCs without PSs. However, after 1 Hz (A) or 5 Hz (B) LPS, the threshold protocol consistently produce reliable LTP, suggesting that the θLTP/LTD shifted to the left. C, D, HPS led to right-shifting of the θLTP/LTD. After 50 Hz (C) or 100 Hz (D) HPS, the threshold protocol consistently produced reliable LTD, suggesting that the θLTP/LTD shifted to the right. Filled arrows refer to the time points when PSs were delivered. Open arrows refer to the time points when the threshold pairing protocols were applied. E, Summary of data comparing normalized EPSCs in cells primed with 1 or 5 Hz LPS with that in interleaved control cells without PSs. *p < 0.05; **p < 0.01. F, Cumulative distributions of normalized EPSCs in cells primed with 1 or 5 Hz LPS and in interleaved control cells. G, Summary of data comparing normalized EPSCs in cells primed with 50 or 100 Hz HPS with that in interleaved control cells without PSs. *p < 0.001. H, Cumulative distributions of normalized EPSCs in cells primed with 50 or 100 Hz HPS and in interleaved control cells. Norm., Normalized; con., control.
PS-induced changes in LTP/LTD induction are correlated with changes in the NR2A/NR2B ratio
There is evidence for altered NMDAR function as a basis for metaplasticity in the visual system (Quinlan et al., 1999a,b; Philpot et al., 2003). We therefore also examined whether any NMDAR function changes were accompanied with above changes in LTP/LTD induction after PSs in CA1 pyramidal neurons. Because previous studies in the visual system demonstrated altered NR2 subunit composition or the NR2A/NR2B ratio after different visual experiences (light deprivation or light reexposure), we examined the NR2A/NR2B ratio at the same time points for detecting LTP/LTD induction after PSs. We used an NR2B-specific antagonist, ifenprodil (3 μm), to distinguish the NR2A and NR2B components. The area between the NMDAR-mediated EPSC current curves before and after ifenprodil application (i.e., the ifenprodil-sensitive component) was considered as the NR2B-mediated current. The area under the remaining current curve was considered to be mediated by the NR2A subunit (Fig. 4A). We found that 600-pulse LPS (1 and 5 Hz) markedly decreased the NR2A/NR2B ratio (1 Hz: 1.8 ± 0.7 compared with control, 2.0 ± 0.1; p < 0.05, ANOVA post hoc LSD test; n = 6; 5 Hz: 1.6 ± 0.1, compared with control, p < 0.001, n = 6). Conversely, 600-pulse HPS (50 and 100 Hz) markedly increased the NR2A/NR2B ratio (50 Hz: 2.5 ± 0.04 compared with control, p < 0.001, n = 6; 100 Hz: 2.6 ± 0.1, p < 0.001, n = 6). PSs at either 10 or 20 Hz failed to exert any influence on subsequent NR2A/NR2B ratios (10 Hz: 2.1 ± 0.1, p > 0.05, n = 6; 20 Hz: 2.1 ± 0.1, p > 0.05, n = 6) (Fig. 4A). Our results suggest that at the same time points at which we observed changes in LTP/LTD threshold, PSs at different frequencies exerted differential influence on subsequent NMDAR subunit components. We also examined whether the amplitude of NMDAR EPSCs changed along with the above changes in NR2A/NR2B ratios. We found that PSs at both 1 and 100 Hz failed to induce any change in the NMDAR EPSC (before 1 Hz, 74.9 ± 5.6 pA; after 1 Hz, 84.5 ± 8.5 pA; p > 0.05; before 100 Hz, 72.1 ± 16.1 pA; after 100 Hz, 74.8 ± 16.4 pA; p > 0.05) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 4.

PS-induced changes in LTP/LTD induction are correlated with changes in the NR2A/NR2B ratio. A, Summary of data comparing the NR2A/NR2B ratio in cells primed with stimulations with that in interleaved control (con.) cells. The 1 and 5 Hz LPS led to a decreased NR2A/NR2B ratio. Conversely, 50 and 100 Hz HPS led to an increased NR2A/NR2B ratio. Stimulations at 10 and 20 Hz failed to induce marked alterations in the NR2A/NR2B ratio. *p < 0.05; ***p < 0.001; ANOVA and post hoc LSD test. Representative traces at the top show measurements of the NR2A/NR2B ratios using the NR2B-specific antagonist ifenprodil (ifen.; 3 μm). This measurement of the NR2A/NR2B ratio was conducted at the same time point that subsequent LTP/LTD was induced with pairing protocols. The NR2B-specific antagonist ifenprodil (3 μm) abolished the EPSC components mediated by the NR2B subunit, which was measured as the difference of area under the EPSC curves before and after ifenprodil application (left). The effect of ifenprodil on NR2B was confirmed by the shortened current decay time after ifenprodil treatment (right). B, The mean (±SEM) effects of PSs of 600 pulses at various frequencies delivered to the Schaffer fibers on the normalized (Norm.) NR2A/NR2B ratio measured in CA1 neurons at the same time point that subsequent LTP/LTD was induced with pairing protocols after PSs. C, Plot of averaged data showing LTP magnitude as a function of the NR2A/NR2B ratio. The potentiation magnitude after LTP production and the level of NR2A/NR2B ratios set by PSs were negatively correlated, which fit with the equation Y = −77.0X + 206.4 (r = −0.93; p < 0.01). D, Plot of averaged data showing the LTD magnitude as a function of the NR2A/NR2B ratio. LTD production and the level of the NR2A/NR2B ratio were positively correlated, which fit with the equation Y = 38.0X − 43.3 (r = 0.97; p < 0.001).
We examined whether the PS-induced changes in LTP/LTD and NR2A/NR2B ratios were correlated. Indeed, we found that changes in the magnitude of potentiation and in the level of the NR2A/NR2B ratio set by PSs were negatively correlated. The correlation could be explicitly interpreted by the equation Y = −77.0X + 206.4 (r = −0.93; p < 0.01) (Fig. 4C). Conversely, the changes in the magnitude of depression and in the level of NR2A/NR2B ratios set by PSs were positively correlated and well interpreted by the equation Y = 38.0X − 43.3 (r = 0.97; p < 0.001) (Fig. 4D). These data analyses suggest that changes in the NR2A/NR2B ratio and LTP/LTD threshold are highly correlated. One possibility is that PSs first lead to changes in the NR2A/NR2B ratio, which in turn causes a shift in the LTP/LTD threshold.
We also examined whether the metaplasticity caused by PS required NMDAR activity. After simultaneous application of AP-5 (50 μm) with PSs, the subsequent NR2A/NR2B ratio showed no marked difference compared with the no-priming control (1 Hz PSs and AP-5: NR2A/NR2B = 2.1 ± 0.1, n = 7; compared with control, NR2A/NR2B = 2.0 ± 0.1, n = 7, p > 0.05, ANOVA post hoc LSD test; 100 Hz PSs and AP-5: 2.1 ± 0.03, n = 6; compared with control, p > 0.05, n = 6). Accordingly, the threshold protocol failed to produce LTP or LTD (1 Hz PSs and AP-5: 92.9 ± 1.7% of baseline, n = 7; compared with no-priming control, p > 0.05; 100 Hz PSs and AP-5, 95.0 ± 7.5% of baseline, n = 6; compared with control, p > 0.05) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These results demonstrated that PS-induced metaplasticity is dependent on NMDAR activity.
Sequential PSs bidirectionally regulate the NR2A/NR2B ratio and LTP/LTD threshold
Practically speaking, the number of prior activity history events could be more than just one, as we observed here. It was unknown how multiple PSs with distinct stimulation parameters delivered at different time points regulate subsequent LTP or LTD production. To address this question, we interleaved 100 Hz HPS between the former 1 Hz LPS and the subsequent threshold protocol, or interleaved 1 Hz LPS between the former 100 Hz HPS and the threshold protocol. The pulse number was fixed at 600. We found that the interleaved 100 Hz HPS could totally reverse the effect on both the NR2A/NR2B ratio and LTP/LTD threshold exerted by the former 1 Hz LPS (former 1 Hz plus subsequent 100 Hz PSs, 101.5 ± 2.1% of baseline, n = 6; compared with 1 Hz, 134.3 ± 5.7% of baseline, n = 7; p < 0.05, ANOVA post hoc LSD test; former 1 Hz plus subsequent 100 Hz PSs, NR2A/NR2B = 2.1 ± 0.1, n = 8; compared with 1 Hz, NR2A/NR2B = 1.7 ± 0.1, n = 7; p < 0.05, ANOVA post hoc LSD test) (Fig. 5A,C,E), as did the 1 Hz LPS on the effect of the former 100 Hz HPS (former 100 Hz plus subsequent 1 Hz PSs, 94.3 ± 9.8% of baseline, n = 7; compared with 100 Hz, 64.7 ± 7.4% of baseline, n = 6; p < 0.05, ANOVA post hoc LSD test; former 100 Hz plus subsequent 1 Hz PSs, NR2A/NR2B = 2.0 ± 0.2, n = 7; compared with 100 Hz, NR2A/NR2B = 2.6 ± 0.1, n = 7; p < 0.01, ANOVA post hoc LSD test) (Fig. 5B,D,F). Therefore, it is likely that the subsequent LPS counteracts the effect of the former HPS, and vice versa.
Figure 5.
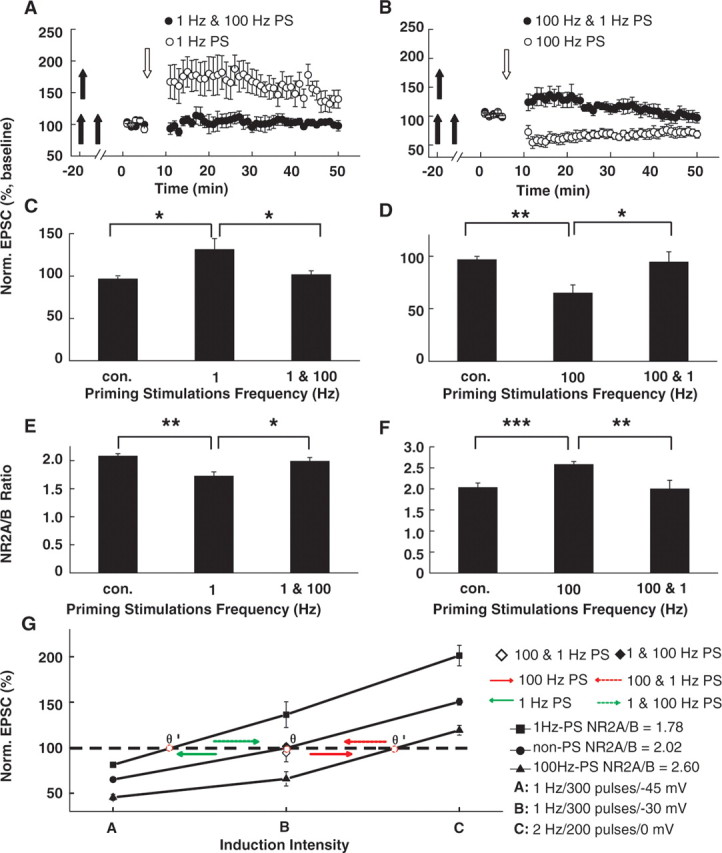
Sequential PSs bidirectionally regulate the NR2A/NR2B ratio and LTP/LTD threshold. A, The effect of 1 Hz LPS on subsequent LTP induction was reversed by the interleaved 100 Hz HPS. Treatment with 1 Hz LPS alone led to the emergence of LTP produced with the threshold protocol. However, the threshold protocol failed to produce LTP after 100 Hz intervened between 1 Hz LPS and threshold protocol stimulations. Double filled arrows refer to sequential PSs at different frequencies delivered at different time points. Open arrows refer to the time points when the threshold pairing protocols were applied. B, The effect of 100 Hz HPS on subsequent LTP induction was reversed by the interleaved 1 Hz LPS. The 100 Hz HPS alone led to the emergence of LTD produced with the threshold protocol. However, the threshold protocol failed to produce LTD after 1 Hz intervened between the first priming and threshold protocol stimulations. C, D, Statistical plot of averaged data showing normalized EPSCs examined after sequential PSs as a function of PS frequencies. The interleaved 1 Hz PSs reversed the effects of the former 100 Hz, and vice versa. *p < 0.05; **p < 0.01; ANOVA post hoc LSD test versus control. E, F, Statistical plot of averaged data showing the normalized NR2A/NR2B ratio as a function of PS frequencies. *p < 0.05; **p < 0.01; ***p < 0.001; ANOVA post hoc LSD test versus control. G, Summary plot showing bidirectional shifting of the θLTP/LTD after sequential PSs at different frequencies. Without PSs, the NR2A/NR2B ratio at the time points of LTP/LTD induction was 2.02. LTD and LTP induction protocols (stimuli A and C) could produce persistent depression and potentiation, respectively. Stimuli B, which was also referred to as the threshold protocol and was used to induce the θLTP/LTD response, failed to induce any persistent change. The 1 Hz priming LPS decreased the NR2A/NR2B ratio to 1.78. Accordingly, the threshold protocol produced LTP, indicating that the θLTP/LTD shifted to the left. Conversely, 100 Hz HPS increased the NR2A/NR2B ratio to 2.60. As a result, the θLTP/LTD shifted to the right. Sequential PSs of 1 and 100 Hz led the θLTP/LTD to slide back to the no-priming control position, as did sequential PSs of 100 and 1 Hz. The red spot refers to the position of the θLTP/LTD. Norm., Normalized; con., control.
We analyzed the normalized EPSC as to the function of induction intensity at different levels of NR2A/NR2B ratios (Fig. 5G). Without PSs, the NR2A/NR2B ratio at the time point of LTP/LTD induction was 2.0. LTD and LTP induction protocols (stimuli A and C) could produce persistent depression and potentiation, respectively, as we show in the above data (Figs. 1, 2). Stimulus B, which was termed as the threshold protocol and used to induce θLTP/LTD response, could elicit neither LTP nor LTD. A 1 Hz PS decreased the NR2A/NR2B ratio to 1.8. Accordingly, the same LTD induction protocol (stimuli A) produced an LTD with decreased magnitude, whereas the LTP induction protocol (stimuli C) produced LTP with an enhanced potentiation level. Conversely, 100 Hz led to an increase in the NR2A/NR2B ratio to 2.6. The LTD induction protocol (stimuli A) produced LTD with increased magnitude, whereas the LTP induction protocol (stimuli C) produced LTP with a decreased potentiation level. Furthermore, interleaved PSs could reverse the effect on the NR2A/NR2B ratio and LTP/LTD induction exerted by the former PSs at different frequencies, strongly suggesting that distinct PSs delivered consecutively could regulate subsequent NR2A/NR2B ratios and LTP/LTD thresholds bidirectionally.
Changes in the NR2A/NR2B ratio led to LTP/LTD threshold shifting
Although our above data suggest that the changes in the NR2A/NR2B ratio and LTP/LTD threshold are highly correlated, we still do not know whether changes in the NR2A/NR2B ratio caused the LTP/LTD threshold shift. To examine this possibility, we used a low concentration of an NR2B-specific antagonist, ifenprodil, to partially block the NR2B-mediated function and thus increase the NR2A/NR2B ratio. Then we observed how the changes in the NR2A/NR2B ratio through this pharmacological manipulation could affect subsequent θLTP/LTD. It is possible that a partial NR2B block could decrease total NMDAR function, which might in turn lead to a change in θLTP/LTD. To rule out this possibility, we first investigated the dose–response relationship of a competitive NMDAR antagonist, AP-5, and chose a concentration (1 μm) that did not display any marked effect on the NR2A/NR2B ratio or synaptic plasticity. Then we adjusted the concentration of ifenprodil to a level that matched the NMDAR inhibition by 1 μm AP-5 (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). We determined that AP-5 at the 1 μm concentration failed to exert any effect on synaptic plasticity induced by the threshold protocol (87.5 ± 8.1% of baseline compared with control, 94.5 ± 4.4%; p > 0.05) (Fig. 6A). Ifenprodil at 0.6 μm could inhibit NMDAR-mediated EPSCs to the same level that 1 μm AP-5 did (72.2 ± 5.2% of control compared with 71.9 ± 5.9% of control; p > 0.05; n = 5) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). However, 0.6 μm ifenprodil applied before and during LTP induction markedly increased the NR2A/NR2B ratio (2.6 ± 0.1 compared with control, 2.0 ± 0.1; p < 0.001, ANOVA post hoc LSD test; n = 6) (Fig. 6F). In sister slices obtained from the same hippocampus with the same concentration, ifenprodil, the threshold protocol, produced LTD (70.6 ± 9.3% of baseline compared with control, 94.5 ± 4.4%; p < 0.05, ANOVA post hoc LSD test; n = 6) (Fig. 6B). Thus, partial NR2B blockade mimicked the effect of 100 Hz PSs.
Figure 6.

Artificially manipulating the NR2A/NR2B ratio with an NR2 subunit antagonist led to θLTP/LTD shifting, suggesting that changes in the NR2A/NR2B ratio govern θLTP/LTD regulation. A, The competitive NMDAR antagonist AP-5 at 1 μm concentration did not display any marked effect on synaptic plasticity induced by the threshold protocol. AP-5 was applied before and during threshold protocol stimulation. B, Partial NR2B inhibition by the specific NR2B antagonist ifenprodil at a concentration of 0.6 μm, which inhibited NMDAR-mediated EPSCs to the same level as 1 μm AP-5 did, led to the emergence of LTD induced by the threshold protocol. C, The 1 μm AP-5 application seems to partially inhibit LTP amplitude, but this effect was not significant (1 Hz and AP-5, 117.3 ± 3.9%, n = 8, vs 1 Hz, 125.7 ± 2.8%, n = 6; p > 0.05, ANOVA post hoc LSD test). D, Comparison of EPSCs after 1 Hz LPS alone with EPSCs after 1 Hz LPS combined with partial NR2B blockade. The 1 Hz LPS led to the emergence of LTP produced with the threshold protocol. In contrast, after both 1 Hz LPS and subsequent 0.6 μm ifenprodil treatment, the threshold protocol failed to induce any persistent change. These data suggest that changes in NR2B contribute to subsequent metaplasticity. E, Summary graphs showing normalized EPSCs induced by the threshold protocol under various treatments including 1 Hz PS (1 Hz), 1 Hz PSs plus ifenprodil treatment (1 Hz & ifen.), 1 Hz PSs plus AP-5 treatment (1 Hz & AP-5), AP-5 treatment (AP-5), ifenprodil treatment, and no-priming control. F, Summary graphs showing changes in NR2A/NR2B ratios under various treatments. *p < 0.05; **p < 0.01; ***p < 0.001; ANOVA post hoc LSD test versus control. Norm., Normalized; con., control; ifen., ifenprodil.
These data suggest that pharmacologically resetting the NR2A/NR2B ratio could lead to a reset of the subsequent LTP/LTD threshold. Next, we asked whether and how this reset of the NR2A/NR2B ratio by pharmacological manipulation could affect changes in subsequent LTP/LTD induction caused by PSs. After 1 Hz LPS priming, we applied 0.6 μm ifenprodil immediately before and during threshold protocol stimulation. This treatment elevated the NR2A/NR2B ratio, which was already decreased by 1 Hz LPS, to the level of the no-priming control (Fig. 6F). As a result, induction with the threshold protocol failed to elicit any persistent change, similar to the control without PSs (Fig. 6D,E). Application of 1 μm AP-5 seemed to partially inhibit the LTP amplitude, but this effect was not significant (1 Hz and AP-5, 117.3 ± 3.9%, n = 8, vs 1 Hz, 125.7 ± 2.8%, n = 6; p > 0.05) (Fig. 6C,E). This observation was at odds with several previous studies, which demonstrated that low concentration of AP-5 tends to either partially or totally block LTP production (Cummings et al., 1996; Nishiyama et al., 2000; Liu et al., 2004). This discrepancy could be caused by the different protocols used to produce LTP in these experiments. The threshold pairing protocol (stimulation with 300 pulses at 1 Hz during depolarization to −30 mV) used in the present study could induce neither LTP nor LTD but was now able to facilitate the production of plasticity by PSs (see above data).
To further examine how changes in the NR2A/NR2B ratio caused by changes in NR2B could regulate LTP/LTD thresholds, we investigated the effect of low-concentration ifenprodil (0.6 μm) on LTP/LTD induction with a conventional pairing protocol. Our results showed that ifenprodil inhibited the induction of LTP by the 2 Hz pairing protocol and enhanced the induction of LTD by pairing 1 Hz synaptic stimulation with depolarization of the postsynaptic cell to −45 mV. These data provide more convincing support to our above results that increases in the ratio of NR2A- to NR2B-containing NMDARs to the synaptic currents underlie the effects of HPS on LTD/LTP induction (Fig. 7).
Figure 7.

The effect of partial NR2B inhibition on LTP and LTD induction produced by the conventional pairing protocol. A, The 0.6 μm ifenprodil application, which significantly increased the NR2A/NR2B ratio (Fig. 6), primarily inhibited the LTP amplitude induced by the LTP-producing pairing protocol (200 synaptic stimuli at 2 Hz during a 2.5 min depolarization to 0 mV). B, In contrast, 0.6 μm ifenprodil application facilitated the LTD level induced by the LTD-producing pairing protocol (300 synaptic stimuli at 1 Hz during depolarization to −45 mV).
The above results could not rule out the possibility that only changes in NR2B contribute to changes in the NR2A/NR2B ratio that play a role in regulating the subsequent LTP/LTD threshold. To test this possibility, we also used a low concentration of NR2A antagonist NVP-AAM077 (10 nm) (Auberson et al., 2002) to partially block the NR2A component of NMDAR-mediated EPSC to the same level as 1 μm AP-5 did (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). It is true that NVP-AAM077 at saturated concentrations has only ∼10 times the selectivity for NR2A over NR2B and is thus considered as a nonspecific NR2A antagonist (Berberich et al., 2005; Neyton and Paoletti, 2006; Morishita et al., 2007). However, at a low concentration like 10 nm, NVP-AAM077 exerts almost no effect on NR2B and therefore has a specific effect on NR2A (Neyton et al., 2006). NVP-AAM077 (10 nm) partially blocked NR2A over NR2B and, as a result, decreased the NR2A/NR2B ratio, which was already increased by 100 Hz HPS (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Accordingly, the subsequent threshold protocol produced LTD with a decreased depression magnitude, suggesting a sliding back of the θLTP/LTD toward the no-priming state. These data confirm that changes in both NR2A and NR2B could contribute to observed changes in NR2A/NR2B, which in turn led to changes in the θLTP/LTD.
Discussion
Prior activity induces bidirectional switching of synaptic NMDAR subunit components and sliding of the LTP/LTD threshold
Recently, it was reported that the LTP-producing pairing protocol could induce shortening of NMDAR-mediated synaptic responses. This observation was demonstrated by the simultaneous increase in synaptic NR2A and decrease of NR2B components in postsynaptic sites, which was reversed by depotentiation of LTP. Therefore, the NMDAR component could be rapidly and bidirectionally regulated by different induction protocols (Bellone and Nicoll, 2007). In the present study, we found that PSs at different frequencies could also bidirectionally change NR2 components represented by the NR2A/NR2B ratio examined ∼20 min later, on which subsequent LTP/LTD induction was conducted. LPS of 600 pulses at 1 or 5 Hz decreased the subsequent NR2A/NR2B ratio. Conversely, 600-pulse 50 or 100 Hz HPS increased the resulting NR2A/NR2B ratio. Thus, it is likely that LPS (1 and 5 Hz) and HPS (50 and 100 Hz) could mimic the effects exerted by light deprivation and reexposure in the visual system, respectively (Quinlan et al., 1999a,b; Philpot et al., 2001; Smith et al., 2008). In addition, the changes caused by 1 Hz LPS in both the NR2A/NR2B ratio and θLTP/LTD could be reversed by subsequent 100 Hz HPS delivered later, and vice versa (Fig. 5). The opposing actions exerted by 100 and 1 Hz PSs were probably caused by distinct underlying molecular mechanisms. High-frequency stimulation at 100 Hz, which is usually used to induce LTP, could activate CaMKII (calcium/calmodulin-dependent protein kinase II), which is critical for LTP production (Malinow et al., 1988; Silva et al., 1992). In contrast, 1 Hz low-frequency stimulation was observed to erase LTP through an NMDAR-mediated activation of protein phosphatases (O'Dell and Kandel, 1994). Thus, sequential prior activities at different frequencies could bidirectionally regulate subsequent plasticity.
Metaplasticity occurs when PSs fail to induce persistent changes
Previous studies confirmed that metaplasticity occurs when PSs lead to persistent changes in the direction or degree of synaptic activity (Christie et al., 1995; Abraham and Bear, 1996; Abraham and Tate, 1997; Abraham, 2008). However, it is still unclear whether prior activity without producing persistent changes could exert influence on subsequent synaptic activity, although a few studies hint that it might occur (Mockett et al., 2002). In the present study, we address this question by setting the intensity to produce one-fifth maximal fEPSPs. PSs at this intensity failed to produce any persistent changes in either fEPSPs or EPSCs (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, prior activity through PSs could still induce reliable changes in subsequent LTP/LTD induction, which as we demonstrated here was caused by accompanying changes in the NR2A/NR2B ratio. These results demonstrated that metaplasticity could also occur when PSs fail to induce persistent changes in synaptic plasticity.
Altered NR2A/NR2B ratio as a mechanism for shifting of θLTP/LTD
Previous studies have proven that altered NMDAR functions are a basis for metaplasticity in the visual cortex (Quinlan et al., 1999a,b; Philpot et al., 2001, 2003). This altered NMDAR function could be caused by changes in visual experience. Light deprivation led to a decrease in the NR2A/NR2B ratio and a slower NMDAR current decay time in neurons in layer 2/3 of visual cortex. In contrast, increases in visual experience caused an increase in the NR2A/NR2B ratio and a faster NMDAR current decay time. Accompanying these changes in the NR2A/NR2B ratio are changes in LTP/LTD induction. These data are consistent with our present observations in hippocampal CA1 neurons, suggesting a strong correlation between changes in the NR2A/NR2B ratio and subsequent LTP/LTD induction. Although a recent investigation suggested that distinct mechanisms may underlie LTD in layer 2/3 visual cortex and CA1 region, both mechanisms could contribute to the changes in NR2A/NR2B (Crozier et al., 2007). Observations through extreme deletion of NR2A in NR2A knock-out animals also supported an obligatory role for NR2A in visual cortex metaplasticity (Philpot et al., 2007). However, it is still unclear whether changes in the NR2A/NR2B ratio or in NR2A or NR2B alone take an active role in inducing LTP/LTD production. In the present study, we manipulated the NR2A/NR2B ratio with low-concentration NR2A or NR2B antagonist that partially attenuated the NR2A or NR2B component. We demonstrated that changes in the NR2A/NR2B ratio are causative factors for subsequent metaplasticity. Priming-induced changes in NR2A/NR2B could be reset by this pharmacological method, and, as a result, the θLTP/LTD could also be reset. These results confirm that changes in either the NR2A or NR2B subtype could contribute to the observed changes in NR2A/NR2B, which governs the threshold for subsequent activity-dependent synaptic modifications.
Then, we asked whether there is an effective range for the NR2A/NR2B ratio to take an active role in metaplasticity. The observation that the absence of NR2A through NR2A knockout failed to enhance the magnitude of subsequent LTP suggests that the effect of changing the NR2A/NR2B ratio could be invalid when this ratio goes beyond a certain range. In our study, after 100 Hz PSs, the NR2A/NR2B ratio was reset from 2.0 ± 0.1 to 2.6 ± 0.1 (p < 0.001; n = 6). Further increasing the NR2A/NR2B ratio with partial NR2B blockade failed to facilitate subsequent LTD induction (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Consistently, decreasing the NR2A/NR2B ratio with partial NR2A blockade after 1 Hz PSs failed to further facilitate subsequent LTP induction. Along with some other recent findings (Lu et al., 2001; Philpot et al., 2007), these results highly support the notion that only when the activity-dependent changes in NR2A/NR2B ratio are within a certain range can they exert influence on subsequent plasticity. The precise mechanism by which the NR2A/NR2B ratio alters the plasticity threshold is currently unknown. It is possible that changes in the NR2A/NR2B ratio regulate Ca2+ influx through the NMDAR (Sobczyk et al., 2005; Sobczyk and Svoboda, 2007), or that the NR2A/NR2B ratio might regulate synaptic plasticity through changing the NR2A- and/or NR2B-associated downstream intracellular signaling molecules (Yashiro and Philpot, 2008).
Although our present findings provide direct evidence for the assumption that activity-dependent changes in the NR2A/NR2B ratio are involved in bidirectional metaplastic regulation of the LTP/LTD threshold (Bear, 2003; Iny et al., 2006; Philpot et al., 2007), we should keep in mind that there are probably many different mechanisms contributing to metaplasticity, as is also the case for synaptic plasticity [e.g., the observation that proportional but delayed NMDA potentiation after AMPA potentiation is the opposite of the prediction by the BCM theory, which predicts that LTP should reduce NMDA currents to raise the threshold for inducing subsequent LTP (Bienenstock et al., 1982; Watt et al., 2004)]. Therefore, although the BCM theory might have more generalized internal representations, the underlying mechanisms for θLTP/LTD sliding could vary on a case-by-case basis. In addition, the metaplasticity that we observed here is homosynaptic in nature. It still remains to be determined whether the activity at the CA1 neurons that we studied could also induce the same affect on subsequent plasticity at neighboring synapses, as is predicted by the BCM theory and supported by recent studies (Holland and Wagner, 1998; Wang and Wagner, 1999; Abraham et al., 2001; Roth-Alpermann et al., 2006).
Footnotes
This work was supported by grants to W.L. from the Chinese National Natural Science Foundation (30500145), The Education Department of Jiangsu Province (08KJA180004), and the Natural Science Foundation of Jiangsu Province (BK2005424). We thank Dr. Gong Chen at Pennsylvania State University (University Park, PA), Dr. Guo-Qiang Bi at the University of Pittsburgh (Pittsburgh, PA), Dr. Wei-Yang Lu at the University of Toronto (Toronto, ON, Canada), and Dr. Yu Tian Wang at the University of British Columbia (Vancouver, BC, Canada) for critical reading and constructive comments on this manuscript. We also thank Novartis for providing us with NR2A antagonist NVP-AAM077.
References
- Abraham, 2008.Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9:387. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- Abraham and Bear, 1996.Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Abraham and Tate, 1997.Abraham WC, Tate WP. Metaplasticity: a new vista across the field of synaptic plasticity. Prog Neurobiol. 1997;52:303–323. doi: 10.1016/s0301-0082(97)00018-x. [DOI] [PubMed] [Google Scholar]
- Abraham et al., 2001.Abraham WC, Mason-Parker SE, Bear MF, Webb S, Tate WP. Heterosynaptic metaplasticity in the hippocampus in vivo: a BCM-like modifiable threshold for LTP. Proc Natl Acad Sci U S A. 2001;98:10924–10929. doi: 10.1073/pnas.181342098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artola et al., 1990.Artola A, Brocher S, Singer W. Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature. 1990;347:69–72. doi: 10.1038/347069a0. [DOI] [PubMed] [Google Scholar]
- Auberson et al., 2002.Auberson YP, Allgeier H, Bischoff S, Lingenhoehl K, Moretti R, Schmutz M. 5-Phosphono-methylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett. 2002;12:1099–1102. doi: 10.1016/s0960-894x(02)00074-4. [DOI] [PubMed] [Google Scholar]
- Bear, 1995.Bear MF. Mechanism for a sliding synaptic modification threshold. Neuron. 1995;15:1–4. doi: 10.1016/0896-6273(95)90056-x. [DOI] [PubMed] [Google Scholar]
- Bear, 2003.Bear MF. Bidirectional synaptic plasticity: from theory to reality. Philos Trans R Soc Lond B Biol Sci. 2003;358:649–655. doi: 10.1098/rstb.2002.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone and Nicoll, 2007.Bellone C, Nicoll RA. Rapid bidirectional switching of synaptic NMDA receptors. Neuron. 2007;55:779–785. doi: 10.1016/j.neuron.2007.07.035. [DOI] [PubMed] [Google Scholar]
- Berberich et al., 2005.Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby O, Kohr G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci. 2005;25:6907–6910. doi: 10.1523/JNEUROSCI.1905-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienenstock et al., 1982.Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen et al., 1999.Chen HX, Otmakhov N, Lisman J. Requirements for LTP induction by pairing in hippocampal CA1 pyramidal cells. J Neurophysiol. 1999;82:526–532. doi: 10.1152/jn.1999.82.2.526. [DOI] [PubMed] [Google Scholar]
- Christie et al., 1995.Christie BR, Stellwagen D, Abraham WC. Reduction of the threshold for long-term potentiation by prior theta-frequency synaptic activity. Hippocampus. 1995;5:52–59. doi: 10.1002/hipo.450050107. [DOI] [PubMed] [Google Scholar]
- Cline et al., 1987.Cline HT, Debski EA, Constantine-Paton M. N-methyl-D-aspartate receptor antagonist desegregates eye-specific stripes. Proc Natl Acad Sci U S A. 1987;84:4342–4345. doi: 10.1073/pnas.84.12.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier et al., 2007.Crozier RA, Wang Y, Liu CH, Bear MF. Deprivation-induced synaptic depression by distinct mechanisms in different layers of mouse visual cortex. Proc Natl Acad Sci U S A. 2007;104:1383–1388. doi: 10.1073/pnas.0609596104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings et al., 1996.Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- Deisseroth et al., 1995.Deisseroth K, Bito H, Schulman H, Tsien RW. Synaptic plasticity: a molecular mechanism for metaplasticity. Curr Biol. 1995;5:1334–1338. doi: 10.1016/s0960-9822(95)00262-4. [DOI] [PubMed] [Google Scholar]
- Holland and Wagner, 1998.Holland LL, Wagner JJ. Primed facilitation of homosynaptic long-term depression and depotentiation in rat hippocampus. J Neurosci. 1998;18:887–894. doi: 10.1523/JNEUROSCI.18-03-00887.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang et al., 1992.Huang YY, Colino A, Selig DK, Malenka RC. The influence of prior synaptic activity on the induction of long-term potentiation. Science. 1992;255:730–733. doi: 10.1126/science.1346729. [DOI] [PubMed] [Google Scholar]
- Iny et al., 2006.Iny K, Heynen AJ, Sklar E, Bear MF. Bidirectional modifications of visual acuity induced by monocular deprivation in juvenile and adult rats. J Neurosci. 2006;26:7368–7374. doi: 10.1523/JNEUROSCI.0124-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood et al., 1996.Kirkwood A, Rioult MC, Bear MF. Experience-dependent modification of synaptic plasticity in visual cortex. Nature. 1996;381:526–528. doi: 10.1038/381526a0. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt et al., 1987.Kleinschmidt A, Bear MF, Singer W. Blockade of “NMDA” receptors disrupts experience-dependent plasticity of kitten striate cortex. Science. 1987;238:355–358. doi: 10.1126/science.2443978. [DOI] [PubMed] [Google Scholar]
- Liu et al., 2004.Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Lu et al., 2001.Lu HC, Gonzalez E, Crair MC. Barrel cortex critical period plasticity is independent of changes in NMDA receptor subunit composition. Neuron. 2001;32:619–634. doi: 10.1016/s0896-6273(01)00501-3. [DOI] [PubMed] [Google Scholar]
- Malinow et al., 1988.Malinow R, Madison DV, Tsien RW. Persistent protein kinase activity underlying long-term potentiation. Nature. 1988;335:820–824. doi: 10.1038/335820a0. [DOI] [PubMed] [Google Scholar]
- Malinow et al., 1989.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC and CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Mockett et al., 2002.Mockett B, Coussens C, Abraham WC. NMDA receptor-mediated metaplasticity during the induction of long-term depression by low-frequency stimulation. Eur J Neurosci. 2002;15:1819–1826. doi: 10.1046/j.1460-9568.2002.02008.x. [DOI] [PubMed] [Google Scholar]
- Morishita et al., 2007.Morishita W, Lu W, Smith GB, Nicoll RA, Bear MF, Malenka RC. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology. 2007;52:71–76. doi: 10.1016/j.neuropharm.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Morris et al., 1986.Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Neyton and Paoletti, 2006.Neyton J, Paoletti P. Relating NMDA receptor function to receptor subunit composition: limitations of the pharmacological approach. J Neurosci. 2006;26:1331–1333. doi: 10.1523/JNEUROSCI.5242-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama et al., 2000.Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–588. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- O'Dell and Kandel, 1994.O'Dell TJ, Kandel ER. Low-frequency stimulation erases LTP through an NMDA receptor-mediated activation of protein phosphatases. Learn Mem. 1994;1:129–139. [PubMed] [Google Scholar]
- Osten et al., 2000.Osten P, Khatri L, Perez JL, Kohr G, Giese G, Daly C, Schulz TW, Wensky A, Lee LM, Ziff EB. Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron. 2000;27:313–325. doi: 10.1016/s0896-6273(00)00039-8. [DOI] [PubMed] [Google Scholar]
- Perez-Otano et al., 2001.Perez-Otano I, Schulteis CT, Contractor A, Lipton SA, Trimmer JS, Sucher NJ, Heinemann SF. Assembly with the NR1 subunit is required for surface expression of NR3A-containing NMDA receptors. J Neurosci. 2001;21:1228–1237. doi: 10.1523/JNEUROSCI.21-04-01228.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot et al., 2001.Philpot BD, Sekhar AK, Shouval HZ, Bear MF. Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron. 2001;29:157–169. doi: 10.1016/s0896-6273(01)00187-8. [DOI] [PubMed] [Google Scholar]
- Philpot et al., 2003.Philpot BD, Espinosa JS, Bear MF. Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. J Neurosci. 2003;23:5583–5588. doi: 10.1523/JNEUROSCI.23-13-05583.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot et al., 2007.Philpot BD, Cho KK, Bear MF. Obligatory role of NR2A for metaplasticity in visual cortex. Neuron. 2007;53:495–502. doi: 10.1016/j.neuron.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan et al., 1999a.Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci. 1999a;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- Quinlan et al., 1999b.Quinlan EM, Olstein DH, Bear MF. Bidirectional, experience-dependent regulation of N-methyl-D-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc Natl Acad Sci U S A. 1999b;96:12876–12880. doi: 10.1073/pnas.96.22.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth-Alpermann et al., 2006.Roth-Alpermann C, Morris RG, Korte M, Bonhoeffer T. Homeostatic shutdown of long-term potentiation in the adult hippocampus. Proc Natl Acad Sci U S A. 2006;103:11039–11044. doi: 10.1073/pnas.0600894103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng et al., 1994.Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- Shouval et al., 2002.Shouval HZ, Bear MF, Cooper LN. A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc Natl Acad Sci U S A. 2002;99:10831–10836. doi: 10.1073/pnas.152343099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva et al., 1992.Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Smith et al., 2008.Smith GB, Heynen AJ, Bear MF. Bidirectional synaptic mechanisms of ocular dominance plasticity in visual cortex [review] Philos Trans R Soc Lond B Biol Sci. 2008;364:357–367. doi: 10.1098/rstb.2008.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobczyk and Svoboda, 2007.Sobczyk A, Svoboda K. Activity-dependent plasticity of the NMDA-receptor fractional Ca2+ current. Neuron. 2007;53:17–24. doi: 10.1016/j.neuron.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Sobczyk et al., 2005.Sobczyk A, Scheuss V, Svoboda K. NMDA receptor subunit-dependent [Ca2+] signaling in individual hippocampal dendritic spines. J Neurosci. 2005;25:6037–6046. doi: 10.1523/JNEUROSCI.1221-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda et al., 2005.Toyoda H, Zhao MG, Zhuo M. Roles of NMDA receptor NR2A and NR2B subtypes for long-term depression in the anterior cingulate cortex. Eur J Neurosci. 2005;22:485–494. doi: 10.1111/j.1460-9568.2005.04236.x. [DOI] [PubMed] [Google Scholar]
- Wagner et al., 1998.Wagner KD, Berenson A, Harding O, Joiner T. Attributional style and depression in pregnant teenagers. Am J Psychiatry. 1998;155:1227–1233. doi: 10.1176/ajp.155.9.1227. [DOI] [PubMed] [Google Scholar]
- Wang and Wagner, 1999.Wang H, Wagner JJ. Priming-induced shift in synaptic plasticity in the rat hippocampus. J Neurophysiol. 1999;82:2024–2028. doi: 10.1152/jn.1999.82.4.2024. [DOI] [PubMed] [Google Scholar]
- Watt et al., 2004.Watt AJ, Sjostrom PJ, Hausser M, Nelson SB, Turrigiano GG. A proportional but slower NMDA potentiation follows AMPA potentiation in LTP. Nat Neurosci. 2004;7:518–524. doi: 10.1038/nn1220. [DOI] [PubMed] [Google Scholar]
- Yashiro and Philpot, 2008.Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao et al., 2008.Zhao J, Peng Y, Xu Z, Chen RQ, Gu QH, Chen Z, Lu W. Synaptic metaplasticity through NMDA receptor lateral diffusion. J Neurosci. 2008;28:3060–3070. doi: 10.1523/JNEUROSCI.5450-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhu et al., 2002.Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]