

Abstract
We hypothesize that microglial chemotactic responses to amyloid-β peptide (Aβ1–42) serve as an early and integral component of inflammatory response in Alzheimer's disease (AD) brain. This study reports a receptor for vascular endothelial growth factor (VEGF), termed VEGF-1 (Flt-1), subserves microglial chemotactic responses induced by Aβ1–42 stimulation, in vivo and in vitro. Expression of Flt-1 was significantly increased in tissue obtained from AD patients [compared with tissue from nondemented (ND) individuals], in Aβ1–42-injected rat hippocampus, and in peptide-stimulated human microglia. Single and double immunohistochemical staining demonstrated marked immunoreactivity, for both Flt-1 and its ligand VEGF, in association with microglia and Aβ deposits in AD, but not ND, brain tissue. Functionally, treatment with anti-Flt-1 antibody was highly effective in inhibiting microglial mobility and chemotactic responses measured in vitro using a transwell migration assay. In vivo, transplanted enhanced green fluorescent protein (EGFP)-labeled microglia exhibited Flt-1-dependent chemotaxis induced by peptide injection with anti-Flt-1 effective in blocking migration of cells. Importantly, anti-Flt-1 reduction of microglial mobility was neuroprotective in peptide-injected hippocampus and associated with a significant increase in numbers of viable hippocampal neurons. The results of this study suggest critical functional roles for Flt-1 in mediating microglial chemotactic inflammatory responses which contribute to pathological conditions in AD brain.
Keywords: VEGF receptor Flt-1, VEGF, microglia, chemotaxis, amyloid-β peptide, Alzheimer's disease, chronic inflammation
Introduction
The Flt-1 subtype of VEGF receptor has been reported as a signaling factor in mediating chemotactic responses of immune responding cells (Barleon et al., 1996; Forstreuter et al., 2002). In addition, the results from several studies have indicated that Flt-1-mediated signaling could contribute to pathological conditions in disease (Sköld et al., 2005). Elevated VEGF expression has been documented in asthma (Hoshino et al., 2001) and eosinophil inflammatory responses are dependent on activation of Flt-1 (Feistritzer et al., 2004). In a model of rheumatoid arthritis (RA), blockade of Flt-1 was effective in reducing symptoms of disease, whereas inhibition of the Flk-1 subtype of VEGF receptor was without effect (De Bandt et al., 2003). Recently, a number of pathological inflammatory responses have been found to be attenuated in Flt-1-deficient mice compared with their wild-type cohorts in a murine model of RA (Murakami et al., 2006). It was concluded that enhanced inflammation in RA was mediated by Flt-1-dependent signaling in activated monocytes/macrophages.
At present, functional roles for Flt-1 in neurodegenerative brain disease have not been addressed. Deposition of amyloid-β peptide (Aβ) is considered a critical inducer of inflammatory responses and neuronal damage in Alzheimer's disease (AD) brain (Combs et al., 2001; Hardy and Selkoe, 2002; Spires et al., 2005). We have reported that intrahippocampal injection of full-length peptide (Aβ1–42) increased inflammatory reactivity including mobility of resident brain microglia and also caused perturbed vasculature including leakiness of blood–brain barrier (BBB) (Ryu and McLarnon, 2006). Furthermore, separate in vivo experiments have shown increased expression of VEGF, neovascularization, and neurodegeneration in inflamed rat brain with double-staining analysis indicating VEGF was primarily associated with microglia (Tran et al., 2005). These data would be consistent with microglia acting both as a source (paracrine signal) and as a responder (autocrine signal via Flt-1) to VEGF.
Overall, our findings suggested the possibility that Flt-1 and VEGF-dependent mobilization of microglia and cellular chemotactic responses could serve critical response functions in AD brain. To test this premise we have used three independent approaches: analysis of Flt-1 in both brain tissue and in microglia obtained from AD patients [with control brain sections and microglia from nondemented (ND) individuals] and following intrahippocampal injection of Aβ1–42 (controls, saline and reverse peptide). In the latter experiments we report the first in vivo effects of pharmacological modulation of Flt-1 (using anti-Flt-1 antibody) to inhibit microglial chemotactic responses to peptide. Our findings also suggest block of microglial chemotaxis as a novel strategy for neuroprotection in AD brain.
Materials and Methods
Preparation of human ND and AD sections.
Entorhinal cortical sections from six ND cases (ages from 40 to 85 years, postmortem intervals, 4–24 h) and seven AD cases (ages from 67 to 87 years, postmortem intervals, 5–10 h) were obtained from the Kinsmen Laboratory brain bank at the University of British Columbia (UBC, Vancouver, British Columbia, Canada). All cases of AD met the clinical criteria and postmortem confirmation for AD. The procedures used to isolate postmortem tissue have been described (Miklossy et al., 2007).
Immunohistochemical staining and analysis in human ND and AD sections.
For single immunofluorescent staining of Flt-1 and VEGF (Arai et al., 2006), free-floating sections (30 μm) from ND and AD tissues were washed in PBS with Triton X-100 (PBST; 0.01 m PBS, pH 7.4, containing 0.3% Triton X-100) and transferred into 5% skim milk in PBST for 1 h. Sections were then incubated for 48 h at 4°C with rabbit anti-human vascular endothelial growth factor receptor-1 (Flt-1, 1:200; R&D Systems) or rabbit anti-VEGF (a marker for the potent angiogenic factor VEGF, 1:200; Santa Cruz Biotechnology) antibody. Sections were then rinsed in PBST and incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:200; Invitrogen) for 1 h at room temperature. After washing in PBST, sections were mounted on glass slides and coverslipped with Prolong Gold anti-fading agent (Invitrogen).
For double-immunofluorescence staining (Guo et al., 2006), free-floating sections were incubated for 48 h at 4°C with a mixture of two primary antibodies: rabbit anti-Flt-1 (1:100; R&D Systems) in combination with mouse anti-HLA-DR (a marker for microglia, 1:500; DakoCytomation) or mouse anti-β-amyloid (Aβ, clone 6F/3D, 1:100; DakoCytomation) and rabbit anti-VEGF (1:100; Santa Cruz Biotechnology) in combination with mouse anti-HLA-DR or mouse anti-β-amyloid. For Aβ staining, 100% formic acid (Sigma) pretreatment for 15 min at room temperature was performed before primary antibody incubation (Guo et al., 2006). After incubation with the indicated primary antibodies, sections were rinsed in PBST and incubated for 1 h at room temperature with a mixture of Alexa Fluor 488 goat anti-rabbit IgG (1:200; Invitrogen) and Alexa Fluor 594 goat anti-mouse IgG secondary antibody (1:200; Invitrogen). To block endogenous lipofuscin autofluorescence, Sudan black (1% w/v) was prepared in 70% methanol and immunostained brain sections were stained with the compound for 10 min as previously described (Guo et al., 2006; Hashemzadeh-Bonehi et al., 2006). Typical Sudan black staining is presented (see supplemental Fig. 1A, available at www.jneurosci.org as supplemental material): also see supplemental text for details of immunostaining controls and immunohistochemical analysis for human tissue.
Preparation of cultured human microglia from ND and AD brains.
Cultured adult human ND and AD microglia were used in several experiments. Following isolation of brains, tissue was enzymatically dissociated and microglia were harvested and placed under culture conditions following standard protocols (Walker et al., 1995; Lue et al., 1996; McLarnon et al., 2005). Microglia were separated from other glial cells by differential adherence and maintained in DMEM with high glucose (5 mg/ml) containing 10% fetal bovine serum (Hyclone) and 50 μg/ml gentamicin. Microglia were used in experiments, after 10–14 d in culture, following previously described procedures (McLarnon et al., 2006). The typical morphological appearance of HLA-DR(+) microglia obtained from AD patients, with swollen cell bodies and retracted processes, has been shown [see McLarnon et al. (2006), their Fig. 2]. ND microglia in culture exhibit a predominant ramified morphology with cells expressing extended processes (not shown). These representative patterns of AD and ND morphology remained constant throughout culture times. Cell purity was assessed using immunocytochemistry with antibodies to CD11b [American Type Culture Collection (ATCC)] to class II major histocompatibility antigen (HLA-DR) (1:800; CN Pharmaceuticals) and to CD68 (1:1000; Accurate Chemical and Scientific). Immunocytochemical results showed purity of microglia was in excess of 98%.
Figure 2.

Association of Flt-1 and VEGF with β-amyloid peptide in AD tissue. The upper panels show representative images of Flt-1 and Aβ ir (left and middle panels) and merged staining of Flt-1/Aβ (right panel) in AD brain tissue. The lower panels show representative results for staining of VEGF with Aβ. Scale bar: 50 μm.
In vivo studies using intrahippocampal injection of β-amyloid Surgical procedures.
All animal procedures were approved by the UBC Animal Care Ethics Committee, with adherence to guidelines of the Canadian Council on Animal Care. Male Sprague Dawley rats weighing 280–300 g (Charles River Laboratories) were used for stereotaxic injection of Aβ1–42, or controls (PBS and reverse peptide Aβ42–1) as previously described (Franciosi et al., 2006; Ryu and McLarnon, 2008). In brief, animals received intraperitoneal injection of an anesthetic mixture (ketamine 100 mg/kg and xylazine 10 mg/kg) and were placed in a stereotaxic apparatus (David Kopf Instruments). Beta-amyloid (Aβ) peptide (2 nmol; California Peptides) was slowly injected (0.2 μl/min) into the dentate gyrus [anteroposterior (AP), −3.3 mm; mediolateral (ML), −1.6 mm; dorsoventral (DV), −3.2 mm] or CA1 layer (AP, −3.3 mm; ML, −1.8 mm; DV, −2.6 mm) of hippocampus.
Immunohistochemical staining of rat brain.
Animals were transcardially perfused with heparinized cold saline followed by 4% paraformaldehyde under ketamine/xylazine anesthesia. Brains were then removed, postfixed, cryoprotected, and cut into 40 μm sections (Ryu and McLarnon, 2008). Free-floating sections were processed for immunohistochemistry as described previously (Franciosi et al., 2006; Ryu and McLarnon, 2008). Briefly, sections were permeabilized in 0.2% Triton X-100, blocked with 10% NGS, and incubated for 48 h at 4°C with the primary antibodies: anti-Flt-1 (1:200; Santa Cruz Biotechnology), anti-neuronal nuclei (NeuN, a marker for neurons, 1:1000; Millipore Bioscience Research Reagents), and anti-ionized calcium binding adapter molecule 1 (Iba-1, a marker for microglia, 1:500; Wako Chemicals). Sections were rinsed in PBS with 0.5% BSA and incubated with secondary antibodies conjugated with Alexa Fluor 488 or 594 (1:200; Invitrogen) for 1 h in the dark.
To detect the Flt-1-expressing cell types in Aβ-injected hippocampus, tissue sections were incubated for 48 h at 4°C with anti-Flt-1, in combination with anti-glial fibrillary acidic protein (GFAP, a marker for astrocytes, 1:1000; Sigma-Aldrich) or anti-CR3R (OX-42, a marker for microglia/macrophages, 1:500; Serotec) as described previously (Choi et al., 2007). Sections were washed and incubated with a mixture of secondary antibodies (Alexa Fluor 488 and 594; 1:100; Invitrogen). Since Iba-1 antibody is raised in rabbit it was not suitable for double staining of microgliosis with either Flt-1 Ab or VEGF Ab. Thus OX-42 (raised in mouse) was used in double staining with Flt-1 and VEGF.
Immunohistochemical analysis of rat brain.
Quantification of immunohistochemical staining was performed on digitized images obtained with a Zeiss Axioplan 2 fluorescent microscope at a magnification of 400× equipped with DVC camera (Diagnostic Instruments) and Northern Eclipse software (Empix Imaging). Quantitative image analysis for the immunostained rat hippocampal sections was performed on three equally spaced sections through the level of the injection site (AP: −3.3 mm). The number of Flt-1 or OX-42/GFAP+Flt-1(+) cells in the superior blade of the dentate granule cell layer was determined for three sections (Franciosi et al., 2006; Ryu and McLarnon, 2008). The number of Iba-1 or NeuN(+) cells in the CA1 region was quantified at three hippocampal sections. To keep the consistency between the selected sections, a rectangular box (0.4 × 0.3 mm) was centered over the CA1 cell layer beginning 1.6 mm lateral to the midline in each image as previously described (Casolini et al., 2007). For quantification of migrating enhanced green fluorescent protein (EGFP)-expressing microglia after transplantation, the number of EGFP(+) microglia were counted within rectangular areas (0.3 × 0.2 mm) for three different regions (centered 600, 1200, and 1800 μm away from the transplantation site). All quantitative analyses were performed in a blinded manner.
Reverse transcription-PCR in microglia from human ND and AD brains.
Adult human microglia were prepared from frontal neocortex of ND and AD brains (Walker et al., 2006). In one set of RT-PCR experiments, expression of Flt-1 and VEGF in microglia from ND and AD brains were examined. In another set of experiments, human microglia from ND samples were plated on 12 well multiplates and incubated in serum-free medium for 48 h (McLarnon et al., 2006; Ryu and McLarnon, 2008) before treatment with PBS or Aβ1–42 (8 h exposure to 5 μm peptide). Our use of RT-PCR followed published protocols (McLarnon et al., 2006; Choi et al., 2007) using specific primers for human Flt-1, VEGF, and GAPDH (see the supplemental text for the primer sequences). A PCR cycle number of 35 was chosen for analysis of product levels on the basis that at this cycle number, densitometric signal was within the amplification phase of PCR (see representative graph in supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Analysis of PCR cycle versus band intensity indicated plateau regions of plots to occur in excess of 40 PCR cycles. Previous studies have indicated 35 cycles of PCR as suitable for amplification of cDNA for a host of inflammatory factors expressed in activated microglia (Choi et al., 2007; Ryu and McLarnon, 2008).
RT-PCR in peptide-injected rat hippocampus. Anesthetized animals were killed by decapitation at 1, 3, and 7 d after peptide injection. The control animals were killed at 7 d after PBS or reverse peptide Aβ42–1 injection. Brains were removed and hippocampal tissues were freshly dissected onto cold metal tissue matrices (Harvard Apparatus) and quickly frozen in liquid nitrogen. RT-PCR was performed using rat Flt-1 and β-actin primers as described previously (Ryu and McLarnon, 2008). The sequences and expected product sizes of the PCR primers are shown in the supplemental text.
Western blot analysis of rat brain.
Detailed procedures for use of Western blot analysis (Ryu et al., 2006) are contained in supplemental text.
Study of APP23 transgenic AD mice
Experiments were performed using APP23 transgenic mice (Sturchler-Pierrat et al., 1997). These animals carry the human APP751 cDNA with the Swedish double mutation under control of the murine Thy-1.2 promoter. APP23 animals were 9 months of age. Littermates of APP23 mice carrying no human Swedish mutant APP751 cDNA were used as wild-type (WT) controls. These studies measured Flt-1 and VEGF expression in APP23 and WT mice. Preparation of sections followed the same protocols as for rat brain sections. Free-floating sections were processed for double-immunofluorescence staining for Flt-1/Aβ and VEGF/Aβ.
Preparation and administration of compounds
β-Amyloid peptide.
Fibrillar Aβ1–42 and reverse peptide Aβ1–42 (California Peptides) were first dissolved in 35% acetonitrile (Sigma) and further diluted to 500 μm with incremental additions of PBS with vortexing. The peptide solution was subsequently incubated at 37°C for 18 h to promote fibrilization and aggregation and stored at −20°C (Lue et al., 2001; Franciosi et al., 2006). We also performed immunohistochemical staining (Thioflavin-S staining and Aβ immunostaining) to examine the aggregation state of Aβ peptide in vivo. The results show that aggregated Aβ peptide was labeled with Thioflavin-S and Aβ antibody at 7 d after injection (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Anti-Flt-1 antibody.
For the inhibition of Flt-1, the neutralizing monoclonal antibody against Flt-1 (R&D Systems) was coadministered at 10 μg with peptides into CA1 layer. Control animals were injected with control antibody rat IgG. The dose and treatment protocol for anti-Flt-1 Ab in this study have been previously reported to be effective in inhibition of Flt-1-mediated cellular responses (Feistritzer et al., 2004; Ambati et al., 2006).
Functional chemotaxis assays
In vivo chemotaxis assay of rat microglia.
An EGFP-lentivirus construct was generated by transfecting HEK 293T cells with three plasmids: the transfer plasmid pHR'-CMV-EGFP, the packaging plasmid PR8–2, and the envelop plasmid PVSVG, as described previously (Skarsgard et al., 2005). Lentiviral particles containing media were collected over 4 d and centrifuged at 27,500 rpm for 3 h to concentrate. A titer of 6.36 × 108 transducing units (TU)/ml was assayed by measuring serial dilutions of the concentrated virus on primary rat cultured cells. Lentiviral vectors were used to infect primary fetal microglia cultures.
Rat cultured microglia were prepared from embryonic day 14 Sprague Dawley rats, as described previously (Yang et al., 2006). Three days after lentiviral infection (with threefold higher titers of lentivirus than the seeded microglial cell density), immunostaining of EGFP-labeled microglia were performed with a mixture of primary antibodies: anti-Iba-1 (1:1000; Wako) and anti-EGFP, (1:200; Invitrogen). Cells were then incubated with appropriate Alexa-labeled secondary antibodies (Invitrogen), followed by DAPI staining.
Rat microglial cultures, prelabeled with EGFP transfection, were incubated with anti-Flt-1 Ab or control Ab (10 μg, R&D Systems) for 30 min before transplantation. Cells (2 μl, 1 × 105 cells per μl) were transplanted (at 0.5 μl/min) under anesthesia (Ryu et al., 2004), 3 d after Aβ1–42 injection (2 nmol, CA1) into the medial corpus callosum at the following coordinates (AP, −3.3; ML, 0.0; DV, −2.8). Animals were perfused at 7 d after cell transplantation and double immunohistochemical procedures were performed as follows. Sections were incubated with primary Ab: anti-EGFP (1:500, Invitrogen) or anti-EGFP in combination with anti-VEGF (1:200, Santa Cruz Biotechnology) or anti-Flt-1 (1:200, Santa Cruz Biotechnology).
In vitro chemotaxis assay of human microglia.
The migration of human microglia in vitro was determined using Transwell inserts (pore size, 8 μm, BD Biosciences Discovery) and 24-well culture plates (Corning Costar) as previously described (Aarum et al., 2003). Cell-free conditioned medium was collected from microglial cultures stimulated with PBS, Aβ1–42 (5 μm), or Aβ42–1 (5 μm) for 24 h and placed in a lower chamber. Human microglia were placed in an upper chamber in serum-free medium and treated (1 h at 37°C) with anti-Flt-1 neutralizing antibody or control IgG (10 μg/ml, R&D Systems). The transwell plates were incubated for 24 h at 37°C. Human microglial migration was quantified by counting the number of cells that migrated through the membrane to the lower chamber using an inverted bright-field microscope (five fields/each well).
Statistical analysis.
Results are presented as mean ± SEM. The statistical analysis was performed using a one-way ANOVA, followed by the Student–Newman–Keuls multiple comparison test or Student's t test (GraphPad Prism 3.0; Graph Pad) with the significance level set at p < 0.05.
Results
Expression of Flt-1 and VEGF in microglia and brain tissue from ND and AD individuals
An initial aspect of this work was the characterization of expressions of Flt-1, and its ligand VEGF, in microglia and in brain sections obtained from ND and AD individuals. Human microglia obtained from ND and AD tissues were initially analyzed for levels of Flt-1 and VEGF. Representative RT-PCR showed little or no expression of either receptor or ligand in human ND microglia (Fig. 1A), however, AD microglia demonstrated markedly increased levels of both factors. Semiquantification of data are shown in Figure 1A. In total (n = 8 ND cases, n = 6 AD cases), Flt-1 was increased 3.2-fold and VEGF was increased 8.2-fold in AD, compared with ND, human microglia.
Figure 1.
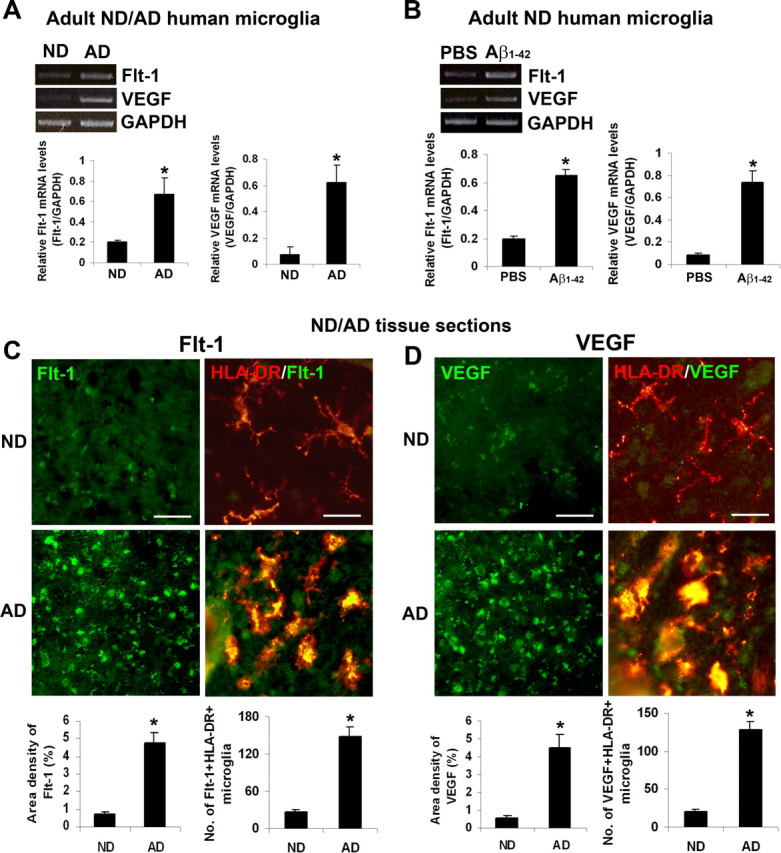
Expression of Flt-1 and VEGF in human ND/AD microglia and in ND/AD tissue. A, Typical RT-PCR for Flt-1 and VEGF expressions in microglia obtained from ND and AD brain tissue. The bar graphs show semiquantitative results for relative levels of mRNA for Flt-1 and VEGF from 8 ND and 6 AD individuals. *p < 0.05. B, Representative expressions of Flt-1 and VEGF in human ND microglia treated with PBS or Aβ1–42 (5 μm for 8 h). The bar graph presents overall semiquantitative results for the two factors (n = 4 independent samples/group) *p < 0.05. C, Marker immunoreactivities (ir) from ND tissue and AD tissue showing representative single staining for Flt-1 and double staining of Flt-1/HLA-DR. Areas of merged double staining are yellow. Scale bar: 150 μm (left panels) and 50 μm (right panels). Overall ir (n = 6 cases from ND, 7 cases from AD) for Flt-1 and numbers of Flt-1(+) microglia. D, Representative results showing single staining for VEGF and double staining for VEGF/HLA-DR in ND and AD tissue. Scale bar: 150 μm (left panels) and 50 μm (right panels). Quantification (n = 6 cases from ND, 7 cases from AD) of VEGF ir and numbers of VEGF(+) microglia. *p < 0.05.
We next examined effects of Aβ1–42 stimulation of human ND microglia. As shown in Figure 1B, low levels of Flt-1 and VEGF were evident in PBS control with a marked increase for both ligand and receptor induced by peptide stimulation (5 μm for 8 h). Semiquantitative analysis is presented in Figure 1B with overall (n = 4 independent samples/group) mRNA levels increased by 3.3-fold (for Flt-1) and by 9-fold (for VEGF) with peptide, compared with control, treatment.
Immunohistochemical staining of Flt-1 and VEGF with microglia in ND and AD brain
We next investigated for association between the VEGF receptor and VEGF itself with microgliosis in ND and AD sections. Representative immunohistochemical staining showed minimal levels of either Flt-1 (Fig. 1C) or VEGF (Fig. 1D) in ND sections. However, both receptor (Fig. 1C) and ligand (Fig. 1D) were highly expressed in AD brain sections.
Representative double staining for HLA-DR(+) microglia with Flt-1 and VEGF are presented (Fig. 1C,D, respectively), for ND and AD tissue. Both Flt-1 and VEGF showed little merged staining with microglia in ND sections. However, AD brain tissue demonstrated marked levels of association of Flt-1/HLA-DR (Fig. 1C) and VEGF/HLA-DR (Fig. 1D).
Single-staining analysis was used to quantify levels of Flt-1 and VEGF and double staining to quantify association of these factors with microglia (n = 6 ND cases, 7 AD cases). The area density of Flt-1 ir was significantly increased by 6.4-fold in AD, relative to ND, tissue (Fig. 1C, lower left graph). A similar magnitude of increase (by 5.6-fold) was measured for the number of microglia expressing Flt-1 (Fig. 1C, lower right graph) in AD versus ND tissue. The area density of VEGF was increased eightfold in AD, compared with ND, brain tissue (Fig. 1D, lower left graph). The corresponding increase in the number of microglia associated with VEGF was 6.2-fold in AD versus ND sections (Fig. 1D, lower right graph).
Double staining of Flt-1 and VEGF with β-amyloid in AD brain tissue
A relevant question was the possible association of Flt-1 and VEGF with peptide deposition. This point was addressed using immunohistochemical staining to determine localization of receptor and ligand with Aβ in AD brain sections. Representative staining patterns for Flt-1 (upper left panel), VEGF (lower left panel), and Aβ (middle panels) in AD tissue are presented in Figure 2 The corresponding double-staining immunoreactivities for Flt-1/Aβ and VEGF/Aβ are also shown in Figure 2 (right panels). Although quantification of double staining was not done, clearly defined areas of Flt-1 and VEGF ir colocalized to peptide were evident. The patterns of staining also showed regions of Flt-1 and VEGF ir which were not associated with peptide deposits.
Expression of Flt-1 and VEGF in transgenic APP23 mice
The findings of association between Flt-1 and VEGF with Aβ deposits in human AD tissue sections (Fig. 2) led us to investigate expression of these factors in APP transgenic mice. These experiments used cortical tissue from transgenic APP23 mice and WT controls (see Materials and Methods). Double immunohistochemical staining was performed on Flt-1 and VEGF with peptide. Representative patterns of double staining are presented in Figure 3A for Flt-1/Aβ and in Figure 3B for VEGF/Aβ for WT controls (upper panels) and APP23 mice (lower panels). The staining results indicate enhanced expression of both Flt-1 and VEGF in transgenic animals compared with the WT mice. The double-staining panels show representative examples of Flt-1 and VEGF ir in proximity to Aβ deposits in the cortex of APP23 mice compared with WT controls. Although we did not quantify extents of changes in expression of Flt-1 and VEGF, or their association with Aβ, between transgenic and WT animals, the patterns of marker ir were consistent with results obtained from analysis of human tissue.
Figure 3.
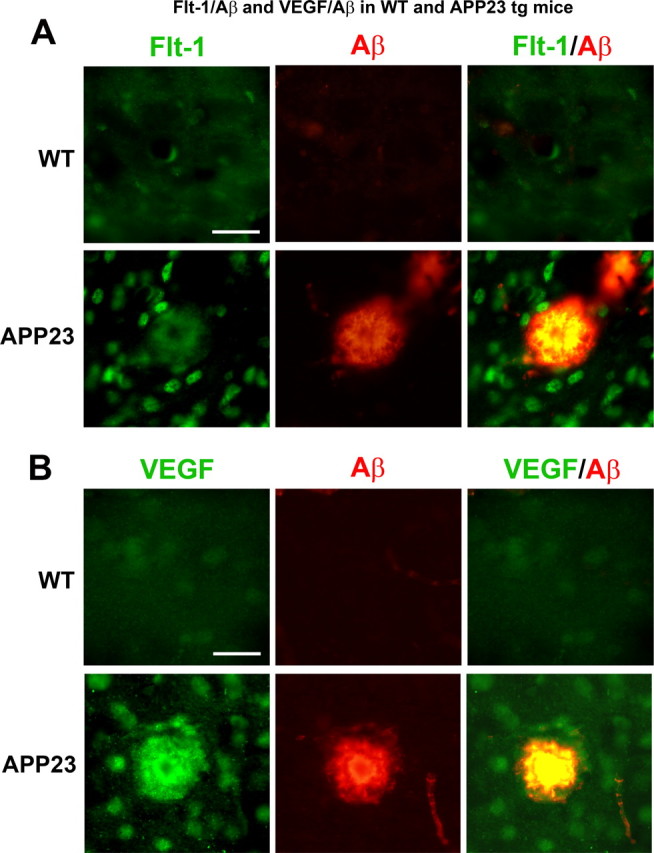
Staining of Flt-1, VEGF, and Aβ in wild-type (WT) and transgenic mice. A, The upper panels show typical Flt-1 and Aβ ir and merged staining in WT mice. The lower panels present representative staining for the markers in transgenic APP23 animals. Scale bar is for 50 μm. B, Upper panels show representative patterns of staining for VEGF, Aβ, and merging of the two factors in WT mice. The lower panels present staining for the same markers in APP23 mice. Scale bar represents 50 μm.
Time dependency of Flt-1 expression in vivo
We initially used RT-PCR to determine time-dependent changes in VEGF receptor-1 (Flt-1) expression following intrahippocampal injection of Aβ1–42. Representative results indicated controls (7 d injections with PBS or Aβ42–1) to exhibit minimal levels of the VEGF receptor-1 (Fig. 4A). However, Aβ1–42 injection was associated with elevated Flt-1, relative to controls, at 1 and 3 d after peptide with a further increase at a duration of 7 d. Semiquantitative RT-PCR is presented in the bar graph of Figure 4A (n = 5 animals/group). Expression of Flt-1 was significantly higher (by 3-fold at 1 d and 2.6-fold at 3 d) for peptide versus PBS control (7 d) injections. The corresponding increase for the longest duration of Aβ1–42 injection (7 d) was a 4.5-fold increase in Flt-1 with peptide compared with PBS.
Figure 4.

Time dependency of Flt-1 expression and production in Aβ1–42-injected rat hippocampus. A, Representative Flt-1 mRNA expression following hippocampal injections with controls [PBS and Aβ42–1 (7 d)] and Aβ1–42 (1, 3, and 7 d). β-actin was used as a reaction standard. The bar graph shows semiquantitative RT-PCR results for relative levels of Flt-1 mRNA expression (n = 5 animals/group). B, Western blot analysis for time-dependent changes (1, 3, and 7 d) in levels of Flt-1 after Aβ1–42 injection, PBS control was for 7 d; β-actin was used as a reaction standard. Quantification of data for PBS and peptide treatments (n = 4 animals/group). *p < 0.05 versus PBS. C, Representative Flt-1 immunoreactivity following hippocampal injection with PBS (7 d), reverse peptide Aβ42–1 (7 d), and Aβ1–42 (1, 3, and 7 d). Scale bar: 50 μm. The bar graph shows number of Flt-1(+) cells for PBS and Aβ42–1 controls (7 d) and the time dependence of Flt-1 expression for the different times of peptide injections (n = 6 animals/group). *p < 0.05 versus PBS.
We also performed Western blot analysis to determine levels of Flt-1 protein following peptide injection (at 1, 3, and 7 d). Representative immunoblots are presented for Flt-1 in Figure 4B with data quantification shown in the accompanying bar graph. Levels of protein production in Aβ1–42-injected hippocampus (n = 4 animals/group) were significantly increased by 46% relative to control PBS injection (7 d after injection).
We also used immunohistochemical staining to examine changes in Flt-1 with duration of peptide injection (1–7 d). Representative staining for controls (7 d injections of either PBS or Aβ42–1) demonstrated little or no immunoreactivity (ir) for Flt-1 (Fig. 4C). However, Aβ1–42 injection caused a progressive time-dependent increase in numbers of Flt-1(+) cells (Fig. 4C). Overall (n = 6 animals/group), significant increases in Flt-1 were determined at 1, 3, and 7 d after peptide injections (by 4-fold, 12-fold, and 30-fold, respectively, relative to PBS 7 d injection) (Fig. 4C, lower right bar graph). These results are consistent with RT-PCR data with the exception that immunostaining analysis showed progressive increases in Flt-1 at early times after peptide injection.
Cell-dependent expression of Flt-1
We next used double staining to examine the specific cells expressing Flt-1. These experiments were performed at a single time point of 7 d after Aβ1–42 injection and used GFAP and OX-42 as respective cell-specific markers for astrocytes and microglia. The results (Fig. 5A, left panels) showed low levels of GFAP/Flt-1 double staining in PBS control with an elevated astrocyte expression of Flt-1 marker in peptide-injected brain. Low levels of merged OX-42/Flt-1 staining were evident in PBS control (Fig. 5A, upper right panel). However, peptide-injected hippocampus demonstrated a considerable proportion of microglia in association with Flt-1 (Fig. 5A, lower right panel). Quantification of data are presented in the right bar graph of Figure 5B (n = 6 animals/group). Relatively low values of Flt-1 associated GFAP(+) astrocytes or OX-42(+) microglia were evident in PBS control. However, following Aβ1–42 injection GFAP/Flt-1 ir was increased by 6.2-fold and OX-42/Flt-1 ir was increased by 15.5-fold, relative to controls. Both increases were significant, however, association of Flt-1 was significantly higher with microglia compared with astrocytes.
Figure 5.

Flt-1 double staining with microglia and astrocytes. A, Upper panels show representative Flt-1 double staining with astrocytes (GFAP marker) and microglia (OX-42 marker) following 7 d injection of PBS. The lower panels are corresponding double-staining images at 7 d after injection of Aβ1–42. Scale bar: 30 μm. B, Quantification of number of Flt-1(+) astrocytes and microglia in PBS and peptide-injected brain (n = 6 animals/group). *p < 0.05 versus PBS; #comparison of OX-42 versus GFAP marker.
Effects of anti-Flt-1 Ab on microglial responses and neuronal viability
The findings of marked increases of Flt-1 levels induced by peptide injection prompted investigation on the effects of receptor modulation on microgliosis and neuronal viability. These experiments used an anti-Flt-1 neutralizing antibody (anti-Flt-1 Ab) in conjunction with Iba-1 (microglial marker) and NeuN (neuronal marker) in Aβ1–42-injected (7 d) rat brain. Nonimmune rat IgG Ab was used as an antibody control in peptide-injected brain.
Representative microglial ir for the different treatments are presented in Figure 6A. Relatively low levels of Iba-1 staining (Fig. 6A, upper panels) were observed in controls (7 d injections of PBS or Aβ42–1). However, the Aβ1–42-injected hippocampus demonstrated a diffuse and high level of Iba-1 staining which was reduced in the presence of anti-Flt-1 Ab with peptide (Fig. 6A, lower panels). Overall (n = 6 animals/group), the number of Iba-1(+) microglia was increased fivefold in Aβ1–42, compared with PBS-injected animals (Fig. 6B). Treatment with anti-Flt-1 Ab significantly reduced microgliosis (by 26%) compared with Aβ1–42 injection alone.
Figure 6.

Effects of anti-Flt-1 antibody on Iba-1 (microglial marker) and NeuN expression in vivo. A, Representative Iba-1 immunoreactivity (ir) following 7 d intrahippocampal injection for the following: PBS, Aβ42–1, Aβ1–42 plus control Ab and Aβ1–42 plus anti-Flt-1 Ab. Scale bar: 50 μm. B, Numbers of Iba-1(+) microglia for the different injections (n = 6 animals/group). *p < 0.05 for comparison of PBS versus Aβ1–42 and # for comparison of Aβ1–42 plus control Ab versus Aβ1–42 plus anti-Flt-1 Ab. C, Representative NeuN ir with 7 d injections for PBS, Aβ42–1, Aβ1–42 plus control Ab, and Aβ1–42 plus anti-Flt-1 Ab. Scale bar: 50 μm. D, Numbers of NeuN cells for the different injections (n = 6 animals/group). *p < 0.05 for PBS versus Aβ1–42; #p < 0.05 for Aβ1–42 versus Aβ1–42 plus anti-Flt-1 Ab.
Representative NeuN ir indicated an intact layer of CA1 pyramidal neurons in controls (PBS or Aβ42–1) (Fig. 6C, top panels). The number of NeuN(+) cells was markedly diminished following Aβ1–42 injection (Fig. 6C, lower left panel). However, if anti-Flt-1 Ab treatment was applied to peptide-injected animals, numbers of NeuN(+) neurons were increased (Fig. 6C, bottom right panel). Overall (n = 6 animals/group), the number of hippocampal neurons was reduced by 37% with Aβ1–42, compared with PBS injection (Fig. 6D). Application of anti-Flt-1 Ab was effective in conferring neuroprotection whereby numbers of neurons were significantly increased (by 23%) compared with control Ab injection.
Microglial chemotactic responses in vitro
The upregulation of Flt-1 in the peptide-injected hippocampus (Fig. 4C) and association with OX-42(+) cells (Fig. 5A,B) could reflect chemotactic responses of microglia. Experiments were designed to examine this possibility in vitro and in vivo. A schematic diagram (Fig. 7A) indicates the method used to measure in vitro migration of human microglia. Microglial cells were initially subjected to 24 h exposure for the following treatments; PBS, Aβ1–42, and reverse peptide Aβ42–1. Supernatants from the treated microglia were then applied to a transwell chamber (lower chamber) for the measurement of migration. Microglia in the upper chamber were preincubated with control Ab or with anti-Flt-1 Ab (10 μg/ml for 1 h), before the chemotactic assay. The bar graph in Figure 7B summarizes the results from n = 5 experiments. Microglial chemotaxis increased 4.6-fold with Aβ1–42 treatment, relative to PBS control. Inclusion of anti-Flt-1 Ab with Aβ1–42 peptide, significantly diminished microglial mobility by 37% compared with the application of peptide alone. Numbers of migrating microglia were similar for controls and for anti-Flt-1 Ab applied alone.
Figure 7.

Migration of human microglia, in vitro. A, Schematic diagram for the treatment of microglia with PBS, Aβ1–42, or Aβ42–1 and supernatant application, in the absence and presence of anti-Flt-1 Ab, in the transwell microglial migration assay. B, The bar graph shows the number of migrating microglia with the different treatments and the effects of anti-Flt-1 Ab on migration (n = 5 samples/group). *p < 0.05 for peptide versus PBS treatment; #p < 0.05 for effects of anti-Flt-1 Ab on migration with peptide application.
Microglial chemotactic responses in vivo
To examine chemotactic responses in vivo, we used the procedures schematically diagrammed in Figure 8A (top panel). Microglia were first isolated from rat brain and then infected with a lentivirus-expressing EGFP. Figure 8A (bottom panels) shows representative in vitro double staining (EGFP and Iba-1) for the cells before injection; colocalization of EGFP with the microglial marker Iba-1 was evident for all cells examined. The EGFP-microglia were incubated with control or anti-Flt-1 Ab (10 μg/ml for 30 min) and then stereotaxically injected into peptide-injected rat brain (at 3 d after peptide injection) at the location shown (Fig. 8A, asterisk). Three areas were designated for the counting of EGFP-microglia (labeled 1–3 in Fig. 8A), at sites between injected EGFP-microglia and peptide.
Figure 8.
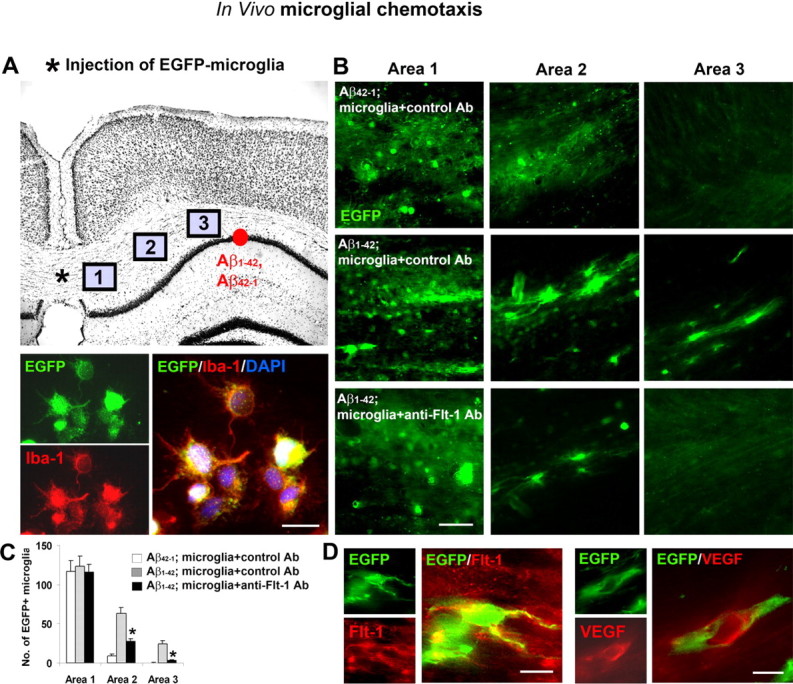
Chemotactic responses of EGFP-labeled microglia, in vivo. A, Upper panel, Schematic diagram showing in vivo locations of EGFP-labeled microglial transplantation (*) and Aβ1–42 or reverse peptide injection (red circle). The three areas chosen for the analysis of EGFP cell migration are shown (refer to text). Lower panels show representative double staining for EGFP and for Iba-1, and merged staining for both markers. Scale bar: 20 μm. B, Representative EGFP microglial staining in the three areas for: Aβ42–1 plus microglia incubated with control Ab, Aβ1–42 plus microglia incubated with control Ab, and Aβ1–42 plus microglia incubated with anti-Flt-1 Ab. Scale bar: 50 μm. C, The bar graph shows the numbers of EGFP-labeled microglia in areas 1–3, for the three protocols (n = 6 animals/group). *p < 0.05 for the effects of anti-Flt-1 Ab on microglial chemotaxis in peptide-injected hippocampus. D, Representative single and double staining for EGFP/Flt-1 and for EGFP/VEGF. Scale bar: 20 μm.
The number of EGFP(+) microglia in the different areas were measured in three groups of animals; Aβ1–42 plus microglia incubated with control Ab; Aβ1–42 plus microglia incubated with anti-Flt-1 Ab or reverse peptide Aβ42–1 plus microglia incubated with control Ab. Animals receiving reverse peptide injection (Fig. 8B, top panels) showed high levels of EGFP-microglia in area 1 adjacent to transplantation site but minimal migration of cells to area 2 or 3 nearer to peptide. Representative immunostaining showed a different pattern of chemotaxis with Aβ1–42 injection (Fig. 8B, middle panels) whereby considerable levels of EGFP(+) microglia were evident in both areas 2 and 3. Animals receiving anti-Flt-1 Ab with peptide (Fig. 8B, bottom panels) exhibited similar levels of EGFP ir in area 1 as for the other treatments. However, anti-Flt-1 Ab was effective in decreasing numbers of migrating microglia in areas more proximal to peptide, compared with animals receiving control antibody treatment.
The quantification for in vivo chemotaxis of EGFP(+) microglia is presented in Figure 8C. Overall (n = 6 animals/group), no differences in numbers of EGFP(+) microglia were measured in area 1 for the different treatments. In area 2, anti-Flt-1 Ab treatment significantly reduced the number of EGFP(+) cells (by 57%) relative to peptide-injection. The corresponding decrease in area 3 was 87%, representing a significant reduction by anti-Flt-1 Ab in the migration of EGFP-labeled microglia induced by peptide stimulation.
Double immunostaining of the EGFP-microglia-injected hippocampus was also examined to determine association between injected cells and markers for Flt-1 and VEGF. Representative double staining for EGFP(+) microglia and Flt-1 are shown in the left panels of Figure 8D, with merged staining indicating considerable regions of overlap of the markers. Similarly, the association of VEGF with EGFP(+) microglia was also demonstrated (Fig. 8D, right panels).
Discussion
Chronic inflammation is integral to AD pathology, however, specific mechanisms underlying inflammatory responses are not well understood (Mrak and Griffin, 2007). A critical and early component of inflammatory response is an increased microglial mobility and chemotactic activity to Aβ peptide deposition in AD brain. Our results suggest that the VEGF receptor subtype Flt-1 serves as a microglial chemotactic receptor to mobilize these immune cells in proximity to peptide deposits yielding localized and amplified inflammatory microenvironments in AD brain. Importantly, such microenvironments may be neurotoxic since we provide evidence that Flt-1-dependent mobilization of microglia is associated with neurodegeneration.
The conclusions stated above are supported by the findings that Flt-1 are upregulated under three independent experimental conditions; in AD cortical brain tissue, following intrahippocampal injection of Aβ1–42 in rat brain and after peptide stimulation of human microglia. This work has directly demonstrated involvement of Flt-1 in mediating microglial chemotactic responses in vivo and in vitro, in response to peptide stimulation. Importantly, anti-Flt-1 Ab was found effective in inhibiting mobility of EGFP-labeled microglia induced by Aβ1–42 injection into rat hippocampus and attenuating mobility of cultured microglia responding to a transwell insert of peptide. Previous work, using the murine microglial cell line (BV-2), has reported increased cell migration in response to VEGF (Forstreuter et al., 2002). Since VEGF is also a potent factor for induction of neovasculature, the present data also suggest microglial chemokine activity may be associated with vascular remodeling in AD brain, a point discussed below.
An important finding was that microglia obtained from AD brain samples exhibited significantly higher expressions and levels of Flt-1 and ligand VEGF relative to ND brain tissue. Double staining showed little or no localization of either Flt-1 or VEGF with HLA-DR(+) microglia in ND sections. A very different result was obtained in AD brain tissue where considerable Flt-1 and VEGF ir was associated with microglia. These data could indicate that in AD, enhanced Flt-1 and VEGF expression contributes to microglial autocrine and paracrine processes. Thus, β-amyloid peptide not only induces chemotaxis in AD brain but also acts to amplify responses resulting in microgliosis.
Our results also demonstrate considerable extents of Flt-1 and VEGF ir colocalized with peptide. Since both factors are associated with microglia (Fig. 1), these data imply clusters of mobile microglial cells in proximity to peptide deposits. The patterns of Flt-1 staining in both AD tissue (Fig. 2) and APP23 transgenic mice (Fig. 3) showed evidence for areas of Flt-1 ir coincident with Aβ peptide deposits in addition to regions with no association between the two markers. It was noteworthy that Flt-1 ir away from deposits appeared discrete, whereas Flt-1 ir in association with plaques exhibited diffuse patterns of staining. This finding was observed in both AD tissue and in transgenic animals. One possible interpretation of these immunostaining data are that a diffuse Flt-1 ir colocalized with Aβ is indicating the accumulation of numbers of microglial cells nearby peptide. In this case the diffuse Flt-1 ir could represent the endpoint of microglial chemotactic responses initiated by deposits of amyloid-β peptide. This explanation would be consistent with the expression and activation of Flt-1 in microglia serving as a mechanism to induce cellular mobility and mobilize and accumulate microglia in the vicinity of plaques. Although speculative at present, Flt-1-mediated microglial reactivity and inflammatory responses could then be linked with, and modulated by, the extents of amyloid deposits in AD brain.
In vivo, intrahippocampal injection of Aβ1–42 caused a time-dependent (1–7 d) increase in expression of Flt-1 as measured using RT-PCR, Western blots, or immunohistochemical staining (Fig. 4). Double-staining analysis demonstrated a predominant association of Flt-1 with microglia, however, a smaller extent of Flt-1 localization to astrocytes was also evident. At 7 d after peptide injection, significant increases of microgliosis and neuronal damage were measured relative to controls (injection of PBS or reverse peptide, Aβ42–1). A novel finding was that the inclusion of anti-Flt-1 Ab with peptide, inhibited microgliosis and conferred partial neuroprotection. These results suggest blocking peptide-induced microglial migration as a rationale strategy to attenuate neuronal damage in AD brain.
To examine functional roles of Flt-1 in vivo, EGFP-labeled microglia were transplanted into rat hippocampus previously injected with Aβ1–42 or reverse peptide. Three areas, differentially located between sites of microglial transplantation and peptide injection, were used to quantify microglial mobility. In the presence of reverse peptide, no net migration of EGFP-microglia away from the transplantation site was evident. However, with Aβ1–42 as a chemotactic stimulus, microglia exhibited increased mobility into parenchymal areas directed toward the peptide-injection site. Treatment of peptide-injected brain with anti-Flt-1 Ab was highly effective in reducing the numbers of migrating EGFP-microglia in regions 2 and 3 between sites of microglial transplantation and peptide injection. Double-staining analysis showed considerable extents in overlaps of EGFP microglial marker with both Flt-1 and VEGF.
Anti-Flt-1 Ab was also effective in inhibiting the migration of human microglia in vitro, where supernatants (conditioned medium) from cells exposed to PBS, Aβ1–42, and Aβ42–1 were used as stimulants. Under control conditions, neither PBS nor reverse peptide were effective in the induction of microglial mobility. With Aβ1–42 as a stimulus, a markedly increased migration was evident (by fourfold) relative to PBS; this enhanced migration was reduced with inclusion of anti-Flt-1 Ab treatment with peptide. However, anti-Flt-1 Ab was only partially effective in the inhibition of microglial chemotactic response. This latter result could reflect that under the experimental conditions of this assay, factors in the conditioned medium, other than VEGF, could also contribute to the increased mobility of microglia. Future studies using Flt-1 knock-out transgenic mice could have utility in characterizing functional roles of this chemotactic receptor in AD brain. However, interpretation of results could be confounded by the possibility that Flt-1 knock-out animals could exhibit deficits in developmental and other functional responses (Fong et al., 1995).
The possibility that accumulation of peptide could serve as foci for the induction of microglial-mediated inflammatory paracrine responses involving VEGF is an important consideration in the pathology of AD. Previous work has reported increased intrathecal levels of VEGF in AD, relative to non-AD, individuals (Tarkowski et al., 2002). This study also reported elevated VEGF was correlated with clinical severity of disease. Since VEGF is a highly potent vascular mediator, our results suggest the possibility of vascular remodeling near sites of microgliosis, elevated VEGF and peptide deposits. It has been pointed out that vascular angiogenic factors are elevated in AD pathology (Vagnucci and Li, 2003; Pogue and Lukiw, 2004), however, despite this finding there is little evidence for formation of new blood vessels in AD diagnosed individuals. Instead, vasculature in AD brain exhibits considerable morphological abnormalities and increased permeability and leakage of BBB (Prat et al., 2001; Fiala et al., 2002; Park et al., 2005; Zlokovic, 2005; Zipser et al., 2007). Our results could help account for this discrepancy. The lack of neovascularization in AD brain, despite elevated levels of angiogenic inducers, may be explained by a progressive and concomitant increase in an inflammatory environment causing newly formed blood vessels to degenerate.
Deposition of amyloid β peptide is a critical stimulus for inflammation in AD brain (Combs et al., 2001; Lue et al., 2001). We postulate that peptide deposits act as foci for assimilating clusters of mobile activated microglia and serving as sites for vascular remodeling in sustaining inflammatory environments. Microglial Flt-1 and VEGF are suggested as primary, but clearly not as sole, factors in maintaining chronic inflammatory responses dealing with removal of unwanted peptide. Although intrahippocampal peptide injection represents an acute insult, the procedure exacerbates inflammatory responses and likely compresses inflammation in this animal model into a much shorter timeframe (days) compared with the slow progression of inflammation (years) evident in AD brain. This point is testable in future studies using transgenic mouse models of AD where it is predicted that aged animals (months) will exhibit Flt-1 and VEGF microglial-dependent chemotactic processes with anti-Flt-1 Ab effective in inhibiting microgliosis and conferring neuroprotection.
Footnotes
This work was supported by grants from the Pacific Alzheimer Research Foundation and Alzheimer's Association (J.G.M.). We thank Dr. Weihong Song (Department of Psychiatry, University of British Columbia) for the use of APP23 transgenic mice and Dr. Patrick McGeer (Department of Psychiatry, University of British Columbia) and Dr. Douglas Walker (Laboratory of Neuroinflammation, Sun Health Research Institute, Sun City, AZ) for AD brain tissue and AD microglia, respectively.
References
- Aarum J, Sandberg K, Haeberlein SL, Persson MA. Migration and differentiation of neural precursor cells can be directed by microglia. Proc Natl Acad Sci U S A. 2003;100:15983–15988. doi: 10.1073/pnas.2237050100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambati BK, Nozaki M, Singh N, Takeda A, Jani PD, Suthar T, Albuquerque RJ, Richter E, Sakurai E, Newcomb MT, Kleinman ME, Caldwell RB, Lin Q, Ogura Y, Orecchia A, Samuelson DA, Agnew DW, St Leger J, Green WR, Mahasreshti PJ, et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature. 2006;443:993–997. doi: 10.1038/nature05249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Miklossy J, Klegeris A, Guo JP, McGeer PL. Thrombin and prothrombin are expressed by neurons and glial cells and accumulate in neurofibrillary tangles in Alzheimer disease brain. J Neuropathol Exp Neurol. 2006;65:19–25. doi: 10.1097/01.jnen.0000196133.74087.cb. [DOI] [PubMed] [Google Scholar]
- Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marmé D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87:3336–3343. [PubMed] [Google Scholar]
- Casolini P, Domenici MR, Cinque C, Alemà GS, Chiodi V, Galluzzo M, Musumeci M, Mairesse J, Zuena AR, Matteucci P, Marano G, Maccari S, Nicoletti F, Catalani A. Maternal exposure to low levels of corticosterone during lactation protects the adult offspring against ischemic brain damage. J Neurosci. 2007;27:7041–7046. doi: 10.1523/JNEUROSCI.1074-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HB, Ryu JK, Kim SU, McLarnon JG. Modulation of the purinergic P2X7 receptor attenuates lipopolysaccharide-mediated microglial activation and neuronal damage in inflamed brain. J Neurosci. 2007;27:4957–4968. doi: 10.1523/JNEUROSCI.5417-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CK, Karlo JC, Kao SC, Landreth GE. β-Amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21:1179–1188. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bandt M, Ben Mahdi MH, Ollivier V, Grossin M, Dupuis M, Gaudry M, Bohlen P, Lipson KE, Rice A, Wu Y, Gougerot-Pocidalo MA, Pasquier C. Blockade of vascular endothelial growth factor receptor I (VEGF-RI), but not VEGF-RII, suppresses joint destruction in the K/BxN model of rheumatoid arthritis. J Immunol. 2003;171:4853–4859. doi: 10.4049/jimmunol.171.9.4853. [DOI] [PubMed] [Google Scholar]
- Feistritzer C, Kaneider NC, Sturn DH, Mosheimer BA, Kähler CM, Wiedermann CJ. Expression and function of the vascular endothelial growth factor receptor Flt-1 in human eosinophils. Am J Respir Cell Mol Biol. 2004;30:729–735. doi: 10.1165/rcmb.2003-0314OC. [DOI] [PubMed] [Google Scholar]
- Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC, Vinters HV. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer's disease brain and damage the blood-brain barrier. Eur J Clin Invest. 2002;32:360–371. doi: 10.1046/j.1365-2362.2002.00994.x. [DOI] [PubMed] [Google Scholar]
- Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- Forstreuter F, Lucius R, Mentlein R. Vascular endothelial growth factor induces chemotaxis and proliferation of microglial cells. J Neuroimmunol. 2002;132:93–98. doi: 10.1016/s0165-5728(02)00315-6. [DOI] [PubMed] [Google Scholar]
- Franciosi S, Ryu JK, Choi HB, Radov L, Kim SU, McLarnon JG. Broad-spectrum effects of 4-aminopyridine to modulate amyloid β1–42-induced cell signaling and functional responses in human microglia. J Neurosci. 2006;26:11652–11664. doi: 10.1523/JNEUROSCI.2490-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JP, Arai T, Miklossy J, McGeer PL. Abeta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:1953–1958. doi: 10.1073/pnas.0509386103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hashemzadeh-Bonehi L, Phillips RG, Cairns NJ, Mosaheb S, Thorpe JR. Pin1 protein associates with neuronal lipofuscin: potential consequences in age-related neurodegeneration. Exp Neurol. 2006;199:328–338. doi: 10.1016/j.expneurol.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Hoshino M, Takahashi M, Aoike N. Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunoreactivity in asthmatic airways and its relationship to angiogenesis. J Allergy Clin Immunol. 2001;107:295–301. doi: 10.1067/mai.2001.111928. [DOI] [PubMed] [Google Scholar]
- Lue LF, Brachova L, Walker DG, Rogers J. Characterization of glial cultures from rapid autopsies of Alzheimer's and control patients. Neurobiol Aging. 1996;17:421–429. doi: 10.1016/0197-4580(96)00006-1. [DOI] [PubMed] [Google Scholar]
- Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM, Jr, Brachova L, Yan SD, Walker DG, Shen Y, Rogers J. Inflammatory repertoire of Alzheimer's disease and nondemented elderly microglia in vitro. Glia. 2001;35:72–79. doi: 10.1002/glia.1072. [DOI] [PubMed] [Google Scholar]
- McLarnon JG, Choi HB, Lue LF, Walker DG, Kim SU. Perturbations in calcium-mediated signal transduction in microglia from Alzheimer's disease patients. J Neurosci Res. 2005;81:426–435. doi: 10.1002/jnr.20487. [DOI] [PubMed] [Google Scholar]
- McLarnon JG, Ryu JK, Walker DG, Choi HB. Upregulated expression of purinergic P2X(7) receptor in Alzheimer disease and amyloid-beta peptide-treated microglia and in peptide-injected rat hippocampus. J Neuropathol Exp Neurol. 2006;65:1090–1097. doi: 10.1097/01.jnen.0000240470.97295.d3. [DOI] [PubMed] [Google Scholar]
- Miklossy J, Qing H, Guo JP, Yu S, Wszolek ZK, Calne D, McGeer EG, McGeer PL. Lrrk2 and chronic inflammation are linked to pallido-ponto-nigral degeneration caused by the N279K tau mutation. Acta Neuropathol. 2007;114:243–254. doi: 10.1007/s00401-007-0230-9. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. Common inflammatory mechanisms in Lewy body disease and Alzheimer disease. J Neuropathol Exp Neurol. 2007;66:683–686. doi: 10.1097/nen.0b013e31812503e1. [DOI] [PubMed] [Google Scholar]
- Murakami M, Iwai S, Hiratsuka S, Yamauchi M, Nakamura K, Iwakura Y, Shibuya M. Signaling of vascular endothelial growth factor receptor-1 tyrosine kinase promotes rheumatoid arthritis through activation of monocytes/macrophages. Blood. 2006;108:1849–1856. doi: 10.1182/blood-2006-04-016030. [DOI] [PubMed] [Google Scholar]
- Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, Carlson GA, Iadecola C. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid β peptide. J Neurosci. 2005;25:1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogue AI, Lukiw WJ. Angiogenic signaling in Alzheimer's disease. Neuroreport. 2004;15:1507–1510. doi: 10.1097/01.wnr.0000130539.39937.1d. [DOI] [PubMed] [Google Scholar]
- Prat A, Biernacki K, Wosik K, Antel JP. Glial cell influence on the human blood-brain barrier. Glia. 2001;36:145–155. doi: 10.1002/glia.1104. [DOI] [PubMed] [Google Scholar]
- Ryu JK, McLarnon JG. Minocycline or iNOS inhibition block 3-nitrotyrosine increases and blood-brain barrier leakiness in amyloid beta-peptide-injected rat hippocampus. Exp Neurol. 2006;198:552–557. doi: 10.1016/j.expneurol.2005.12.016. [DOI] [PubMed] [Google Scholar]
- Ryu JK, McLarnon JG. Thalidomide inhibition of perturbed vasculature and glial-derived tumor necrosis factor-α in an animal model of inflamed Alzheimer's disease brain. Neurobiol Dis. 2008;29:254–266. doi: 10.1016/j.nbd.2007.08.019. [DOI] [PubMed] [Google Scholar]
- Ryu JK, Kim J, Cho SJ, Hatori K, Nagai A, Choi HB, Lee MC, McLarnon JG, Kim SU. Proactive transplantation of human neural stem cells prevents degeneration of striatal neurons in a rat model of Huntington disease. Neurobiol Dis. 2004;16:68–77. doi: 10.1016/j.nbd.2004.01.016. [DOI] [PubMed] [Google Scholar]
- Ryu JK, Choi HB, McLarnon JG. Combined minocycline plus pyruvate treatment enhances effects of each agent to inhibit inflammation, oxidative damage, and neuronal loss in an excitotoxic animal model of Huntington's disease. Neuroscience. 2006;141:1835–1848. doi: 10.1016/j.neuroscience.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Skarsgard ED, Huang L, Reebye SC, Yeung AY, Jia WW. Lentiviral vector-mediated, in vivo gene transfer to the tracheobronchial tree in fetal rabbits. J Pediatr Surg. 2005;40:1817–1821. doi: 10.1016/j.jpedsurg.2005.08.047. [DOI] [PubMed] [Google Scholar]
- Sköld MK, von Gertten C, Sandberg-Nordqvist AC, Mathiesen T, Holmin S. VEGF and VEGF receptor expression after experimental brain contusion in rat. J Neurotrauma. 2005;22:353–367. doi: 10.1089/neu.2005.22.353. [DOI] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkowski E, Issa R, Sjögren M, Wallin A, Blennow K, Tarkowski A, Kumar P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer's disease and vascular dementia. Neurobiol Aging. 2002;23:237–243. doi: 10.1016/s0197-4580(01)00285-8. [DOI] [PubMed] [Google Scholar]
- Tran KC, Ryu JK, McLarnon JG. Induction of angiogenesis by platelet-activating factor in the rat striatum. Neuroreport. 2005;16:1579–1583. doi: 10.1097/01.wnr.0000179073.24412.7b. [DOI] [PubMed] [Google Scholar]
- Vagnucci AH, Jr, Li WW. Alzheimer's disease and angiogenesis. Lancet. 2003;361:605–608. doi: 10.1016/S0140-6736(03)12521-4. [DOI] [PubMed] [Google Scholar]
- Walker DG, Kim SU, McGeer PL. Complement and cytokine gene expression in cultured microglia derived from post-mortem brains. J Neurosci Res. 1995;40:478–493. doi: 10.1002/jnr.490400407. [DOI] [PubMed] [Google Scholar]
- Walker DG, Link J, Lue LF, Dalsing-Hernandez JE, Boyes BE. Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J Leukoc Biol. 2006;79:596–610. doi: 10.1189/jlb.0705377. [DOI] [PubMed] [Google Scholar]
- Yang MS, Ji KA, Jeon SB, Jin BK, Kim SU, Jou I, Joe E. Interleukin-13 enhances cyclooxygenase-2 expression in activated rat brain microglia: implications for death of activated microglia. J Immunol. 2006;177:1323–1329. doi: 10.4049/jimmunol.177.2.1323. [DOI] [PubMed] [Google Scholar]
- Zipser BD, Johanson CE, Gonzalez L, Berzin TM, Tavares R, Hulette CM, Vitek MP, Hovanesian V, Stopa EG. Microvascular injury and blood-brain barrier leakage in Alzheimer's disease. Neurobiol Aging. 2007;28:977–986. doi: 10.1016/j.neurobiolaging.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]