

Abstract
Here we report that a serotonergic neuron evokes two distinct neuromodulatory actions with different state, timing, and firing pattern dependencies. These neuromodulatory actions may have important behavioral functions. In the mollusc, Tritonia diomedea, EPSCs evoked by ventral swim interneuron B (VSI) exhibited intrinsic plasticity; after a spike train, EPSC amplitude increased from a basal state to a potentiated state, which usually lasted >10 min. While the synapse was in a potentiated state, stimulation of a serotonergic dorsal swim interneuron (DSI) decreased VSI synaptic strength, returning it to a basal state. The extent of the DSI-evoked decrement was strongly correlated with the magnitude of the homosynaptic potentiation. This synaptic reset, or depotentiation, by DSI was blocked by the serotonin receptor antagonist methysergide and mimicked by a serotonin puff. In contrast to this state-dependent neuromodulatory action, we found that a previously described DSI-evoked transient enhancement of VSI synaptic strength was state-independent, producing the same multiplicative increase in EPSC amplitude regardless of whether the synapse was in a potentiated or basal state. These two actions also differed in their dependencies on the firing pattern of DSI and VSI action potentials. Results suggest that state-independent synaptic enhancement by DSI may play a short-term role during a swim motor pattern, whereas state-dependent actions may have longer-lasting consequences, resetting VSI synaptic strength after a swim bout. Thus, differences in two neuromodulatory actions at one synapse may allow a serotonergic neuron to play distinct roles at different stages of a motor pattern.
Keywords: serotonin, synaptic plasticity, posttetanic, potentiation, depression, presynaptic facilitation, presynaptic mechanisms, synaptic transmission, central pattern generator, mollusc
Introduction
Neuromodulation has been defined as an effect of one neuron on the cellular and/or synaptic properties of another neuron (Katz, 1999). There may be a temptation to infer from this that neuromodulatory actions are imposed unilaterally on other cells and synapses. However, neuromodulatory signaling may also be contingent on the state of the neuron or synapse being modulated. For example, in rat prefrontal cortex, the basal level of protein kinase A activity can determine whether serotonin [5-hydroxytryptamine (5-HT)] will enhance or reduce the amplitude of GABA-evoked currents (Cai et al., 2002). Similarly, the level of neuronal activity determines the direction of change produced by dopaminergic modulation of calmodulin-dependent protein kinase II (Gu and Yan, 2004). Determining whether such state-dependent neuromodulatory effects occur in neuronal circuits requires the ability to monitor and perturb the neuromodulating neuron as well as the state of the neuron that it modulates.
Such experimental control can be obtained using the neuronal circuit underlying the escape swim response of the mollusc, Tritonia diomedea. In this preparation, it has been shown that identified serotonergic neurons called the dorsal swim interneurons (DSIs) (http://NeuronBank.org/Tri0001043) evoke short-term neuromodulatory actions on other swim interneurons (Katz and Frost, 1995; Sakurai and Katz, 2003). In particular, DSI stimulation transiently enhances the synaptic strength of ventral swim interneuron B (VSI) (http://NeuronBank.org/Tri0002436). This heterosynaptic enhancement decays within 15 s after the end of a DSI spike train, making it contingent on the timing of spikes in the DSI and VSI.
Here, we found that the VSI synapses can undergo activity-dependent changes in synaptic output from a stable basal state to a potentiated state and that the DSI-evoked transient enhancement of VSI synaptic strength is independent of its current potentiation state. We also determined that DSI evokes an additional state-dependent neuromodulatory action; when the VSI synapse is in the potentiated state, DSI stimulation can depotentiate it, returning it to a basal state. The efficacies of these two neuromodulatory actions differ depending on the pattern of DSI and VSI spiking activity. The recognition that a single neuron can evoke both state-dependent and state-independent actions at the same synapse and that these actions can have different spike-timing dependencies could have important implications for understanding the roles of neuromodulation and plasticity in the nervous system.
Materials and Methods
Preparation.
Specimens of the nudibranch, T. diomedea, were obtained from Living Elements. All experiments were performed on the isolated brain preparation. Briefly, the brain, consisting of the fused cerebropleural and pedal ganglia, was removed from the animal and immediately pinned to the bottom of a Sylgard-lined chamber (1 ml), where it was superfused with saline at 4°C. Physiological saline composition was (in mm): 420 NaCl, 10 KCl, 10 CaCl2, 50 MgCl2, 10 d-glucose, and 10 HEPES, pH 7.4. The cell bodies of the neurons were exposed by removing the connective tissue sheath from the surface of the ganglia (Willows et al., 1973). Suction electrodes, made from polyethylene tubing, were placed on pedal nerves including pedal nerves 3, 5, and 6. Then the preparation was left overnight, superfused in saline at 8–10°C to recover from possible damage during the dissection.
Neurons were identified by soma location, coloration, synaptic connectivity, and activity pattern at rest and during the swim motor program as previously described (Getting, 1981, 1983). For simplicity, we will refer to VSI-B as VSI for the remainder of this study. To identify neurons, the swim motor program was evoked by stimulating pedal nerve 3 with a train of voltage pulses (5–15 V, 1.5 ms) at 5 Hz for 1.5 s via a suction electrode. After identifying the interneurons, the bathing medium was switched to saline containing a high concentration of divalent cations (Hi-Di saline), which raises the threshold for spiking and reduces spontaneous neural firing. The composition of the Hi-Di saline was (in mm): 285 NaCl, 10 KCl, 25 CaCl2, 125 MgCl2, 10 d-glucose, and 10 HEPES, pH 7.4 (Sakurai and Katz, 2003). All experiments were conducted in the Hi-Di saline superfused at 2 ml/min at 10°C.
5-HT (Sigma) was dissolved in Hi-Di saline (100 μm) just before use and puff-applied via glass pipette using a Picospritzer (General Valve). The puffs were given for 5–10 ms duration, one to three times at 1–2 Hz. Methysergide (Sigma) was dissolved in dimethyl sulfoxide (Sigma) at 20 mm and diluted to 50 μm in Hi-Di saline just before use, and bath-applied by switching the superfusion paths (Sakurai and Katz, 2003).
Electrophysiological recordings and stimulations.
Neurons were impaled with glass microelectrodes filled with 3 m KCl (resistance, 8–15 MΩ). Axoclamp-2B amplifiers (Molecular Devices) were used for all electrophysiological experiments. To examine changes in the synaptic strength of VSI, orthodromic action potentials were evoked by injecting current pulses (4–15 nA, 20 ms) into the soma through a second electrode, or through the recording electrode with a balanced bridge circuit. Either a ventral flexion neuron (VFN) or an unidentified VSI follower cell in the ipsilateral pedal ganglion had its membrane potential held at −50 mV under two-electrode voltage-clamp mode to measure VSI-evoked EPSCs.
Action potentials in DSI were elicited by injecting 20 ms current pulses (7–15 nA) through a recording electrode via a balanced bridge circuit. Each current pulse evoked a single spike, allowing the spike frequency to be controlled precisely. To bring the VSI synapse to the potentiated state (compare Fig. 1), VSI was stimulated at 5 Hz for 15 s in most of the present experiments (Sakurai et al., 2007). The frequency 5 Hz was chosen because the stimulus at higher rate often caused failure in generating action potential one-to-one to the stimulus current pulses. The time 15 s was chosen because the VSI action potentials started to fail after 15 s.
Figure 1.
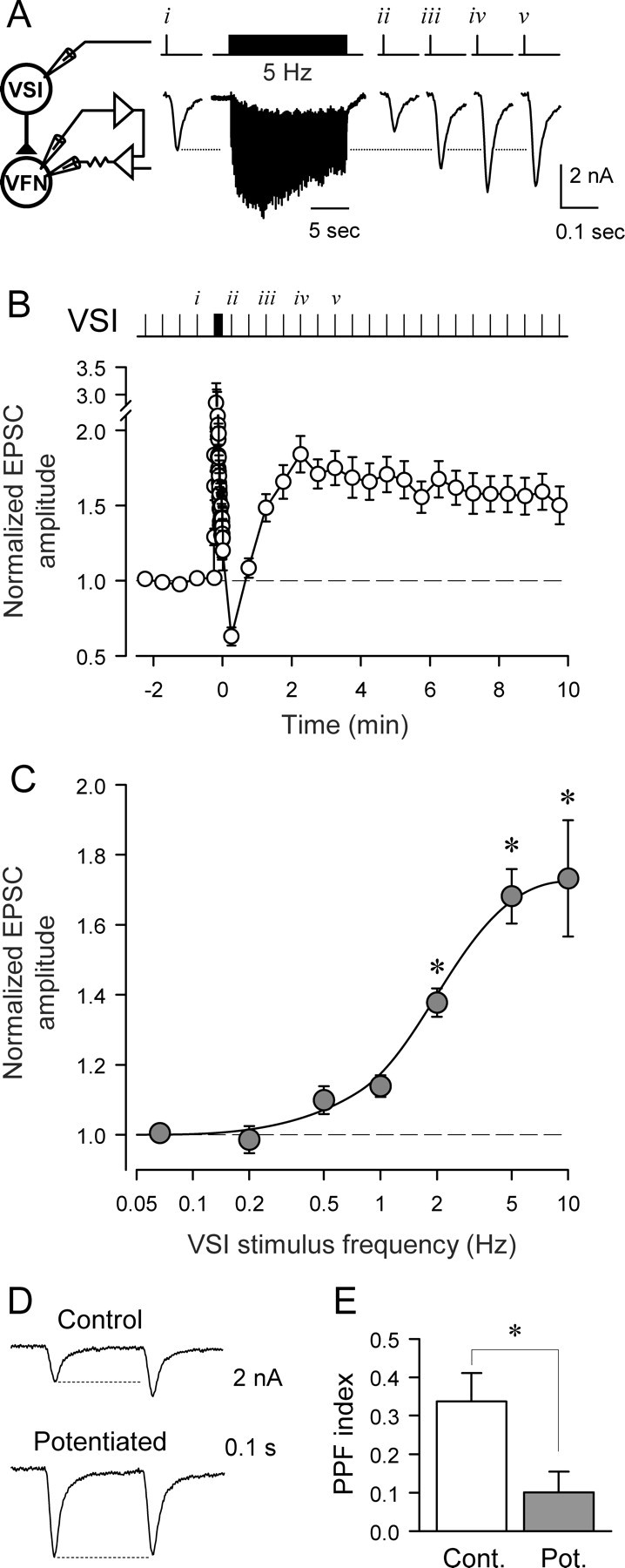
Homosynaptic potentiation of the VSI-VFN synapse. A, The experimental paradigm for inducing the potentiation state. In Hi-Di saline, VSI was stimulated with intracellular current pulses (20 ms, 7–10 nA) to fire one action potential every 30 s while recording EPSCs from VFN under voltage clamp. After the establishment of baseline EPSC amplitude by 3–5 stimuli, VSI was stimulated at 5 Hz for 15 s (76 pulses), and then the interval was set back to 30 s again. B, Averaged data show a sustained increase in the amplitude of EPSCs after the 5 Hz VSI stimulation (n = 22). Time 0 is set as the time of the last VSI action potential in the 5 Hz train. After the VSI spike train, the EPSC showed a transient decrease followed by sustained potentiation state. The EPSC amplitudes were normalized to the mean amplitude of three EPSCs recorded before the VSI spike train. C, The magnitude of the potentiation increased with frequency of VSI spiking (F(6,81) = 30.2, p < 0.001, by one-way ANOVA). All trains were 15 s in duration except for 0.5 Hz, which was 16 s. Asterisks indicate a significant difference from the baseline amplitude at 0.067 Hz (30 s interval) (p < 0.05 by pairwise multiple comparison by Student–Newman–Keuls method). There was no significant difference between 5 Hz and 10 Hz (p = 0.63 by pairwise multiple comparison by Student–Newman–Keuls method). D, The potentiation state affects frequency facilitation. Paired pulse stimuli (0.2 s interval) were given before (Control, upper trace) and after establishment of the potentiated state (lower trace). The extent of facilitation was markedly reduced when the synapse was in the potentiated state. E, A bar graph showing the paired pulse facilitation (PPF) index, which was calculated by dividing the change in the amplitude of EPSC by the amplitude of the first EPSC, showing the relative increase in the second EPSC (p < 0.05, paired t test, n = 10).
To calculate the extent of potentiation state (p), the extent of decrement (d), and the extent of DSI-evoked enhancement (e) (see Figs. 3 and 5), we first calculated the basal EPSC amplitudes by averaging three EPSCs before the VSI spike train. Then, the potentiated EPSC amplitude was calculated by averaging three EPSCs before the DSI stimulation. The p value is the difference between the basal and potentiated EPSC amplitudes shown as a percentage of the basal EPSC amplitude. The d value was calculated by subtracting the potentiated EPSC amplitude from the average of three EPSCs evoked after the DSI spike train, and shown as a percentage change of the potentiated EPSC amplitude. The e value was calculated by subtracting the average of three EPSCs before the DSI spike train from the EPSC evoked 5 s after the DSI spike train, and shown as a percentage change. The depotentiation (see Figs. 6B,D, 8Aiii,Biii) is the DSI-evoked decrement shown as a percentage change of the magnitude of the potentiation state, which is the difference between the basal and potentiated EPSC amplitudes.
Figure 3.
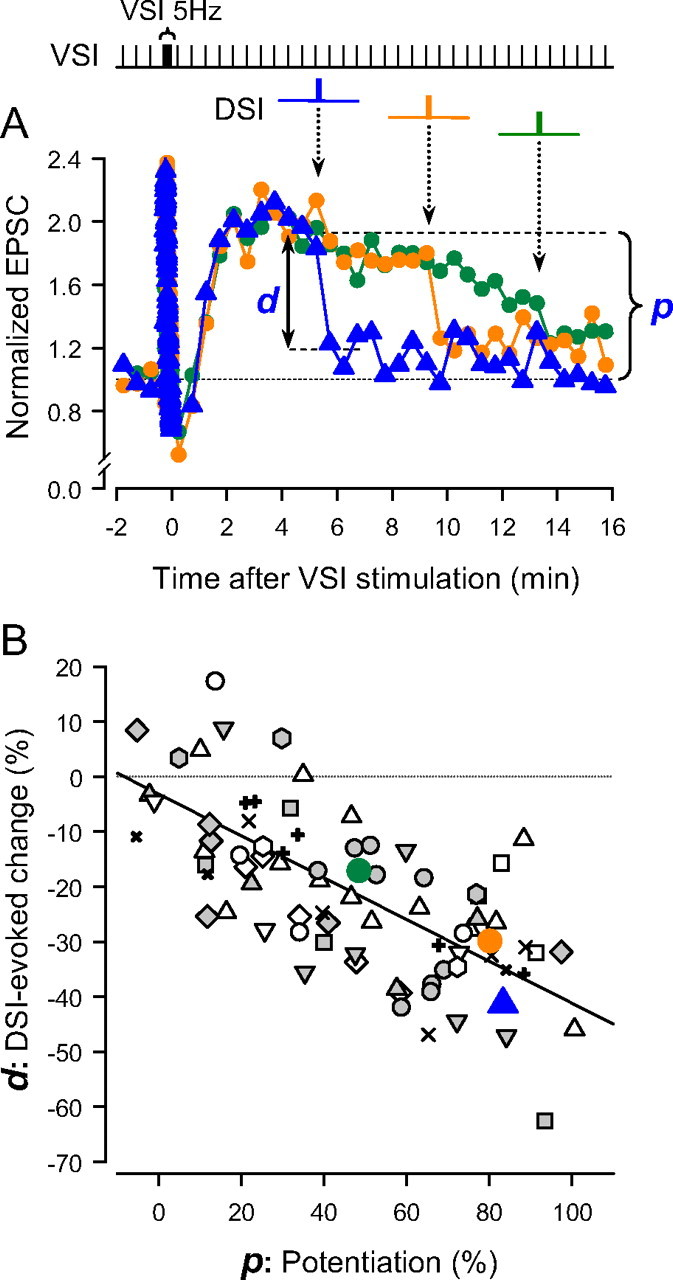
The extent of DSI-evoked decrement is dependent on the magnitude of the potentiation. A, Overlaid plots of the normalized EPSCs against time (seconds). Time 0 is set as the time of the last action potential in the VSI spike train (5 Hz, 15 s). In each plot, a DSI spike train was stimulated at three different times with respect to the end of the VSI train (blue, 315 s; orange, 555 s; green, 795 s). The amplitudes were normalized to the average of 3 EPSCs before the VSI tetanus. The bracket and the arrow indicate the extent of potentiation (p) at the time of DSI stimulation and decrement (d) produced by DSI for the trial indicated in blue. B, The extent of DSI-evoked decrement is correlated to the amount of the potentiation state (R2 = 0.47). The DSI-evoked decrement (%) was plotted as a function of the magnitude of potentiation (%). The p values were shown as a percentage change from the basal EPSC amplitude, whereas the d values as a percentage change from the potentiated EPSC amplitude (see Materials and Methods for details). Data from 15 preparations are plotted; each symbol represents data from a single preparation. There was a negative-slope, linear relationship between the extent of the decrement and the degree of potentiation (R2 = 0.47, F(1,76) = 59.4, p < 0.001 by ANOVA).
Figure 5.
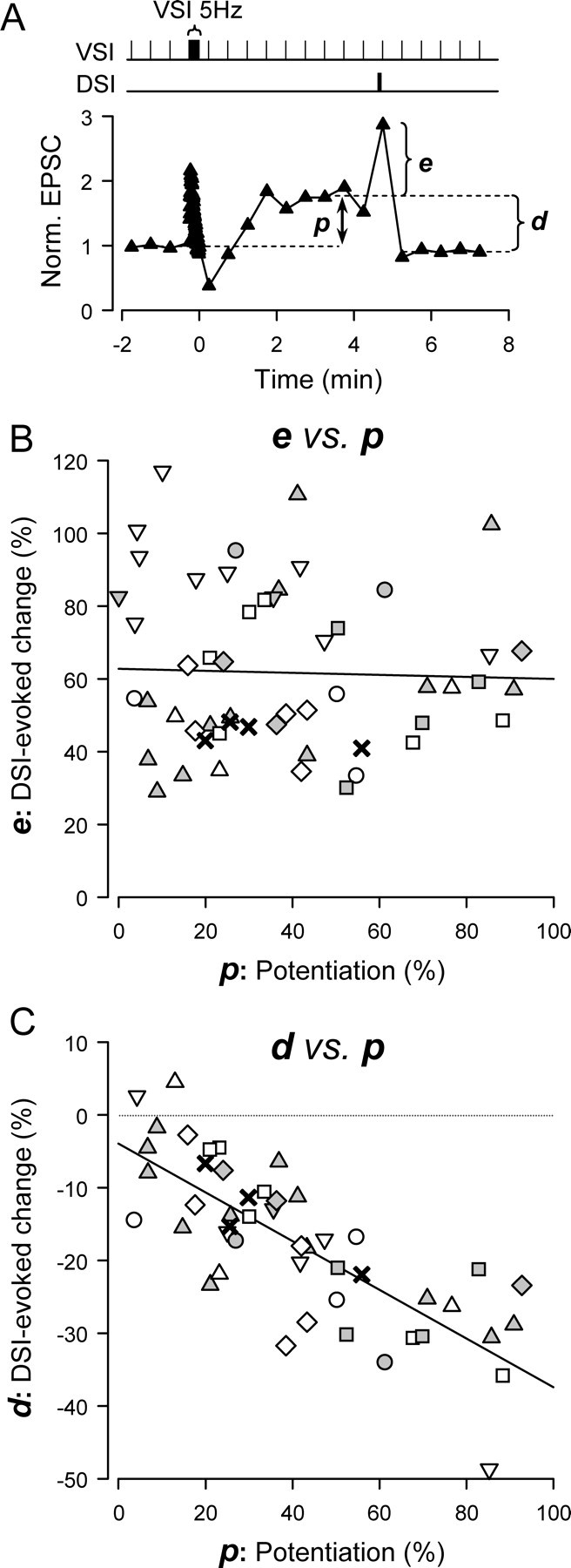
DSI stimulation caused a state-independent transient increase and a state-dependent decrease of VSI EPSC amplitude. A, A plot of a single trial showing the normalized EPSC amplitude over time. VSI was stimulated to fire one action potential every 30 s with a single train of action potentials (5 Hz, 15 s) inserted into the paradigm. Time 0 is set as the time of the last action potential in the VSI spike train. The EPSC amplitude increased to the potentiated state (p). DSI stimulation (10Hz, 5 s) ending 5 s before the next VSI spike caused a transient enhancement of the EPSC amplitude (e) and the subsequent decrement (d). The EPSCs were normalized to the average of 3 EPSCs before the VSI train. B, C, Plots from 59 individual trials across nine preparations showing the relationships of enhancement (e) and decrement (d) to the extent of potentiation (p) in each trial. Each symbol represents data from a single preparation. B, The extent of DSI-evoked transient enhancement was not correlated with the extent of potentiation. The p values are shown as a percentage change from the basal EPSC amplitude, whereas the e values are shown as a percentage change from the potentiated EPSC amplitude (see Materials and Methods for details). The regression line is almost flat (R2 = 0.001, F(1,52) = 0.06, p = 0.81 by ANOVA). C, The magnitude of the DSI-evoked decrement after the initial enhancement was correlated with the extent of potentiation (R2 = 0.61, F(1,46) = 70.7, p < 0.001 by ANOVA). Calculations for p and d values are the same as in Figure 3.
Figure 6.

5-HT mediates the DSI-evoked depotentiation. A, The DSI-evoked depotentiation was blocked by a 5-HT receptor blocker, methysergide. VSI-evoked EPSC amplitudes from two trials in the same preparation are plotted; one was recorded in normal high divalent cation saline (Normal Hi-Di, white circles) and the other in the presence of 50 μm methysergide (black triangles). In both trials, VSI was stimulated to fire a train (5 Hz, 15 s) to put the synapse in a potentiated state and DSI was subsequently stimulated (10 Hz, 5 s) 25 s before the next VSI spike. Depotentiation of the synapse occurred in normal Hi-Di saline, but this depotentiation was reduced significantly by methysergide. B, Methysergide significantly reduced the extent of depotentiation caused by DSI stimulation. The extent of depotentiation was shown as a percentage change of the amplitude from the potentiation state (n = 4). C, A brief 5-HT puff mimicked the effect of a DSI spike train. EPSC amplitudes from two trials from the same preparation are plotted. In both trials, a VSI spike train (5 Hz, 15 s) caused the synapse to enter the potentiated state. The control trace (white circles) shows the time course of potentiation in this preparation. A pressure-applied 5-HT puff (100 μm, 10 ms) produced a large transient increase and subsequent fall of the EPSC amplitude, bringing it down to the original level (gray triangles). D, 5-HT puffs caused significantly more decrement than occurred in control trials with no puff (n = 4). In A and C, time 0 is set as the time of the last action potential in the VSI spike train (5 Hz, 15 s). In B and D, asterisks indicate significant differences (p < 0.01 by paired t test).
Figure 8.

DSI-evoked enhancement and depotentiation differ in their integration times. A, The effect of holding the duration of a DSI spike train constant and varying spike frequency (i). The magnitudes of both the transient enhancement (ii) and the depotentiation (iii) increased with frequency (or number) of DSI spikes (ii, F(4,33) = 20.4, p < 0.001; iii, F(4,19) = 48.2, p < 0.001, by one-way ANOVA). B, The effect of holding the number of DSI spikes constant and varying spike frequency (i). The magnitudes of the transient enhancement (ii) increased with DSI spike frequency (F(4,44) = 27.9, p < 0.001, by one-way ANOVA). The extent of the DSI-evoked depotentiation (iii) also increased with increasing spike frequency (F(4,17) = 153.6, p < 0.001, by one-way ANOVA), but a significant change from control was seen even at low spike frequencies (0–2 Hz, p < 0.05 by pairwise multiple comparison by Student–Newman–Keuls method). No further increase in the extent of depotentiation was seen at higher DSI spike frequencies (2–10 Hz, p > 0.23 by pairwise multiple comparison by Student–Newman–Keuls method).
Recordings were digitized at 3–20 kHz with a 1401plus A/D converter from Cambridge Electronic Design (CED). For data analysis and plots, the amplitudes of EPSCs were normalized to the averaged amplitude of three EPSCs before the test stimulation. Data acquisition and analysis were performed with Spike2 software (CED) and SigmaPlot (Jandel Scientific). Statistical comparisons were made using a paired t test or a one-way ANOVA with pairwise multiple comparison by the Student–Newman–Keuls method. In all cases, p < 0.05 was considered significant. Results are expressed as the mean ± SEM.
Results
VSI synapses could enter a long-lasting potentiation state
VSI exhibited a number of forms of intrinsic or homosynaptic plasticity, i.e., changes in the synaptic output of VSI that depended on its own firing history. Baseline VSI-evoked EPSC amplitudes recorded in a postsynaptic VFN were stable when VSI was stimulated with intracellular current pulses (20 ms, 7nA) to fire one action potential every 30 s. However, after a 15 s, 5 Hz train of action potentials, the VSI-evoked EPSCs switched into a prolonged potentiation state (Fig. 1. During the 5 Hz train, the initial EPSCs rapidly facilitated before depressing to a steady-state amplitude as previously reported for this synapse (Sakurai et al., 2007) (Fig. 1A). For 15–30 s after the end of the train, the EPSC amplitude was strongly depressed relative to the baseline before the train (Fig. 1A,B). However, over the course of ∼90 s, the EPSC amplitude increased above baseline levels to a potentiated state, where it remained, in most preparations, for ∼5–15 min. The exact duration of the potentiation state was variable between preparations. Of the 22 preparations tested, at least 15 remained potentiated for >10 min, with one lasting more than 1 h.
The magnitude of the potentiation state was dependent on the VSI spike frequency during the train (Fig. 1C). Analysis with one-way ANOVA shows that there was a progressive increase in the extent of potentiation as VSI was made to spike at higher frequencies (Fig. 1C legend). The maximal potentiation occurred when VSI was stimulated to fire at 5 Hz or above. The VSI spike frequency needed to produce a half-maximal response was 2.1 Hz. During normal swim motor patterns, VSI fired at frequencies ranging from 4.3 to 12.0 Hz with a median of 8.9 Hz (n = 13). Thus, the firing rates needed to cause potentiation are well within the physiological range.
To test whether the potentiation state is caused by presynaptic changes in neurotransmitter release from VSI, we examined its effect on paired-pulse facilitation (Fig. 1D), a measure typically associated with changes in synaptic release (Zucker and Regehr, 2002). In the unpotentiated state, the paired-pulse facilitation (PPF) index for VSI-evoked EPSCs with a 0.2 s interval was 0.34 ± 0.07 (n = 10). This ratio decreased significantly to 0.10 ± 0.05 (n = 10) when the synapse was in the potentiated state (Fig. 1E), suggesting that the potentiation state at least involves presynaptic changes at the synapse.
DSI stimulation caused state-dependent synaptic depotentiation
DSI stimulation caused a marked decrement of VSI synaptic strength, but only when the synapse was already in the potentiated state. When the VSI to VFN synapse was in the basal, unpotentiated state, stimulating DSI to fire a train of action potentials (10 Hz, 5 s) that ended 25 s before a VSI test spike had no effect on subsequent VSI-evoked EPSCs recorded in VFN (Fig. 2 Bi,Ci). However, when the VSI to VFN synapse was in the potentiated state after a VSI spike train, DSI stimulation caused a large decrement of the EPSCs that returned the amplitudes of VSI-evoked EPSCs to the baseline level recorded before the VSI spike train (Fig. 2Bii, 3Cii).
Figure 2.

Heterosynaptic depotentiation by a DSI spike train. DSI reduced the VSI-VFN EPSC amplitude only when the synapse was in the potentiated state. A, Experimental protocol. Intracellular electrodes were placed in DSI and VSI for eliciting action potentials. The membrane potential of the postsynaptic VFN was held constant by two-electrode voltage clamp. VSI was stimulated to fire one action potential every 30 s. DSI was stimulated to fire a train of action potentials (10 Hz, 5 s) that ended 25 s before the next VSI spike. B, DSI stimulus trials were delivered once before (i) and twice after (ii, iii) a VSI spike train (5 Hz, 15 s). B, EPSC amplitudes from two recording sessions in the same neurons, one without DSI stimulation (white circles) and one showing three trials of DSI stimulation (i, ii, iii, black triangles:). The first DSI spike train (i) had no effect on EPSC amplitude. The 5 Hz VSI spike train shifted the EPSCs into a potentiated state. In the potentiated state, a DSI spike train depotentiated the EPSCs (ii). A subsequent DSI train had little additional effect (iii). C, The mean amplitudes of VSI-evoked EPSCs normalized to the basal state are shown from three conditions: i, basal state (before VSI spike train, n = 8); ii, potentiated state (first trial after VSI train, n = 12); iii, basal state (second trial after VSI train, n = 8). The DSI spike train is represented by the arrow. The insets show the EPSC recordings before and 25 s after the DSI stimulation.
In 73% of the preparations, the EPSC decrement was apparently permanent; the EPSC remained at the nonpotentiated level for at least 10 min (n = 9 in 12 preparations), and additional DSI stimuli had no further effect (Fig. 2Biii, Ciii). However, in 3 of 12 preparations, the EPSCs partially reverted to the potentiated level after the first DSI stimulation, but a second DSI spike train again reset the EPSC amplitude to the baseline level.
Analysis with one-way ANOVA shows that DSI-evoked decrement in the EPSC size was significant when the VSI synapse was in a potentiated state (ii, F(12,142) = 15.42, p < 0.001, n = 12). Pairwise multiple comparisons by the Student–Newman–Keuls method showed significant differences between the EPSCs before the DSI stimulation and those after it (p < 0.001). In contrast, DSI had little effect when the synapse was in the basal state before the VSI spike train (i, F(12,77) = 0.44, p = 0.94, n = 8), or by the second DSI stimulation after the VSI spike train (iii, F(12,87) = 0.91, p = 0.54, n = 8). Thus, the ability of DSI to cause VSI synaptic decrement was dependent on the state of the VSI synapse.
The extent of the DSI-evoked decrement of VSI synaptic strength was correlated with the magnitude of the potentiation state exhibited at the time of the DSI stimulation (Fig. 3). As the potentiation state slowly decayed, the synaptic decrement caused by DSI stimulation correspondingly decreased (Fig. 3A, d), rarely exceeding the magnitude of the remaining potentiation (Fig. 3A, p). Indeed, there was a linear relation between the extent of the decrement and the degree of potentiation (Fig. 3B). These results suggest that the DSI-evoked decrement was a negation of the potentiation state rather than a scaling of synaptic strength.
DSI stimulation caused a state-independent synaptic enhancement
We previously showed that DSI stimulation causes a transient enhancement of VSI synaptic strength that lasts up to 15 s (Sakurai and Katz, 2003). This enhancement is not seen in Figures 2 and 3 because VSI was stimulated 25 s or more after the end of the DSI train. To test whether the transient enhancement was affected by the potentiation state of the VSI synapse, we shifted the time of the DSI spike train so that it ended just 5 s before the next VSI test spike (Fig. 4, inset). We found that this stimulus paradigm produced a transient enhancement of VSI synaptic strength regardless of whether the synapse was in a potentiated state or not (Fig. 4B,C).
Figure 4.

DSI produces both a transient enhancement and a state-dependent decrement of VSI-evoked synaptic currents. A, The experimental protocol is the same as in Figure 2 except that the DSI spike trains ended 5 s before the next VSI spike. VSI was stimulated to fire one spike every 30 s. B, EPSC amplitudes from two recording sessions in the same neurons, one without DSI stimulation (white circles) and one showing three trials of DSI stimulation (black triangles: i, ii, iii). In each trial, DSI stimulation increased the amplitude of the EPSC evoked 5 s after the DSI train, but not 35 s later. The 5 Hz VSI spike train shifted the EPSCs into a potentiated state. In the potentiated state, a DSI spike train transiently increased the EPSC and then caused a prolonged depotentiation (ii). A subsequent DSI train again caused a transient increase, but did not produce any further decrease in EPSC amplitude (iii). C, Plots showing the averaged effects of DSI on the EPSC amplitude in the basal state (i, n = 10), the potentiated state (ii, n = 9), and the depotentiated state (iii, n = 7). In all three cases, there was a significant increase in the EPSC amplitude 5 s after the DSI stimulus (p < 0.001, by paired t test). Insets show example EPSCs before DSI stimulation and 5 and 35 s after DSI stimulation. D, A bar graph showing averaged percentage changes in the amplitude of the EPSCs evoked by VSI spikes 5 s after a DSI spike train. Bars (i, ii, iii) represent data obtained in basal state (i, white), the potentiated state (within 5 min after a VSI spike train) (ii, black), and the depotentiated state (iii, gray). There are no significant differences in the magnitude of changes in the EPSC size among the three (see text). E, A bar graph showing mean values of percentage changes in the amplitude of the EPSCs evoked 35 s after a DSI spike train. Bars (i, ii, iii) are from the same states as in D. The extent of the DSI-evoked decrement was significantly greater during the potentiation state (ii) than during the other two conditions (i, iii) (see text). Asterisks indicate significant differences from the other two bars (p < 0.001 by pairwise multiple comparison by Student–Newman–Keuls method).
Unlike DSI-evoked depotentiation, the magnitude of DSI-evoked transient enhancement was independent of the state of the VSI-VFN synapse; DSI stimulation caused an equivalent enhancement of VSI synaptic strength regardless of whether the synapse was in the basal state, the potentiated state, or the depotentiated state (Fig. 4D). The mean values of the increases were not significantly different from each other (F(2,29) = 0.27, p = 0.77 by one way ANOVA). The DSI-evoked transient enhancement in the EPSC size was 64.6 ± 7.9% (N = 13) in the unpotentiated state (i), 58.1 ± 4.4% (n = 12) in the potentiated state (ii), and 61.2 ± 7.2% (n = 8) in the depotentiated state (iii). Furthermore, the magnitude of the transient enhancement did not correlate with the magnitude of the potentiated state (Fig. 5A, B). Thus, the DSI-evoked enhancement produced an equivalent multiplicative scaling of VSI synaptic strength regardless of whether the synapse was in the basal state, the potentiated state, or the depotentiated state.
In contrast, only if the VSI synapse was already in the potentiated state, was the DSI-evoked transient enhancement followed by a strong decrement (35 s after the DSI train), returning the amplitude of EPSCs to the basal level (Fig. 4Bii,Cii), as we had observed with the 25 s test interval (Fig. 2). Once the synapse was depotentiated, additional DSI stimuli produced only the transient enhancement without any further decrement (Fig. 4Biii,Ciii). The DSI-evoked decrease in EPSP amplitude was 3.5 ± 3.5% (n = 13) in the basal state (i) and 6.0 ± 3.3% (n = 8) in the depotentiated state (iii), whereas in the potentiated state (ii) the mean decrease was 37.5 ± 2.5% (n = 12) (Fig. 4E). The magnitude of the DSI-evoked decrement was significantly greater during the potentiated state (F(2,29) = 39.9, p < 0.001 by one-way ANOVA). Moreover, the extent of decrement showed a correlation with the extent of the potentiation state (Fig. 5C), consistent with the results obtained with the first protocol using a long interval between DSI and VSI (Figs. 2, 3). Thus, unlike the state-independent multiplicative enhancement produced by the same protocol, the DSI-evoked synaptic decrement depended on the state of VSI synapse at the time when DSI was stimulated.
The DSI actions are mediated by serotonin
To determine whether the termination of the potentiation phase is caused by 5-HT released from DSI, we tested the effect of the 5-HT receptor antagonist, methysergide. Methysergide was previously shown to be effective at blocking DSI-evoked transient enhancement (Katz and Frost, 1995; Sakurai and Katz, 2003). In this study, bath-applied methysergide (50 μm) significantly reduced the magnitude of DSI-evoked enhancement from 94.3 ± 9.8% down to 16.4 ± 3.7% (p < 0.01 by paired t test, n = 4). Methysergide also significantly reduced the extent of the DSI-evoked depotentiation from −75.9 ± 2.3% (control) to −21.0 ± 4.0% (methysergide) (p < 0.05 by paired t test, n = 4) (Fig. 6A,B). In contrast, methysergide did not affect either the basal amplitude of VSI-evoked EPSPs (1.76 ± 0.53 nA in control saline, 1.75 ± 0.47 nA in methysergide; p = 0.98 by paired t test, n = 4) or the magnitude of homosynaptic potentiation (62.6 ± 16.8% increase in control saline, 52.5 ± 16.3% increase in methysergide; p = 0.61 by paired t test, n = 4). These results support the hypothesis that 5-HT released from DSI is responsible for the depotentiation.
We found that exogenous serotonin can mimic both the DSI-evoked transient enhancement and the depotentiation. By carefully probing the surface of the ganglion with a pipette containing 100 μm 5-HT, we could locate a spot in which a brief 5-HT puff (5–10 ms) produced a transient enhancement of the VSI-to-VFN EPSC. The enhancement of VSI synapses by puff-applied 5-HT lasted >30 s, longer than the DSI-evoked enhancement (10 s). This is presumably because it took longer for the clearance of puff-applied 5-HT than for 5-HT released by a DSI burst. As a result of this longer time course, we could not examine the effect of 5-HT on depotentiation without also causing transient enhancement with our VSI stimulus paradigm.
To determine whether puffed 5-HT could mimic the DSI depotentiating effect, we stimulated VSI to fire a train of action potentials (5 Hz, 15 s) to cause homosynaptic potentiation and applied a single puff of 5-HT. This caused a transient enhancement followed by depotentiation of the VSI-evoked EPSC to basal levels (Fig. 6C). The extent of depotentiation 90–180 s after the puff was −72.4 ± 8.7%, which is significantly greater than the spontaneous decline of the potentiated state (−22.7 ± 9.6%) that occurred at equivalent times (p < 0.05 by paired t test, n = 4) (Fig. 6D). Thus, puffs of 5-HT could mimic both the transient enhancement and the state-dependent depotentiation of VSI synaptic strength caused by DSI stimulation. Together with the methysergide results, it suggests that 5-HT released by a DSI spike train mediates both state-dependent and state-independent effects.
Timing dependence of depotentiation
After a VSI spike train, it takes ∼90 s for the EPSC amplitude to reach the fully potentiated state. We tested whether DSI stimulation before the full onset of potentiation would be effective at causing depotentiation (Fig. 7). We found that when DSI was stimulated after the end of the VSI train, but during the period of VSI synaptic depression, it did not prevent the subsequent expression of the potentiation state (Fig. 7Ai). By increasing the delay after the end of the VSI train, DSI stimulation became more effective at causing depotentiation (Fig. 7Aii–iv,B). Thus, the ability of DSI to depotentiate the VSI synapse was not only dependent on the state of the VSI synapse, but also on the timing of the VSI spike train with respect to the DSI train. Of course, this apparent time dependence could simply be another manifestation of the state dependence in that it appears to take 90 s for the synapse to reach the potentiated state.
Figure 7.
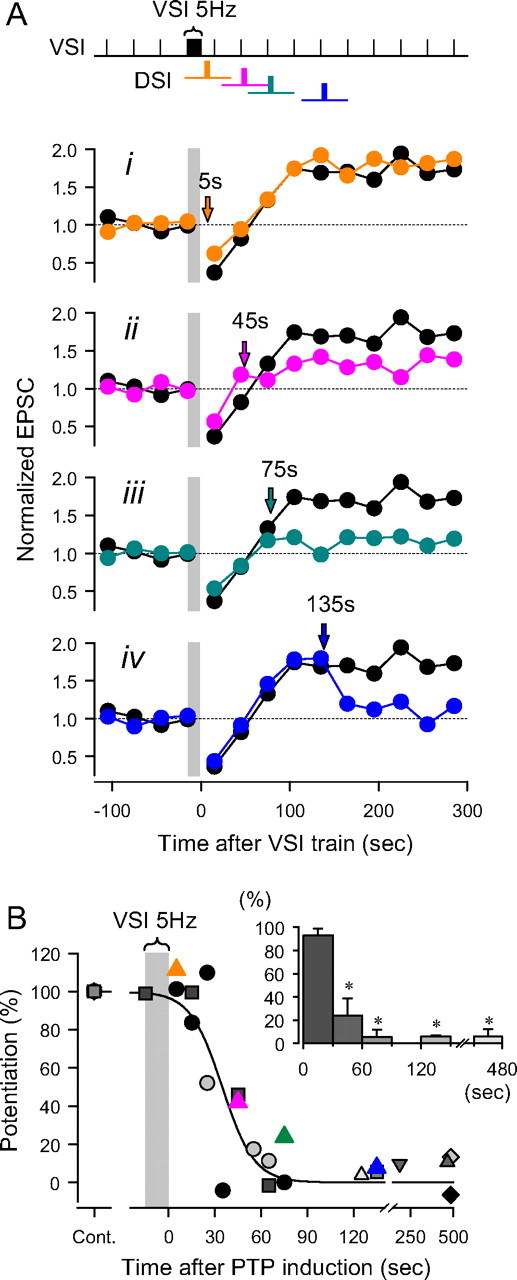
Timing dependence of DSI-evoked depotentiation of VSI synaptic strength. A, The time of DSI stimulation was varied with respect to the end of the VSI spike train. For comparison a control trial is shown in which DSI was not stimulated (black circles). i, At 5 s after the VSI tetanus, DSI had little effect on potentiation. ii, At 45 s, DSI partially reduced the magnitude of the potentiation. iii, After 75 s, DSI stimulation almost completely reduced the magnitude of potentiation. iv, At 135 s, the potentiation was already maximum and DSI stimulation caused depotentiation. EPSC amplitudes during the VSI 5 Hz spike train are omitted and shown by a gray bar. B, Results from nine preparations show the time course over which DSI stimulation becomes effective at depotentiating the synapse. The extent of potentiation after a DSI spike train is shown as percentage of the amplitude of the control potentiation without DSI stimulation, and is plotted against the time of DSI stimulation. DSI stimulation must follow the VSI spike train by at least 60 s to be maximally effective at depotentiation. The plot was fit by a sigmoidal curve, f(t) = a/(1 + exp(−(t − t0)/b)), where t is the time after the end of VSI spike train, a = 1, b = −10.4, t0 = 34.8 (R2 = 0.82). Inset, A bar graph showing the extent of potentiation binned in 30 s intervals after a VSI spike train (5 Hz). Each bar shows mean ± SEM. Asterisks indicate significant difference in the magnitude of potentiation with respect to the data at 0–30 s (F(4,10) = 21.8, p < 0.001 by one-way ANOVA with pairwise multiple comparison by Student–Newman–Keuls method).
DSI-evoked depotentiation integrates over long time periods
To begin to determine the functional significance of DSI-evoked enhancement and depotentiation, we examined their sensitivity to parameters of the DSI spike train (spike frequency, spike number and train duration). We stimulated DSI at various spike frequencies either for a fixed duration (Fig. 8A) or for a fixed number of spikes (Fig. 8B). When we held the duration of the DSI spike train at 5 s (Fig. 8Ai), the magnitude of both the transient enhancement and the depotentiation increased as a function of DSI spike frequency (Fig. 8Aii,iii). However, when we held the number of DSI spikes constant at 51 pulses and varied the frequency and duration of the DSI spike train (Fig. 8Bi), we found that the enhancement and depotentiation behaved differently (Fig. 8Bii,iii). For depotentiation, a significant change was seen even at low spike frequencies and no further increase was seen at higher DSI spike frequencies (see Fig. 8B legend), whereas the transient enhancement increased gradually as a function of DSI spike frequency. These results suggest that the magnitude of the DSI-evoked transient enhancement, which lasts just a short time, has a short integration period, whereas the magnitude of the depotentiation depends more on the absolute number of DSI spikes, allowing the depotentiation to be integrated over a long time period.
Functional significance of the DSI action
Both DSI and VSI are the members of the central pattern generator circuit underlying the escape swim response of T. diomedea (Fig. 9Ai). During the swim motor pattern, DSI and VSI fire bursts of action potentials at 8–20 Hz in an alternating manner with a cycle period of 4–12 s (Fig. 9Aii) (Getting, 1981, 1983). The swim motor pattern terminates after a VSI burst, and then DSI continues to fire tonically at 1–2 Hz, whereas VSI stays silent unless the animal receives tactile stimulation (Popescu and Frost, 2002). We tested the effect of DSI on VSI synaptic output that would occur during and after a swim bout by mimicking this firing pattern and recording the VSI-evoked EPSCs in a VFN.
Figure 9.
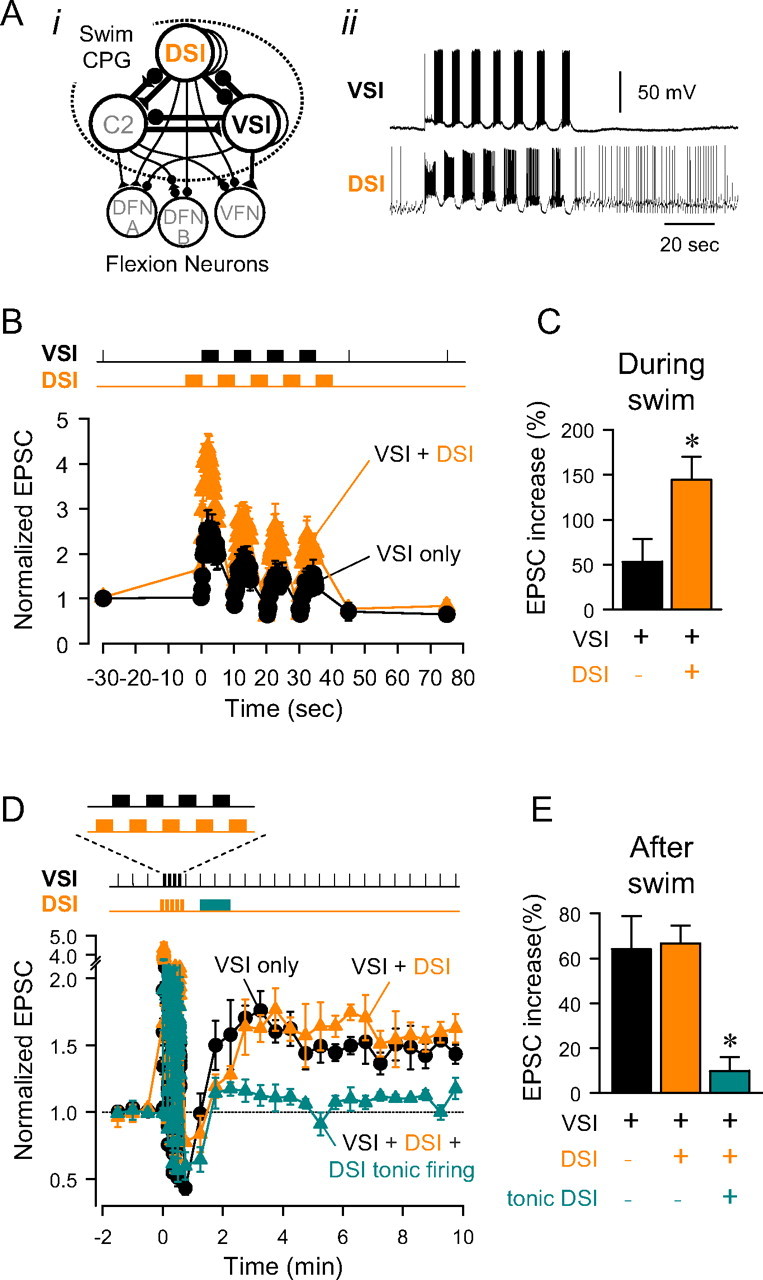
Functional significance of state- and timing-dependent neuromodulation. A, A schematic diagram of the neural circuit underlying the swim motor pattern (i). Filled triangles represent excitatory synapses and filled circles represent inhibitory synapses. Combinations of triangles and circles are multicomponent synapses. C2, cerebral cell 2; DFN-A,B, dorsal flexion neurons. Based on Getting et al. (1980), Getting (1981), Hume and Getting (1982). All neurons have a contralateral homolog that is not represented. DSI and VSI show alternating bursting activity during a swim motor pattern (ii). After the swim bout, DSI fires at an elevated tonic firing rate of 1–2 Hz. B, Testing the role of DSI bursts using an imposed swim-like firing pattern. Bursts of action potentials were evoked repeatedly every 10 s either in VSI only (5 Hz for 5 s) or in both VSI (5 Hz, 5 s) and DSI (10 Hz, 5 s) in alternating manner, mimicking the swim motor pattern. Normalized amplitudes of the VSI-evoked EPSCs were plotted against time. The graph shows the changes in the amplitudes of VSI-evoked EPSCs when VSI was stimulated alone (black) and when both VSI and DSI were stimulated in alternating bursts (orange). Imposing DSI bursts between VSI bursts increased the EPSC amplitudes. C, There was a significant increase in the amplitudes of VSI-evoked EPSCs when VSI was stimulated in alternation with DSI (p < 0.01 by paired t test, n = 4). The EPSC amplitude during the artificial swim motor pattern was determined by averaging the amplitude of all EPSCs during the motor pattern. D, The role of the tonic firing of DSI after the swim. VSI-evoked EPSCs were tested every 30 s. Normalized EPSC amplitudes are plotted before and after a series VSI spike trains (mimicking the swim motor pattern). When VSI was stimulated alone (black), the EPSPs entered the potentiation state after the artificial swim. When VSI was stimulated in alternation with DSI during the artificial swim, the EPSCs still potentiated after the artificial swim bout (orange). Adding tonic DSI spiking (2 Hz, for 60 s) after the artificial swim greatly reduced the magnitude of the postswim potentiation (green). E, Repeated bursts of VSI (black) caused an increase in EPSC amplitude. Firing VSI and DSI in an altering swim-like firing pattern (orange) did not significantly affect the percentage increase in EPSC amplitude. Addition of tonic DSI firing (green) significantly reduced the postswim potentiation. Asterisks indicates a significant difference (C, p < 0.05 by paired t test, n = 4; E, p < 0.01 by one-way ANOVA with pairwise multiple comparison by Student–Newman–Keuls method).
When VSI was stimulated alone in a repeated bursting pattern, mimicking its firing during a swim motor pattern, the EPSCs that it evoked exhibited short term facilitation and depression (Fig. 9B, black plot). Immediately after the VSI spike trains, the EPSCs evoked by test VSI spikes were depressed. Over the course of 2 min, those EPSCs increased in amplitude until they reached a potentiated level that was stable for >10 min (Fig. 9D, black plot), similar to when VSI was stimulated to fire a single 5 Hz, 15 s train (compare Fig. 1B).
When DSI was stimulated in alternation with VSI bursts, as would occur during a swim motor pattern, the VSI-evoked EPSC amplitudes increased, presumably because of the DSI transient enhancement (Fig. 9B, orange plot). The average EPSC amplitude during the artificial swim showed a significant increase when DSI bursts were imposed (Fig. 9C). After the artificial swim bout, VSI continued to exhibit a prolonged period of synaptic potentiation that was not significantly different from when VSI was stimulated alone (Figs. 9D,E, orange). Thus, DSI firing during the swim caused short-term enhancement of VSI synaptic strength but did not affect the potentiation after the swim.
However, when DSI tonic firing was added after an artificial swim motor pattern (2 Hz for 1 min, from 35 to 95 s after the end of the swim pattern), it significantly reduced the magnitude of the following long-lasting potentiation (Fig. 9D,E, green). Thus, the tonic DSI firing that occurs after the swim motor pattern might help reset VSI synaptic strength by depotentiating the EPSCs from the potentiated state that is induced by the swim motor pattern. The long integration time of the depotentiation allows low frequency DSI firing to be effective without also causing short term enhancement.
Discussion
In this study, we found that a serotonergic neuron (DSI) evokes distinct state-dependent and state-independent neuromodulatory actions on the strength of synapses made by another neuron (VSI). As a result, the net effect of the serotonergic neuron is dependent on its firing frequency, its burst duration, the timing of its spikes with respect to those of the neuron being modulated, and the state of the synapse being modulated (summarized in Fig. 10). The differences in state- and time-dependency allow the two heterosynaptic actions to play different roles at different stages in the production of a behavior.
Figure 10.

Schematic diagram showing state-, timing-, and pattern-dependent neuromodulation of the strength of the VSI-VFN synapse by a DSI spike train. In A and B, a VSI spike train (top trace), the synaptic strength (middle trace), and DSI spike trains (bottom trace) evoked at three different times are shown. A VSI spike train produces frequency facilitation and a subsequent long-lasting potentiation of synaptic strength (Pot.). Changes in synaptic strength caused by a DSI spike train are indicated by dashed lines. A, When the strength of VSI-VFN synapse is in basal state, a DSI spike train (i) at high frequency (e.g., 5 s, 10 Hz) produces only a transient enhancement (TE). In contrast, when a DSI spike train is imposed during the potentiated state (iii), it both causes a transient enhancement and depotentiates (De-pot.) the synapse. When a DSI spike train is imposed after a VSI spike train but before the induction of the potentiated state (ii), it partially reduces the amplitude of the after-potentiated state, depending on the timing of the DSI spike train. B, When a DSI is stimulated at a lower frequency for a longer duration, producing the same number of spikes (i), it produces a very weak synaptic enhancement, but still evokes a strong depotentiation when the synapse is in the potentiated state (iii) or even in the beginning of the potentiated state (ii) (compare Fig. 9D).
State-dependent synaptic depotentiation
The state of a synapse can be determined by intrinsic or homosynaptic plasticity, which is dependent on the presynaptic neuron's own firing history. In this study, we found that the synapse from VSI to VFN enters a potentiation state after a spike train. This potentiated state was of intermediate term (∼10 min) and seemed to involve a presynaptic change in VSI because it interfered with pair-pulse facilitation (another presynaptic mechanism). Thus, it may be similar to post-tetanic potentiation or augmentation studied in various model systems (Zucker and Regehr, 2002). Only when VSI synapses were in a potentiated state did DSI stimulation decrease the amplitude of VSI EPSCs. The extent of the decrease rarely exceeded the extent of the potentiation. In this way, DSI appeared to depotentiate the synapse, resetting it to its basal state. This suggests that the homosynaptic potentiation and the heterosynaptic depotentiation share a common site of action.
The ability of neuromodulatory inputs to reset the synaptic strength to a “basal” state has implications for synaptic release mechanism. It supports a model suggesting that some forms of homosynaptic plasticity are added to a basal state and thus can be taken away, leaving the original state of the synapse. For example, different pools of vesicles have been suggested to underlie different forms of homosynaptic plasticity in Aplysia (Jiang and Abrams, 1998). In Tritonia, neither heterosynaptic depotentiation nor homosynaptic potentiation appeared to affect the basal release of the VSI synapse.
Our finding that heterosynaptic neuromodulation can negate the effect of homosynaptic plasticity appears to be unique in the literature. Although there have been studies that described interactions of homo- and hetero-synaptic plasticity (Levy and Steward, 1979; Doyère et al., 1997; Chen et al., 2001; Huang et al., 2008), the term “heterosynaptic” in these studies was taken to mean multiple synapses of the same type rather than neuromodulatory input as in this study. Resetting or depotentiation through low frequency homosynaptic mechanisms has been studied extensively in hippocampal slice preparations (Kulla et al., 1999; Chen et al., 2001; Huang and Hsu, 2001; Huang et al., 2001; Manahan-Vaughan and Kulla, 2003). But to our knowledge, there have been no reports of neuromodulatory actions resetting homosynaptic plasticity.
There are however, studies describing neuromodulatory effects on the induction of homosynaptic plasticity, in which the threshold and the magnitude of the synaptic plasticity is altered by neuromodulatory input (Qian and Delaney, 1997; Bristol et al., 2001; Koh and Weiss, 2005; Froemke et al., 2007; Giocomo and Hasselmo, 2007; Seol et al., 2007). Such “metaplasticity” has been considered as important for determining the threshold for induction of LTP and LTD (Abraham and Bear, 1996; Philpot et al., 1999, 2007; Abraham et al., 2001). Homosynaptic depotentiation also exhibits metaplasticity in that neuromodulatory substances have been shown to change the spike rate threshold for depotentiation (Huang et al., 1999, 2002; Kulla and Manahan-Vaughan, 2002, 2008; Zho et al., 2002; Manahan-Vaughan and Kulla, 2003).
It is tempting to speculate about the potential importance of a heterosynaptic reset mechanism for systems that exhibit activity-dependent synaptic plasticity. State-dependent neuromodulatory actions have been reported in a number of systems (Cai et al., 2002; Gu and Yan, 2004). In Aplysia, the one of the effects of serotonin is dependent on whether the sensory to motor synapse is in a depressed state or not (Byrne and Kandel, 1996). This may play a role at the behavioral level in distinguishing dishabituation from sensitization (Goldsmith and Abrams, 1991). Resetting synapses to a basal state could potentially play a role in rapidly clearing working memory (Mongillo et al., 2008).
State-independent synaptic enhancement
In contrast to synaptic depotentiation, the ability of DSI to enhance VSI synaptic strength is independent of the state of the synapse. Regardless of whether the synapse was in the basal state or in the potentiated state, DSI stimulation produced approximately a 60% increase in the EPSC amplitude (Fig. 5B). This suggests that the transient enhancement acts as a multiplicative scaling factor on the current output state of the synapse. This is consistent with our previous results, which showed that DSI enhanced VSI synaptic strength to an equivalent degree even when the amount of transmitter release was increased by spike broadening (Sakurai et al., 2006). The value of the scaling factor exhibited a great deal of variability within and between preparations, ranging from a 30% increase to a 120% increase (see Fig. 5B). The source of that variability is not known. However, it is clear that it does not correlate with the potentiation state of the synapse.
The mechanism for the transient enhancement appears to be distinct from the mechanism for the activity-dependent, intrinsic potentiation at this synapse. We previously showed that the DSI-evoked transient enhancement is caused by an increase in the fraction of the readily releasable pool of neurotransmitter (Sakurai et al., 2007). Although in this study, we found that the transient enhancement is independent of the potentiation state, it is still dependent on the release state of the synapse; depleting the size of the readily releasable pool impairs the ability of 5-HT to increase release (Sakurai et al., 2007).
In other systems, it has been suggested that an increased fraction of release plays major roles in some forms of homosynaptic potentiation (Zucker and Regher, 2002). If that is the case in VSI, then there would likely be two separate pathways for increasing the fraction of release. In some systems the recruitment of release sites and/or increased pool size are suggested to be at least partly involved in synaptic potentiation (Byrne and Kandel, 1996; Atwood and Wojtowicz, 1999; Rosenmund et al., 2002; Poncer, 2003; Reid et al., 2004; Zhao and Klein, 2004; Habets and Borst, 2007), and these mechanisms may also play a role in VSI homosynaptic potentiation. Further studies are needed to elucidate the mechanisms of these two types of synaptic enhancement.
Spike timing-dependent neuromodulation
Previously, we described DSI modulation of VSI synaptic strength as spike timing-dependent in which the timing of the VSI spikes with respect to DSI determines the polarity of the DSI action; when VSI is stimulated within 15 s of a DSI spike train, its synaptic output is enhanced, whereas it is diminished when VSI is stimulated at longer latencies (up to 2 min) (Sakurai and Katz, 2003). However, those studies did not take into account the firing history of VSI and therefore the potentiation state of its synapse. Having now found that VSI exhibits homosynaptic plasticity that changes the state of its synaptic output, we recognize that what we previously described as DSI-evoked synaptic decrease is really depotentiation from a potentiated state. When the VSI synapse is not in a potentiated state, DSI stimulation does not decrease VSI synaptic strength.
We infer from our data that the timing dependencies of the neuromodulatory actions arise from the time courses of independent intracellular signaling mechanisms induced by the DSI input and the VSI spike train. The synaptic enhancement produced by a DSI spike train lasts no longer than 15 s. This has consequences for the time duration over which the DSI-evoked enhancement can summate; DSI spikes spread out over a longer time period are less effective at enhancing VSI synaptic strength than a brief train with an equal number of spikes (Fig. 8Bii). Conversely, because the depotentiation is a negation of an altered state, it can integrate over longer periods of time; each DSI spike subtracting from the current state until the baseline is reached (Fig. 8Biii).
The DSI-evoked synaptic depotentiation has its own timing dependence; there is a window of time after a VSI spike train and before the onset of the potentiation state, when DSI spiking neither prevents the potentiation nor diminishes it. This timing-sensitivity presumably arises from the intracellular processes after a VSI spike train that lead to the potentiated state; DSI cannot depotentiate the synapse until the potentiation state is achieved.
Functional significance of timing- and state-dependent neuromodulation
It has been shown in many studies that short-term synaptic plasticity plays crucial roles in configuration of neural networks for motor pattern generation (Katz and Frost, 1995; Calabrese, 1998; Katz, 1999; Sanchez and Kirk, 2000; Sakurai and Katz, 2003; Koh and Weiss, 2005; Giocomo and Hasselmo, 2007; Briggman and Kristan, 2008; Huang et al., 2008), and sensory processing (Nagai et al., 1996; Mercer, 1999; Billimoria et al., 2006; Schneggenburger and Forsythe, 2006). Previously, we suggested that the DSI transient enhancement of VSI synaptic strength serves to boost the synaptic output during the swim motor program, whereas the synaptic decrement would suppress spurious synaptic inputs made by VSI after the cessation of swimming (Sakurai and Katz, 2003). In this study, we found that repeated VSI bursts by themselves can produce a significant potentiation of VSI synapse, which would keep VSI synapses in a potentiated state for >10 min after a swim motor pattern. We further found that DSI firing during the swim motor pattern would not suppress this potentiation, whereas mimicking the tonic DSI firing that occurs after a swim motor pattern suppresses the potentiation and resets the synapse to its basal state. This is likely to have important behavioral consequences for the animal. It has been shown that VSI spiking inhibits crawling after a swim (Popescu and Frost, 2002). Thus, depotentiating these synapses after the swim would make them less effective at inhibiting crawling. In this way, the different actions of a serotonergic neuron have different functions at different stages in the production of the behavior.
More generally, our results demonstrate that neuromodulatory actions of neurons can have state, timing, and pattern dependencies. It is not possible to predict the effect of the neuromodulatory neuron without knowing when it is firing with respect to the neuron that it modulates and what the pattern of activity is. In the case of state-dependent neuromodulatory actions, predicting the effect of the neuromodulatory neuron would also require knowing the state of the neuron or synapse being modulated. Similar contingent actions of neuromodulatory neurons are likely to play important roles in controlling the dynamics of any neuronal circuit that receives phasic neuromodulatory input.
Footnotes
This work was supported by National Institutes of Health Grant R21 MH76753.
References
- Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Mason-Parker SE, Bear MF, Webb S, Tate WP. Heterosynaptic metaplasticity in the hippocampus in vivo: a BCM-like modifiable threshold for LTP. Proc Natl Acad Sci U S A. 2001;98:10924–10929. doi: 10.1073/pnas.181342098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood HL, Wojtowicz JM. Silent synapses in neural plasticity: current evidence. Learn Mem. 1999;6:542–571. doi: 10.1101/lm.6.6.542. [DOI] [PubMed] [Google Scholar]
- Billimoria CP, DiCaprio RA, Birmingham JT, Abbott LF, Marder E. Neuromodulation of spike-timing precision in sensory neurons. J Neurosci. 2006;26:5910–5919. doi: 10.1523/JNEUROSCI.4659-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggman KL, Kristan WB. Multifunctional pattern-generating circuits. Annu Rev Neurosci. 2008;31:271–294. doi: 10.1146/annurev.neuro.31.060407.125552. [DOI] [PubMed] [Google Scholar]
- Bristol AS, Fischer TM, Carew TJ. Combined effects of intrinsic facilitation and modulatory inhibition of identified interneurons in the siphon withdrawal circuitry of Aplysia. J Neurosci. 2001;21:8990–9000. doi: 10.1523/JNEUROSCI.21-22-08990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne JH, Kandel ER. Presynaptic facilitation revisited: state and time dependence. J Neurosci. 1996;16:425–435. doi: 10.1523/JNEUROSCI.16-02-00425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Flores-Hernandez J, Feng J, Yan Z. Activity-dependent bidirectional regulation of GABAA receptor channels by the 5-HT4 receptor-mediated signalling in rat prefrontal cortical pyramidal neurons. J Physiol. 2002;540:743–759. doi: 10.1113/jphysiol.2001.013391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese RL. Cellular, synaptic, network, and modulatory mechanisms involved in rhythm generation. Curr Opin Neurobiol. 1998;8:710–717. doi: 10.1016/s0959-4388(98)80112-8. [DOI] [PubMed] [Google Scholar]
- Chen YL, Huang CC, Hsu KS. Time-dependent reversal of long-term potentiation by low-frequency stimulation at the hippocampal mossy fiber-CA3 synapses. J Neurosci. 2001;21:3705–3714. doi: 10.1523/JNEUROSCI.21-11-03705.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyère V, Srebro B, Laroche S. Heterosynaptic LTD and depotentiation in the medial perforant path of the dentate gyrus in the freely moving rat. J Neurophysiol. 1997;77:571–578. doi: 10.1152/jn.1997.77.2.571. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Merzenich MM, Schreiner CE. A synaptic memory trace for cortical receptive field plasticity. Nature. 2007;450:425–429. doi: 10.1038/nature06289. [DOI] [PubMed] [Google Scholar]
- Getting PA. Mechanisms of pattern generation underlying swimming in Tritonia. I. Neuronal network formed by monosynaptic connections. J Neurophysiol. 1981;46:65–79. doi: 10.1152/jn.1981.46.1.65. [DOI] [PubMed] [Google Scholar]
- Getting PA. Mechanisms of pattern generation underlying swimming in Tritonia. III. Intrinsic and synaptic mechanisms for delayed excitation. J Neurophysiol. 1983;49:1036–1050. doi: 10.1152/jn.1983.49.4.1036. [DOI] [PubMed] [Google Scholar]
- Getting PA, Lennard PR, Hume RI. Central pattern generator mediating swimming in Tritonia. I. Identification and synaptic interactions. J Neurophysiol. 1980;44:151–164. doi: 10.1152/jn.1980.44.1.151. [DOI] [PubMed] [Google Scholar]
- Giocomo LM, Hasselmo ME. Neuromodulation by glutamate and acetylcholine can change circuit dynamics by regulating the relative influence of afferent input and excitatory feedback. Mol Neurobiol. 2007;36:184–200. doi: 10.1007/s12035-007-0032-z. [DOI] [PubMed] [Google Scholar]
- Goldsmith BA, Abrams TW. Reversal of synaptic depression by serotonin at Aplysia sensory neuron synapses involves activation of adenylyl cyclase. Proc Natl Acad Sci U S A. 1991;88:9021–9025. doi: 10.1073/pnas.88.20.9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Yan Z. Bidirectional regulation of Ca2+/calmodulin-dependent protein kinase II activity by dopamine D4 receptors in prefrontal cortex. Mol Pharmacol. 2004;66:948–955. doi: 10.1124/mol.104.001404. [DOI] [PubMed] [Google Scholar]
- Habets RLP, Borst JG. Dynamics of the readily releasable pool during post-tetanic potentiation in the rat calyx of Held synapse. J Physiol. 2007;581:467–478. doi: 10.1113/jphysiol.2006.127365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Hsu KS. Progress in understanding the factors regulating reversibility of long-term potentiation. Rev Neurosci. 2001;12:51–68. doi: 10.1515/revneuro.2001.12.1.51. [DOI] [PubMed] [Google Scholar]
- Huang CC, Liang YC, Hsu KS. A role for extracellular adenosine in time-dependent reversal of long-term potentiation by low-frequency stimulation at hippocampal CA1 synapses. J Neurosci. 1999;19:9728–9738. doi: 10.1523/JNEUROSCI.19-22-09728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Liang YC, Hsu KS. Characterization of the mechanism underlying the reversal of long term potentiation by low frequency stimulation at hippocampal CA1 synapses. J Biol Chem. 2001;276:48108–48117. doi: 10.1074/jbc.M106388200. [DOI] [PubMed] [Google Scholar]
- Huang CC, Chen YL, Liang YC, Hsu KS. Role for cAMP and protein phosphatase in the presynaptic expression of mouse hippocampal mossy fibre depotentiation. J Physiol. 2002;543:767–778. doi: 10.1113/jphysiol.2002.025668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yasuda H, Sarihi A, Tsumoto T. Roles of endocannabinoids in heterosynaptic long-term depression of excitatory synaptic transmission in visual cortex of young mice. J Neurosci. 2008;28:7074–7083. doi: 10.1523/JNEUROSCI.0899-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume RI, Getting PA. Motor organization of Tritonia swimming. II. Synaptic drive to flexion neurons from premotor interneurons. J Neurophysiol. 1982;47:75–90. doi: 10.1152/jn.1982.47.1.75. [DOI] [PubMed] [Google Scholar]
- Jiang XY, Abrams TW. Use-dependent decline of paired-pulse facilitation at Aplysia sensory neuron synapses suggests a distinct vesicle pool or release mechanism. J Neurosci. 1998;18:10310–10319. doi: 10.1523/JNEUROSCI.18-24-10310.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz PS. Beyond neurotransmission: neuromodulation and its importance for information processing. Oxford: Oxford UP; 1999. [Google Scholar]
- Katz PS, Frost WN. Intrinsic neuromodulation in the Tritonia swim CPG: the serotonergic dorsal swim interneurons act presynaptically to enhance transmitter release from interneuron C2. J Neurosci. 1995;15:6035–6045. doi: 10.1523/JNEUROSCI.15-09-06035.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh HY, Weiss KR. Peptidergic contribution to posttetanic potentiation at a central synapse of Aplysia. J Neurophysiol. 2005;94:1281–1286. doi: 10.1152/jn.00073.2005. [DOI] [PubMed] [Google Scholar]
- Kulla A, Manahan-Vaughan D. Modulation by serotonin 5-HT4 receptors of long-term potentiation and depotentiation in the dentate gyrus of freely moving rats. Cereb Cortex. 2002;12:150–162. doi: 10.1093/cercor/12.2.150. [DOI] [PubMed] [Google Scholar]
- Kulla A, Manahan-Vaughan D. Modulation by group 1 metabotropic glutamate receptors of depotentiation in the dentate gyrus of freely moving rats. Hippocampus. 2008;18:48–54. doi: 10.1002/hipo.20366. [DOI] [PubMed] [Google Scholar]
- Kulla A, Reymann KG, Manahan-Vaughan D. Time-dependent induction of depotentiation in the dentate gyrus of freely moving rats: involvement of group 2 metabotropic glutamate receptors. Eur J Neurosci. 1999;11:3864–3872. doi: 10.1046/j.1460-9568.1999.00807.x. [DOI] [PubMed] [Google Scholar]
- Levy WB, Steward O. Synapses as associative memory elements in the hippocampal formation. Brain Res. 1979;175:233–245. doi: 10.1016/0006-8993(79)91003-5. [DOI] [PubMed] [Google Scholar]
- Manahan-Vaughan D, Kulla A. Regulation of depotentiation and long-term potentiation in the dentate gyrus of freely moving rats by dopamine D2-like receptors. Cereb Cortex. 2003;13:123–135. doi: 10.1093/cercor/13.2.123. [DOI] [PubMed] [Google Scholar]
- Mercer AR. Changing the way we perceive things: sensory systems modulation. In: Katz PS, editor. Beyond neurotransmission: neuromodulation and its importance for information processing. Oxford: Oxford UP; 1999. pp. 198–240. [Google Scholar]
- Mongillo G, Barak O, Tsodyks M. Synaptic theory of working memory. Science. 2008;319:1543–1546. doi: 10.1126/science.1150769. [DOI] [PubMed] [Google Scholar]
- Nagai T, Kim DJ, Roper SD. Neuromodulation of transduction and signal processing in the end organs of taste. Chem Senses. 1996;21:353–365. doi: 10.1093/chemse/21.3.353. [DOI] [PubMed] [Google Scholar]
- Philpot BD, Bear MF, Abraham WC. Metaplasticity: The plasticity of synaptic plasticity. In: Katz PS, editor. Beyond neurotransmission: neuromodulation and its importance for information processing. Oxford: Oxford UP; 1999. pp. 160–197. [Google Scholar]
- Philpot BD, Cho KK, Bear MF. Obligatory role of NR2A for metaplasticity in visual cortex. Neuron. 2007;53:495–502. doi: 10.1016/j.neuron.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poncer JC. Hippocampal long term potentiation: silent synapses and beyond. J Physiol Paris. 2003;97:415–422. doi: 10.1016/j.jphysparis.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Popescu IR, Frost WN. Highly dissimilar behaviors mediated by a multifunctional network in the marine mollusk Tritonia diomedea. J Neurosci. 2002;22:1985–1993. doi: 10.1523/JNEUROSCI.22-05-01985.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian SM, Delaney KR. Neuromodulation of activity-dependent synaptic enhancement at crayfish neuromuscular junction. Brain Res. 1997;771:259–270. doi: 10.1016/s0006-8993(97)00812-3. [DOI] [PubMed] [Google Scholar]
- Reid CA, Dixon DB, Takahashi M, Bliss TV, Fine A. Optical quantal analysis indicates that long-term potentiation at single hippocampal mossy fiber synapses is expressed through increased release probability, recruitment of new release sites, and activation of silent synapses. J Neurosci. 2004;24:3618–3626. doi: 10.1523/JNEUROSCI.3567-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Sigler A, Augustin I, Reim K, Brose N, Rhee JS. Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron. 2002;33:411–424. doi: 10.1016/s0896-6273(02)00568-8. [DOI] [PubMed] [Google Scholar]
- Sakurai A, Katz PS. Spike timing-dependent serotonergic neuromodulation of synaptic strength intrinsic to a central pattern generator circuit. J Neurosci. 2003;23:10745–10755. doi: 10.1523/JNEUROSCI.23-34-10745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai A, Darghouth NR, Butera RJ, Katz PS. Serotonergic enhancement of a 4-AP-sensitive current mediates the synaptic depression phase of spike timing-dependent neuromodulation. J Neurosci. 2006;26:2010–2021. doi: 10.1523/JNEUROSCI.2599-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai A, Calin-Jageman RJ, Katz PS. Potentiation phase of spike timing-dependent neuromodulation by a serotonergic interneuron involves an increase in the fraction of transmitter release. J Neurophysiol. 2007;98:1975–1987. doi: 10.1152/jn.00702.2007. [DOI] [PubMed] [Google Scholar]
- Sanchez JA, Kirk MD. Short-term synaptic enhancement modulates ingestion motor programs of Aplysia. J Neurosci. 2000;20:RC85. doi: 10.1523/JNEUROSCI.20-14-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Forsythe ID. The calyx of Held. Cell Tissue Res. 2006;326:311–337. doi: 10.1007/s00441-006-0272-7. [DOI] [PubMed] [Google Scholar]
- Seol GH, Ziburkus J, Huang S, Song L, Kim IT, Takamiya K, Huganir RL, Lee HK, Kirkwood A. Neuromodulators control the polarity of spike-timing-dependent synaptic plasticity. Neuron. 2007;55:919–929. doi: 10.1016/j.neuron.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willows AO, Dorsett DA, Hoyle G. The neuronal basis of behavior in Tritonia. I. Functional organization of the central nervous system. J Neurobiol. 1973;4:207–237. doi: 10.1002/neu.480040306. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Klein M. Changes in the readily releasable pool of transmitter and in efficacy of release induced by high-frequency firing at Aplysia sensorimotor synapses in culture. J Neurophysiol. 2004;91:1500–1509. doi: 10.1152/jn.01019.2003. [DOI] [PubMed] [Google Scholar]
- Zho WM, You JL, Huang CC, Hsu KS. The group I metabotropic glutamate receptor agonist (S)-3,5-dihydroxyphenylglycine induces a novel form of depotentiation in the CA1 region of the hippocampus. J Neurosci. 2002;22:8838–8849. doi: 10.1523/JNEUROSCI.22-20-08838.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]