

Abstract
In animal models of diet-induced obesity, the activation of an inflammatory response in the hypothalamus produces molecular and functional resistance to the anorexigenic hormones insulin and leptin. The primary events triggered by dietary fats that ultimately lead to hypothalamic cytokine expression and inflammatory signaling are unknown. Here, we test the hypothesis that dietary fats act through the activation of toll-like receptors 2/4 and endoplasmic reticulum stress to induce cytokine expression in the hypothalamus of rodents. According to our results, long-chain saturated fatty acids activate predominantly toll-like receptor 4 signaling, which determines not only the induction of local cytokine expression but also promotes endoplasmic reticulum stress. Rats fed on a monounsaturated fat-rich diet do not develop hypothalamic leptin resistance, whereas toll-like receptor 4 loss-of-function mutation and immunopharmacological inhibition of toll-like receptor 4 protects mice from diet-induced obesity. Thus, toll-like receptor 4 acts as a predominant molecular target for saturated fatty acids in the hypothalamus, triggering the intracellular signaling network that induces an inflammatory response, and determines the resistance to anorexigenic signals.
Keywords: obesity, inflammation, hypothalamus, cytokine, nutrition, feeding
Introduction
The consumption of fat-rich diets is among the most important environmental factors predisposing to obesity in modern societies (Stein and Colditz, 2004; Freire et al., 2005; Moreno and Rodríguez, 2007). In animal models of genetic and diet-induced obesity, the activation of an inflammatory response in the hypothalamus leads to the molecular and functional resistance to the adipostatic hormones, leptin and insulin, resulting in a defective control of food intake and energy expenditure (Carvalheira et al., 2003; Howard et al., 2004; De Souza et al., 2005). Reversal of these effects can be achieved by distinct genetic and pharmacological approaches, aimed at inhibiting inflammatory signaling (Howard et al., 2004; De Souza et al., 2005).
Recent studies have provided strong evidence for the contribution of toll-like receptor (TLR) activation and endoplasmic reticulum stress (ER stress) induction as mechanisms linking the consumption of high-fat diets and obesity to insulin resistance and type 2 diabetes mellitus (Ozcan et al., 2004, 2006; Shi et al., 2006; Tsukumo et al., 2007). TLRs are highly conserved members of the interleukin-1 receptor superfamily that respond to microbial signature motifs, leading to the activation of innate immune responses (Akira, 2003; Akira et al., 2006). Four members of the TLR family, TLR1, 2, 4, and 6, are known to recognize lipid-containing motifs; TLR1/2 dimmers recognize diacyl lipopeptides, TLR2/6 dimmers recognize triacyl lipopeptides and TLR4 recognizes lipopolysaccharides (LPS). The activation of TLR signaling leads to the coordinated induction of cytokine and other immune-related genes expression (Shimazu et al., 1999; Takeuchi et al., 2001; Akira, 2003; Akira et al., 2006).
The ER is the organelle responsible for the synthesis and processing of membrane and secretory proteins (Xu et al., 2005). Under certain harmful conditions, the ER homeostasis is disrupted, leading to the accumulation of misfolded and unfolded proteins in the ER lumen (Schröder and Kaufman, 2005; Xu et al., 2005). To deal with this condition, the affected cells activate a complex signaling system known as the unfolded protein response (UPR), aimed at preserving cell integrity while the harmful condition persists (Schröder and Kaufman, 2005; Xu et al., 2005). One of the outcomes of the activation of UPR is the induction of the expression of cytokines and proteins involved in immune surveillance (Krappmann et al., 2004; Marciniak and Ron, 2006). In addition to classical activation by viruses and bacteria (Watowich et al., 1991; Pahl and Baeuerle, 1995), ER stress can be induced by metabolic and nutritional factors such as high levels of glucose and lipids (Ozcan et al., 2004; Nakatani et al., 2005; Ozawa et al., 2005).
Cytokines, induced either by TLR activation and/or by ER stress, can play a pathogenetic role in the development of insulin resistance in peripheral tissues (Ozcan et al., 2004, 2006; Shi et al., 2006; Tsukumo et al., 2007). To explore the hypothesis that fatty acids can trigger an inflammatory response in the hypothalamus by inducing TLR activation and/or ER stress, we determined the molecular and functional outcomes of intracerebroventricular injection of fatty acids in rodents. Our results show that long-chain saturated fatty acids act predominantly through TLR4 and suggest that ER stress is a downstream event in the cascade that ultimately leads to inflammatory activation in the hypothalamus.
Materials and Methods
Antibodies, chemicals, and buffers.
Antibodies against TNF-α (s.c.-1347, goat polyclonal and s.c.-8301, rabbit polyclonal), IL-1β (s.c.-1252, goat polyclonal and s.c.-7884 rabbit polyclonal), IL-6 (s.c.-1266, goat polyclonal and s.c.-7920, rabbit polyclonal), IL-10 (s.c.-1783, goat polyclonal), eIF2α (s.c.-11386, rabbit polyclonal), RNA-dependent protein kinase-like endoplasmic reticulum kinase (PERK) (s.c.-13073, rabbit polyclonal), phosphor[Thr981]-PERK (pPERK, s.c.-32577, rabbit polyclonal), GRP78 (s.c.-13968, rabbit polyclonal), c-Jun N-terminal protein kinase (JNK) (s.c.-46009, goat polyclonal), phosphor[Thr183/Tyr185] JNK (pJNK, sc12882, rabbit polyclonal), F4/80 (s.c.-25830, rabbit polyclonal), proopiomelanocortin (POMC) (s.c.-20148, rabbit polyclonal), agouti-related protein (AgRP) (s.c.-50299, rabbit polyclonal), TLR2 (s.c.-10739, rabbit polyclonal and s.c.-16237, goat polyclonal), TLR4 (s.c.-16240 goat polyclonal and s.c.-13591, rat monoclonal), and MyD88 (s.c.-11356, rabbit polyclonal) were purchased from Santa Cruz Biotechnology. The anti-phosphor[Ser52] eIF2α (peIF2α, #9721 s, rabbit polyclonal) was purchased from Cell Signaling Technology. All the reagents for SDS-PAGE and immunoblotting were from Bio-Rad. HEPES, phenylmethylsulfonyl fluoride, aprotinin, dithiothreitol, Triton X-100, Tween 20, glycerol, collagenase, oleic acid/C18:1 (O-1383–1G), linoleic acid/C18:2 (L-1012), linolenic acid/C18:3 (L-2376), palmitic acid/C16:0 (P-5177), stearic acid/C18:0 (5376), arachidic acid/C20:0 (A-3881), behenic acid/C22:0 (B-3271), bovine serum albumin (fraction V), bovine serum albumin fatty acid free (A-6003), 5-phenylbutyric acid, (PBA, #P21005), and HBP (2-hydroxypropyl-β-cyclodextrin, #01816LD) were purchased from Sigma-Aldrich. Sodium thiopental was from Lilly, recombinant leptin and highly purified Escherichia coli LPS were from Calbiochem (Merck; KGaA). All the chemicals used in the real-time PCR and XBP-1 splicing experiments and 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) used in immunofluorescence staining were purchased from Invitrogen and Applied Biosystems.
Diets and fatty acids.
Mice and rats were fed either a standard rodent chow (CD) containing 4.0% (wt/wt) (g%) fat, a high-fat chow (HF) containing 36.0 g% fat from animal source, or an unsaturated fat-rich chow [oleic acid-rich (OL)] containing 36.0 g% fat from olive oil. Fatty acids for intracerebroventricular injection were always diluted in ultrapure water containing HBP detergent (0.1%) and fatty acid free BSA (75 μm). The volumes injected were always 2.0 μl/dose. The final concentration of fatty acids was always 225 μm. In some experiments, rats were intracerebroventricularly treated with oleic acid alone (225 μm) in parallel with a mixture of fatty acids containing palmitic, stearic, arachidic, and behenic acids (25% each, to a final concentration of 225 mm), referred to as the saturated mixture (SM); or SM (20%) plus oleic (20%), linoleic (30%), and linolenic (30%) acids (to a final concentration of 225 mm), referred to as the vegetable mixture (VM). LPS contamination of the fatty acids, HBP and BSA preparations were evaluated by Limulus amoebocyte lysate assay produced by Associates of Cape Cod. LPS in the diets were evaluated by HPLC as described below. Only trace amounts of LPS (ranging from 0.026 to 0.075 EU/nmol) were detected in the reagents. According to a previously study (Weinstein et al., 2008), these levels of LPS do not interfere with the results. Nevertheless, to assure that signal transduction through TLR2 and TLR4 would not be activated by these amounts of LPS, rats were intracerebroventricularly treated with 2.0 μl solution containing 3.0 or 300 ng LPS (corresponding to the amounts of LPS equivalent to 0.075 and 7.5 EU/nmol, respectively), and signal transduction was determined by immunoblot, as described below and presented in Figure 2A.
Figure 2.

A, Immunoprecipitation/immunoblot (IP/IB) analysis of the associations of TLR2 and TLR4 with MyD88 in hypothalamic protein extracts obtained from rats treated intracerebroventricularly with diluent (DL), 3.0 ng LPS (LPS3), or 300 ng LPS (LPS300) for 3 d. B, Immunoblot (IB) analysis of expression of TNF-α, IL-1β, IL-6, and IL-10 in hypothalamic protein extracts obtained from rats treated intracerebroventricularly with diluent (DL), oleic acid (C18:1), VM (as presented in Materials and Methods), or SM (as presented in Materials and Methods). C, Real-time PCR determination of IL-6 and IL-10 mRNA expression in hypothalamic samples obtained from rats treated intracerebroventricularly with diluent (DL), oleic acid (C18:1), VM or SM. D, Immunoblot analysis of expressions of TNF-α, IL-1β, IL-6, and IL-10 in hypothalamic protein extracts obtained from rats treated intracerebroventricularly with diluent (DL), palmytic acid (C16:0), stearic acid (C18:0), linoleic acid (C18:2), linolenic acid (C18:3), arachidic acid (C20:0), and behenic acid (C22:0). In all experiments, n = 5; in B and D, results are presented as arbitrary scanning units (ASU) ±SEM, *p < 0.05 versus respective control; in C, results are presented as transcript amount, and different letters refer to significant differences between groups, p < 0.05.
Animal models and experimental protocols.
Male Wistar rats and male TLR4 loss-of-function mutant (C3H/HeJ) mice and their respective controls (C3H/HeN) were used in the experiments. The investigation followed the University guidelines for the use of animals in experimental studies and conforms to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (NIH publication No. 85–23 revised 1996). The animals were maintained on a 12 h light/dark cycle and housed in individual cages. For intracerebroventricular cannulation, 8-week-old rats with a body mass of 250–300 g or mice with a body mass of 25–30 g were used. For evaluation of the effects of distinct diets on metabolic and inflammatory parameters, rats were fed from the 4th to 20th weeks of life on CD or HF diets (experiments presented in Fig. 1) or on CD, HF, or OL diets from the 6th to 14th weeks of life (experiments presented in Figs. 8, 9). C3H/HeJ and C3H/HeN mice were fed on HF diet from the 8th to 16th weeks of life (experiments presented in Fig. 8). Pharmacological inhibition of TLR2 and TLR4 was achieved by a daily intraperitoneal injection of 100 μl solution containing 1.0 μg IgG of anti-TLR2 or anti-TLR4 antibodies (see Fig. 8), or by a daily intracerebroventricular injection of 2.0 μl solution containing 0.4 μg IgG of anti-TLR2 or anti-TLR4 antibodies (see Figs. 4, 6, 9). Pharmacological inhibition of endoplasmic reticulum stress was achieved by a daily intraperitoneal injection of 1.0 g/kg PBA (see Fig. 8) or by a daily intracerebroventricular injection of 1.0 mg PBA (see Figs. 5, 6).
Figure 1.

A, Immunoblot (IB) analysis of expression of TNF-α, IL-1β, IL-6, and IL-10 in hypothalamic protein extracts obtained from 4 (4 w)- and 20 (20 w)-week old rats fed on CD and HF diets for 16 weeks, starting at 4 weeks of age. B, Immunofluorescence staining of F4/80 in the CNS of rats fed on CD and HF diets for 16 weeks; representative microphotographs were obtained from hypothalamic periventricular (3v) zone, in the first and second panels from the left; dorsal third ventricular (D3v) zone, in the third panel from the left; dentate gyrus zone (DG), in the fourth panel from the left; and granular layer of cerebellum, in the fifth panel from the left. In A, n = 5; results are presented as arbitrary scanning units (ASU) ±SEM, *p < 0.05 versus respective control. B, Microphotographs are representative of three distinct experiments; nuclei are stained with DAPI.
Figure 8.
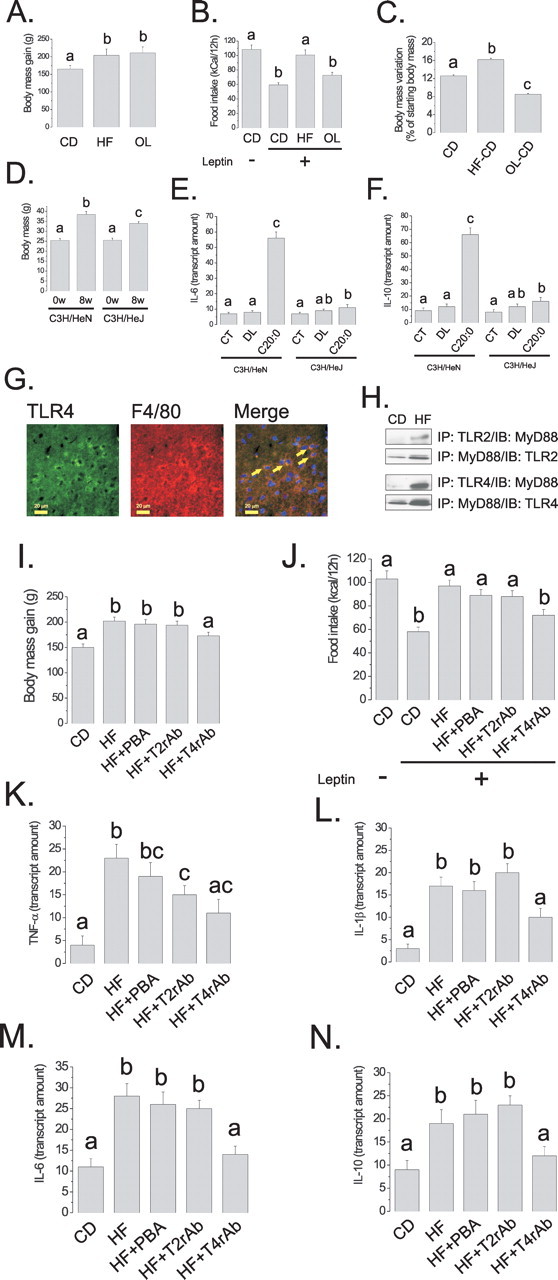
A–C, Rats were fed on CD, HF, or OL diets for 8 weeks; body mass variation (A) was determined during the experimental period. At the end of the experimental period, rats were treated intraperitoneally with saline (100 μl) (−) or a similar volume of leptin (10−6 m) (+), and spontaneous food intake was determined over 12 h (B). After 8 weeks on either diet, HF and OL rats were reallocated to CD diet for a further 4 weeks, and body mass variation was determined (C). D, C3H/HeN and C3H/HeJ mice were fed on a saturated-rich high-fat diet for 8 weeks, and body mass was determined in the first (0w) and last days (8w) of the experimental periods. E, F, Real-time PCR determination of IL-6 (E) and IL-10 (F) mRNA expressions in hypothalamic samples obtained from C3H/HeN and C3H/HeJ mice nonintracerebroventricularly canullated (CT), or treated intracerebroventricularly with diluent (DL) or arachidic acid (C20:0). G, Double-immunofluorescence staining of TLR4 and F4/80 in the arcuate nucleus of rats fed on high-fat diet for 8 weeks, the arrows depict double-positive cells. H, Immunoprecipitation (IP) analysis of the associations of TLR2 and TLR4 with MyD88 in hypothalamic protein extracts obtained from rats fed on CD or HF diets for 8 weeks. I, J, Rats were fed on CD or HF diets for 8 weeks. Throughout the period, the rats were treated with a daily intraperitoneal dose of PBA (HF+PBA), TLR2-inhibiting antibody (HF+T2rAb), or TLR4-inhibiting antibody (HF+T4rAb). Body mass variation (I) was determined. At the end of the experimental period, rats were treated intraperitoneally with saline (100 μl) (−) or a similar volume of leptin (10−6 m) (+), and spontaneous food intake was determined over 12 h (J). K–N, Real-time PCR determination of TNF-α (K), IL-1β (L), IL-6 (M) and IL-10 (N) mRNA expressions in hypothalamic samples obtained from rats treated according to the same protocol as for I, J. In all experiments, except G, n = 5; different letters mean significant differences between groups, p < 0.05. G, Microphotographs are representative of three distinct experiments; nuclei are stained with DAPI. The depicted blots are representative of five distinct experiments.
Figure 9.

A, Rats were fed on high-fat diet for 8 weeks. During the last 7 d of the protocol, the rats were intracerebroventricularly treated with saline (HF), TLR2-inhibiting antibody (HF+T2rAb), or TLR4-inhibiting antibody (HF+T4rAb). Body mass change was measured over the 7 d (A). At the end of the experimental period, hypothalami were obtained for real-time PCR analysis of TNF-α (B), IL-1β (C), IL-6 (D), and IL-10 (E) mRNA determination. In all experiments, n = 5; different letters mean significant differences between groups, p < 0.05.
Figure 4.

A, Immunoprecipitation (IP) analysis of the associations of TLR2 and TLR4 with MyD88 in hypothalamic protein extracts obtained from rats not intracerebroventricularly cannulated (CT), and rats treated intracerebroventricularly with diluent (DL), arachidic acid (C20:0), TLR2 receptor antibody plus arachidic acid (T2rAb+C20:0) or TLR4 receptor antibody plus arachidic acid (T4rAb+C20:0) for 3 d. B, Immunoblot (IB) analysis of expression of pJNK, JNK, pPERK, PERK, GRP78 and peIF2α in hypothalamic protein extracts. C, PCR analysis of spliced/total XBP-1 transcripts in hypothalamic samples. In A and B, the depicted blots are representative of five distinct experiments; in C, n = 5; different letters mean significant differences between groups, p < 0.05.
Figure 6.

Real-time PCR determination of TNF-α (A), IL-1β (B), IL-6 (C), and IL-10 (D) mRNA expressions in hypothalamic samples obtained from rats nonintracerebroventricularly canulated (CT), or treated intracerebroventricularly with diluent (DL), arachidic acid (C20:0), TLR4 receptor antibody plus arachidic acid (T4rAb+C20:0), TLR2 receptor antibody plus arachidic acid (T2rAb+C20:0) or PBA plus arachidic acid (PBA+C20:0) for 3 d. The results are presented as transcript amount, n = 5, and different letters mean significant differences between groups, p < 0.05.
Figure 5.

A, Immunoblot (IB) analysis of expression of pJNK, JNK, pPERK, PERK, GRP78, and peIF2α in hypothalamic protein extracts obtained from rats not intracerebroventricularly cannulated (CT), and from rats treated intracerebroventricularly with diluent (DL), arachidic acid (C20:0), or PBA plus arachidic acid (PBA+C20:0) for 3 d. B, Immunoprecipitation (IP) analysis of the associations of TLR2 and TLR4 with MyD88 in hypothalamic protein extracts obtained from rats treated as described in A. The depicted blots are representative of five distinct experiments.
Primary culture of macrophages and flow cytometry.
Eight-week-old C3H/HeJ and C3H/HeN mice, fed on control diet, were injected intraperitoneally with a single dose of 2% (w/v) sodium thioglycolate (Sigma-Aldrich), and after 2 d, the animals received a lethal dose of anesthetics (sodium thiopental 20 mg/kg) followed by 2.0 ml PBS into the peritoneal cavity to recover the macrophages. Primary cultures were obtained by plating the macrophages at an initial density of 105 cells/ml Roswell Park Memorial Institute 1640, in 1.5 cm culture dishes. After 30 min, the macrophages were treated, for 16 h, with one of the following conditions: 22.5 mm arachidic acid; 22.5 mm arachidic acid plus 5.0 mm PBA or diluent. At the end of the incubation period, the macrophages were harvested and resuspended in PBS containing 10% fatty acid free BSA. For F4/80 labeling, the cells were not submitted to any fixation and permeabilization protocol. For the remainder of the proteins (p-JNK, p-PERK, p-elF2α, and GRP78), cells were fixed in 4% paraformaldehyde and permeabilized with 0.1% saponin. Primary antibodies were used in a final concentration of 2.5 μg/ml and the secondary, FITC conjugated, antibody was used in a final dilution of 1:100. Signal detection was performed in a FACSCalibur flow cytometer equipped with an argon laser, and analysis of data were performed with the CellQuest software (BD Biosciences). Differences are presented as percent variation of control.
HPLC.
Determination of lipids and LPS in diets was performed using a method described previously (Martins et al., 2004). Briefly, fatty acids were derivatized with 4-bromomethyl-7-coumarin and the analysis performed in a Shimadzu model LC-10A liquid chromatographer. The samples were eluted using a C8 column (25 cm × 4.6 id, 5 μm of particles) with a C8 precolumn (2.5 cm × 4.6 id, 5 μm of particles), 1.0 ml/min of acetonitrile/water (77%/23%, v/v) flow and fluorescence detector (325 nm excitation and 395 nm emission). For quantification of fatty acids, the capacity factor, elution sequence, linearity, recovery, precision, interference, and limit of detection were determined. The lower limit of detection was 1.0 pg. Highly purified LPS was used as a tracer.
Hypothalamus histology.
Hydrated, 5.0 μm sections of paraformaldehyde-fixed, paraffin-embedded CNS specimens were obtained from rats treated for 8 or 16 weeks with CD or HF diets. The expression of F4/80 and the coimmunolocalization of TLR4 with F4/80, AgRP, and POMC were evaluated by indirect immunofluorescence staining, as described previously (Bertelli et al., 2006). The expression of F4/80 was evaluated in hypothalamus, frontal, parietal, occipital and cerebellar cortexes, thalamus and hippocampus. The coimmunolocalizations were determined in the hypothalamus.
Reverse-transcription PCR and XBP-1 splicing.
Standard reverse-transcription PCR was performed using total RNA from hypothalamic samples as described previously (Bertelli et al., 2006). The primers used for the amplification of XBP-1 (GenBank accession number, AF443192) were as follows: forward, 5′-AAA CAG AGT AGC AGC GCA GAC TGC-3′; reverse, 5′-GGA TCT CTA AAA CTA GAG GCT TGG TG-3′. Products were digested with PstI, and the products were separated on agarose gels and visualized by Cyber Gold staining.
Real-time PCR.
IL-6, IL-10, TNF-α, and IL-1β mRNAs were measured in the hypothalami of rats treated with CD or HF diets and in intracerebroventricular cannulated rats treated with arachidic acid in the presence or absence of the TLR2 or TLR4 receptor-inhibiting antibodies. Alternatively, these procedures were performed in the presence or absence of the ER stress inhibitor, PBA. In some experiments, C3H/HeJ and C3H/HeN mice were intracerebroventricularly cannulated and treated with arachidic acid or diluent for 3 d. Intron-skipping primers were obtained from Applied Biosystems: TNF-α, Rn00562055_m1; IL-1β, Rn00580432_m1; IL-6, Rn00561420; IL-10, Rn00563409 for rats; or, TNF-α, Mm00443258_m1; IL-1β, Mm00434228_m1; IL-6, Mm99999064; IL-10, Mm00439615 for mice. Glyceraldehyde-3-phosphate dehydrogenase primers (Applied Biosystems) were used as control: #4352338E for rats, and #4352339E for mice. Real-time PCR analysis of gene expression was performed in an ABI Prism 7700 sequence detection system (Applied Biosystems). The optimal concentration of cDNA and primers, as well as the maximum efficiency of amplification, were obtained through five-point, twofold dilution curve analysis for each gene. Each PCR contained 3.0 ng of reverse-transcribed RNA, 200 nm of each specific primer, SYBR SAFE PCR master mix, and RNase free water to a 20 μl final volume. Real-time data were analyzed using the Sequence Detector System 1.7 (Applied Biosystems).
Immunoprecipitation and immunoblotting.
For evaluation of cytokine expression, TLR activation and ER stress induction, the hypothalami of anesthetized rats were excised and immediately homogenized in solubilization buffer at 4°C. Aliquots of the resulting protein extract containing 2.0 mg of total protein were used for immunoprecipitation with antibodies against TLR2, TLR4, and MyD88 at 4°C overnight, followed by SDS-PAGE transfer to nitrocellulose membranes and blotting with anti-TLR2, anti-TLR4, or anti-MyD88 antibodies. In direct immunoblot experiments, 0.2 mg of protein extracts were separated by SDS-PAGE, transferred to nitrocellulose membranes, and blotted with anti-TNF-α, anti-IL-1β, anti-IL-6, anti-IL-10, anti-pJNK, anti-JNK, anti-pPERK, anti-PERK, anti-GRP78, and anti-peIF2α. Specific bands were detected by chemiluminescence, and visualization was performed by exposure of the membranes to RX-films.
Statistical analysis.
Specific protein bands present in the blots and cDNA bands in agarose gels were quantified by digital densitometry (ScionCorp). Mean values ± SEM obtained from densitometry scans and from real-time PCR, XBP-1 splicing measurements, body mass determination, and food intake were compared using Tukey–Kramer test (ANOVA) or Student's t test, as appropriate; p < 0.05 was accepted as statistically significant.
Results
High-fat diet induces inflammatory protein expression in hypothalamus
In the first part of the study, Wistar rats were treated either with a CD containing 4.0 g% total fat (2.0 g% saturated fat, as determined by HPLC), or an HF, containing 36.0 g% total fat (5.0 g% saturated fat, as determined by HPLC). Consumption of the HF diet for 16 weeks led to a significant increase in the expression of the inflammatory cytokines TNF-α, IL-1β and IL-6, but not of the anti-inflammatory cytokine, IL-10, in the hypothalamus (Fig. 1A). These effects were accompanied by an increased expression of the F4/80 antigen in cells present mostly in the medial eminence and arcuate nucleus of the hypothalamus, but not in all other anatomical sites of the brain examined (frontal, parietal, occipital and cerebellar cortexes, thalamus and hippocampus) (Fig. 1B).
Long-chain saturated fatty acids exert the most potent inflammatory stimulus in hypothalamus
To determine the direct effect of different fatty acids in the expression of inflammatory proteins in hypothalamus, rats were intracerebroventricularly cannulated and treated, initially, with the monounsaturated fatty acid, oleic acid or with fatty acid mixtures containing predominantly fatty acids present in VM, or in animal fat (SM). First, to evaluate if the trace amounts of LPS detected in the reagents could interfere with the results by activating TLR2/4 signaling, we treated rats for 3 d with a dose of LPS corresponding to the highest contaminating level determined in our reagents. As depicted in Figure 2A, 3.0 ng LPS, corresponding to 0.075 EU/nmol endotoxin activity, produced no effect on TLR2 or TLR4 signal transduction. Three days intracerebroventricular oleic acid treatment produced significant increases in the expression of IL-6 and IL-10, as determined by immunoblot (Fig. 2B) and real-time PCR (Fig. 2C). VM produced a significant increase of TNF-α and IL-1β, as determined by immunoblot (Fig. 2B), and significant increases in IL-6 and IL-10, as determined by real-time PCR (Fig. 2C). SM produced significant increases in the expressions in TNF-α, IL-1β and IL-6, as determined by immunoblot (Fig. 2B), and IL-6 as determined by real-time PCR (Fig. 2C). To further understand the role of each individual fatty acid type to induce cytokine expression in the hypothalamus, rats were intracerebroventricularly treated for 3 d with palmitic, stearic, linoleic, linolenic, arachidic, or behenic acids. As depicted in Figure 2D, the long-chain saturated fatty acids, mostly stearic, arachidic and behenic, induced the expression of inflammatory cytokines. In the case of stearic and arachidic, some stimulus for the expression of IL-10 was also detected.
ER stress and TLR signaling are induced by long-chain saturated fatty acids
To evaluate the effect of long-chain saturated fatty acids on the induction of ER stress, rats were intracerebroventricularly treated from 1 to 3 d with two daily doses of arachidic acid and the expressions of proteins induced during the UPR were determined by immunoblot. As depicted in Figure 3A, UPR was rapidly induced in the hypothalamus of arachidic acid-treated rats. The expressions of the phosphorylated forms of JNK, PERK and eIF2α and the protein amount of GRP78 were clearly increased after 1–2 d treatment, reaching the highest levels on day 3 (23 ± 4 vs 203 ± 18* for pJNK, 14 ± 3 vs 265 ± 21* for pPERK, 12 ± 4 vs 205 ± 16 for GRP78, and 19 ± 8 vs 229 ± 21 for peIF2α; n = 5; *p < 0.05). No induction of UPR was detected in the hypothalamus of control rats. In addition, the increased ratios of spliced/total (0.61 ± 0.07 vs 0.84 ± 0.09; n = 5; p < 0.05) and spliced/nonspliced XBP-1 (0.49 ± 0.04 vs 0.81 ± 0.07; n = 5; p < 0.05) transcripts further confirmed the ability of arachidic acid to induce ER stress. The capacity of arachidic acid to induce signal transduction through TLR2 and TLR4 in the hypothalamus was tested in rats intracerebroventricularly treated for 3 d with this fatty acid. As shown in Figure 3B, both TLR2/MyD88 and TLR4/MyD88 associations/activations were induced by the long-chain saturated fatty acid. The greatest effect was seen on TLR4/MyD88 association, which was 5.6-fold (1234 ± 102 vs 222 ± 34 arbitrary scanning units; n = 5; p < 0.05) more stimulated than TLR2/MyD88, suggesting that TLR4 is the main receptor engaged by the long-chain saturated fatty acid.
Figure 3.

A, Immunoblot (IB) analysis of the expressions of pJNK, JNK, pPERK, PERK, GRP78, and peIF2α in hypothalamic protein extracts obtained from rats treated intracerebroventricularly with diluent (DL) or arachidic acid (C20:0) for 1–3 d; some rats were intracerebroventricularly cannulated but received no treatment (−). B, Immunoprecipitation (IP) analysis of the associations of TLR2 and TLR4 with MyD88 in hypothalamic protein extracts obtained from nonintracerebroventricular cannulated (CT) rats, and rats treated intracerebroventricularly with DL or C20:0 for 3 d. The depicted blots are representative of five distinct experiments.
Activation of TLR4 is the main event linking long-chain saturated fatty acid to cytokine expression in the hypothalamus
To determine the impact of TLR2, TLR4, and ER stress activation, by long-chain saturated fatty acids, on induction of an inflammatory response in the hypothalamus, specific inhibitors of TLR2, TLR4, and ER stress were used. The inhibition of TLR2 was achieved by intracerebroventricularly treating the rats with a daily dose of an inhibitory TLR2 antibody. This treatment completely blunted arachidic acid-induced activation of TLR2 signaling (Fig. 4A) but promoted no modulation of arachidic acid-induced activation of TLR4 (Fig. 4A) or induction of ER stress (Fig. 4B). Conversely, the inhibition of TLR4 by a daily intracerebroventricular dose of an inhibitory TLR4 antibody completely abolished TLR4 activation (Fig. 4A) and ER stress induction (Fig. 4B), including the inhibition of the arachidic acid-induced increase in the spliced form of XBP-1 (Fig. 4C). No significant modulation of TLR2 activation was obtained by this approach (Fig. 4A). When rats were intracerebroventricularly treated with a daily dose of the chaperone PBA to inhibit ER stress, only the reversal of arachidic acid-induced ER stress was obtained (Fig. 5A), with no impact on TLR2 and TLR4 activation (Fig. 5B). Furthermore, the inhibition of TLR4 signaling by the TLR4-inhibiting antibody completely restrained the capacity of arachidic acid to induce TNF-α, IL-1β, IL-6 and IL-10 expression (Fig. 6A–D). However, PBA treatment only partially inhibited arachidic acid-induced expression of these cytokines (Fig. 6A–D), suggesting that ER stress induction mediates only part of the signals generated by the long-chain saturated fatty acid toward cytokine expression.
TLR4 loss-of-function mutation protects macrophages from saturated fatty acid-induced ER stress activation
To evaluate if the phenomenon described above, in the hypothalamus, could be reproduced in isolated cells, peritoneal macrophages prepared from control (C3H/HeN) and TLR4 loss-of-function mutation (C3H/HeJ) mice were treated for 16 h with arachidic acid, and the activation of markers of ER stress was determined by flow cytometry. Initially, the specificity of the method was tested by treating the cells from C3H/HeN mice with PBA to chemically inhibit ER stress induction. As shown in Figure 7A, PBA was unable to inhibit arachidic acid-induced F4/80 expression but almost completely blunted fatty acid-induced activation of p-JNK (89 ± 6%; p < 0.05), p-eIF2α (93 ± 4%; p < 0.05), p-PERK (79 ± 5%; p < 0.05) and GRP78 (93 ± 4%; p < 0.05). When macrophages from C3H/HeJ mice were submitted to a similar treatment, an almost complete inhibition of arachidic acid-induced expression of F4/80 (86 ± 4%; p < 0.05), p-JNK (88 ± 5%; p < 0.05), and GRP78 (95 ± 4%; p < 0.05), a partial inhibition of arachidic acid-induced expression of p-eIF2α (58 ± 9%; p < 0.05) and p-PERK (69 ± 5, p < 0.05), were observed (Fig. 7B).
Figure 7.
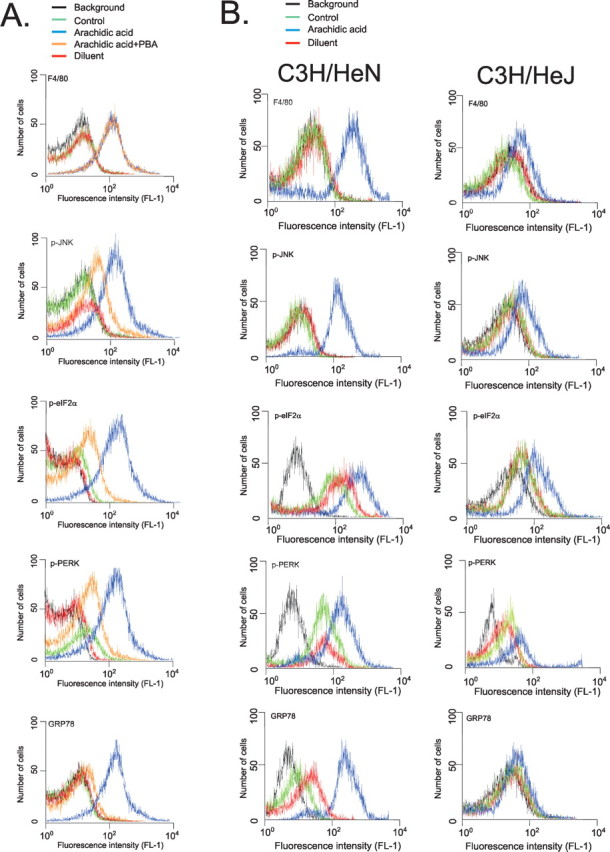
Flow cytometry analysis of expressions of F4/80, p-JNK, p-elf2α, p-PERK, and GRP78 in isolated macrophages. A, Macrophages from C3H/HeN mice were plated and incubated for 16 h in the presence of arachidic acid (blue), arachidic acid plus PBA (orange) or diluent (red). After harvesting, the cells were incubated with specific primary antibodies and then labeled with secondary conjugated antibody. Signal detection was performed by flow cytometry. B, Macrophages from C3H/HeN and C3H/HeJ mice were plated and incubated for 16 h in the presence of arachidic acid (blue) or diluent (red). After harvesting, the cells were incubated with specific primary antibodies and then labeled with secondary conjugated antibody. Signal detection was performed by flow cytometry. Graphs are representative of n = 5. Background counts (black); control, not added primary antibody (green).
Saturated, but not unsaturated, fatty acid-rich diet leads to resistance to anorexigenic hormone action
As one of the main molecular mechanisms linking the consumption of fat-rich diets to insulin and leptin resistance in peripheral and hypothalamic tissues is the induction of an inflammatory response and the expression/action of inflammatory cytokines, we tested the hypothesis that only the consumption of a saturated fatty acid-rich, and not of an isocaloric unsaturated fatty acid-rich diet, would promote resistance to anorexigenic signals in the hypothalamus. For this, Wistar rats were treated for 8 weeks with CD, HF, or OL diets and evaluated for metabolic parameters and response to intracerebroventricularly injected leptin. The OL diet is isocaloric with HF but contains less saturated fat, as determined by HPLC (HF contains 5.0 g% saturated fat, whereas OL contains 2.8 g% saturated fat). As shown in Figure 8, the consumption of either HF or OL diets led to similar changes in body mass (Fig. 8A) and mean daily caloric intake (data not shown). However, whereas in HF fed rats intracerebroventricular leptin promoted no suppression of food intake, in OL rats, the effect of leptin was similar to controls (Fig. 8B), leading to a 38% reduction in spontaneous food intake over 12 h. In addition, when rats fed for 8 weeks on either HF or OL diets were moved to CD diet for a further 4 weeks, the rate of body mass gain was significantly reduced in previously OL fed rats, whereas in previously HF fed rats, the rate was still higher than control (Fig. 8C).
TLR4 loss-of-function mutation protects from diet-induced body mass gain and from fatty acid-induced hypothalamic cytokine expression
To test the hypothesis that TLR4 mediates most of the effect of dietary saturated fatty acids toward hypothalamic inflammation and impairment of anorexigenic signals, TLR4 loss-of-function mutant mice were fed on an HF diet for 8 weeks and metabolic parameters were determined. As depicted in Figure 8D, whereas control mice presented a 50% body mass gain during the period, the body mass of mutant mice increased by only 30% in the same period. This effect was independent of any significant change in food intake. In addition, TLR4 loss-of-function mutant mice failed to induce a remarkable increase in the expression of IL-6 and IL-10 in hypothalamus, after a 3 d intracerebroventricular treatment with arachidic acid (Fig. 8E,F).
Most TLR4 expression occurs in activated microglia
The cellular distribution of TLR4 in the hypothalamus was evaluated by double-immunofluorescence staining of hypothalamic sections obtained from Wistar rats fed on HF diet for 8 weeks. Most TLR4-positive cells were microglia cells expressing the F4/80 protein (Fig. 8G). AgRP and POMC neurons expressed virtually no TLR4 (data not shown).
TLR4 and TLR2 signal transductions are constitutively activated in the hypothalamus of rats fed on high-fat diet
The associations of TLR2 and TLR4 with MyD88 were significantly increased in the hypothalamus of rats fed on HF diet from 8 weeks. As depicted in Figure 8H, HF diet led to 123 ± 14% (p < 0.05) increase in the association TLR2/MyD88 and 321 ± 23% (p < 0.05) increase in the association TLR4/MyD88.
Pharmacological inhibition of TLR4, but not of TLR2 and ER stress, impairs diet-induced body mass gain and leptin resistance
To determine the impact of TLR2/4 activity and ER stress on diet-induced obesity and hypothalamic leptin resistance, Wistar rats were fed on a HF diet for 8 weeks and treated with daily intraperitoneal doses of TLR2-inhibiting antibody, TLR4-inhibiting antibody, or PBA. As shown in Figure 8I, TLR4, but not TLR2 or ER stress inhibitions, completely abolished diet-induced body mass gain independently of significant changes in food intake (data not shown). In addition, TLR4, but not TLR2 and ER stress, inhibition reversed diet-induced leptin resistance in the hypothalamus (Fig. 8J). These effects were accompanied by significant reductions of hypothalamic TNF-α (Fig. 8K), IL-1β (Fig. 8L), IL-6 (Fig. 8M), and IL-10 (Fig. 8N) expressions by the inhibition of TLR4. Similar results were obtained by inhibiting TLR4 only in the hypothalamus. For that, Wistar rats were fed on HF diet for 8 weeks. During the last 7 d of the protocol, the rats were treated with a daily intracerebroventricular dose of TLR4- or TLR2-inhibiting antibodies. As shown in Figure 9A, only the inhibition of TLR4 was capable of significantly reducing body mass variation. This was accompanied by significant reductions of TNF-α (Fig. 9B), IL-1β (Fig. 9C), IL-6 (Fig. 9D), and IL-10 (Fig. 9E) expressions.
Discussion
Molecular and functional resistance to anorexigenic/thermogenic signaling in the hypothalamus is a common feature of most animal models of obesity (Carvalheira et al., 2003; Howard et al., 2004; De Souza et al., 2005; Münzberg and Myers, 2005; Enriori et al., 2007). The main molecular events determining this phenotype are the systemic and local production of cytokines (Howard et al., 2004; De Souza et al., 2005) and the activation of inflammatory pathways in cells of restricted areas of the hypothalamus involved in the control of feeding and thermogenesis (Howard et al., 2004; De Souza et al., 2005; Prada et al., 2005; Araújo et al., 2007). Although it is generally accepted that consumption of fat-rich diets is among the most important environmental factors leading to obesity, little is known about the mechanisms linking dietary fats to the activation of inflammatory response and impairment of anorexigenic/thermogenic signaling in the hypothalamus. Recently, the activation of TLR signaling and the induction of ER stress have been implicated in the pathogenesis of diet- and obesity-related insulin resistance and diabetes mellitus. In this study, we evaluated the capacity of fatty acids to induce TLR signaling and ER stress in the hypothalamus (Ozcan et al., 2004, 2006; Shi et al., 2006; Tsukumo et al., 2007).
Initially, we determined the ability of different fatty acids to induce the expression of cytokines in the hypothalamus. The HF diet contained as much as twofold more saturated fat than the control diet. This is consistent with the amount of saturated fat present in most Western diets and is much beyond that recommended by the guidelines of diabetes, nutrition, and cardiovascular international societies (Shekelle et al., 1981; McKeigue et al., 1985; Dougherty et al., 1988). The rats fed on the HF diet expressed high levels of TNF-α, IL-1β, and IL-6 in the hypothalamus, and this was accompanied by the increased presence of F4/80-expressing cells, predominantly in the medium eminence and arcuate nucleus. These data reinforce the concept that consumption of a fat-rich diet induces an inflammatory response in hypothalamus and suggests that the activation of an innate immune response is implicated in the process (De Souza et al., 2005). Moreover, by ruling out the expression of F4/80 in other regions of the brain, we provide further support for the anatomic specificity of the phenomenon (Howard et al., 2004; De Souza et al., 2005; Enriori et al., 2007).
To evaluate whether fatty acids, acting directly in the hypothalamus, can reproduce the effect of HF diet, rats were intracerebroventricularly treated with different mixtures of fatty acids, and the expression of cytokines was determined. The compositions of the mixtures were chosen based on the expected compositions of vegetable oils and animal fat, particularly soybean oil (Fedeli and Jacini, 1971) and pork lard (Brooks, 1971), respectively. Oleic acid, used as a control, induced only the expression of IL-10 and IL-6, both known to possess anti-inflammatory actions, and therefore, reinforcing the alleged anti-inflammatory property of this fatty acid (Martínez-Dominguez et al., 2001; Yoneyama et al., 2007). Conversely, both the vegetable and the saturated fatty acid mixtures provided consistent stimuli for the expression of inflammatory cytokines, however, with the greater effect induced by the saturated mixture. When pure saturated and unsaturated fatty acids were tested separately, there was a clear difference with greatest inflammatory effect produced by long-chain saturated fatty acids. Although some previous studies have evaluated the abilities of fatty acids to induce cytokine expression, this is the first time the phenomenon has been shown in the hypothalamus (de Pablo and Alvarez de Cienfuegos, 2000; Pompéia et al., 2000).
Arachidic acid is a 20-carbon saturated fatty acid present in triacylglycerols from animal fat and some vegetable oils (Hlongwane et al., 2001). We decided to employ this fatty acid in the remainder of the experiments because it produced the highest stimulus for TNF-α expression in the hypothalamus and because some previous reports have shown its property to be transported across the blood–brain barrier (Strosznajder et al., 1996). When intracerebroventricularly injected in the hypothalamus, arachidic acid rapidly induced the activation of ER stress and TLR2 and TLR4 signaling. These were specific events, because no effect was seen with the intracerebroventricular injection of the diluent BSA, and complete inhibition was achieved using specific inhibitors for each pathway tested. Interestingly, in the experiments performed to evaluate TLR activation, the bands obtained when immunoprecipitation was performed with anti-TLRs and blotting with anti-MyD88 antibodies, were different in density compared with bands produced by anti-MyD88 immunoprecipitation and anti-TLRs blotting. Although we have no current explanation for this fact, we suspect that it can be attributable to differences in stoichiometric ratio between the receptors and the adaptor protein, because other adaptor proteins can bind to these receptors (Tanimura et al., 2008).
In previous studies, when TLR signaling and ER stress were evaluated in the context of diet- and genetic-induced obesity, insulin resistance was produced by the activation of intracellular inflammatory signaling through JNK and/or IKK, and further enhanced by the induction of proinflammatory cytokine expression (Ozcan et al., 2004, 2006; Shi et al., 2006; Tsukumo et al., 2007). However, the possible interaction between both mechanisms was never tested. Here, to explore the hypothesis that TLR signaling and ER stress can interact, at the intracellular level, and to evaluate the possibility that one of these mechanisms could in fact lead to or potentiate the other, we performed specific inhibitions of TLR2, TLR4, and ER stress and evaluated the capacity of arachidic acid to induce activation of each one. Our results show that the inhibition of one of the TLRs does not interfere with the activation of the other. However, only the inhibition of TLR4 completely blunts arachidic acid-induced ER stress, suggesting that TLR4 activation precedes and determines ER stress induction. This was further confirmed by studding isolated macrophages from TLR loss-of-function mutation mice. Conversely, the inhibition of ER stress fails to modulate TLR signaling. In addition, when comparing the activation of both TLRs, it is clear that arachidic acid produces a much stronger signal for the activation of TLR4 than TLR2, which is consistent with the fact that TLR2/1 and TLR2/6 dimmers recognize preferentially diacyl and triacyl lipopeptides, respectively, whereas TLR4 recognizes fatty acids contained in LPS and also free fatty acids (Shimazu et al., 1999; Takeuchi et al., 2001; Akira, 2003; Akira et al., 2006). Finally, the in situ inhibition of TLR4 or the genetic disarrangement of this receptor, as it occurs in the TLR4 loss-of-function mutant, completely inhibits arachidic acid induction of cytokine expression, whereas PBA treatment only partially modulates this response. Thus, at the cellular level, the long-chain saturated fatty acid acts predominantly through TLR4, leading to an inflammatory response that can be potentiated by the activation of ER stress, which is also dependent on TLR4 engagement.
The specificity of the response to saturated fatty acids was further investigated by feeding rats isocaloric hyperlipidic diets rich in saturated or unsaturated fat. Although food intake and body mass gain is the same, independently of the diet composition, the anorexigenic response to leptin is preserved in OL rats while completely blunted in HF rats. In addition, the replacement of hyperlipidic diets by normolipidic diet shows that previous consumption of HF diet acts as an imprint that predisposes to continuous high rate body mass gain while a significant decrease in the rate of body mass gain occurs in previously OL fed rats. Thus, the induction of resistance to anorexigenic signaling depends more on diet composition than on caloric value.
In the last part of the study, we show that a functional TLR4 signaling is required for the induction of dietary obesity. Initially, we show that most TLR4 present in the hypothalamus of rats fed on HF diet is expressed in microglia, placing the innate immune system in the center of this process. TLR4 loss-of-function mutation does not affect food intake but protects mice from obesity induced by hypercaloric-hyperlipidic diet, whereas TLR4, but not ER stress pharmacological inhibition, preserves the anorexigenic response to leptin and inhibits diet-induced obesity. In a previous study, some of these questions were addressed, and the protection from diet-induced obesity in C3H/HeJ was mostly related to energy expenditure than to control of feeding, which seem to be the case here, because no change in food intake was seen (Tsukumo et al., 2007). In addition, inhibition of ER stress by chemical means promoted no change of body mass in ob/ob mice (Ozcan et al., 2006).
In conclusion, saturated fatty acids activate TLR2 and TLR4 signaling and ER stress in hypothalamus. The greatest signal is delivered through TLR4, which determines ER stress induction (Fig. 10). The presence of a functional TLR4 signaling is required for the development of hyperlipidic dietary obesity. Thus, in addition to its important role in the development of diet- and obesity-related insulin resistance and diabetes mellitus, TLR4 is an important mediator of hypothalamic dysfunction during the development of obesity. As hypothalamic resistance to anorexigenic signaling is a very incipient phenomenon during the installation of obesity (Prada et al., 2005), TLR4 is an attractive target for therapeutics of this epidemic condition.
Figure 10.

Proposed mechanism for saturated fatty acid (SFA)-induced activation of inflammatory response in hypothalamus. Initially, SFA activates predominantly TLR4, engaging MyD88. The classical downstream signaling through IRAK/TRAF6/NFkB (not tested in the present study) leads to cytokine expression. Through a hitherto unknown mechanism, TLR4 activation induces ER stress, which boosts cytokine expression enhancing the inhibitory signals of anorexigenic hormone action.
Footnotes
This work was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo and Conselho Nacional de Desenvolvimento Científico e Tecnológico. We thank Dr. Nicola Conran for English grammar review and Gerson Ferraz for technical support.
References
- Akira S. Toll-like receptor signaling. J Biol Chem. 2003;278:38105–38108. doi: 10.1074/jbc.R300028200. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Araújo EP, De Souza CT, Ueno M, Cintra DE, Bertolo MB, Carvalheira JB, Saad MJ, Velloso LA. Infliximab restores glucose homeostasis in an animal model of diet-induced obesity and diabetes. Endocrinology. 2007;148:5991–5997. doi: 10.1210/en.2007-0132. [DOI] [PubMed] [Google Scholar]
- Bertelli DF, Araújo EP, Cesquini M, Stoppa GR, Gasparotto-Contessotto M, Toyama MH, Felix JV, Carvalheira JB, Michelini LC, Chiavegatto S, Boschero AC, Saad MJ, Lopes-Cendes I, Velloso LA. Phosphoinositide-specific inositol polyphosphate 5-phosphatase IV inhibits inositide trisphosphate accumulation in hypothalamus and regulates food intake and body weight. Endocrinology. 2006;147:5385–5399. doi: 10.1210/en.2006-0280. [DOI] [PubMed] [Google Scholar]
- Brooks CC. Fatty acid composition of pork lipids as affected by basal diet, fat source and fat level. J Anim Sci. 1971;33:1224–1231. doi: 10.2527/jas1971.3361224x. [DOI] [PubMed] [Google Scholar]
- Carvalheira JB, Ribeiro EB, Araújo EP, Guimarães RB, Telles MM, Torsoni M, Gontijo JA, Velloso LA, Saad MJ. Selective impairment of insulin signalling in the hypothalamus of obese Zucker rats. Diabetologia. 2003;46:1629–1640. doi: 10.1007/s00125-003-1246-x. [DOI] [PubMed] [Google Scholar]
- de Pablo MA, Alvarez de Cienfuegos G. Modulatory effects of dietary lipids on immune system functions. Immunol Cell Biol. 2000;78:31–39. doi: 10.1046/j.1440-1711.2000.00875.x. [DOI] [PubMed] [Google Scholar]
- De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJ, Velloso LA. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146:4192–4199. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- Dougherty RM, Fong AK, Iacono JM. Nutrient content of the diet when the fat is reduced. Am J Clin Nutr. 1988;48:970–979. doi: 10.1093/ajcn/48.4.970. [DOI] [PubMed] [Google Scholar]
- Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, Grove KL, Cowley MA. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007;5:181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Fedeli E, Jacini G. Lipid composition of vegetable oils. Adv Lipid Res. 1971;9:335–382. doi: 10.1016/b978-0-12-024909-1.50014-4. [DOI] [PubMed] [Google Scholar]
- Freire RD, Cardoso MA, Gimeno SG, Ferreira SR. Dietary fat is associated with metabolic syndrome in Japanese Brazilians. Diabetes Care. 2005;28:1779–1785. doi: 10.2337/diacare.28.7.1779. [DOI] [PubMed] [Google Scholar]
- Hlongwane C, Delves IG, Wan LW, Ayorinde FO. Comparative quantitative fatty acid analysis of triacylglycerols using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and gas chromatography. Rapid Commun Mass Spectrom. 2001;15:2027–2034. doi: 10.1002/rcm.462. [DOI] [PubMed] [Google Scholar]
- Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjørbaek C, Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med. 2004;10:734–738. doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- Krappmann D, Wegener E, Sunami Y, Esen M, Thiel A, Mordmuller B, Scheidereit C. The IkappaB kinase complex and NF-kappaB act as master regulators of lipopolysaccharide-induced gene expression and control subordinate activation of AP-1. Mol Cell Biol. 2004;24:6488–6500. doi: 10.1128/MCB.24.14.6488-6500.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- Martínez-Domínguez E, de la Puerta R, Ruiz-Gutiérrez V. Protective effects upon experimental inflammation models of a polyphenol-supplemented virgin olive oil diet. Inflamm Res. 2001;50:102–106. doi: 10.1007/s000110050731. [DOI] [PubMed] [Google Scholar]
- Martins EF, Miyasaka CK, Newsholme P, Curi R, Carpinelli AR. Changes of fatty acid composition in incubated rat pancreatic islets. Diabetes Metab. 2004;30:21–27. doi: 10.1016/s1262-3636(07)70085-x. [DOI] [PubMed] [Google Scholar]
- McKeigue PM, Marmot MG, Adelstein AM, Hunt SP, Shipley MJ, Butler SM, Riemersma RA, Turner PR. Diet and risk factors for coronary heart disease in Asians in northwest London. Lancet. 1985;2:1086–1090. doi: 10.1016/s0140-6736(85)90684-1. [DOI] [PubMed] [Google Scholar]
- Moreno LA, Rodríguez G. Dietary risk factors for development of childhood obesity. Curr Opin Clin Nutr Metab Care. 2007;10:336–341. doi: 10.1097/MCO.0b013e3280a94f59. [DOI] [PubMed] [Google Scholar]
- Münzberg H, Myers MG., Jr Molecular and anatomical determinants of central leptin resistance. Nat Neurosci. 2005;8:566–570. doi: 10.1038/nn1454. [DOI] [PubMed] [Google Scholar]
- Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, Ozawa K, Ogawa S, Hori M, Yamasaki Y, Matsuhisa M. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem. 2005;280:847–851. doi: 10.1074/jbc.M411860200. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Miyazaki M, Matsuhisa M, Takano K, Nakatani Y, Hatazaki M, Tamatani T, Yamagata K, Miyagawa J, Kitao Y, Hori O, Yamasaki Y, Ogawa S. The endoplasmic reticulum chaperone improves insulin resistance in type 2 diabetes. Diabetes. 2005;54:657–663. doi: 10.2337/diabetes.54.3.657. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Görgün CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL, Baeuerle PA. A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by transcription factor NF-kappa B. EMBO J. 1995;14:2580–2588. doi: 10.1002/j.1460-2075.1995.tb07256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompéia C, Lopes LR, Miyasaka CK, Procópio J, Sannomiya P, Curi R. Effect of fatty acids on leukocyte function. Braz J Med Biol Res. 2000;33:1255–1268. doi: 10.1590/s0100-879x2000001100001. [DOI] [PubMed] [Google Scholar]
- Prada PO, Zecchin HG, Gasparetti AL, Torsoni MA, Ueno M, Hirata AE, Corezola do Amaral ME, Höer NF, Boschero AC, Saad MJ. Western diet modulates insulin signaling, c-Jun N-terminal kinase activity, and insulin receptor substrate-1ser307 phosphorylation in a tissue-specific fashion. Endocrinology. 2005;146:1576–1587. doi: 10.1210/en.2004-0767. [DOI] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- Shekelle RB, Shryock AM, Paul O, Lepper M, Stamler J, Liu S, Raynor WJ., Jr Diet, serum cholesterol, and death from coronary heart disease. The Western Electric study. N Engl J Med. 1981;304:65–70. doi: 10.1056/NEJM198101083040201. [DOI] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein CJ, Colditz GA. The epidemic of obesity. J Clin Endocrinol Metab. 2004;89:2522–2525. doi: 10.1210/jc.2004-0288. [DOI] [PubMed] [Google Scholar]
- Strosznajder J, Chalimoniuk M, Strosznajder RP, Albanese V, Alberghina M. Arachidonate transport through the blood-retina and blood-brain barrier of the rat during aging. Neurosci Lett. 1996;209:145–148. doi: 10.1016/0304-3940(96)12624-0. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Kawai T, Mühlradt PF, Morr M, Radolf JD, Zychlinsky A, Takeda K, Akira S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol. 2001;13:933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- Tanimura N, Saitoh S, Matsumoto F, Akashi-Takamura S, Miyake K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem Biophys Res Commun. 2008;368:94–99. doi: 10.1016/j.bbrc.2008.01.061. [DOI] [PubMed] [Google Scholar]
- Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, Araújo EP, Vassallo J, Curi R, Velloso LA, Saad MJ. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56:1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- Watowich SS, Morimoto RI, Lamb RA. Flux of the paramyxovirus hemagglutinin-neuraminidase glycoprotein through the endoplasmic reticulum activates transcription of the GRP78-BiP gene. J Virol. 1991;65:3590–3597. doi: 10.1128/jvi.65.7.3590-3597.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JR, Swarts S, Bishop C, Hanisch UK, Möller T. Lipopolysaccharide is a frequent and significant contaminant in microglia-activating factors. Glia. 2008;56:16–26. doi: 10.1002/glia.20585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama S, Miura K, Sasaki S, Yoshita K, Morikawa Y, Ishizaki M, Kido T, Naruse Y, Nakagawa H. Dietary intake of fatty acids and serum C-reactive protein in Japanese. J Epidemiol. 2007;17:86–92. doi: 10.2188/jea.17.86. [DOI] [PMC free article] [PubMed] [Google Scholar]