

Abstract
Understanding the modulation of the neural circuitry of fear is clearly one of the most important aims in neurobiology. Protein phosphorylation in response to external stimuli is considered a major mechanism underlying dynamic changes in neural circuitry. TrkB (Ntrk2) neurotrophin receptor tyrosine kinase potently modulates synaptic plasticity and activates signal transduction pathways mainly through two phosphorylation sites [Y515/Shc site; Y816/PLCγ (phospholipase Cγ) site]. To identify the molecular pathways required for fear learning and amygdalar synaptic plasticity downstream of TrkB, we used highly defined genetic mouse models carrying single point mutations at one of these two sites (Y515F or Y816F) to examine the physiological relevance of pathways activated through these sites for pavlovian fear conditioning (FC), as well as for synaptic plasticity as measured by field recordings obtained from neurons of different amygdala nuclei. We show that a Y816F point mutation impairs acquisition of FC, amygdalar synaptic plasticity, and CaMKII signaling at synapses. In contrast, a Y515F point mutation affects consolidation but not acquisition of FC to tone, and also alters AKT signaling. Thus, TrkB receptors modulate specific phases of fear learning and amygdalar synaptic plasticity through two main phosphorylation docking sites.
Introduction
Emotional fear-related disorders are among the most common human psychiatric disorders. Determining the molecular mechanisms underlying the development and manifestation of such disorders is essential. Pavlovian fear conditioning (FC), a simple form of associative learning, involves learning that certain environmental stimuli predict aversive events and is considered to be a model system to examine the neurobiological basis of learning and memory in the mammalian brain. The amygdala is central for the acquisition, storage, and expression of conditioned fear memory (Lavond et al., 1993; LeDoux, 1996; Davis, 1997; Fendt and Fanselow, 1999); it consists of different nuclei, including the lateral (LA), basolateral (BL), and basal medial (BM) nuclei, which together form the basolateral complex (BLA), and the central nucleus (CeA) (Swanson and Petrovich, 1998). The BLA receives synaptic inputs from different sensory structures, and lesions of these structures impair pavlovian FC (Gale et al., 2004; Koo et al., 2004). Synaptic inputs to the LA from the thalamic medial geniculate nucleus and from the auditory cortex are essential for conditioning with an auditory conditioned stimulus (CS) (LeDoux et al., 1990; Romanski and LeDoux, 1992; Campeau and Davis, 1995), whereas projections from the hippocampus to the BLA support conditioning with contextual CSs (Kim and Fanselow, 1992; Phillips and LeDoux, 1992; Maren et al., 1997). FC also induces behavioral long-term potentiation (LTP) in the amygdala, and synaptic responses measured in the amygdala of a behaving rat (Rogan et al., 1997) and in amygdalar slices obtained from previously conditioned rats (McKernan and Shinnick-Gallagher, 1997) are similar. Interestingly, behavioral LTP exhibits properties common to tetanic LTP in both preparations. TrkB has been genetically shown to play an important role in hippocampal synaptic plasticity and learning (Minichiello et al., 1999; Xu et al., 2000). BDNF/NT4 binding to TrkB activates three main intracellular signaling cascades. The Ras/mitogen-activated protein kinase (MAPK) pathway and the phosphoinositide 3-kinase (PI3K) pathway are activated primarily through Shc/FRS-2 binding to Y515, whereas the calcium/calmodulin kinase pathway is activated through phospholipase Cγ (PLCγ) recruitment at Y816. We have shown that the Ras/MAPK and PLCγ/CaMK/CREB (cAMP response element-binding protein) pathways act in parallel downstream of TrkB and that the PLCγ docking site is necessary for TrkB-mediated hippocampal synaptic plasticity (Minichiello et al., 2002). Moreover, a TrkB Y816F but not Y515F point mutation impairs the acquisition of an associative learning task and in vivo hippocampal LTP (Gruart et al., 2007). Experiments based on temporal changes in BDNF gene expression and the intra-amygdala infusion of a lentiviral vector expressing a dominant-negative TrkB isoform have established an essential role for TrkB in pavlovian FC acquisition (Rattiner et al., 2004). However, the molecular pathways downstream of TrkB contributing to fear learning and amygdalar synaptic plasticity were not identified. The use of genetic mouse models carrying a point mutation at either the Shc or the PLCγ docking site of TrkB (trkBSHC or trkBPLC, respectively) allows examination of the physiological relevance of pathways activated through these sites during pavlovian FC. We found that in vivo, pathways activated through the TrkB/PLCγ site modulate the acquisition of conditioned fear responses to contextual and auditory stimuli, as well as, synaptic plasticity at the lateral–basolateral (LA–BL) and the thalamic–lateral (thalamic–LA) amygdala synapses. In contrast, the TrkB/Shc site through its downstream effector(s) mainly modulates the consolidation but not acquisition of fear learning to tone and the amygdalar synaptic plasticity at the thalamic–LA synapses but not at the LA–BL synapses, indicating that TrkB Y816 and Y515 contribute to specific phases of fear learning and amygdalar synaptic plasticity.
Materials and Methods
Ethics statement
All animal procedures were conducted in accordance with the rules pertaining to animal well-being and were performed in agreement with national and international laws and policies [European Economic Community Council Directive 86/609, OJ L 358, 1, December 12, 1987; National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals, NIH Publication No. 85-23, 1985, revised in 1995). All initiatives to minimize animal experimentation and to alleviate animal pain, suffering, and distress were taken into account.
Mouse strains
The mouse strains used in this study have been previously described (Minichiello et al., 1998, 2002); however, a summary of the strategies used and validation of the point mutations are reported in the supplemental Methods, available at www.jneurosci.org as supplemental material.
Behavioral procedures
Fear conditioning
Adult mice (2–4 months of age) were trained and tested in an operant chamber (18.5 × 18 × 21.5 cm) possessing aluminum sidewalls and Plexiglas rear and front walls (Coulbourn Instruments). The auditory cue (tone) emanated from a loudspeaker located in the sidewall, 15 cm from the floor. The activity of animals was recorded directly on a computer through a single camera located at the top of the chamber. The presentation of tone and shock stimuli in all training and testing sessions was controlled by GraphicState software (Coulbourn Instruments). The chamber was cleaned with ethanol and dried between mice after each training or testing session.
Conditioning.
Mice were allowed to acclimate to the training chamber for 2 min, and then a tone (CS) of 2800 Hz frequency and 85 dB intensity was presented for 30 s and coterminated in the last 2 s with a mild footshock (0.5 mA) [unconditioned stimulus (US)]. Two minutes later, another CS–US pairing was presented, and the mice were returned to their home cages 30 s later. To facilitate the acquisition of conditioned fear in trkBPLC/+ mutants, five CS–US pairings were used instead of two CS–US pairings.
Behavioral testing.
To test for a conditioned fear response to context, the mice were placed in the same chamber used for the conditioning trial 24 h after conditioning and allowed to explore for 4 min without presentation of the auditory CS or footshock. Freezing behavior, defined as complete absence of voluntary movements except for respiratory movements, was scored every 1 s. Twenty-four hours after the contextual conditioning test, the mice were tested for cue memory by returning them to the same chamber, which was modified by the addition of various shapes and designs on the walls and a vanilla scent. Freezing behavior was scored for 2 min before delivery of the tone and then for 2 min in the presence of a continuous auditory cue. Freezing behavior was measured with an automated system whereby continuous video data were analyzed with the aid of Freeze-frame software (Coulbourn Instruments). The software measures the variance in pixel intensity across successive video frames (taken at 1 Hz) and computes its SD. A threshold is then applied to the data to yield a percentage freezing score. Two-way ANOVA tests (with genotype as a between-subjects factor and context or tone tests as a within-subject factor) were used for statistical analysis with p < 0.05 set as the criterion for statistical significance. Post hoc group comparisons were performed with Fisher's LSD test.
Radial maze
The radial maze test was performed essentially as described previously (Minichiello et al., 1999). Briefly, adult male mice (3–6 months of age) were singly housed with water provided ad libitum and gradually reduced to 85% of their free-feeding body weight, which was maintained for the duration of the experiment by providing the mice with a premeasured amount of chow each day. The maze was constructed from gray plastic and was comprised of eight identical arms radiating 37 cm from a central starting platform (perimeter was 7 × 8 cm). At the end of each arm 20 mg of food pellets was placed in a cup. All arms were baited only once and at the beginning of each daily session. Animals received one trial per day for 10 consecutive training days. Each daily trial terminated when 8 correct choices were made (maximum 15 choices) or 15 min elapsed. An arm choice was defined as placement of all paws on a maze arm. An error was scored when the mouse entered a previously explored arm. The percentage of errors made over 15 trials, the number of correct arm choices in the first 8 trials, and the occurrence of first error were considered. For statistical analysis, mean values were compared using a two-way ANOVA with the factors genotype (2 levels = mutants and controls) and learning (10 levels = training days), in which the numbers of correct arm choices out of the first 8 trials, the percentages of errors out of 15 trials, or the occurrences of the first error on each training day were compared. Individual between-group comparisons, when appropriate, were performed using post hoc tests (Duncan multiple-range test).
Electrophysiology
Coronal slices (400 μm thick) containing the amygdala were prepared and maintained using standard procedures [artificial CSF (ACSF) medium: 124 mm NaCl, 3 mm KCl, 1.25 mm KH2PO4, 2 mm MgSO4, 26 mm NaHCO3, 1.8 mm CaCl2, and 10 mm glucose; temperature, 28°C; submerged recording). To evoke field EPSPs (fEPSPs), electric pulses were applied by a bipolar electrode placed either in the LA nucleus near the BL nucleus, in which recording glass electrodes were located, or in the ventral striatum, just medial to the LA, to activate fibers originating, at least in part, in the auditory thalamus. The recording glass electrodes were located in the LA nucleus (Weisskopf et al., 1999). Synaptic field potentials were elicited at a frequency of 0.1 Hz. The slope of the recorded field was calculated and used as a measurement of synaptic strength. LTP was induced with a theta-burst stimulation (TBS) of 10 bursts of four pulses each (100 Hz, 100 ms duration, 200 ms interburst interval) with the same strength as the test stimulus. This TBS was repeated three times at intervals of 10 s. Alternatively, LTP was induced with a tetanus of 3 × 30 pulses (100 Hz, 100 ms duration, at intervals of 10 s) with the same strength as the test stimulus. Strong tetanic stimulation consisted of three series of 100 pulses (100 Hz) with pulse lengths of 100 ms and delivered at intervals of 5 min. Poststimulation recordings were continued for 60 min for early LTP (E-LTP) and for 180 min for late LTP (L-LTP). E-LTP or L-LTP was considered successfully induced when the average of the fEPSP slope size (measured at 55–60 min or 175–180 min, respectively, after TBS or tetanus) showed an increase of at least 20% above baseline (100%) values. Routinely, paired-pulse facilitation (PPF) was tested at intervals of 10, 30, 40, 50, 100, and 200 ms. All measurements were performed and analyzed in a strictly blind manner. The genotypes of the animals were revealed only after the electrophysiological experiments and their evaluation were complete. To analyze the functionality of the NMDA and AMPA/kainate receptors, fEPSPs were recorded initially for 20 min (baseline recording) and then after the addition of 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) in low-Mg2+ (0.5 mm) ACSF applied to the bath solution. After 15 min, dl-2-amino-5-phosphonovalerate (AP-5) (50 mm) together with CNQX was applied for 30 min to the same slice in low-Mg2+ ACSF. Afterward, normal ACSF was used for washout. The results were analyzed for statistical significance using two-tailed, unpaired Student's t tests.
Anatomical/biochemical experiments
Conditioning
Adult mice (2–4 months) were trained in a polymodal operant chamber (see above). Before training, they were allowed to acclimate for 2 min, and then two or five conditioning trials were applied consisting of a 30 s, 2800 Hz, and 85 dB tone that coterminated with a 2 s, 0.5 mA footshock; intertrial intervals were 120 s. Mice were returned to their home cages and 45 min later they were either immediately killed (when analyzed for pAKT and pMAPKs) or first tested for both context (3 min) and cue (2 min tone) and then killed immediately thereafter (when analyzed for pCaMKII). Brains were taken for histological and biochemical analysis. Control animals were not exposed to tones or shocks.
Brain sample collection
For biochemical analysis, mice were killed by cervical dislocation, and the brains were removed from the skull and (without olfactory bulbs) positioned in a cold matrix with which 3 mm coronal slices were collected, followed by the isolation of amygdala nuclei from the basolateral portions of the slice. Samples were frozen in liquid nitrogen and stored at −80°C until use.
Biochemistry
Brain samples were homogenized using micropestles (Eppendorf) and lysed on ice in NP-40 complete lysis buffer [10% glycerol, 50 mm HEPES/NaOH (pH 8.0), 100 mm KCl, 1% NP-40, 2 mm EDTA, 2 mm DTT, 10 mm NaF, 1 mm Na3VO4, 1 mm NaPP, 5 mm benzamidine, 10 μg/ml leupeptine, 14 μg/ml aprotinin]. Cellular debris were removed by centrifugation at 16,000 rpm for 20 min at 4°C. Cortical neurons were washed twice with cold PBS and then lysed on ice for 15 min with 0.6 ml of 1% Triton X-100 lysis buffer [50 mm Tris (pH 7.5), 120 mm NaCl, 10 mm NaF, 1 mm Na3VO4, and protease inhibitors as described above]. Insoluble material was removed from the protein extracts by centrifugation at 13,000 rpm for 15 min at 4°C. The protein content in supernatant fractions was quantified (Bio-Rad DC protein assay kit) and aliquots were frozen in liquid nitrogen and stored at −80°C until use. Protein levels were analyzed by Western blot as previously described (Minichiello et al., 1998, and supplemental Methods, available at www.jneurosci.org as supplemental material). The antibodies (Abs) used were anti-pAKT [Cell Signaling, catalog no. (Cat.) 9271; 1:1000], anti-AKT (Cell Signaling, Cat. 9272; 1:1000), anti-pMAPK (New England Biolabs, clone E10; 1:2000), and anti-ERK1 (extracellular signal-regulated kinase 1) (Zymed 13-8600; 1:3000). The relative levels of proteins were quantified as described in the supplemental Methods, available at www.jneurosci.org as supplemental material.
Synaptosome preparation
Brain samples were processed according to the synaptosomal preparation described by Nagy and Delgado-Escueta (1984). Briefly, the amygdalae of each animal were homogenized in 2 ml of buffer [0.32 m sucrose, 1 mm EDTA, 1 mg/ml BSA, 5 mm HEPES (pH 7.4, reached using 1 m NaOH)] by use of a Dounce homogenizer (10 strokes and 520 rpm). Thirty microliters of crude homogenate was removed and served as the whole-tissue lysate sample and the remaining homogenate was centrifuged for 10 min at 3000 × g. The supernatants were subsequently centrifuged for 12 min at 14,000 × g and resultant pellets were resuspended in 220 μl of Krebs–Ringer buffer [140 mm NaCl, 5 mm KCl, 10 mm HEPES, 1 mm EDTA, 5 mm glucose (pH 7.4, reached using 1 m NaOH)] and 180 μl of Percoll (Sigma-Aldrich, P7828). Samples were then centrifuged for 2 min at 14,000 × g. The synaptosome fraction of the gradient was removed, washed twice with 500 μl of Krebs–Ringer buffer, and centrifuged for 30 s at 14,000 × g. The final pellets were resuspended in 40 μl of Krebs–Ringer buffer. All steps of the procedure were performed at 4°C using ice-cold buffers. Samples were separated by SDS-PAGE using 10% acrylamide and transferred onto nitrocellulose membranes. The blots were probed using anti-pCaMKII (ProMega, V1111; 1:1000), anti-CaMKII (Santa Cruz, H-300 s.c.-13082; 1:1000), anti-synapsin I/II (Synaptic Systems, 106 002; 1:5000), and anti-PSD95 (Upstate Biotechnology, cloneK28/43 05-494; 1:200,000). The secondary antibodies used were a goat anti-rabbit HRP-conjugated (Jackson ImmunoResearch, 115-035-003; 1:5000) and a goat anti-mouse HRP-conjugated (Jackson ImmunoResearch 115-035-146; 1:5000) IgG.
Histology
Mice were anesthetized with avertin (20 mg/kg, i.p.) and transcardially perfused with ice-cold PBS followed by 3% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PB). The perfusion was performed using a peristaltic pump and a flow rate of 1 ml/min. The brains were removed from the skull and postfixed in 3% PFA–PB overnight at 4°C, cryoprotected in 30% sucrose–PB at 4°C for 48 h, and then frozen in OCT Tissue Tek compound (Bayer) by immersion in liquid nitrogen-cooled isopentane. Immunohistochemistry was performed as described by Minichiello et al. (1999). Briefly, 30-μm-thick coronal free-floating sections through the amygdala were obtained using a cryostat. Selected sections were immunostained using an anti-pMAPK Ab (Cell Signaling, 9106; 1:4000). More details of the tissue stainings are reported in the supplemental Methods, available at www.jneurosci.org as supplemental material.
Neuronal cultures
Cerebral cortices were dissected from embryonic day 15.5 mouse embryos generated from breedings of control mice or trkB point mutants. Cortical neurons were dissociated and plated onto poly-l-lysine hydrobromide (Sigma) in the presence of 10% heat-inactivated horse serum for 3 h. Cultures were maintained for 16 h before ligand stimulation in N2–MEM at 37°C in 5% pCO2. Cells were stimulated with 5 or 50 ng/ml of purified recombinant BDNF (Genentech and Regeneron Pharmaceuticals) as described previously (Minichiello et al., 2002).
Imaging
Immunofluorescence
Sections were imaged on a Leica TCS SP5 confocal microscope using a Leica 40×/1.4 NA Plan Apochromat oil objective.
Immunohistochemistry
Imaging of stained sections was performed using a Leica bright-field DMRA microscope.
Results
Mouse models
To determine which site-specific activated pathway(s) downstream of the TrkB receptor contributes to fear-related synaptic plasticity in the amygdala, we used genetic mouse models carrying a point mutation at the Shc or the PLCγ docking site (trkBSHC or trkBPLC) of the TrkB receptor [these two mouse strains were previously described by Minichiello et al. (1998, 2002); also see supplemental Methods, available at www.jneurosci.org as supplemental material]. To avoid concerns about developmental abnormalities, we focused mainly on heterozygous point mutants of both strains, as they do not display relevant developmental phenotypes (supplemental Methods and Figs. S1, S2, available at www.jneurosci.org as supplemental material) and thus allow for behavioral analysis.
Differential regulation of fear-related learning by the PLCγ and Shc site in TrkB
Analysis of behavior in the open-field test and elevated plus maze revealed no significant difference in terms of locomotor, nonlocomotor, grooming and object exploration activity, and anxiety-like behavior between all groups of mice analyzed [trkB+/+, trkBSHC/+, trkB wild-type knock-in (trkBWT/+), and trkBPLC/+ mice] (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). We then used an associative learning paradigm (classical fear conditioning) to assess aversive learning and memory. On the conditioning day, mice were given two pairings of a tone (CS) and a footshock (US) (Fig. 1A,D). The trkBPLC/+ mice showed impaired associative learning of the CS–US pairing during the training phase compared with the control mice (trkBWT/+) (Fig. 1A), as evidenced by a similar amount of freezing to the second CS presentation (CS2) as to the first CS presentation (CS1) (CS2 vs CS1; F(4.83) = 2.5; p = 0.29). In contrast, the control mice (trkBWT/+) showed normal acquisition during the training phase (CS2 vs CS1; F(6.83) = 19.1; p < 0.001) and an overall significantly higher number of freezing responses compared with the trkBPLC/+ mice (F(2.648) = 9.398; p < 0.001). At 24 h, control (trkBWT/+) and mutant (trkBPLC/+) mice were tested for contextual fear conditioning. As expected, the trkBPLC/+ mice were unable to recognize the context and displayed almost no freezing responses compared with their controls (trkBWT/+ mice) (F(7.827) = 26.336; p < 0.001) (Fig. 1B). Twenty-four hours after the test of contextual conditioning, the mice were placed in a novel chamber for 2 min before the tone was delivered for 2 min. Whereas the control mice showed a significantly higher number of freezing responses at the tone onset (CS onset vs pre-CS; F(10.40) = 33.54; p < 0.001), the trkBPLC/+ mutants elicited a very low number of freezing responses (CS onset vs pre-CS; F(7.359) = 8.89; p = 0.02) (Fig. 1C) and significantly less compared with control mice (F(6.373) = 20.31; p < 0.001). However, similar amounts of freezing were observed between the trkBPLC/+ mutants and control mice before CS onset (trkBPLC/+ pre-CS vs trkBWT/+ pre-CS; F(9.013) = 7.98; p = 0.08). In contrast, the trkBSHC/+ mutants showed normal acquisition during the training phase when compared with the control mice (trkB+/+), and no significant difference was observed between the two groups (F(3.085) = 0.369; p = 0.081) (Fig. 1D). Twenty-four hours after conditioning the trkBSHC/+ mutants together with the control mice were tested first for contextual fear conditioning. Both groups of mice were able to recognize the context and displayed comparable levels of freezing (p > 0.05) (Fig. 1E). However, when later tested for auditory fear conditioning, whereas significantly increased levels of freezing elicited by the auditory conditioned stimulus were observed in the control group (CS onset vs pre-CS; F(11.28) = 27.99; p < 0.001), the trkBSHC/+ mutants showed a significantly reduced number of freezing responses compared with the control mice (F(11.28) = 16.80; p = 0.005) (Fig. 1F). Similar amounts of freezing were observed for the trkBSHC/+ mutants and control mice before CS onset (trkBSHC/+ pre-CS vs trkB+/+ pre-CS; F(11.28) = 5.79; p = 0.3). To verify whether the deficits observed at 48 h after conditioning in the trkBSHC/+ point mutant mice were specifically related to consolidation processes, new groups of mice (trkBSHC/+ and control) were tested for cued fear conditioning 30 min after training. In this test, the trkBSHC/+ mutants showed freezing responses (44.5 ± 3%; n = 5) to the cue similar to those of the control group (39 ± 10%; n = 4) (p = 0.5; t test), and similar levels of freezing were displayed by the two groups before the onset of the CS (p = 0.3) (supplemental Fig. S4D,E, available at www.jneurosci.org as supplemental material), suggesting that the deficit observed 48 h after training is caused by a failure in the trkBSHC/+ mutants to consolidate the auditory fear-related memory. A similar test was performed with the trkBPLC/+ and control mice. As shown in supplemental Figure S4F–H, available at www.jneurosci.org as supplemental material, trkBPLC/+ mice exhibited a deficit in freezing responses to both context and cue compared with control mice 30 min after training (p = 0.05 and p < 0.0001, respectively; t test), suggesting that the reduced level of conditioned fear responses seen at 24 and 48 h is caused mainly by an impaired acquisition. To directly assess memory consolidation in the trkBPLC/+ mice, we attempted to facilitate the acquisition phase by increasing the number of CS–US pairings to five during training. We found that with this protocol trkBPLC/+ mice showed improved acquisition, which was comparable with that of control mice (p = 0.5) (supplemental Fig. S5A, available at www.jneurosci.org as supplemental material), as well as improved memory consolidation when tested 24 h later for contextual fear conditioning and 48 h later for cued fear conditioning (p = 0.3 and p = 0.2 for CTX1 and CTX2, respectively, compared with controls) (supplemental Fig. S5B, available at www.jneurosci.org as supplemental material) and CS onset (p = 0.4 compared with controls) (supplemental Fig. S5C, available at www.jneurosci.org as supplemental material).
Figure 1.

Associative CS–US pairing is impaired in trkBPLC/+ mutants but normal in trkBSHC/+ mutants. A, The percentage of freezing of control (trkBWT/+) and mutant (trkBPLC/+) mice from a typical training trial. Whereas the control mice (trkBWT/+) showed normal acquisition during the training phase, the trkBPLC/+ mutants showed impaired or highly reduced CS–US association (ANOVA; p < 0.001). B, C, The control (trkBWT/+) and the mutant (trkBPLC/+) group of mice were tested 24 h later for contextual fear conditioning (B) followed by cued fear conditioning (C). D, Freezing of trkBSHC/+ mutants during the acquisition phase was similar to that of control mice (trkB+/+). E, F, Twenty-four hours after conditioning, both groups of mice were tested first for contextual fear conditioning (E) and then for cued fear conditioning (F). n = number of mice per group. Bars represent mean ± SEM. **Statistically significant (see Results), p < 0.001; ns, not significant; ITI, intertrial interval; Hb, habituation.
Pain sensitivity is normal in both trkBPLC/+ and trkBSHC/+ point mutants
To rule out the possibility that differences in freezing behavior were attributable to altered pain sensitivity in the trkB point mutant mice, we measured the current threshold of flinch movements, vocalization, and jump movements caused by the nociceptive shock. We also checked for fecal boli and micturition (supplemental Table S1, available at www.jneurosci.org as supplemental material). Using Fisher's exact probability test (two tailed), no differences in pain sensitivity were found between genotypes (trkBWT/+ vs trkBPLC/+, p = 0.8; trkB+/+ vs trkBSHC/+, p = 0.9). Moreover, no differences were found between the control groups (trkBWT/+ vs trkB+/+, p = 0.54) or between the two point mutants (trkBPLC/+ vs trkBSHC/+, p = 0.54).
trkBPLC but not trkBSHC phosphorylation mutants presented impaired early LTP at the BL amygdalar synapses
To characterize the molecular basis for the impaired fear conditioning learning to both context and an auditory cue that was observed in the trkBPLC/+ mutants, and of impaired consolidation for cued fear conditioning in the trkBSHC/+ mutants, we obtained field potential recordings from neurons of the BL nucleus of the amygdala by stimulating neurons in the LA nucleus in brain slices of adult mice. To prevent an underestimation of the requirements of TrkB signaling in fear learning, both homozygous and heterozygous point mutant mice were used for the in vitro electrophysiology experiments. Both types of stimulation, TBS and tetanus, produced LTP. As shown in supplemental Figure S6A, available at www.jneurosci.org as supplemental material, C57BL/6 or trkB+/+ control mice showed LTP in 91.7% of the cases (24 slices from nine mice) or 80% of the cases (5 slices from three mice), respectively; trkBWT/+ or trkBWT/WT control mice showed LTP in 83.3% of the cases (6 slices from three mice) or 100% of the cases (4 slices from three mice), respectively; trkBSHC/+ or trkBSHC/SHC mutant mice showed LTP in 87.5% of the cases (8 slices from three mice) or 100% of the cases (6 slices from three mice), respectively; whereas trkBPLC/+ or trkBPLC/PLC mutant mice showed LTP in 10% of the cases (10 slices from three mice) or 20% of the cases (10 slices from three mice), respectively. The magnitude of the potentiation, expressed as the mean percentage of fEPSP slope with respect to baseline (set at 100%), quantified at 55–60 min after stimulation, was 167 ± 12% of baseline (n = 5) for the trkB+/+ control mice and 162 ± 16% of baseline (n = 8) for the trkBSHC/+ mutants (Fig. 2B). Similar results were observed in trkBSHC/SHC mutant slices (164 ± 9% of baseline; n = 6) (supplemental Fig. S6B, available at www.jneurosci.org as supplemental material), suggesting that ablation of signaling from the Shc site of TrkB does not interfere with this form of synaptic activity. In contrast, E-LTP was abolished in trkBPLC/+ mice (102 ± 3% of baseline; n = 10) compared with control mice (trkBW/+, 160 ± 8.5% of baseline; n = 6) (Fig. 2C). The difference between trkBPLC/+ mutants and control mice (trkBWT/+) was statistically significant (p < 0.01). Similar results were observed in trkBPLC/PLC mutant slices (109 ± 7% of baseline; n = 10) (supplemental Fig. S6B, available at www.jneurosci.org as supplemental material).
Figure 2.

Synaptic plasticity at the LA–BL synapses is impaired in trkBPLC/+ but not in trkBSHC/+ mutants. A, Schematic representation of stimulating and recording electrodes. B, C, Symbols represent average responses plotted every 2 min. After 20 min of control recordings, an HFS train was presented, marked by the arrow. Data are represented as the mean ± SEM. SLA, Stimulating (lateral amygdala); RBL, recording (basolateral amygdala). B, Normal E-LTP in trkBSHC/+ point mutant mice compared with control trkB+/+ mice (p > 0.1). The number of slices analyzed were five for control mice (trkB+/+) and eight for trkBSHC/+ mice. C, E-LTP is abolished in trkBPLC/+ mice. The difference between trkBPLC/+ mutants and control mice (trkBWT/+) is statistically significant (p < 0.01). Traces show EPSP before (1) and 60 min after (2) LTP protocol was applied. Calibration: 5 ms, 0.5 mV. The number of slices analyzed were 6 for control mice (trkBWT/+) and 10 for trkBPLC/+ mice.
Synaptic transmission in trkBPLC mice is indistinguishable from that of control mice
A number of control experiments were performed to rule out the possibility that the failure to induce LTP in the trkBPLC/+ mutants was attributable to impaired synaptic transmission at the BL synapses. First, the presynaptic fiber volley (PSFV), which is proportional to the number of presynaptic neurons recruited by stimulation, was compared with the slope of the field potential, and this comparison revealed that basal synaptic transmission was normal in trkBPLC/+ mutants. The EPSP slope/PSFV ratio was 1.6 ± 0.21 for controls, 1.6 ± 0.3 for trkBPLC/+ mice, and 1.5 ± 04 for trkBPLC/PLC. Second, using an interstimulus interval (ISI) from 10 to 200 ms, the PPF was found to be normal in all genotypes analyzed (Fig. 3A), including the homozygous point mutants for both strains (trkBPLC/PLC, 10 ms: 108 ± 6; 30 ms: 160 ± 8; 40 ms: 168 ± 8; 100 ms: 128 ± 8; 200 ms: 105 ± 5. trkBSHC/SHC, 10 ms: 110 ± 8; 30 ms: 164 ± 9; 40 ms: 164 ± 9; 100 ms: 126 ± 7; 200 ms: 106 ± 4). Although the induction of LTP failed in trkBPLC/+ mutants, all slices showed post-tetanic potentiation when the TBS or tetanus was applied (data not shown). We next tested glutamate receptor-mediated transmission by measuring the NMDA receptor component of the fEPSP under low-Mg2+ conditions (Fig. 3B). In the presence of the AMPA/kainate receptor antagonist CNQX, amygdalar slices from all genotypes showed a comparable NMDA receptor component of the EPSP. The fEPSP slope was reduced to 24.2 ± 3.9% (baseline = 100%) in trkBPLC/+ mice, compared with 23.5 ± 3.1% in trkBWT/+ control mice and 24 ± 2% in trkBSHC/+ mutants (Fig. 3B,C). Thus, there was no statistically significant difference between the three genotypes (p > 0.1). Similar results were obtained for the homozygous point mutants of both strains (trkBPLC/PLC, 24.6 ± 5%; trkBSHC/SHC, 23.9 ± 4%). The NMDA component of the fEPSP was in all cases reduced to zero when the NMDA receptor antagonist AP-5 was added to the bath in the presence of CNQX, indicating that the remaining EPSP component after CNQX application is indeed the NMDA component of the fEPSP. The AMPA/kainate component of the fEPSP from trkBPLC/+ and trkBSHC/+ mutants was also comparable with that of control mice. Finally, to test whether LTP blockade was attributable to impaired fiber function or to altered neurotransmitter release probabilities, we analyzed the EPSP slope during theta-burst stimulation. Synaptic fatigue during high-frequency stimulation (HFS) was observed in all genotypes. To quantify these effects we compared the fourth and first responses in a burst of four stimuli and calculated the ratio for burst 1, burst 2, and burst 3 (B1, B2, and B3). There were no statistically significant differences between all genotypes analyzed (p > 0.1; Student's t test) (Fig. 3D–F), including the homozygous point mutants for both strains (trkBPLC/PLC, B1: 78 ± 6%, B2: 60 ± 5%, B3: 53 ± 6%; trkBSHC/SHC, B1: 76 ± 6%, B2: 57 ± 5%, B3: 54 ± 6%). Collectively, these experiments demonstrate that basal synaptic transmission is normal in all the genotypes analyzed but synaptic strengthening is impaired in trkBPLC/+ mutants.
Figure 3.

Normal basal synaptic transmission at BL–LA synapses of trkBPLC/+ mice. A, PPF was measured to determine whether this aspect of synaptic transmission was normal in trkBPLC/+ mice compared with controls. The percentages denote the ratio of the second fEPSP slope to the first fEPSP slope. PPF was tested at 10, 30, 40, 50, 100, and 200 ms ISIs. There was no significant difference between genotypes. Error bars correspond to the SEM. B, AMPA/kainate receptor (CNQX) and NMDA receptor (AP-5) antagonists were used to analyze whether NMDA and AMPA/kainate receptors are functional in trkBPLC/+ mice. Shown are single traces of extracellular recordings for trkBWT/+, trkBPLC/+, and trkBSHC/+ genotypes. Arrows indicate baseline EPSP 15 min after CNQX application or 15 min after AP-5-plus-CNQX application. C, Summary data for all pharmacological experiments. Plotted is the ratio of the EPSP slope 15 min after CNQX application in comparison with baseline recordings. There was no statistically significant difference between genotypes (p > 0.1; Student's t test). D, E, Synaptic fatigue during high-frequency stimulation. Single traces collected during theta-burst stimulation from trkBWT/+ control animal (D) and from trkBPLC/+ mutant mouse (E). The response to the fourth stimulus (indicated by 4) was compared with the response to the first one (indicated by 1). F, Data corresponding to controls and to trkBSHC/+ and trkBPLC/+ mutants during high-frequency stimulation. Plotted is the ratio of the fourth to the first response in a burst of four stimuli during theta-burst stimulation. For LTP induction, three theta bursts were applied, and data for all three are shown. Synaptic fatigue can be observed in all genotypes, and there is no statistically significant difference between control, trkBSHC/+, and trkBPLC/+ mice (p > 0.1; Student's t test).
Long-lasting LTP is strongly impaired at BL synapses in trkBPLC mutants
As trkBPLC/+ mutants showed impaired long-term contextual and auditory-cued fear memory, and the trkBSHC/+ mutants showed impaired long-term memory (LTM) for cued FC, we also measured L-LTP, which like LTM requires new protein synthesis. Repeated theta bursts or tetanic stimuli were applied to induce L-LTP [three series of 100 pulses (100 Hz) for a pulse length of 100 μs, 5 min ISI]. As illustrated in supplemental Figure S6C, available at www.jneurosci.org as supplemental material, slices from control and trkBSHC/+ mice showed L-LTP in 100% and 90% of the cases, respectively (5–10 slices from three mice per group), and similar results were observed in the trkBSHC/SHC mutants (100%, 7 slices from three mice). Conversely, the trkBPLC/+ mutants did not show L-LTP (0% of cases, zero of five slices from three mice; similar results were obtained in trkBPLC/PLC mutants). In this analysis, only slices that showed E-LTP were included. The application of a stronger stimulus (normally used to induce L-LTP) partially rescued the E-LTP in the trkBPLC/+ mutants; the magnitude of the potentiation increased from 102 ± 3 (Fig. 2C) to 135 ± 9 (p < 0.01) (five slices of nine) after LTP induction (Fig. 4C). Nonetheless L-LTP still failed in trkBPLC/+ mutants; the magnitude of the potentiation (expressed as the mean fEPSP slope percentage of the baseline at 175–180 min after TBS) was 167 ± 8% for the trkB+/+ control mice, 171 ± 12% for the trkBSHC/+ mutants, 161 ± 9% for the trkBWT/+ controls, and 105 ± 6% for the trkBPLC/+ mutant mice (Fig. 4B,C). Differences between the controls and the trkBPLC/+ mutant were statistically significant (p < 0.05). These results indicate that the signaling activated through the PLCγ site of the TrkB receptor is necessary for the acquisition of the pavlovian FC paradigm as well as for LTP generated at synapses in the BL nucleus of the amygdala. In contrast, the TrkB/Shc site-activated signaling is dispensable for plasticity at BL synapses.
Figure 4.

Long-lasting LTP is strongly impaired in BL–LA synapses of trkBPLC/+ mutants. A, Schematic representation of stimulating and recording electrodes. SLA, Stimulating (lateral amygdala); RBL, recording (basolateral amygdala). B, C, Slope size of fEPSP recordings before and after the TBS stimulus was plotted. Symbols represent average responses plotted every 4 min. After 20 min of control recordings, HFSs were presented at the time marked by the arrow. Data are represented as the mean ± SEM. B, L-LTP is not significantly different in trkBSHC/+ mutants compared with controls (trkB+/+) (p > 0.1). The number of slices was 5 for control mice (trkB+/+) and 10 for trkBSHC/+ mice. C, L-LTP is abolished in trkBPLC/+ mice. The difference between trkBPLC/+ mutants and control mice (trkBWT/+) was statistically significant (p < 0.01). Traces show EPSP before (1) and 180 min after (2) LTP protocol was applied. Calibration: 5 ms, 0.5 mV. The number of slices was four for control mice (trkBWT/+) and five for trkBPLC/+ mice.
E-LTP is impaired at the LA synapses in both trkBPLC and trkBSHC phosphorylation mutants
The dorsal subdivision of the LA is the primary target of thalamic inputs (Rodrigues et al., 2004a). Thus, field recordings were obtained in the neurons of the dorsal LA nucleus in brain slices of adult mice by stimulating fibers emerging from the internal capsule in the ventral striatum, which has been shown to carry, in part, efferents from the medial geniculate nucleus to the LA (Weisskopf et al., 1999). This form of LTP is essential for auditory FC in mice (LeDoux, 2000). We used TBS as well as tetanus stimulation. Both protocols evoked LTP: C57BL/6 or trkB+/+ control mice showed LTP in 100% of the cases (16 slices or 4 slices from eight or three mice each group, respectively), trkBWT/+ control mice showed LTP in 80% of the cases (5 slices from three mice), trkBSHC/+ mice showed LTP in 25% of the cases (4 slices from three mice), and trkBPLC/+ mice showed LTP in 40% of the cases (5 slices from three mice) (supplemental Fig. S7A, available at www.jneurosci.org as supplemental material). The magnitude of the potentiation, expressed as the mean percentage of fEPSP slope with respect to baseline (set at 100%) was quantified at 55–60 min after stimulation as follows: trkB+/+ control mice, 190 ± 16%, trkBSHC/+ mice, 121 ± 7%, trkBWT/+ controls, 172 ± 10%, and trkBPLC/+ mice, 119 ± 5% (Fig. 5B,C). The differences between controls and trkBSHC/+ mutants, as well as between controls and trkBPLC/+ mice, were significant (p < 0.01) (Student's t test, two tailed). Similar results were obtained for the trkBSHC/SHC and the trkBPLC/PLC homozygous point mutants (supplemental Fig. S7B, available at www.jneurosci.org as supplemental material). Thus, both trkBSHC/+ and trkBPLC/+ mice showed reduced E-LTP in the auditory input pathways to the LA. Control experiments suggested that the reduced E-LTP observed in both trkBSHC/+ and trkBPLC/+ mutants was not caused by impaired synaptic transmission, as it was indistinguishable from that of control mice at LA synapses. PPF was normal in all genotypes analyzed (Fig. 5D). To test whether poor LTP induction at LA synapses in trkBSHC/+ and trkBPLC/+ mice was attributable to impaired fiber function or to altered neurotransmitter release probabilities, we analyzed the EPSP slope during theta-burst stimulation. Synaptic fatigue during high-frequency stimulation was observed in all genotypes. Comparing the fourth and first responses in a burst of four stimuli, and calculating this ratio for the three theta bursts given, revealed no statistically significant differences between all genotypes analyzed (p > 0.1; Student's t test) (Fig. 5E). These experiments indicated that basal synaptic transmission is normal in all the genotypes analyzed, but synaptic strengthening is reduced at the LA synapses in trkBSHC/+ and trkBPLC/+ point mutants. Of note is that short-term memory (STM) in trkBSHC/+ mutants is intact 30 min after training, whereas LTP in amygdala slices is decayed to baseline within 30 min. This suggests that the electrical pattern of stimulation necessary to induce LTP in vitro could be quantitatively different from the natural pattern of activation in the LA of behaving mice during CS–US pairings. In addition, the preparation of the brain slices for the in vitro experiments could disrupt modulatory inputs that are normally present in vivo (Schafe et al., 2001).
Figure 5.

Synaptic plasticity at the thalamic–lateral amygdala synapses is impaired in both trkBPLC/+ and trkBSHC/+ mutants. A, Schematic representation of stimulating (S) and recording (R) electrodes. B, C, Symbols represent average responses plotted every 2 min. After 20 min of control recordings, an HFS was presented, as marked by the arrow. Data are represented as the mean ± SEM. B, E-LTP was significantly reduced in trkBSHC/+ mutant mice. p < 0.01 compared with control mice (trkB+/+). C, E-LTP was also significantly reduced in trkBPLC/+ mutants. p < 0.01 compared with control mice (trkBWT/+). Traces show EPSP before (1) and 60 min after (2) LTP protocol was applied. Calibration: 5 ms, 0.5 mV. The number of slices was four for trkB+/+ and trkBSHC/+ mice and five for trkBWT/+ and trkBPLC/+ mice. D, PPF was measured to determine whether this aspect of synaptic transmission was normal in trkBSHC/+ and trkBPLC/+ mice compared with controls (trkB+/+ and trkBWT/+, respectively). The percentages denote the ratio of the second fEPSP slope to the first fEPSP slope. PPF was tested at 10, 30, 40, 50, 100, and 200 ms ISI. There was no significant difference between genotypes. Error bars correspond to the SEM. E, Data corresponding to controls and to trkBSHC/+ and trkBPLC/+ mutants during high-frequency stimulation. Plotted is the ratio of the fourth to the first response in a burst of four stimuli during theta-burst stimulation. For LTP induction, three theta bursts were applied, and data for all three are shown. Synaptic fatigue can be observed in all genotypes with no statistically significant difference between controls and trkBSHC/+ and trkBPLC/+ mice (p > 0.1; Student's t test).
L-LTP is reduced at the LA synapses in trkBSHC and trkBPLC mutants
We then looked at expression of LTP at the thalamic–LA synapses of both trkBSHC/+ and trkBPLC/+ mutants by using the L-LTP paradigm induced with a stronger stimulus protocol (Fig. 6B). As illustrated in supplemental Figure S7C, available at www.jneurosci.org as supplemental material, slices from control mice (trkB+/+ or trkBWT/+) showed L-LTP in 100% of cases (four or five slices, respectively, from three mice), whereas trkBSHC/+ showed L-LTP in 33.3% of cases (six slices from three mice), and similar results were observed in trkBSHC/SHC mice (42.8%) (seven slices from three mice). The trkBPLC/+ mutants showed L-LTP in 40% of cases (five slices from three mice), and similar results were obtained in trkBPLC/PLC mutants. All of the slices analyzed showed E-LTP. In particular, with the stronger stimulus protocol the EPSP slope size increased from 121 ± 7 to 152 ± 14 (p < 0.01) (six slices of nine) in trkBSHC/+ mutants and from 119 ± 5 to 148 ± 12 (p < 0.01) (five slices of eight) in trkBPLC/+ mutants. However, slices from both the trkBSHC/+ and the trkBPLC/+ mutants presented reduced L-LTP; the magnitude of the potentiation (expressed as the mean fEPSP slope percentage of the baseline at 175–180 min after TBS) was as follows: for the trkB+/+ control mice, 175 ± 10%; for the trkBSHC/+ mice, 122 ± 6%; for the trkBWT/+ mice, 169 ± 12%; and for the trkBPLC/+ mice, 119 ± 10% (Fig. 6B,C). The differences between controls and trkBSHC/+ mutants as well as controls and trkBPLC/+ mice were significant (p < 0.01) (Student's t test, two tailed). Similar results were obtained for the trkBSHC/SHC and the trkBPLC/PLC homozygous point mutants (supplemental Fig. S7D, available at www.jneurosci.org as supplemental material).
Figure 6.

Long-lasting LTP is also reduced in trkBSHC/+ and trkBPLC/+ mutants at thalamic–lateral amygdala synapses. A, Schematic representation of stimulating (S) and recording (R) electrodes. B, C, Slope size of fEPSP recordings before and after the TBS stimulus was plotted. Symbols represent average responses plotted every 4 min. After 20 min of control recordings, HFSs were presented at the time marked by the arrow. Data are represented as the mean ± SEM. B, L-LTP was significantly reduced in trkBSHC/+ mutants compared with control mice (trkB+/+); p < 0.01. C, L-LTP was significantly reduced also in trkBPLC/+ mice compared with control mice (trkBWT/+); p < 0.01. Traces show EPSP before (1) and 180 min after (2) LTP protocol was applied. Calibration: 5 ms, 0.5 mV. Four slices were analyzed for trkB+/+ mice, six for trkBSHC/+ mice, and five for trkBWT/+ and trkBPLC/+ mice.
Point mutation at the Shc site in TrkB impairs specific amygdala function but not hippocampal function
It has been suggested that the amygdala is essential for the conditioning of fear responses to both a cue and a context, whereas the hippocampus is involved in contextual but not auditory-cued fear conditioning (Phillips and LeDoux, 1992). The differential involvement of the Shc and the PLCγ phosphorylation sites of TrkB in fear learning and amygdalar synaptic plasticity indicates that the hippocampal formation, contrary to what is seen for the trkBPLC/+ mice, is intact in the trkBSHC/+ mutants, as they showed specific impairment in fear responses to cue stimuli but not to context. Indeed, we have previously shown by in vitro and in vivo electrophysiology that the PLCγ docking site, but not the Shc docking site, is necessary to mediate the TrkB-dependent hippocampal late LTP (Minichiello et al., 2002; Gruart et al., 2007). However, we have also shown by in vivo electrophysiology an enhancement of TrkB-dependent hippocampal early LTP evoked in alert behaving trkBSHC/+ point mutant mice (Gruart et al., 2007). The latter results are partially in agreement with results describing an enhancement of hippocampal early LTP by in vitro electrophysiology in ShcC mutant mice; ShcC is a member of the Shc family of adapter molecules, predominantly expressed in the nervous system (Miyamoto et al., 2005). To further test the functioning of the hippocampus in relation to behavior, we subjected the trkBPLC/+ and the trkBSHC/+ mutant and control groups to the eight-arm radial maze task, which depends on an intact hippocampus (Olton et al., 1978; Rossi-Arnaud and Ammassari-Teule, 1998; Minichiello et al., 1999) and allows for the assessment of spatial learning and memory in rodents. In this paradigm, rodents are trained to visit a number of arms to receive a food reward. To properly perform the task, animals must use spatial information to remember which arm(s) of the maze was already visited. Our analysis of the number of correct arm choices (during the first eight choices) showed that the trkBPLC/+ mutants (compared with the trkBWT/+ control mice) and the trkBSHC/+ mutants (compared with the trkB+/+ control mice) learned the radial maze (F(9.243) = 7.61; p < 0.0001; F(9.297) = 9.29; p < 0.0001, respectively) (Fig. 7A,B). However, the trkBPLC/+ mutants learned significantly less than the trkBWT/+ controls (F(1.27) = 5.65; p < 0.05) (Fig. 7A), whereas the trkBSHC/+ mice performed similarly to controls (F(1.33) = 0.009; p > 0.05) (Fig. 7B). The analysis of the error scores of 15 trials (maximum permitted number of trials) revealed significantly higher error scores for the trkBPLC/+ mutants compared with controls (trkBWT/+) (F(1.27) = 6.53; p < 0.05) (Fig. 7C). Interestingly, as previously observed in adult mice lacking TrkB in all principal neurons (the trkBcre mice) (Minichiello et al., 1999), the percentage of errors committed by the trkBPLC/+ mice was significantly higher than that of trkBWT/+ mice (#p < 0.005) during the last days of the test, whereas the trkBSHC/+ mice and trkB+/+ mice committed very few errors during the same period of the session and were statistically indistinguishable (Fig. 7C,D). When the occurrence of the first error was considered, trkBSHC/+ mice performed significantly better than trkB+/+ mice during the first 6 d of training (*p < 0.05). However, the performance of trkBSHC/+ and trkB+/+ animals became statistically indistinguishable during the last 2 d of training (supplemental Fig. S8A,B, available at www.jneurosci.org as supplemental material). Together, these results show a clear impairment in both short- and long-term spatial memory in mice carrying a point mutation at the PLCγ site of the TrkB receptor but not at the Shc site.
Figure 7.

Spatial learning is severely impaired in trkBPLC/+ but not in trkBSHC/+ point mutants. A–D, Mice were trained for 10 consecutive days in a fully baited eight-arm radial maze. Learning performance is expressed as the mean number of correct arm choices in the first 8 trials, which is a measure of spatial working memory (A, B), and the percentage of errors until eight correct choices were observed or after the maximal permitted number of trials (15), which reflect long-term spatial memory (C, D). A, C, The performance of the trkBPLC/+ mice was significantly impaired compared with the trkBWT/+ control mice in all measures considered. B, D, In contrast, the performance of the trkBSHC/+ mice was very similar to that of trkB+/+ control mice in both the correct arm choices in the first eight trials and the percentage of errors. *p < 0.05; #p < 0.005.
CaMKII phosphorylation is weak in amygdala synapses of trkBPLC point mutants
It has been suggested that the phosphorylation at Thr286 of CaMKIIα is critically involved in synaptic plasticity and the acquisition and/or initial formation of short-term memory during fear conditioning in the LA. CaMKIIα is found to be distributed throughout the LA and to be postsynaptic to thalamic inputs. In addition, inhibition of CaMKII in the amygdala impairs the acquisition of auditory and contextual fear conditioning (Rodrigues et al., 2004b). BDNF has been shown to induce the phosphorylation of this kinase in neurons (Finkbeiner et al., 1997), and we have previously shown that CaMKII phosphorylation is impaired in trkBPLC/PLC but not trkBSHC/SHC mutant synaptosomal preparations isolated from adult mouse cerebral cortices and stimulated with BDNF (Minichiello et al., 2002). Here, we found that point mutation at the PLCγ site of TrkB results in impaired fear acquisition and impaired amygdalar synaptic plasticity at the LA and the BL synapses. To further examine the mechanism by which the PLCγ site of TrkB regulates conditioned fear responses and LTP at amygdalar synapses, we measured the activation (p-Thr286) of CaMKII in untrained (control) versus fear-conditioned mice. Since it has been reported that fear conditioning increases CaMKII phosphorylation at the lateral amygdala spines (Rodrigues et al., 2004b), and we found that impairments in the acquisition of conditioned fear responses by trkBPLC/+ mutant mice are revealed with a milder (two CS–US pairings), but not an intense (five CS–US pairings), conditioning trial, we asked whether the degree of CaMKII phosphorylation at amygdalar synapses correlated with the acquisition of conditioned fear in the trkBPLC/+ mutants. No differences in pCaMKII levels were found in synaptosomes isolated from trkBPLC/+ mice that were either unconditioned or trained using two CS–US pairings (ANOVA, two CS–US versus control, p = 0.16); however, a significant increase in pCaMKII levels was seen in amygdalar synaptosomes from trkBPLC/+ mice trained with five CS–US pairings (ANOVA; five CS–US versus control, p = 0.01; five CS–US versus two CS–US, p = 0.03) (Fig. 8B). The pCaMKII levels in synaptosomes from trkBPLC/PLC mice either untrained (control) or conditioned with two CS–US or five CS–US pairings were similarly affected (ANOVA; two CS–US versus control, p = 0.09; five CS–US versus control, p = 0.0007; five CS–US versus two CS–US, p = 0.0002) (Fig. 8C). Conversely, amygdalar synaptosomes isolated from trkB+/+ mice trained with either two CS–US or five CS–US pairings possessed similar levels of pCaMKII that were significantly greater than the level seen in untrained trkB+/+ mice (ANOVA; two CS–US versus control, p = 0.047; five CS–US versus control, p = 0.0059; two CS–US versus five CS–US, p = 0.3) (Fig. 8A). Collectively, these results suggest that impaired fear acquisition and amygdalar synaptic plasticity downstream of the TrkB/PLCγ site correlate with weak pCaMKII at the amygdalar synapses.
Figure 8.

Weak pCaMKII at amygdala synapses in trkBPLC point mutants correlates with impaired fear acquisition. A–C, Immunoblots showing pCaMKII (Thr286) levels in synaptosomal preparations from amygdala tissues of trkB+/+ untrained (control, n = 4) versus trained (2 CS–US, n = 5; or 5 CS–US, n = 5), trkBPLC/+ (control, n = 3; 2 CS–US, n = 3; or 5 CS–US, n = 3) and trkBPLC/PLC (control, n = 3; 2 CS–US, n = 3; or 5 CS–US, n = 3) mice. Blots were reprobed with CaMKIIα Abs to visualize CaMKIIα levels. Top, Quantification of pCaMKII levels A, **, ANOVA, five CS–US versus control, p = 0.0059; *, two CS–US versus control, p = 0.047. B, *, ANOVA, five CS–US versus control, p = 0.01; five CS–US versus two CS–US, p = 0.03. C, ***, ANOVA, five CS–US versus control, p = 0.0007; five CS–US versus two CS–US, p = 0.0002. D–F, Immunoblots of PSD95 and synapsin I/II confirmed that the synaptosomal preparation from amygdala tissues contained both post- and presynaptic fractions, respectively.
AKT but not MAPK phosphorylation is affected in amygdala synapses of trkBSHC point mutants
Previously, we showed normal MAPK activation and nuclear translocation after BDNF stimulation in dissociated cortical neurons from trkBPLC/PLC mice (Minichiello et al., 2002). In contrast, in cortical neurons from trkBSHC/SHC mutants the activation of MAPK by BDNF is transient and reduced in amplitude (Minichiello et al., 1998). Thus, we analyzed MAPK phosphorylation in response to BDNF in cultured cortical neurons from trkBSHC/+ mutants and found similar activation kinetics compared with BDNF-treated cultures from trkB+/+ control mice (5 min trkBSHC/+ versus 5 min trkB+/+, p = 0.7; 30 min trkBSHC/+ versus 30 min trkB+/+, p = 0.5) (Fig. 9A). As expected, trkBPLC/+ mutant cortical neurons showed normal activation of MAPKs in response to BDNF (data not shown). Moreover, to elucidate the contribution of MAPK to TrkB/Shc site-dependent amygdala function, MAPK phosphorylation was examined in the amygdala of untrained or fear-conditioned trkBSHC/+ mice. Immunostaining of amygdalae for pMAPKs produced similar results for trkBSHC/+ and trkB+/+ mice (Fig. 9B–E). We have previously shown that the phosphorylation of AKT in response to BDNF is not affected in cortical neurons of trkBPLC/PLC mutants but is significantly reduced in trkBSHC/SHC mutants (Postigo et al., 2002; Medina et al., 2004). This prompted us to analyze the kinetics of PI3K/AKT activation in cortical neurons from trkBSHC/+ mutants. Remarkably, AKT phosphorylation after BDNF treatment was significantly reduced compared with controls (5 min trkBSHC/+ versus 5 min trkB+/+, p = 0.02; 30 min trkBSHC/+ versus 30 min trkB+/+, p = 0.02) (Fig. 9F). Similar results were observed in amygdala tissue lysate from conditioned versus naive animals. Whereas fear conditioning increased pAKT levels in trkB+/+ amygdalar tissue (p = 0.01), no significant difference was observed in the amygdala tissue of conditioned versus untrained trkBSHC/+ mice (p = 0.4) (Fig. 9G). These results suggested that the consolidation of cued fear conditioning and the maintenance of TrkB-dependent LTP in the LA mainly require activation of the PI3K/AKT pathway through the Shc site.
Figure 9.
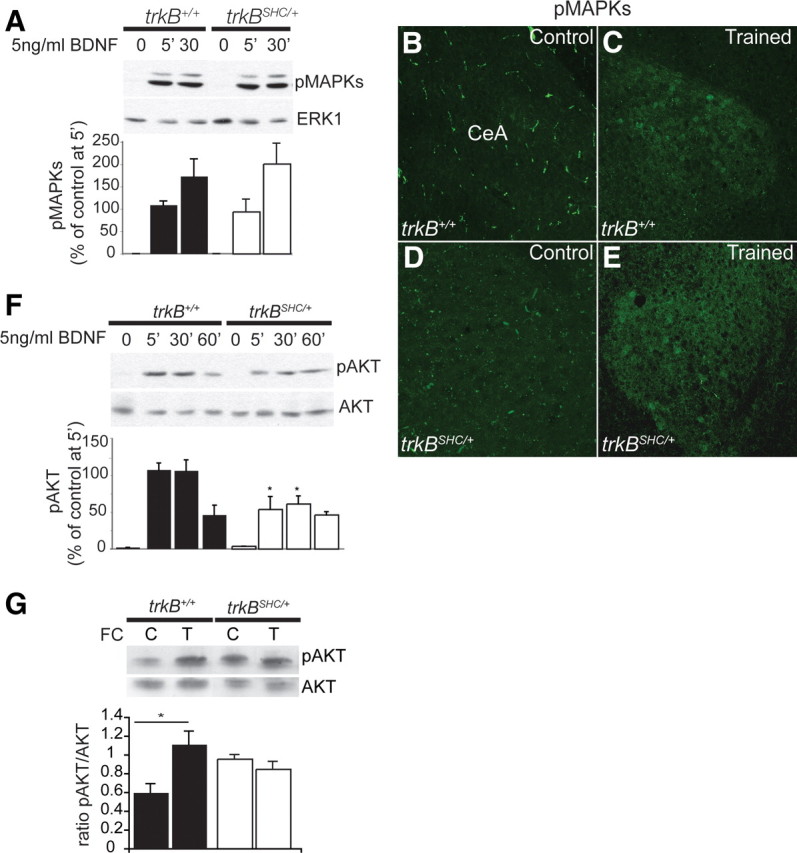
Decreased pAKT but not pMAPKs in trkBSHC/+ point mutants. A, Top, Immunoblot showing the time course of MAPK phosphorylation after BDNF stimulation of control and trkBSHC/+ mutant cortical primary neurons using Abs against the phosphorylated forms of ERK1/2, p42/44 (pMAPKs). To control for protein loading, the blots were reprobed with anti-ERK1 Abs. Bottom, Quantification of pMAPK levels. Densitometric analysis was performed on two different blots for each antibody (n = 3 cortices per time point). B–E, Immunofluorescence analysis of amygdala tissues from fear-conditioned (n = 3) versus untrained (n = 2) trkB+/+ and trkBSHC/+ mice by use of pMAPKs Abs. F, Top, Immunoblot showing the time course of AKT phosphorylation after BDNF stimulation of control and trkBSHC/+ mutant cortical primary neurons by using pAKT antibodies. To control for protein loading, blots were reprobed with anti-AKT Abs. Bottom, Quantification of pAKT levels. *, pAKT in trkBSHC/+ versus trkB+/+, p = 0.02, ANOVA. Densitometric analysis was performed on two different blots for each antibody (n = 3 cortices per time point). G, Top, Immunoblot analysis of pAKT in amygdala lysates from trkB+/+ untrained (control) (n = 2) versus trained (fear-conditioned) (n = 3) amygdala lysate and from trkBSHC/+ mice (control) (n = 2) versus trained (fear-conditioned) (n = 3) amygdala lysate. To control for protein loading, blots were reprobed with anti-AKT Abs. Bottom, Quantification of pAKT levels. *p = 0.01. C, Control (untrained); T, trained.
Discussion
Ntrk2/TrkB receptors are essential for synaptic plasticity and learning in different paradigms, including pavlovian fear conditioning. However, the molecular pathways required for fear learning and amygdalar synaptic plasticity downstream of the TrkB receptor have not yet been identified. The brain structure central to the fear memory circuits is the amygdala, in particular substructures that form the BLA complex (LA, BL, BM) and the output nucleus CeA (LeDoux, 2000; Maren, 2001). It is clear that the LA plays an essential role in the acquisition and maintenance of fear memories. However, latest evidence suggests that LA, BL, and CeA perform certain associative functions in parallel (Everitt et al., 2003; Paré et al., 2004). It was established that both FC-induced neuronal plasticity and LTP at amygdala synapses share common mechanisms of induction and expression (McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997). Thus, to determine whether the molecular mechanisms for pavlovian FC downstream of the TrkB receptor are also required for the induction and expression of long-term synaptic plasticity, such as LTP, we tested highly defined genetic mouse models that carry a point mutation at specific phosphorylation docking sites of the TrkB receptor (trkBSHC, trkBPLC) in a pavlovian FC paradigm (by using heterozygous point mutants, trkBSHC/+, and trkBPLC/+) as well as acquired field recordings from neurons of different amygdala nuclei, including the BL nucleus, by stimulating the LA nucleus, and the LA nucleus by stimulating fibers originating in the auditory thalamus, in brain slices of adult heterozygous (trkBSHC/+ and trkBPLC/+) and homozygous (trkBSHC/SHC and trkBPLC/PLC) point mutant mice and control animals. Remarkably, the results obtained indicate a differential regulation of fear learning and amygdalar synaptic plasticity by the two phosphorylation sites of TrkB receptor. In particular, a point mutation in the TrkB/PLCγ site impairs E- and L-LTP in the amygdalar circuits analyzed and also impairs the acquisition of conditioned fear to both context and an auditory cue. It was previously suggested that the amygdala and the hippocampus differentially contribute to cue and contextual FC. Lesion experiments in rats revealed that whereas the amygdala is involved in the conditioning of fear-related responses to a context and cue, the hippocampus instead modulates conditioning to context but not to a cue (Phillips and LeDoux, 1992). We previously showed that the PLCγ docking site, but not the Shc site, of TrkB is necessary for hippocampal synaptic plasticity, as well as for the acquisition of associative learning (classical conditioning of eyelid responses), in vivo LTP at CA3-CA1 hippocampal synapses (Minichiello et al., 2002; Gruart et al., 2007), and spatial learning (this study). Thus, impaired hippocampal plasticity in trkBPLC mutants likely contributes to the impaired fear conditioning to context. In contrast, the trkBSHC mutants show normal acquisition of contextual and cued FC but impaired consolidation of the conditioned fear response to an auditory cue, as well as a specific impairment in amygdalar synaptic plasticity at the thalamic–LA synapses (observed in both the trkBSHC/+ and the trkBSHC/SHC mutants). We also obtained recordings within the LA (the ventral LA) by stimulation of the dorsal lateral nucleus and found impaired plasticity in the trkBSHC mutants (data not shown), suggesting a specific impairment in plasticity in the LA of the trkBSHC mutants. This is supported by the fact that hippocampal functions are mainly spared in these mutants. Of note is that we have reported by in vivo electrophysiology an enhancement of TrkB-dependent hippocampal early LTP evoked in alert behaving trkBSHC/+ mice (Gruart et al., 2007). Consistent with these results, we now also show a better performance of the trkBSHC mutants in a specific phase of a spatial learning task (namely, the radial maze). Although these results are similar to some of the results reported for the ShcC mutant mice, the mechanisms by which ShcC regulates hippocampal synaptic plasticity seem to be independent of the TrkB/Shc site and apparently via modulation of the NMDA receptor function (Miyamoto et al., 2005). In addition, transgenic mice overexpressing the full-length TrkB receptor show selective activation of the PLCγ site as well as increased spatial learning and improved contextual fear conditioning, whereas signaling via the Shc site remains unaltered (Koponen et al., 2004).
Regarding the biochemical mechanisms underlying pavlovian FC, Rodrigues et al. (2004b) suggested that the phosphorylation of Thr286 of CaMKIIα is critically involved in synaptic plasticity and the acquisition and/or initial formation of STM during fear conditioning in the LA. Here, we show that a point mutation at the PLCγ site of TrkB (trkBPLC/+) results in impaired fear acquisition and impaired amygdalar synaptic plasticity at the LA and the BL synapses, likely attributable to the reduced phosphorylation of CaMKII at Thr286, as seen in amygdalar synapses of FC trkBPLC mice (both trkBPLC/+ and trkBPLC/PLC mutants) trained with two CS–US pairings, indicating that TrkB receptors through the Y816 typically modulate CaMKII phosphorylation and acquisition of FC, as well as amygdalar LTP. However, an intense training protocol (five CS–US pairings) induces normal CaMKII phosphorylation in trkBPLC amygdala synapses (observed in both the trkBPLC/+ and trkBPLC/PLC mutants). These results were consistent with the fact that training trials using five CS–US pairings rescued the memory acquisition deficit in the trkBPLC/+ mice and that application of a stronger stimulus partially rescued the E-LTP in these mutants. Under conditions of stronger stimulation, classical pathways still activated downstream of the mutant TrkB receptor may compensate for the signaling lost from the PLCγ site. Alternatively, recent studies have shown that neurotrophin activation of TrkB can rapidly mediate the gating of sodium channels, resulting in an increase in intracellular calcium concentration and cellular responses independent of PLCγ recruitment to TrkB (Blum and Konnerth, 2005). It is also possible that the effects observed in the trkBPLC/+ mutants under more intense inputs involve the activation of undefined signaling mechanisms directly or indirectly implicated in fear conditioning. The normal phosphorylation of CaMKII seen in the amygdalae of trkBSHC/+ point mutants (data not shown) correlates with their normal acquisition of fear conditioning. Moreover, mice heterozygous for a point mutation at Y515 or Y816 of TrkB possess normal brain levels of BDNF (supplemental Fig. S8, available at www.jneurosci.org as supplemental material), supporting the conclusion that the fear conditioning and the synaptic activity in amygdalar nuclei of these mutants are not affected by changes in the levels of this TrkB ligand.
Gean and colleagues have reported that the PI3K/AKT pathway contributes to amygdalar LTP and the consolidation of amygdala-dependent cued fear conditioning in rats (Lin et al., 2001). Pharmacological studies by the same laboratory revealed that BDNF is required for the acquisition of fear learning via recruitment of the Shc adapter protein to TrkB and the activation of MAPK and PI3K (Ou and Gean, 2006). As discussed above, by using very precise mouse genetic models we have determined in this study that it is primarily the PLCγ site of TrkB that mediates the TrkB-dependent acquisition of FC and amygdalar synaptic plasticity at the LA and the BL synapses through modulation of CaMKII at synapses, whereas the Shc site contributes mainly to consolidation of amygdala-dependent cued fear conditioning and LTP at the LA synapses predominantly through AKT activation.
A concern could be raised that the mice used in this study possess the mutated form of the receptor in all TrkB-expressing cells and this precludes conclusions regarding the role of TrkB in a specific brain region such as the amygdala. However, we have extensively analyzed the PNS and CNS development of the heterozygous point mutants of both strains [shown in previous work (Minichiello et al., 1998, 2002; Medina et al., 2004; Gruart et al., 2007)] (supplemental Figs. S1, S2, available at www.jneurosci.org as supplemental material) and have not identified any relevant abnormalities. This, combined with the fact that the animals display normal basic behavior (supplemental Fig. S3, available at www.jneurosci.org as supplemental material), suggests that the phenotypes observed in fear conditioning are not caused by developmental defects in the amygdala. Experiments based on the intra-amygdala infusion of a lentiviral vector expressing a dominant-negative TrkB isoform (TrkB.T1) in adult rats have also established that the TrkB neurotrophin receptor plays an essential role in the acquisition of pavlovian fear conditioning triggered in the amygdala nuclei (Rattiner et al., 2004). Moreover, we find that basal synaptic activities are similar between the different genotypes analyzed (Fig. 3). Animals possessing developmental defects would show changes especially in basal synaptic transmission. The electrophysiological recordings produced identical results for heterozygous and homozygous point mutants (supplemental Figs. S7–10, available at www.jneurosci.org as supplemental material), suggesting that one copy of a wild-type TrkB allele is sufficient to prevent the developmental PNS phenotypes observed in the homozygous point mutants but is not sufficient to support TrkB-dependent learning in the adult brain (Minichiello et al., 1999, 2002). Similar results were obtained by biochemical analysis of synaptosomal preparations from amygdala tissues of untrained versus trained trkBPLC/+ and trkBPLC/PLC point mutants (Fig. 8). Together, these findings uphold the notion that the genetic tools of the present study are valid and support our main conclusions. However, future work focused on a cell-type-specific ablation of TrkB will be necessary to understand the contribution of TrkB to fear learning and amygdalar LTP in the different neuronal populations.
Footnotes
This work was supported in part by grants from the European Union (EU) [EU Sixth Framework Programme (FP6) Project Memories Grant 037831; EU FP6 StemStroke Grant 037526] to L.M., Grants BFU2005-01024 and BFU2006-14155 from the Spanish Ministry of Education and Science to J.M.D.-G. and A.R.-M., respectively, and Deutsche Forschungsgemeinschaft Grant SFB636/A5 to O.v.B.
References
- Blum R, Konnerth A. Neurotrophin-mediated rapid signaling in the central nervous system: mechanisms and functions. Physiology. 2005;20:70–78. doi: 10.1152/physiol.00042.2004. [DOI] [PubMed] [Google Scholar]
- Campeau S, Davis M. Involvement of subcortical and cortical afferents to the lateral nucleus of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J Neurosci. 1995;15:2312–2327. doi: 10.1523/JNEUROSCI.15-03-02312.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. Neurobiology of fear responses: the role of the amygdala. J Neuropsychiatry Clin Neurosci. 1997;9:382–402. doi: 10.1176/jnp.9.3.382. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Cardinal RN, Parkinson JA, Robbins TW. Appetitive behavior: impact of amygdala-dependent mechanisms of emotional learning. Ann N Y Acad Sci. 2003;985:233–250. [PubMed] [Google Scholar]
- Fendt M, Fanselow MS. The neuroanatomical and neurochemical basis of conditioned fear. Neurosci Biobehav Rev. 1999;23:743–760. doi: 10.1016/s0149-7634(99)00016-0. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- Gale GD, Anagnostaras SG, Godsil BP, Mitchell S, Nozawa T, Sage JR, Wiltgen B, Fanselow MS. Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J Neurosci. 2004;24:3810–3815. doi: 10.1523/JNEUROSCI.4100-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruart A, Sciarretta C, Valenzuela-Harrington M, Delgado-García JM, Minichiello L. Mutation at the TrkB PLC{gamma}-docking site affects hippocampal LTP and associative learning in conscious mice. Learn Mem. 2007;14:54–62. doi: 10.1101/lm.428307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Koo JW, Han JS, Kim JJ. Selective neurotoxic lesions of basolateral and central nuclei of the amygdale produce differential effects on fear conditioning. J Neurosci. 2004;24:7654–7662. doi: 10.1523/JNEUROSCI.1644-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koponen E, Võikar V, Riekki R, Saarelainen T, Rauramaa T, Rauvala H, Taira T, Castrén E. Transgenic mice overexpressing the full-length neurotrophin receptor trkB exhibit increased activation of the trkB-PLCgamma pathway, reduced anxiety, and facilitated learning. Mol Cell Neurosci. 2004;26:166–181. doi: 10.1016/j.mcn.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Lavond DG, Kim JJ, Thompson RF. Mammalian brain substrates of aversive classical conditioning. Annu Rev Psychol. 1993;44:317–342. doi: 10.1146/annurev.ps.44.020193.001533. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. The emotional brain: the mysterious underpinnings of emotional life. New York: Simon and Schuster; 1996. [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- LeDoux JE, Cicchetti P, Xagoraris A, Romanski LM. The lateral amygdaloid nucleus: sensory interface of the amygdala in fear conditioning. J Neurosci. 1990;10:1062–1069. doi: 10.1523/JNEUROSCI.10-04-01062.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Lin CH, Lu KT, Leu TH, Chang WC, Gean PW. A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron. 2001;31:841–851. doi: 10.1016/s0896-6273(01)00433-0. [DOI] [PubMed] [Google Scholar]
- Maren S. Neurobiology of pavlovian fear conditioning. Annu Rev Neurosci. 2001;24:897–931. doi: 10.1146/annurev.neuro.24.1.897. [DOI] [PubMed] [Google Scholar]
- Maren S, Aharonov G, Fanselow MS. Neurotoxic lesions of the dorsal hippocampus and pavlovian fear conditioning in rats. Behav Brain Res. 1997;88:261–274. doi: 10.1016/s0166-4328(97)00088-0. [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- Medina DL, Sciarretta C, Calella AM, Von Bohlen Und Halbach O, Unsicker K, Minichiello L. TrkB regulates neocortex formation through the Shc/PLCgamma-mediated control of neuronal migration. EMBO J. 2004;23:3803–3814. doi: 10.1038/sj.emboj.7600399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Casagranda F, Tatche RS, Stucky CL, Postigo A, Lewin GR, Davies AM, Klein R. Point mutation in trkB causes loss of NT4-dependent neurons without major effects on diverse BDNF responses. Neuron. 1998;21:335–345. doi: 10.1016/s0896-6273(00)80543-7. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron. 2002;36:121–137. doi: 10.1016/s0896-6273(02)00942-x. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Chen L, Sato M, Sokabe M, Nabeshima T, Pawson T, Sakai R, Mori N. Hippocampal synaptic modulation by the phosphotyrosine adapter protein ShcC/N-Shc via interaction with the NMDA receptor. J Neurosci. 2005;25:1826–1835. doi: 10.1523/JNEUROSCI.3030-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A, Delgado-Escueta AV. Rapid preparation of synaptosomes from mammalian brain using nontoxic isoosmotic gradient material (Percoll) J Neurochem. 1984;43:1114–1123. doi: 10.1111/j.1471-4159.1984.tb12851.x. [DOI] [PubMed] [Google Scholar]
- Olton DS, Walker JA, Gage FH. Hippocampal connections and spatial discrimination. Brain Res. 1978;139:295–308. doi: 10.1016/0006-8993(78)90930-7. [DOI] [PubMed] [Google Scholar]
- Ou LC, Gean PW. Regulation of amygdala-dependent learning by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol-3-kinase. Neuropsychopharmacology. 2006;31:287–296. doi: 10.1038/sj.npp.1300830. [DOI] [PubMed] [Google Scholar]
- Paré D, Quirk GJ, Ledoux JE. New vistas on amygdala networks in conditioned fear. J Neurophysiol. 2004;92:1–9. doi: 10.1152/jn.00153.2004. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Postigo A, Calella AM, Fritzsch B, Knipper M, Katz D, Eilers A, Schimmang T, Lewin GR, Klein R, Minichiello L. Distinct requirements for TrkB and TrkC signaling in target innervation by sensory neurons. Genes Dev. 2002;16:633–645. doi: 10.1101/gad.217902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattiner LM, Davis M, French CT, Ressler KJ. Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J Neurosci. 2004;24:4796–4806. doi: 10.1523/JNEUROSCI.5654-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Schafe GE, LeDoux JE. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 2004a;44:75–91. doi: 10.1016/j.neuron.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Rodrigues SM, Farb CR, Bauer EP, LeDoux JE, Schafe GE. Pavlovian fear conditioning regulates Thr286 autophosphorylation of Ca2+/calmodulin-dependent protein kinase II at lateral amygdala synapses. J Neurosci. 2004b;24:3281–3288. doi: 10.1523/JNEUROSCI.5303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, Stäubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Romanski LM, LeDoux JE. Equipotentiality of thalamo-amygdala and thalamo-cortico-amygdala circuits in auditory fear conditioning. J Neurosci. 1992;12:4501–4509. doi: 10.1523/JNEUROSCI.12-11-04501.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi-Arnaud C, Ammassari-Teule M. What do comparative studies of inbread mice add to current investigations on the neural basis of spatial behaviors? Exp Brain Res. 1998;123:36–44. doi: 10.1007/s002210050542. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Nader K, Blair HT, LeDoux JE. Memory consolidation of pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci. 2001;24:540–546. doi: 10.1016/s0166-2236(00)01969-x. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Petrovich GD. What is the amygdala? Trends Neurosci. 1998;21:323–331. doi: 10.1016/s0166-2236(98)01265-x. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Bauer EP, LeDoux JE. L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J Neurosci. 1999;19:10512–10519. doi: 10.1523/JNEUROSCI.19-23-10512.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci. 2000;20:6888–6897. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]