

Abstract
Transient receptor potential vanilloid subtype 1 (TRPV1) and metabotropic glutamate receptor 5 (mGluR5) located on peripheral sensory terminals have been shown to play critical roles in the transduction and modulation of pain sensation. To date, however, very little is known regarding the significance of functional expression of mGluR5 and TRPV1 on the central terminals of sensory neurons in the dorsal horn of the spinal cord. Here we show that TRPV1 on central presynaptic terminals is coupled to mGluR5 in a membrane-delimited manner, thereby contributing to the modulation of nociceptive synaptic transmission in the substantia gelatinosa neurons of the spinal cord. Further, our results demonstrate that TRPV1 is involved in the pain behaviors induced by spinal mGluR5 activation, and diacylglycerol produced by the activation of mGluR5 mediates functional coupling of mGluR5 and TRPV1 on the presynaptic terminals. Thus, mGluR5–TRPV1 coupling on the central presynaptic terminals of nociceptive neurons may be an important mechanism underlying central sensitization under pathological pain conditions.
Introduction
Peripheral transient receptor potential vanilloid subtype 1 (TRPV1) is activated not only by capsaicin (CAP), heat, and acid (Caterina et al., 1997; Tominaga et al., 1998; Szallasi and Blumberg, 1999; Clapham, 2003) but also by inflammatory mediator-related molecules including the products of lipoxygenases, anandamide, and other endocannabinoids (Hwang et al., 2000; Julius and Basbaum, 2001; Ralevic et al., 2001; Di Marzo et al., 2002; van der Stelt et al., 2005). Multiple inflammatory mediators have been shown to heighten the sensitivity of nociceptive sensory neurons after binding to their respective G-protein-coupled receptors (GPCRs) (Scholz and Woolf, 2002), leading to inflammation-induced thermal hyperalgesia via TRPV1. Group I metabotropic glutamate receptors (mGluRs) (especially mGluR5) are expressed together with TRPV1 in dorsal root ganglion (DRG) neurons (Walker et al., 2001b) and are also involved in peripheral sensitization of sensory neurons via G-protein-mediated TRPV1 modulation (Hu et al., 2002).
Notably, both mGluR5 and TRPV1 are expressed on the central terminals of primary afferents in the superficial lamina of the spinal dorsal horn, the key site for the transmission of pain sensation (Jia et al., 1999; Valtschanoff et al., 2001). Capsaicin potently increases the frequency, but not the amplitude, of miniature EPSCs (mEPSCs) in a DRG–dorsal horn neuronal coculture system (Sikand and Premkumar, 2007), as well as in a spinal slice condition (Yang et al., 1998), suggesting that TRPV1-mediated neurotransmitter release from presynaptic terminals of nociceptive neurons contributes to nociceptive transmission. Despite these findings, however, the underlying mechanisms for the activation of central presynaptic TRPV1 under pathophysiological conditions remain unknown (Patapoutian et al., 2009).
It is well known that the modulation of synaptic transmissions in the superficial dorsal horn contributes to the pathophysiology of chronic pain conditions (Woolf and Salter, 2000). Indeed, both spinal mGluR5 and TRPV1 have been demonstrated to contribute to pain hypersensitivity under pathological pain conditions (Caterina et al., 2000; Walker et al., 2001a; Zhu et al., 2005; Kanai et al., 2006). Whereas a functional role for mGluR5 in superficial dorsal horn neurons (i.e., postsynaptic neurons) has recently been demonstrated (Hu et al., 2007), relatively little is known about how presynaptic mGluR5 contributes to nociceptive synaptic transmissions in the spinal dorsal horn.
In the present study, we hypothesized that mGluR5 and TRPV1 are coupled on the central presynaptic terminals of nociceptive neurons, thereby contributing to the pain transmission processing activity exerted by TRPV1. We attempted to elucidate the underlying mechanisms of TRPV1 modulation by mGluR5 activation, as this will likely provide insight into the functional significance of TRPV1 expression in the CNS.
Materials and Methods
All surgical and experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the School of Dentistry, Seoul National University, Seoul, Korea.
Behavioral studies.
All animals were placed in an observation chamber (60 × 100 × 60 mm each) and allowed to habituate. A mirror was positioned behind the observation chamber to provide an unobstructed view. Spontaneous pain behaviors were assessed by measuring the time each animal spent flinching, licking, and/or biting its hindpaws or tail. The cumulative time spent flinching, licking, or biting hindpaws or tails during a 5 min period was recorded immediately before drug administration and then again up to 210 min after drug administration. For mechanical sensitivity (von Frey filament) testing, mice were brought from the animal colony and placed in transparent plastic boxes (60 × 100 × 60 mm) on a metal mesh floor (3 × 3 mm mesh). The mice were then left alone for at least 20 min to allow them to acclimate before testing. To assess mechanical sensitivities, the withdrawal threshold of the hindpaw was measured using a series of von Frey filaments [0.20, 0.69, 1.57, 3.92, 5.88, 9.80, 19.60, and 39.20 mN, Stoelting; equivalent to (in grams) 0.02, 0.07, 0.16, 0.40, 0.60, 1.0, 2.0, and 4.0]. The 50% withdrawal threshold was determined using the up–down method as previously described (Chaplan et al., 1994). A brisk hindpaw lift in response to von Frey filament stimulation was regarded as a withdrawal response. The 0.4 g filament was the first stimulus to be used, and, when a withdrawal response was obtained, the next-weakest filament was used. This process was repeated until no response was obtained, at which time the next-strongest filament was administered. Interpolation of the 50% threshold was then performed using the method of Dixon (1980). All behavioral testing was performed by an investigator who was blind to the genetic background of the mice.
Intrathecal injection of drug.
(R,S)-3,5-Dihydroxyphenylglycine (DHPG) was dissolved in 0.9% saline using an ultrasonic washer and applied intrathecally. The dose used in this study was 15 nmol. Intrathecal administration was performed as described previously (Hylden and Wilcox, 1980). Briefly, under slight enflurane anesthesia (2% in 95% O2), the vertebral column of mouse was held using the thumb and middle finger of the left hand and the drug was injected intrathecally into each mouse at approximately the lumbar enlargement level of the spinal cord by use of a 25 μl Hamilton syringe fitted with a 31 gauge needle. The injection volume was 5 μl and the injection sites were verified by injecting a similar volume of 1% methylene blue solution and determining the distribution of the injected dye in the spinal cord. Before conducting experiments, the injection method was practiced until the success rate was consistently >95%.
DRG preparation.
DRG neurons obtained from 4–7-d-old neonatal rats were prepared as previously described (Oh et al., 2001). Briefly, animals were decapitated, and DRGs were rapidly removed under aseptic conditions and placed in HBSS (Welgene). DRGs were digested in 1 mg/ml collagenase A (Roche) and 2.4 U/ml dispase II (Roche) in HBSS for 10 min, respectively, followed by 20 min in 0.125% trypsin (Sigma), all at 37°C. The DRGs were then washed in DMEM (Welgene) three times and resuspended in F12 medium supplemented with 10% FBS (Invitrogen) and 1% penicillin/streptomycin (Sigma). DRGs were then mechanically dissociated using fire-polished glass pipettes, centrifuged (800 rpm, 5 min), resuspended in F12 medium supplemented with 5% FBS (Invitrogen), 20 ng/ml NGF (Invitrogen), 1× N-2 supplement (Invitrogen), and 1% penicillin/streptomycin (Invitrogen), and plated on 0.5 mg/ml poly-l-ornithine (Sigma)-coated glass coverslips. Cells were maintained at 37°C in a 5% CO2 incubator.
Cell culture and transient transfection.
Human embryonic kidney cells (HEK293 cells, American Type Culture Collection) were maintained according to the supplier's recommendations. For transient transfections, cells were seeded in 12-well plates. The next day, the cells were transfected with 1 μg/well of pcDNA expression vectors for TRPV1, TRPV1 mutants, or mGluR5 using the Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's suggested protocol. After 18–24 h, cells were trypsinized and used for experiments.
Ca2+ imaging.
We performed fura-2 AM-based (Molecular Probes) Ca2+ imaging experiments as previously described (Park et al., 2006). Briefly, the HEK293 cells and DRG neurons prepared were loaded with fura-2 AM (2 μm) for 40 min at 37°C in a balanced salt solution [BSS; containing (in mm) 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulfonic acid] (HEPES), 10 glucose, adjusted to pH 7.3 with NaOH]. Then, the cells were rinsed with BSS and incubated in BSS for an additional 30 min to de-esterify the dye. Cells on slides were placed onto an inverted microscope and illuminated with a 175 W xenon arc lamp; excitation wavelengths (340/380 nm) were selected by a monochromator wavelength changer. Intracellular calcium concentrations ([Ca2+]i) were measured by digital video microfluorometry with an intensified charge-coupled-device camera (CasCade, Roper Scientific) coupled to the microscope and a computer with Metafluor software (Universal Imaging). All drugs were applied via bath perfusion at a flow rate of 5 ml/min.
Electrophysiology.
Whole-cell patch-clamp recordings from DRG neurons and spinal substantia gelatinosa (SG) neurons were performed at room temperature (23 ± 1°C) in normal Tyrode's solution as previously described (Oh et al., 2001; Jung et al., 2006). Whole-cell currents from DRG neurons were recorded from 4–7-d-old Sprague Dawley rats (OrientBio). Whole-cell currents were recorded using an EPC-10 amplifier and Pulse 8.30 software (both from HEKA). Patch pipettes were made from borosilicate glass and had resistances of 3–5 MΩ when filled with standard intracellular solutions. For whole-cell recordings in DRG neurons, we used an external bath solution (normal Tyrode's solution) of the following composition (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, adjusted to pH 7.4 with NaOH. The pipette solution contained (in mm) 126 K–gluconate, 10 NaCl, 1 MgCl2, 10 EGTA, 2 NaATP, and 0.1 Mg-GTP, adjusted to pH 7.3 with KOH, and 295–300 mOsm. All drug solutions were applied to cells by local perfusion through a capillary tube (1.1 mm inner diameter) positioned near the cell of interest. The solution flow was driven by gravity (flow rate, 4–5 ml/min) and controlled by miniature solenoid valves (The Lee Company). For slice patch-clamp recordings, Sprague Dawley rats of both sexes aged 8–12 d were used. Before decapitation, the animals were deeply anesthetized with halothane. The spinal cord was exposed by a dorsal laminectomy and dissected out. The lumbosacral segment of spinal cord was placed into ice-cold artificial CSF (aCSF) and was attached to agarose block (3% in aCSF). Transverse slices (350–400 μm thick) of the lumbar spinal cord were obtained (Vibratome 1000 Plus) and then transferred in aCSF containing the following (in mm): 130 NaCl, 3 KCl, 2.5 CaCl2, 1.5 MgSO4, 1.25 NaH2PO4, 25 NaHCO3, 1.25 HEPES, 10 glucose, and 20 sucrose, adjusted to pH 7.3 (310–315 mOsm, equilibrated with 95% O2 and 5% CO2) for recovery period of at least 1 h and then maintained at room temperature in aCSF. The membrane currents were recorded using an EPC-10 amplifier and Pulse 8.30 software. A single slice was placed in a perfusion chamber (0.5 ml volume) and continuously superfused with extracellular solution (3 ml/min) saturated with 95% O2 and 5% CO2. The recording electrodes were filled with a solution containing the following (in mm): 126 K–gluconate, 10 NaCl, 1 MgCl2, 10 EGTA, 2 NaATP, and 0.1 Mg-GTP, adjusted to pH 7.3 with KOH, and 295–300 mOsm. Whole-cell patch-clamp recordings were made with thin-walled borosilicate unpolished glass pipettes (5–7 MΩ) from visually identified SG neurons in the spinal cord slice by using a fixed-stage microscope (BX50WI, Olympus) with Nomarski optics and a 40× water immersion objective. All recordings were performed at a holding potential (Vh) of −60 mV. mEPSCs were recorded in the presence of 0.5 μm tetrodotoxin (TTX), 5 μm bicuculline, and 2 μm strychnine to block voltage-dependent Na+ channels, activity-dependent sEPSCs, and synaptic inhibition mediated by GABAA and glycine receptors, respectively. Stretches of data of 4 min were used for mEPSC frequency/amplitude analysis. The amplitude threshold for detection of mEPSCs was set above the noise level (5 pA) and events were subsequently verified visually. No attempt was made to group the events by the rise time. The Kolmogorov–Smirnov test was used to assess the effects of the DHPG and capsazepine (CZP) on amplitude and interevent interval. For cell-attached patch-clamp recordings, the recording pipette (6–7 MΩ) contained bath solution containing the following (in mm): 140 NaCl, 5 KCl, 1 MgCl2, 10 glucose, 2 EGTA, and 10 HEPES, adjusted to pH 7.4 with NaOH. The recordings were performed at a command potential of +40 mV. Drugs were applied into recording pipette or through bath solution. All data were analyzed using single-channel analysis program QuB software (www.qub.buffalo.edu).
Constructs.
An expression vector for TRPV1 containing the point mutation Y511A was produced using a two-step PCR approach based on a TRPV1 construct generated in our laboratory (Yang et al., 2003). After mutagenesis, the sequence of the final constructs was confirmed by DNA sequencing. pcDNA3.1(+)/mGluR5 were also generated in our laboratory following conventional methods.
Single-cell reverse transcription-PCR.
Single-cell reverse transcription-PCR (RT-PCR) was performed as previously described (Park et al., 2006). Entire single cells were aspirated into a patch pipette by using negative pressure under visual control. The inner and outer primers used in our study are listed in the supplemental table, available at www.jneurosci.org as supplemental material. Negative controls were obtained from pipettes that did not harvest any cell contents but were submerged in the bath solution.
Electron microscopy.
Three male Sprague Dawley rats (weight, 300–320 g) were used in this study. Tissue samples were prepared as previously described (Bae et al., 2004). We used an anti-TRPV1 antibody (SC-12498 (P-19), lot L1302, Santa Cruz). To verify the specificity of the antibody, preabsorption controls with a blocking peptide completely abolished the staining (data not shown).
Drugs.
DHPG, (RS)-2-chloro-5-hydroxyphenylglycine (CHPG), 5′-iodoresiniferatoxin (IRTX), 6-iodonordihydrocapsaicin, 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt), 2-methyl- 6-(phenylethynyl)pyridine hydrochloride (MPEP), staurosporine, and TTX were purchased from Tocris Bioscience. Capsaicin, capsazepine, thapsigargin, bicuculline, stychinin, bisindolylmaleimide (BIM), 1-oleoyl- 2-acetyl-sn-glycerol (OAG), 1,6-bis(cyclohexyloximinocarbonylamino) hexane (RHC80267), 1-[6-[((17β)-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione (U73122), and 1-[6-[((17β)-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-2,5-pyrrolidinedione (U73343) were purchased from Sigma.
Statistical analysis.
Data are expressed as mean ± SEM. For behavioral testing, statistical analyses of the data obtained from the drug tests were conducted with one-way repeated-measures ANOVA followed by a pairwise comparison of pain behaviors before and after the injection, using Bonferroni's t test method. Student's t test was used for the comparison between the knock-out and wild-type (WT) mice and p < 0.05 was considered statistically significant. For other studies, results were compared using Student's t test and p < 0.05 was considered statistically significant.
Results
Coupling of group I mGluRs and TRPV1 on central terminals of sensory neurons contributes to pain behaviors
It has been previously demonstrated that spontaneous pain responses are produced by the activation of spinal group I mGluRs (Fisher and Coderre, 1998; Karim et al., 2001; Hu et al., 2007). We first investigated whether spinal TRPV1 is associated with the spontaneous pain behavior induced by the activation of spinal group I mGluRs by using TRPV1 knock-out and wild-type mice. With the intrathecal injection of the vehicle alone, we did not see detectable changes in pain behavior compared with the preinjection baseline (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), suggesting marginal effects of general anesthetics on the pain behaviors observed in our behavioral study. In agreement with previous reports (Fisher and Coderre, 1998; Karim et al., 2001; Hu et al., 2007), a single intrathecal injection of 15 nmol DHPG, a selective group I mGluR (mGluR1/5) agonist, induced an immediate and robust increase in wild-type mice in the time spent either flinching, licking, or biting hindpaws or tails (*p < 0.05, **p < 0.001 vs preinjection baseline; one-way repeated-measures ANOVA followed by Bonferroni's t test) (Fig. 1A). We interpreted these responses as signs of spontaneous pain, which persisted up to 120 min after the injection (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). TRPV1−/− mice lacked this early manifestation of spontaneous pain behaviors after administration of DHPG (Fig. 1A). Indeed, the induction of spontaneous pain behavior was delayed so markedly that the display of pain behaviors took 20 min after injection to become statistically significant (*p < 0.05 vs preinjection baseline; one-way repeated-measures ANOVA followed by Bonferroni's t test). Further, spontaneous pain was significantly reduced in TRPV1−/− mice in the first 15 min after injection, compared with the wild-type mice (#p < 0.05, ##p < 0.001 vs the wild-type mice; Student's t test) but did not differ thereafter (Fig. 1A; supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). When we examined mechanical sensitivity of hindpaws in response to von Frey filaments after intrathecal DHPG injection, mechanical hypersensitivity was persistent throughout the 3.5 h observation period in wild-type mice but was maintained for only 2 h in TRPV1−/− mice (*p < 0.05 vs preinjection baseline; one-way repeated-measures ANOVA followed by Bonferroni's t test) (Fig. 1B). In addition, mechanical hypersensitivity in TRPV1−/− mice was lower than that of wild-type mice (#p < 0.05 vs wild-type mice; Student's t test). Together, these results demonstrated that spinal TRPV1 is associated with DHPG-induced pain behavior, such as spontaneous pain behaviors and mechanical allodynia.
Figure 1.
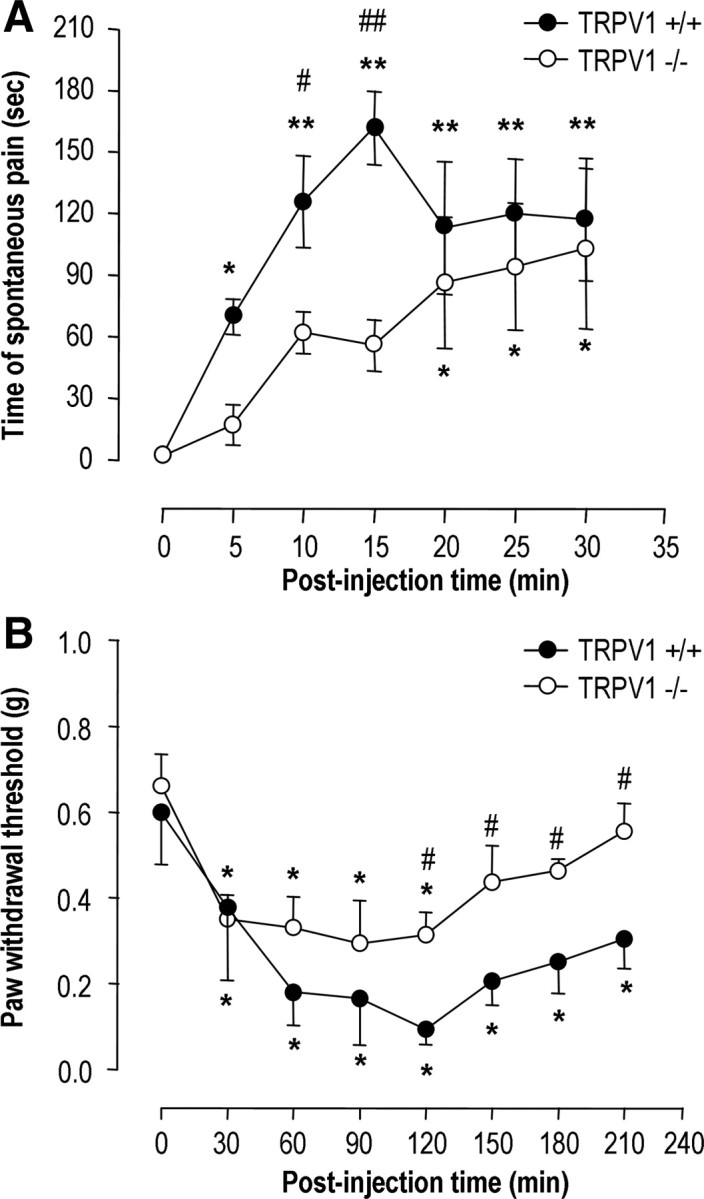
Activation of spinal mGluR5 induces pain behavior. Effects of DHPG (15 nmol, 5 μl, intrathecal) on spontaneous pain behavior (n = 5; each group) (A) and mechanical sensitivity to von Frey filaments (n = 6–8; each group) (B) in both TRPV1 knock-out (open circles) and wild-type (closed circles) mice. *p < 0.05, **p < 0.001 versus preinjection baseline (one-way repeated-measures ANOVA followed by Bonferroni's t test); #p < 0.05, ##p < 0.001 versus wild-type mice (Student's t test).
To determine the mechanisms underlying the interactions between group I mGluR receptors and TRPV1 in the spinal cord, we examined whether DHPG regulates TRPV1 activity in synaptic transmission of the spinal dorsal horn. We measured mEPSCs of SG neurons from spinal cord slice by using whole-cell patch-clamp recording (Fig. 2a). In this approach, DHPG had little effect on the amplitude of mEPSCs (97.52 ± 0.93%; n = 4; p = 0.14; and 89.90 ± 4.04%; n = 3; p = 0.13) (Fig. 2c,d); however, the frequency of mEPSCs was significantly increased by DHPG (137.87 ± 2.10%; n = 4; p < 0.001; and 152.84 ± 1.19%; n = 3; p < 0.001) (Fig. 2c,d), and this was blocked by 5 μm capsazepine, a TRPV1-competitive antagonist (102.56 ± 2.08%; n = 4; p = 0.16) (Fig. 2b,c), and 25 μm 6-iodonordihydrocapsaicin, another structurally different TRPV1 antagonist (101.05 ± 2.07%; n = 3; p = 0.83) (Fig. 2d). In addition, as shown previously (Tominaga et al., 1998; Hwang et al., 2004), EM analysis demonstrated that TRPV1 was clearly expressed on the perisynaptic region of the presynaptic terminals of sensory neurons in SG (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Therefore, we felt it was reasonable to hypothesize that group I mGluRs and TRPV1 coupled on the presynaptic terminals of primary afferents neurons may regulate neurotransmitter releases, thereby contributing to DHPG-induced pain behaviors.
Figure 2.

mGluR5 and TRPV1 are coupled on central presynaptic terminals. a, Effects of DHPG on glutamatergic mEPSCs in spinal SG neurons. Traces from a cell before treatment (left), during treatment with 100 μm DHPG (middle, 1 min after the onset of DHPG application), and during treatment with 5 μm CZP with 100 μm DHPG (right, 5 min pretreatment of CZP and 1 min after the onset of DHPG application). b, Normalized cumulative probability distributions of the amplitude and interevent intervals of mEPSCs. c, d, Bar graphs illustrate the percentage change in mean amplitude and frequency of mEPSCs. *p < 0.001; paired t test versus control; n = 4 and n = 3, respectively. Results are presented as the mean ± SEM.
Group I mGluRs drive DHPG-induced Ca2+ entry through TRPV1 channels in sensory neurons
Next, we examined how DHPG modulates TRPV1 on presynaptic nociceptive neurons using fura-2 AM-based ratiometric Ca2+ imaging. DHPG induced a Ca2+ response in subpopulations of DRG neurons, acutely isolated from neonatal rats. TRPV1-expressing nociceptive neurons were identified by their responsiveness to a 10 s application of 200 nm capsaicin at the end of each experiment. In a subpopulation of capsaicin-sensitive neurons (35.74%; n = 89/249), a 20 s application of 100 μm DHPG induced a transient increase in [Ca2+]i that produced little desensitization during repetitive application of DHPG (93.90 ± 5.83%; n = 12; p = 0.31) (Fig. 3Aa). DHPG-induced Ca2+ transients were abolished by pretreatment with 0 mm Ca2+ in the bath solution (2.44 ± 0.74%; n = 10; p < 0.05) (Fig. 3Ab) but not by 1 μm thapsigargin (93.17 ± 10.61%; n = 15; p > 0.5) (Fig. 3Ac), suggesting that DHPG-induced Ca2+ transients are mostly because of an influx of extracellular Ca2+ rather than Ca2+ release from intracellular Ca2+ stores. We next tested whether DHPG-induced Ca2+ transients were associated with TRPV1. DHPG-induced Ca2+ transients were completely blocked by the pretreatment with 10 μm capsazepine (2.03 ± 0.18%; n = 14; p < 0.005) (Fig. 3Ad) and 100 nm 5′-iodoresiniferatoxin (0.75 ± 0.82%; n = 5; p < 0.005) (Fig. 3Ae), which indicated that Ca2+ transients can indeed be attributed to Ca2+ influx mainly through TRPV1. Further, Ca2+ transients were absent from TRPV1−/− mice (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 3.
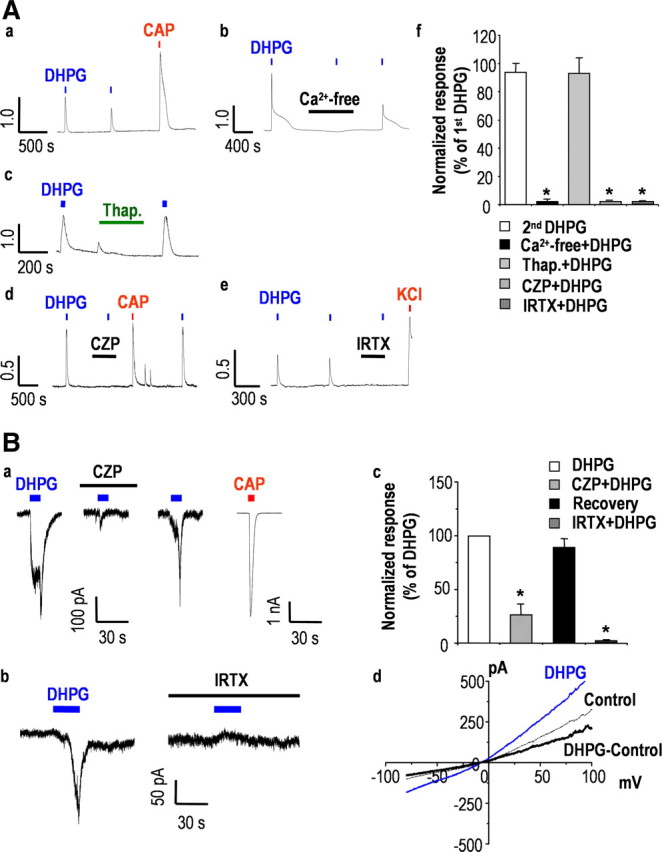
DHPG induces Ca2+ transients through TRPV1 in nociceptive sensory neurons. A, Ca2+ transients evoked by sequential application of 100 μm DHPG (n = 12) (a) were blocked by extracellular Ca2+-free condition (n = 10) (b) but not by 1 μm thapsigargin (n = 15) (c). DHPG-induced Ca2+ transients were also abolished by 10 μm CZP (n = 14) (d) and 100 nm IRTX (n = 5) (e). f, Summary of Ca2+ response relative to peak amplitude of first DHPG response (*p < 0.05; paired t test versus first DHPG response). Results are presented as the mean ± SEM. B, Representative DHPG-induced current traces from CAP-sensitive DRG neurons at a holding potential of −60 mV. DRG neurons were repetitively exposed to 100 μm DHPG at 10 min intervals. DHPG-induced currents were abolished by 10 μm CZP (n = 4) (a) and 100 nm IRTX (n = 5) (b). c, Summary of the current responses as measured by peak amplitude current, relative to peak amplitude of first DHPG response (*p < 0.05; paired t test versus first DHPG response). Results are presented as the mean ± SEM. d, I–V relationship obtained by a voltage ramp protocol from −80 to +100 mV during DHPG-induced current (gray) and before DHPG application (control) exhibited a reversal potential of ∼0 mV and a slight outward rectification.
Given that TRPV1 is a nonselective cation channel with high permeability to both Ca2+ and Na+ (Clapham, 2003), we next examined whether DHPG induces TRPV1-mediated inward currents by using whole-cell patch-clamp recording. When capsaicin-sensitive DRG neurons were exposed to 100 μm DHPG for 10 s at a holding potential of −60 mV, inward currents were clearly evoked; this effect was readily reversible and blocked by 10 μm capsazepine (26.47 ± 9.78%; n = 4; p < 0.05) (Fig. 3Ba) and 100 nm 5′-iodoresiniferatoxin (12.77 ± 2.21%; n = 5; p < 0.001) (Fig. 3Bb). Further, the I–V relationship indicated that DHPG-induced currents have the characteristics of TRPV1-mediated currents with a reversal potential of ∼0 mV and a slight outward rectification (Fig. 3Bd).
mGluR5 drives DHPG-induced Ca2+ entry through TRPV1 channels in sensory neurons
DHPG is a group I mGluRs agonist, and the two subtypes of group I mGluRs, mGluR1 and mGluR5, are both expressed in DRG neurons (Bhave et al., 2001). Thus, we determined the relative contribution of each subtype of mGluRs to DHPG-induced activation of TRPV1. To isolate only TRPV1-mediated Ca2+ responses produced by the application of DHPG, we eliminated the contribution of intracellular Ca2+ stores with 1 μm thapsigargin. Whereas CPCCOEt (50 μm), an mGluR1-specific antagonist, had a slight inhibitory effect (80.52 ± 3.80%; n = 13; p > 0.002) (Fig. 4Aa), MPEP (50 μm), an mGluR5-specific antagonist, clearly blocked DHPG-induced Ca2+ transients (9.15 ± 2.27%; n = 11; p < 0.001) (Fig. 4Ab). Furthermore, CHPG, an mGluR5-specific agonist, induced Ca2+ transients that were similar to DHPG-induced Ca2+ transients (74.93 ± 5.85%; n = 10; p > 0.002) (Fig. 4Ac,Ad). Single-cell RT-PCR analysis revealed that all of the DHPG-responsive cells expressed both mGluR5 and TRPV1, whereas mGluR1 expression was homogenous throughout the cells analyzed and was not correlated with the DHPG-induced Ca2+ transients in DRG neurons (Fig. 4B). These results indicated that DHPG elicits Ca2+ influx via TRPV1, primarily through the activation of mGluR5 in capsaicin-sensitive nociceptors.
Figure 4.

mGluR5 mediates DHPG-induced Ca2+ transients in nociceptive sensory neurons. A, DHPG-induced Ca2+ transients were not abolished by the mGluR1-specific antagonist 50 μm CPCCOEt (n = 13) (a) but were abolished by the mGluR5-specific antagonist 50 μm MPEP (n = 11) (b). c, [Ca2+]i transients were also induced by the mGluR5-specific agonist 300 μm CHPG (n = 10). d, Summary of Ca2+ responses relative to the first DHPG response (*p < 0.001; paired t test versus first DHPG response). Results are presented as the mean ± SEM. B, Whole-tissue RT-PCR analysis indicated expression of mGluR1 (145 bp), mGluR5 (202 bp), and TRPV1 (330 bp) in DRG neurons. Combination of single-cell RT-PCR after Ca2+ imaging (n = 20) revealed an association between the coexpression of mGluR5 and TRPV1 and the responsiveness to DHPG and CAP. DRG neurons responsive to both DHPG and capsaicin expressed mGluR1, mGluR5, and TRPV1 (1) (n = 8/8), whereas DRG neurons responsive only to capsaicin and not to DHPG expressed mGluR1 and TRPV1 (2) (n = 8/8). DRG neurons unresponsive to DHPG only expressed mGluR1 (3) (n = 4/4). The control obtained in bath solution without harvesting cells was negative for all of the tested primers (data not shown). The three images represent fura-2 ratio images taken before and during DHPG and CAP applications. Numbers indicate each cell shown in single-cell RT-PCR results. Traces show Ca2+ transients in response to DHPG or CAP application.
DHPG-induced Ca2+ response results from direct activation of TRPV1 by diacylglycerol
Next, we addressed the underlying mechanism for DHPG-induced activation of TRPV1. Since mGluR5 is Gαq/11-coupled receptors linked to phospholipase C (PLC), the activation of which results in the hydrolysis of PtdIns(4,5)P2 to diacylglycerol (DAG) and inositol triphosphate (IP3) (Hermans and Challiss, 2001), we hypothesized that one of the signaling molecules in this pathway was involved in the TRPV1 activation by DHPG. Whereas DHPG-induced Ca2+ transients were abolished by 2 μm U73122 (1.74 ± 0.13%; n = 12; p < 0.001), a specific PLC inhibitor (2 μm U73343, the inactive form, had no effect) (Fig. 5Aa), DHPG-induced Ca2+ transients persisted in the presence of 1 μm staurosporine (89.10 ± 4.20%; n = 9; p > 0.01), a nonspecific protein kinase inhibitor (Rüegg and Burgess, 1989), or 1 μm BIM (83.50 ± 2.98%; n = 9; p > 0.001), a specific PKC inhibitor, and with 1 μm RHC80267, a DAG lipase inhibitor (Fig. 5Ab). These results indicated that downstream signaling molecules of PLC mediated DHPG-induced Ca2+ transients and that these effects were a result of a protein kinase- and DAG lipase-independent activation of TRPV1, implying DAG as the most plausible candidate molecule. Indeed, we have recently shown that OAG, a membrane-permeable analog of DAG, directly activates TRPV1 (Woo et al., 2008). We further found in the present study that bath application of OAG produced a Ca2+ transient that was blocked by capsazepine (2.68 ± 0.73%; n = 12; p < 0.001), but not by staurosporine and RHC80267 (80.85 ± 3.32%; n = 12; p > 0.001), in DRG neurons (Fig. 5Ac,Ad). We also examined whether OAG could induce inward currents via TRPV1 in DRG neurons. In capsaicin-responsive DRG neurons, 100 μm OAG evoked inward currents (0.20 ± 0.05 nA; n = 6) that were completely blocked by 10 μm capsazepine (Fig. 5Ba). Further, the I–V relationship exhibited a typical nonselective cationic current of the TRPV1 response with a reversal potential of ∼0 mV and a slight outward rectification (Fig. 5Bb). In addition, when we performed cell-attached patch-clamp recordings, single-channel activities were elicited by 100 μm DHPG only when applied through the pipette solution and not when applied through the bath solution. Capsaicin (1 μm) applied either through the pipette or the bath solution elicited higher single-channel activities compared with 100 μm DHPG (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). These results suggest that the effect of DHPG on TRPV1 is mediated by a membrane-delimited pathway but not by a diffusible molecule.
Figure 5.
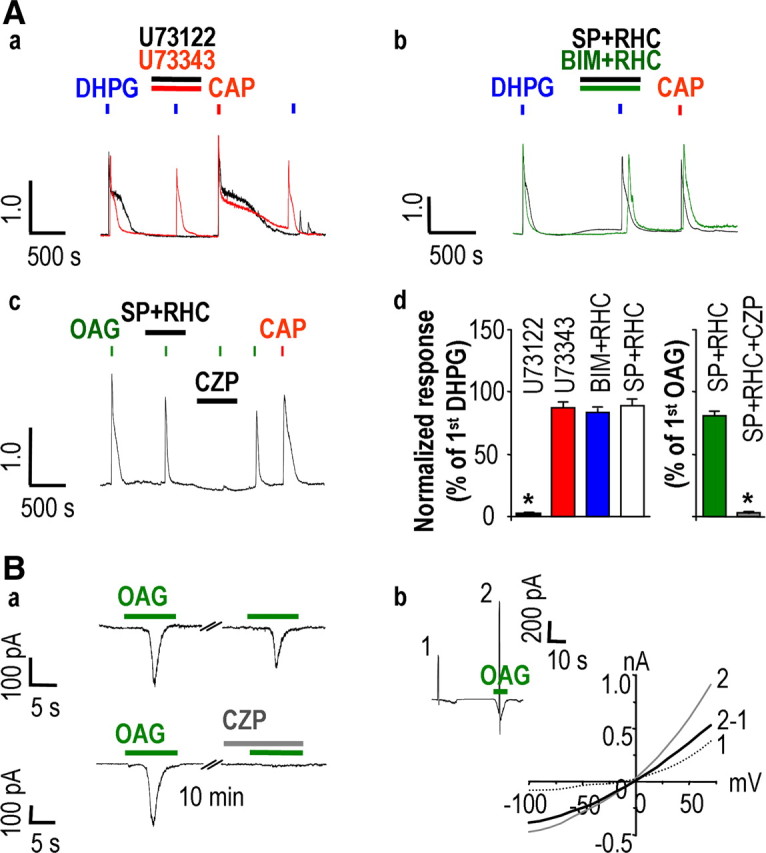
DHPG-induced Ca2+ response results from direct activation of TRPV1 produced by DAG. Aa, DHPG-induced Ca2+ responses were abolished by 2 μm U73122 (n = 12) but not by 2 μm U73343 (n = 12). Ab, Treatment with either 1 μm staurosporine (SP) or 1 μm BIM with 1 μm RHC80267 (n = 9 each) had no effect on [Ca2+]i transients by DHPG. Ac, [Ca2+]i transients evoked by 100 μm OAG were not affected by 1 μm SP and 1 μm RHC80267 (n = 12) but were abolished by pretreatment with 10 μm CZP (n = 12). Ad, Summary of Ca2+ responses relative to the first DHPG or OAG response (*p < 0.001; paired t test versus first DHPG or OAG response). Results are mean ± SEM. Ba, Representative current traces from CAP-sensitive DRG neurons at a holding potential of −60 mV. DRG neurons were exposed to 100 μm OAG twice at 10 min intervals (n = 6). OAG-induced currents were abolished by CZP (n = 6). Bb, I–V relationship obtained by a voltage ramp protocol from −100 to +60 mV during OAG-induced current (2) and before OAG application (1) exhibited a reversal potential of ∼0 mV and a slight outward rectification.
DAG directly activates the TRPV1 channel in a membrane-delimited manner
We also demonstrated the mechanisms by which DAG activates TRPV1 using whole-cell patch-clamp recording in a heterologous expression system (Woo et al., 2008). To provide further evidences on the membrane-delimited activation of TRPV1, we used Ca2+ imaging in the present study. As indicated in our previous study (Woo et al., 2008), one possible mechanism of this pathway would be direct interaction between DAG and TRPV1. To investigate this possibility, we used Y511A mutants of TRPV1 (Jordt and Julius, 2002) transiently expressed in HEK293 cells. This mutant failed to respond to OAG as well as to capsaicin (n = 10) (Fig. 6A), suggesting that capsaicin and OAG share the Y511 binding site for activation of TRPV1. Alternatively, DAG might produce its effects by replacing PtdIns(4,5)P2 from an inhibitory site on TRPV1 (Prescott and Julius, 2003). Thus, we also examined whether DAG activates TRPV1 in Δ774–838 mutants of TRPV1, which lack a PtdIns(4,5)P2 binding site (786–828) (Ferrer-Montiel et al., 2004). OAG-induced Ca2+ transients remained in the Δ774–838 mutant, which suggested that this site might not be critical for the activation of TRPV1 by DAG. Normal response to acid stimulus (pH 5.5) of naive TRPV1, Y511A, and Δ774–838 mutants of TRPV1 was used to demonstrate normal functional expression of these constructs in our experimental system (Fig. 6B).
Figure 6.

DAG and CAP share their binding site at TRPV1. A, Both OAG and CAP transiently increased [Ca2+]i in both WT TRPV1-expressing (n = 10) and Δ774–838 mutant-expressing (n = 10) HEK293 cells but not in Y511A mutant-expressing HEK293 cells (n = 10). Results are presented as the mean ± SEM. B, Representative images obtained from fura-2-based Ca2+ imaging experiments in response to CAP, OAG, and pH 5.5 from HEK293 cells transiently transfected with the WT TRPV1, the Δ774–838 mutant, and the Y511A mutant.
We further verified the modulation of TRPV1 by mGluR5 activation in HEK293 cells. To isolate only TRPV1-mediated responses, we examined DHPG-induced Ca2+ transients again in the presence of 1 μm thapsigargin. We found that mGluR5 behaved differently depending on the coexpression of TRPV1. DHPG failed to elicit any Ca2+ response in either mGluR5-expressing or TRPV1-expressing HEK293 cells. In contrast, DHPG elicited Ca2+ response (1.75 ± 0.08 ratio; n = 10) that was abolished by capsazepine (0.16 ± 0.01 ratio; n = 10; p < 0.01) in HEK293 cells transiently transfected with both mGluR5 and TRPV1 (Fig. 7a,b). Similar response patterns were observed with 100 μm glutamate as an mGluR5 agonist (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). These results clearly demonstrated that TRPV1 is trans activated by mGluR5 to induce Ca2+ influx rather than Ca2+ mobilization from intracellular Ca2+ stores. We also confirmed normal functioning of mGluR5 by evaluating Ca2+ mobilization from intracellular stores, observing either Ca2+ oscillation or Ca2+ transients produced by DHPG without the pretreatment with thapsigargin (Fig. 7a, inset).
Figure 7.

TRPV1 is trans activated by mGluR5 in HEK293 cells. a, In the presence of 1 μm thapsigargin, mGluR5-expressing HEK293 cells had no response to either 100 μm DHPG or 200 nm CAP, whereas TRPV1-expressing HEK293 cells responded only to 200 nm CAP. In contrast, mGluR5/TRPV1-coexpressing HEK293 cells were responsive to both 100 μm DHPG and 200 nm CAP, and this effect was abolished by 10 μm CZP. DHPG induced typical Ca2+ responses when the cells were not pretreated with thapsigargin (inset). n = 10; each data set. b, Summary of DHPG-induced Ca2+ responses in HEK293 cells as measured by peak amplitude of ratio for each transient (*p < 0.01; paired t test versus first DHPG response). Results are presented as the mean ± SEM.
Discussion
We revealed that TRPV1 and mGluR5 are linked in a membrane-delimited manner on the central presynaptic terminals of nociceptive neurons, together serving as presynaptic modulators of nociceptive transmission in the spinal SG area. Spinal TRPV1 is involved in pain behaviors produced by the onset of activation of spinal group I mGluR activation. Given that the enhancement of the mEPSC frequency, but not of the amplitude, induced by DHPG was significantly reduced by TRPV1 antagonism, it was believed that coupling of TRPV1 and group I mGluRs on presynaptic terminals could contribute to DHPG-induced pain responses. Our in vitro results further demonstrated that mGluR5, rather than mGluR1, is coupled to TRPV1, and that DAG produced by the activation of mGluR5 is responsible for mediating the functional coupling of mGluR5 and TRPV1 on presynaptic terminals. Based on our observations, it was thought that cellular mechanisms of spinal presynaptic mGluR5 might be distinct from those of both peripheral mGluR5 and spinal postsynaptic mGluR5. In addition to TRPV1's well known action as a transducer for pain sensation on the peripheral sensory terminals, it is also highly likely that presynaptic TRPV1, coupled with mGluR5, acts as a Ca2+ regulator for synaptic transmission in the SG area.
We found that the effects of mGluR5 activation observed in the calcium imaging and electrophysiology experiments were mediated by TRPV1. Since capsazepine, at micromolar concentrations, is known to produce inhibitory effects on voltage-gated calcium currents (Docherty et al., 1997) in addition to its well known TRPV1-antagonistic effect, we verified TRPV1-dependent actions after mGluR5 activation by additionally using structurally different TRPV1 antagonists such as 5′-iodoresiniferatoxin and 6-iodonordihydrocapsaicin. Notably, we also found that the effect of 5–10 μm capsazepine per se on mEPSCs was negligible, suggesting the limited effects of capsazepine on voltage- gated calcium currents under our experimental conditions. Absence of Ca2+ transients in response to mGluR5 activation in TRPV1−/− mice further revealed TRPV1-specific effects of mGluR5 activation.
Given that the DHPG response was not blocked by the combination of a nonspecific protein kinase inhibitor/or PKC inhibitor and DAG lipase inhibitor, DAG produced a response comparable with DHPG, DHPG applied only into the pipette and not to the bath solution generated single-channel activity, and DAG-induced Ca2+ transients were not observed in the capsaicin-binding site TRPV1 mutant, it seems clear that TRPV1 activation by mGluR5 activation occurs via a membrane-delimited generation of DAG. The generation of DAG may be associated with the cellular mechanism for synaptic transmission of nociceptive information in the SG area in the spinal dorsal horn. Although lipid metabolite products such as hydroperoxyeicosatetraenoic acid and anandamide have been suggested as candidate molecules for mediating GPCR activation of TRPV1 (Hwang et al., 2000; van der Stelt et al., 2005), the identities of endogenous ligands for TRPV1 at the pathophysiological conditions, especially in the central neurons, are still debated. We have recently demonstrated that DAG is a novel endogenous ligand of TRPV1 (Woo et al., 2008). In the present study, we provide further strong evidences that group l mGluR5 activates the TRPV1 channel in a membrane-delimited manner and DAG mediates functional coupling of group l mGluR5 and TRPV1 on central presynaptic terminals of sensory neurons.
Mechanisms for direct induction of TRPV1 activity by mGluR5 activation in the central presynaptic terminals, which are investigated in this study, seem to be different from those of the enhancement of peripheral TRPV1 function by mGluR5 activation. A previous study demonstrated that activation of mGluR5 on peripheral sensory terminals sensitizes and enhances TRPV1 via PKA and cyclo-oxygenase pathways (Hu et al., 2002). It is difficult at this point to link these two distinctive in vitro results with the appropriate behavioral experiments. However, our finding may reflect differential functional roles of peripheral mGluR5 and central presynaptic mGluR5 in nociceptive signaling. Whereas glutamate, as a key inflammatory mediator released in the periphery after inflammation, acts on mGluR5 for a relatively long period (deGroot et al., 2000), glutamate released from presynaptic terminals may act rapidly on mGluR5 because of the recycling of glutamate to astrocytes via glutamate transporters (Oliet et al., 2001). Therefore, we thought that application of DHPG for short periods of time (20 s for Ca2+ imaging, 10 s for electrophysiology) rather than 3 min as in a previous study (Hu et al., 2002) would be more appropriate for elucidating the mechanisms of spinal presynaptic mGluR5. Application of DHPG for long periods could mask the effects of membrane-delimited activation of TRPV1 by mGluR5 activation.
Additionally, as previously suggested (Delmas et al., 2002), it is possible that the spatial proximity of receptors (i.e., mGluR5) and target channels (i.e., TRPV1) may determine the specificity of Ca2+ responses produced by Gαq/11-coupled receptors which could then recruit either membrane-delimited pathways or downstream pathways inside the cytosol. It remains to be elucidated whether geometrical distances between mGluR5 and TRPV1 are different on peripheral and central nociceptive terminals, and how preferential functional couplings between different GPCRs and TRP channels are determined at the molecular level.
Group I mGluRs, namely, mGluR1 and mGluR5, are distributed on peripheral unmyelinated sensory afferents in both DRG neurons (Bhave et al., 2001) and spinal dorsal horn (Alvarez et al., 2000). These receptors function such that the activation of peripheral and spinal mGluR1/mGluR5 is sufficient to evoke pain hypersensitivity (Lesage, 2004). However, mGluR1 and mGluR5 may have different cellular mechanisms for their nociceptive signaling. Recent reports demonstrate that mGluR5 is the predominant group I mGluR mediating DHPG-induced responses both in cultured mouse DRG neurons (Hu et al., 2002) and in spinal dorsal horn neurons (Hu et al., 2007). Our results also demonstrate that mGluR5 is highly likely to be the predominant group I mGluR on the central presynaptic terminals. Immunohistochemical analysis also revealed double labeling of DRG neurons with mGluR5 and TRPV1 (supplemental Fig. 6, available at www.jneurosci.org as supplemental material), which is consistent with the results of a previous report (Walker et al., 2001b).
SG neurons are the first central neurons to relay input from primary afferent neurons (Sugiura et al., 1986). Thus, synaptic modulation of primary afferent neurons in the SG is believed to play an important role not only in acute nociceptive transmission but also in central sensitization associated with chronic pain (Lu and Perl, 2005). Recently, it has been demonstrated that mGluR5 modulates nociceptive plasticity via Kv4.2 signaling in postsynaptic spinal dorsal horn neurons (Hu et al., 2007). Our results suggest that mGluR5 on the central presynaptic terminals of nociceptive neurons may also play an important role for the central sensitization under pathological pain conditions by membrane-delimited coupling with TRPV1. Whereas presynaptic mGluR5 might be silent in normal physiological pain transmission, mGluR5 present in the perisynaptic area (Jia et al., 1999; Pitcher et al., 2007) could be activated by glutamate spilled over the synaptic cleft after excessive release from central terminals of nociceptive neurons under pathological conditions such as peripheral inflammation and nerve injury. The subsequent Ca2+ influx via TRPV1 induced by mGluR5 activation may lead to further glutamate release from central terminals, thereby providing a positive feedback cycle via autocrine function. It has been reported that activation of spinal TRPV1 induced release of substance P (Marvizón et al., 2003), which is necessary for central sensitization of dorsal horn neurons (Ikeda et al., 2003). This observation may be associated with the induction of DHPG-induced pain hypersensitivity documented in our behavioral study.
Our results suggest a plausible cellular mechanism for the contribution of central presynaptic mGluR5 and TRPV1 to nociceptive transmission in the spinal cord. The direct activation of TRPV1 and strong Ca2+ signaling induced by DAG after mGluR5 activation implies a previously unknown significant role for TRPV1 as an integrator of multiple Gαq/11-coupled receptors in other areas of the CNS as well as on the central terminals of nociceptive neurons.
Footnotes
This research was supported by Korea Research Foundation Grant R0A-2008-000-20101-0 from the National Research Laboratory Program and Grant M103KV10015-08K2201-01510 from the Brain Research Center of the 21st Century Frontier Research Program funded by the Ministry of Science and Technology, Republic of Korea, to S.B.O. We thank Dr. Uhtaek Oh of Seoul National University, Korea, for his kind gift of the Δ774-838 mutant and also thank Dr. Lishuang Cao of the University of Manchester, UK, for his advice on single-channel analysis.
References
- Alvarez FJ, Villalba RM, Carr PA, Grandes P, Somohano PM. Differential distribution of metabotropic glutamate receptors 1a, 1b, and 5 in the rat spinal cord. J Comp Neurol. 2000;422:464–487. doi: 10.1002/1096-9861(20000703)422:3<464::aid-cne11>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Bae YC, Oh JM, Hwang SJ, Shigenaga Y, Valtschanoff JG. Expression of vanilloid receptor TRPV1 in the rat trigeminal sensory nuclei. J Comp Neurol. 2004;478:62–71. doi: 10.1002/cne.20272. [DOI] [PubMed] [Google Scholar]
- Bhave G, Karim F, Carlton SM, Gereau RW., 4th Peripheral group I metabotropic glutamate receptors modulate nociception in mice. Nat Neurosci. 2001;4:417–423. doi: 10.1038/86075. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- deGroot J, Zhou S, Carlton SM. Peripheral glutamate release in the hindpaw following low and high intensity sciatic stimulation. Neuroreport. 2000;11:497–502. doi: 10.1097/00001756-200002280-00014. [DOI] [PubMed] [Google Scholar]
- Delmas P, Wanaverbecq N, Abogadie FC, Mistry M, Brown DA. Signaling microdomains define the specificity of receptor-mediated InsP(3) pathways in neurons. Neuron. 2002;34:209–220. doi: 10.1016/s0896-6273(02)00641-4. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Blumberg PM, Szallasi A. Endovanilloid signaling in pain. Curr Opin Neurobiol. 2002;12:372–379. doi: 10.1016/s0959-4388(02)00340-9. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Docherty RJ, Yeats JC, Piper AS. Capsazepine block of voltage-activated calcium channels in adult rat dorsal root ganglion neurones in culture. Br J Pharmacol. 1997;121:1461–1467. doi: 10.1038/sj.bjp.0701272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Montiel A, García-Martínez C, Morenilla-Palao C, García-Sanz N, Fernández-Carvajal A, Fernández-Ballester G, Planells-Cases R. Molecular architecture of the vanilloid receptor. Insights for drug design. Eur J Biochem. 2004;271:1820–1826. doi: 10.1111/j.1432-1033.2004.04083.x. [DOI] [PubMed] [Google Scholar]
- Fisher K, Coderre TJ. Hyperalgesia and allodynia induced by intrathecal (RS)-dihydroxyphenylglycine in rats. Neuroreport. 1998;9:1169–1172. doi: 10.1097/00001756-199804200-00038. [DOI] [PubMed] [Google Scholar]
- Hermans E, Challiss RA. Structural, signalling and regulatory properties of the group I metabotropic glutamate receptors: prototypic family C G-protein-coupled receptors. Biochem J. 2001;359:465–484. doi: 10.1042/0264-6021:3590465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HJ, Bhave G, Gereau RW., 4th Prostaglandin and protein kinase A-dependent modulation of vanilloid receptor function by metabotropic glutamate receptor 5: potential mechanism for thermal hyperalgesia. J Neurosci. 2002;22:7444–7452. doi: 10.1523/JNEUROSCI.22-17-07444.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HJ, Alter BJ, Carrasquillo Y, Qiu CS, Gereau RW., 4th Metabotropic glutamate receptor 5 modulates nociceptive plasticity via extracellular signal-regulated kinase-Kv4.2 signaling in spinal cord dorsal horn neurons. J Neurosci. 2007;27:13181–13191. doi: 10.1523/JNEUROSCI.0269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SJ, Burette A, Rustioni A, Valtschanoff JG. Vanilloid receptor VR1-positive primary afferents are glutamatergic and contact spinal neurons that co-express neurokinin receptor NK1 and glutamate receptors. J Neurocytol. 2004;33:321–329. doi: 10.1023/B:NEUR.0000044193.31523.a1. [DOI] [PubMed] [Google Scholar]
- Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, Cho S, Min KH, Suh YG, Kim D, Oh U. Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci U S A. 2000;97:6155–6160. doi: 10.1073/pnas.97.11.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Heinke B, Ruscheweyh R, Sandkühler J. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science. 2003;299:1237–1240. doi: 10.1126/science.1080659. [DOI] [PubMed] [Google Scholar]
- Jia H, Rustioni A, Valtschanoff JG. Metabotropic glutamate receptors in superficial laminae of the rat dorsal horn. J Comp Neurol. 1999;410:627–642. [PubMed] [Google Scholar]
- Jordt SE, Julius D. Molecular basis for species-specific sensitivity to “hot” chili peppers. Cell. 2002;108:421–430. doi: 10.1016/s0092-8674(02)00637-2. [DOI] [PubMed] [Google Scholar]
- Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- Jung SJ, Kim SJ, Park YK, Oh SB, Cho K, Kim J. Group I mGluR regulates the polarity of spike-timing dependent plasticity in substantia gelatinosa neurons. Biochem Biophys Res Commun. 2006;347:509–516. doi: 10.1016/j.bbrc.2006.06.134. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Hara T, Imai A. Participation of the spinal TRPV1 receptors in formalin-evoked pain transduction: a study using a selective TRPV1 antagonist, iodo-resiniferatoxin. J Pharm Pharmacol. 2006;58:489–493. doi: 10.1211/jpp.58.4.0008. [DOI] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau RW., 4th Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage ASJ. Role of group I metabotropic glutamate receptors mGlu1 and mGlu5 in nociceptive signalling. Curr Neuropharmacol. 2004;2:363–393. [Google Scholar]
- Lu Y, Perl ER. Modular organization of excitatory circuits between neurons of the spinal superficial dorsal horn (laminae I and II) J Neurosci. 2005;25:3900–3907. doi: 10.1523/JNEUROSCI.0102-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvizón JC, Wang X, Matsuka Y, Neubert JK, Spigelman I. Relationship between capsaicin-evoked substance P release and neurokinin 1 receptor internalization in the rat spinal cord. Neuroscience. 2003;118:535–545. doi: 10.1016/s0306-4522(02)00977-6. [DOI] [PubMed] [Google Scholar]
- Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ. Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J Neurosci. 2001;21:5027–5035. doi: 10.1523/JNEUROSCI.21-14-05027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science. 2001;292:923–926. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- Park CK, Kim MS, Fang Z, Li HY, Jung SJ, Choi SY, Lee SJ, Park K, Kim JS, Oh SB. Functional expression of thermo-transient receptor potential channels in dental primary afferent neurons: implication for tooth pain. J Biol Chem. 2006;281:17304–17311. doi: 10.1074/jbc.M511072200. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Tate S, Woolf CJ. Transient receptor potential channels: targeting pain at the source. Nat Rev Drug Discov. 2009;8:55–68. doi: 10.1038/nrd2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher MH, Ribeiro-da-Silva A, Coderre TJ. Effects of inflammation on the ultrastructural localization of spinal cord dorsal horn group I metabotropic glutamate receptors. J Comp Neurol. 2007;505:412–423. doi: 10.1002/cne.21506. [DOI] [PubMed] [Google Scholar]
- Prescott ED, Julius D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science. 2003;300:1284–1288. doi: 10.1126/science.1083646. [DOI] [PubMed] [Google Scholar]
- Ralevic V, Kendall DA, Jerman JC, Middlemiss DN, Smart D. Cannabinoid activation of recombinant and endogenous vanilloid receptors. Eur J Pharmacol. 2001;424:211–219. doi: 10.1016/s0014-2999(01)01153-0. [DOI] [PubMed] [Google Scholar]
- Rüegg UT, Burgess GM. Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends Pharmacol Sci. 1989;10:218–220. doi: 10.1016/0165-6147(89)90263-0. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci. 2002;5:1062–1067. doi: 10.1038/nn942. [DOI] [PubMed] [Google Scholar]
- Sikand P, Premkumar LS. Potentiation of glutamatergic synaptic transmission by protein kinase C-mediated sensitization of TRPV1 at the first sensory synapse. J Physiol. 2007;581:631–647. doi: 10.1113/jphysiol.2006.118620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura Y, Lee CL, Perl ER. Central projections of identified, unmyelinated (C) afferent fibers innervating mammalian skin. Science. 1986;234:358–361. doi: 10.1126/science.3764416. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Blumberg PM. Vanilloid (capsaicin) receptors and mechanisms. Pharmacol Rev. 1999;51:159–212. [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Valtschanoff JG, Rustioni A, Guo A, Hwang SJ. Vanilloid receptor VR1 is both presynaptic and postsynaptic in the superficial laminae of the rat dorsal horn. J Comp Neurol. 2001;436:225–235. [PubMed] [Google Scholar]
- van der Stelt M, Trevisani M, Vellani V, De Petrocellis L, Schiano Moriello A, Campi B, McNaughton P, Geppetti P, Di Marzo V. Anandamide acts as an intracellular messenger amplifying Ca2+ influx via TRPV1 channels. EMBO J. 2005;24:3026–3037. doi: 10.1038/sj.emboj.7600784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker K, Bowes M, Panesar M, Davis A, Gentry C, Kesingland A, Gasparini F, Spooren W, Stoehr N, Pagano A, Flor PJ, Vranesic I, Lingenhoehl K, Johnson EC, Varney M, Urban L, Kuhn R. Metabotropic glutamate receptor subtype 5 (mGlu5) and nociceptive function. I. Selective blockade of mGlu5 receptors in models of acute, persistent and chronic pain. Neuropharmacology. 2001a;40:1–9. doi: 10.1016/s0028-3908(00)00113-1. [DOI] [PubMed] [Google Scholar]
- Walker K, Reeve A, Bowes M, Winter J, Wotherspoon G, Davis A, Schmid P, Gasparini F, Kuhn R, Urban L. mGlu5 receptors and nociceptive function II. mGlu5 receptors functionally expressed on peripheral sensory neurones mediate inflammatory hyperalgesia. Neuropharmacology. 2001b;40:10–19. doi: 10.1016/s0028-3908(00)00114-3. [DOI] [PubMed] [Google Scholar]
- Woo DH, Jung SJ, Zhu MH, Park CK, Kim YH, Oh SB, Lee CJ. Direct activation of transient receptor potential vanilloid 1(TRPV1) by diacylglycerol (DAG) Mol Pain. 2008;4:42. doi: 10.1186/1744-8069-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Yang BH, Piao ZG, Kim YB, Lee CH, Lee JK, Park K, Kim JS, Oh SB. Activation of vanilloid receptor 1 (VR1) by eugenol. J Dent Res. 2003;82:781–785. doi: 10.1177/154405910308201004. [DOI] [PubMed] [Google Scholar]
- Yang K, Kumamoto E, Furue H, Yoshimura M. Capsaicin facilitates excitatory but not inhibitory synaptic transmission in substantia gelatinosa of the rat spinal cord. Neurosci Lett. 1998;255:135–138. doi: 10.1016/s0304-3940(98)00730-7. [DOI] [PubMed] [Google Scholar]
- Zhu CZ, Hsieh G, Ei-Kouhen O, Wilson SG, Mikusa JP, Hollingsworth PR, Chang R, Moreland RB, Brioni J, Decker MW, Honore P. Role of central and peripheral mGluR5 receptors in post-operative pain in rats. Pain. 2005;114:195–202. doi: 10.1016/j.pain.2004.12.016. [DOI] [PubMed] [Google Scholar]