

Abstract
Presenilin 1 (PS1) mutations are responsible for a majority of early onset familial Alzheimer's disease (FAD) cases, in part by increasing the production of Aβ peptides. However, emerging evidence suggests other possible effects of PS1 on synaptic dysfunction where PS1 might contribute to the pathology independent of Aβ. We chose to study the L286V mutation, an aggressive FAD mutation which has never been analyzed at the electrophysiological and morphological levels. In addition, we analyzed for the first time the long term effects of wild-type human PS1 overexpression. We investigated the consequences of the overexpression of either wild-type human PS1 (hPS1) or the L286V mutated PS1 variant (mutPS1) on synaptic functions by analyzing synaptic plasticity and associated spine density changes from 3 to 15 months of age. We found that mutPS1 induces a transient increase observed only in 4- to 5-month-old mutPS1 animals in NMDA receptor (NMDA-R)-mediated responses and LTP compared with hPS1 mice and nontransgenic littermates. The increase in synaptic functions is concomitant with an increase in spine density. With increasing age, however, we found that the overexpression of human wild-type PS1 progressively decreased NMDA-R-mediated synaptic transmission and LTP, without neurodegeneration. These results identify for the first time a transient increase in synaptic function associated with L286V mutated PS1 variant in an age-dependent manner. In addition, they support the view that the PS1 overexpression promotes synaptic dysfunction in an Aβ-independent manner and underline the crucial role of PS1 during both normal and pathological aging.
Introduction
According to the “amyloid cascade” hypothesis, the key event initiating Alzheimer's disease (AD) is the abnormal processing of amyloid precursor protein (APP) by the γ-secretase complex (Selkoe, 2002). The abnormal processing of APP yields β-amyloid (Aβ) peptides, which form small neurotoxic assemblies that may impair synaptic function independent of plaque formation. A growing body of evidence suggests that the study of excitatory synaptic transmission (Kamenetz et al., 2003; Snyder et al., 2005; Chang et al., 2006) is essential to understand early events that contribute to the development of AD before neurodegeneration occurs. Dendritic spine remodeling is correlated with changes in the strength of excitatory synaptic transmission. Structural reorganization of spines is thought to be important for cognitive processes such as learning and memory (Yuste and Bonhoeffer, 2001; Nimchinsky et al., 2002; Segal, 2005). Modifications of dendritic spine morphology could thus be critical for understanding synaptotoxicity involved in AD. Most familial autosomal dominant (FAD) forms of AD are related to missense mutations in the presenilin (PS) 1 gene, which codes for the transmembrane protein PS1, a subunit of the γ-secretase complex. However, the role of PS1 is not well understood. Young adult mice expressing different mutations of PS1 (Parent et al., 1999; Barrow et al., 2000; Zaman et al., 2000; Dewachter et al., 2008) exhibit an increase in long term potentiation (LTP), suggesting a gain of synaptic function. This increase occurs despite an increase in the Aβ42/Aβ40 ratio, which is known to greatly decrease LTP. Presenilin-dependent γ-secretase is involved in intramembranous proteolysis of many type I membrane proteins, which suggests that PS1 may have Aβ-independent roles in regulating synaptic function (Vetrivel et al., 2006).
To better understand the pathogenic mechanisms of the PS1 FAD mutations during aging, we analyzed the L286V variant (Sherrington et al., 1995; Kamino et al., 1996) which is an aggressive mutation and has never been studied using the combination of electrophysiological and morphological analysis. In addition, wild-type PS1 overexpression alone, which does not lead to Aβ hypersecretion, has never been reported to have major pathological manifestations in young animals. Although the long term effects of wild-type human PS1 overexpression have never been analyzed, a growing body of evidence suggests that PS1 expression level during both normal and pathological aging may play an important role (Miller et al., 2008; Kumar et al., 2009).
A relevant approach is to study (1) synaptic function during abnormal aging, i.e., with overexpression of wild-type human PS1 (hPS1) or a mutated PS1 L286V variant (mutPS1), (2) synaptic changes which occur during normal aging in wild-type mice (WT), and (3) the possible morphological changes associated in hippocampal CA1 neurons.
By adopting this approach, we demonstrate for the first time that mutPS1 induces a transient increase in spine density, NMDA-R-mediated responses, and LTP compared with hPS1 and WT mice, which is observed only at 4–5 months of age.
With increasing age, however, we found that the overexpression of human wild-type PS1 decreased NMDA-R-mediated synaptic transmission and LTP without neurodegeneration.
Materials and Methods
Transgenic mice.
Transgenic mice overexpressing normal human PS1 (hPS1) and mutant human PS1 (mutPS1) used in this study were described previously and express the PS1 transgene at the same level (Citron et al., 1997). Heterozygous mutPS1 and hPS1 mice, as well as their respective WT littermates, were originally the second backcross of FVB/N and C57BL/6 mouse strains. These mice were maintained on a common homogeneous genetic background (C57BL/6) after 10 backcrosses. Control mice are the nontransgenic littermates of both the hPS1 line and the mutPS1 line. Mice were genotyped by PCR using the following primers: 5′-AGATGAGCCACGCAGTCC-3′ and 5′-TACCAGAAGATACCGAGACT-3′. Mice were bred and housed in our animal facility, with 12 h/12 h light/dark cycle and ad libitum access to food and water. The brains from mice expressing mutant PS1, even those older than 9 months, were free from any Alzheimer's disease hallmarks (i.e., plaques and tangles) (Janus et al., 2000).
Western blot.
All animal procedures were performed according to the regulations of the Comité National d'Ethique pour les Sciences de la Vie et de la Santé, which are in accordance with the European Communities Council Directive (86/609/EEC). Mice were decapitated after brief isoflurane anesthesia. Samples of the cortex were quickly homogenized in ice-cold homogenization buffer (25 mm HEPES, 1 mm EDTA, 250 mm sucrose). The homogenate was centrifuged at 3000 × g for 5 min and then the supernatant was collected and centrifuged again at 100,000 × g for 1 h. For membrane protein extraction, the membrane pellet was resuspended in lysis buffer (Bonny et al., 2001) using a manual homogenizer apparatus and homogenates were centrifuged at 13,000 rpm for 10 min at 4°C. Supernatants were collected and protein concentrations were determined by the Lowry method (kit DC Protein assay Bio-Rad). Samples were stored at −80°C until use. Equal amounts of protein (30 μg/lane) were separated by 12% SDS-PAGE and transferred to a nitrocellulose membrane. Incubation with primary antibodies was performed overnight at 4°C using rabbit anti-C-terminal fragment of human PS1 (1:500, Cell Signaling Technology), or mouse monoclonal anti-β-Actin (1:5000, Sigma). After treatment for 1 h with the corresponding horseradish peroxidase-coupled secondary antibodies (Goat anti-rabbit IgG 1:1000, Perox Conjugated, Pierce, catalog #31460; or anti-mouse 1:25,000, Jackson ImmunoResearch), the protein bands were detected by ECL reagent (SuperSignal West Dura Extended Duration Substrate, Pierce). All blots were normalized against actin level and quantified by densitometry analysis (Scion Image software).
Electrophysiology.
Male mice were used at the age of 2–3 months, 4–5 months, 8–10 months, and 13–14 months. Mice were decapitated after a brief isoflurane anesthesia. The brain was rapidly dissected and hippocampal slices (400 μm) were collected in ice-cold dissection buffer containing in mm: 252 sucrose, 3 KCl, 7 MgCl2, 0.5 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 25 glucose (Lipton et al., 1995). Slices were placed at 30°C for 2 h in artificial CSF (ACSF) containing in mm: 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 25 glucose. ACSF and dissection buffer were bubbled with 95% O2/5% CO2. For recording, slices were placed in a submersion-recording chamber, maintained at 30°C, and perfused with ACSF. Monopolar stimulation electrodes (2 MΩ, filled with 200 mm NaCl) were placed in the stratum radiatum of the CA1 region. Schaeffer collateral/commissural fibers were stimulated with 100 μs voltage pulses delivered to the pathway at 15 s intervals. Field EPSPs (fEPSPs) were monitored using low-resistance glass pipettes (2 MΩ, filled with 200 mm NaCl) placed in the stratum radiatum of CA1. For each group, the range of maximal fEPSPs was measured to subsequently stimulate at an intensity that yielded 50% of the maximal fEPSP amplitude. The variability was low, with maximal fEPSP amplitude for all groups of 1.29 ± 0.11 mV. Thus, in each LTP experiment, the stimulus intensity was first adjusted to evoke a response of 0.6 mV in amplitude.
Input/output curves were constructed to assess the AMPA/kainate-mediated synaptic responses to electrical stimulation in the presence of the NMDA receptor antagonist d-AP5 (50 μm, Tocris, Bristol, UK). Similar experiments were conducted in the presence of the AMPA receptor antagonist NBQX (10 μm, Sigma). Picrotoxin (10 μm, Sigma) was added to block inhibitory transmission and the CA3 region was cut to avoid epileptiform activity. Schaeffer collateral/commissural fibers were stimulated with 200 μs voltage pulses delivered to the pathway at 15 s intervals. fEPSPs recorded under these conditions are mediated by NMDA receptor (Massey et al., 2004). For I/O curves, the amplitude of four averaged presynaptic fiber volleys and the slope of fEPSPs were plotted as a function of stimulation intensity. For the Paired-Pulse Facilitation (PPF) protocol, intervals between the two pulses were 50, 100, and 150 ms, and stimulus pairs were delivered every 15 s. PPF was calculated as the ratio of the second field potential slope to the first field potential slope.
After the PPF study, slices were subjected to a 15 min period of pre-LTP baseline measures in which stimuli were elicited at 15 s intervals. Baseline measures of fEPSPs were then recorded and LTP was induced by a theta burst protocol, that involved delivering 4 stimulations of 0.1 ms duration at 200 ms intervals, 10 times, at 100 Hz. Stimulus intensity in the theta burst protocol was the same as that used during baseline recordings. Field potentials were recorded for at least 60 min after initiation of the theta burst protocol.
All data consist of fEPSPs slopes and are presented as mean ± SEM. We assessed differences by two-way ANOVA (age and genotype) and post hoc Scheffé comparisons for AMPA-R-mediated transmission and PPF. For NMDA-R-mediated transmission, homogeneous variance was not observed and comparisons were made with the nonparametric Kruskal–Wallis test. For the LTP study, we used multiple-way ANOVA (age, genotype, and time) analysis and post hoc Scheffé comparisons. We considered differences to be significant at the 5% level (p < 0.05). Data were presented as mean ± SEM.
Golgi-Cox impregnation procedure.
For Golgi-Cox impregnation, male mice were deeply anesthetized with chloral hydrate and were perfused transcardially with NaCl 0.9%. Brains were dissected from the skull and immediately placed in Golgi-Cox fixative for 4 weeks in the dark. The fixative consisted of 5% potassium dichromate, 5% mercuric chloride, and 5% potassium chromate in distilled water. After impregnation, brains were preserved in 30% sucrose solution and serial sagittal sections of hippocampus were cut at 100 μm using a sliding vibratome in 6% sucrose solution (Gibb and Kolb, 1998). Processing of the sections involved development in ammonium hydroxide (30%), fixation in photographic fixative (Kodak), counterstain with methylgreen, dehydration, and clearing in xylene. Sections were mounted onto gelatinized glass slides and coverslipped with poly butyl methacrylate-co-methyl methacrylate (Eukitt).
Analysis of spine density and shape.
We examined spines on apical dendrites of CA1 pyramidal neurons in the stratum radiatum using an Imager microscope with a motorized stage and focusing (Zeiss) and Optronics camera connected to a computer. Spines were classified as “mushroom” if the spine head was appreciably wider than the width of the neck (supplemental Fig. 1, available at www.jneurosci.org as supplemental material); “thin” if spine length was greater than the width of the neck and head (supplemental Fig. 1, available at www.jneurosci.org as supplemental material); and “ramified” if two spine heads emerged from a shared neck (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Spine density and shape analysis was conducted blind to the experimental conditions. For each age and genotype, 6 Golgi-impregnated sections per animal were studied from each of 8–10 animals (83 brains were investigated in total), by the optical dissector stereological method. Masked spines protruding above or beneath the dendritic shaft were not sampled. We outlined the stratum radiatum under 2.5× magnification and selected the region containing secondary or tertiary dendritic segments of pyramidal neurons to avoid the smooth primary dendrites. We classified two types of dendrites according to their thickness (<1 μm or >1 μm) to separate spines located on secondary (thick) or tertiary (thin) dendritic segments, more appropriate for analysis of morphological correlation of plasticity. With Stereoinvestigator (Microbrightfield Inc., Williston, USA), we made systematic random sampling inside each selected zone of the sections using frames of 150 × 120 μm2. For each section, the counting was performed in 14 boxes having a sampling volume of 15 × 15 × 15 μm3. The program provided upper and lower guard zones as well as exclusion and inclusion boundaries for focusing within each volume. Spine counting required focusing in and out with the fine adjustment of the microscope using 100× neofluar objective and oil immersion (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Only spines that were in focus in the top and absent from the bottom of the sampling volume were counted. For dendrites, only the length within the sampling volume was measured. To minimize density distorsions, we used nonembedded tissue, cut with a sliding vibratome, and mounted on gelatinized glass slides. The preparation of 100 μm thick sections is thus probably free of the density distortions in the z dimension that can be induced by the sliding procedure in paraffin or plastic embedded sections as a result of the compression by the knife blade (von Bartheld, 1999). We also used 20 μm upper guard zones to avoid the variations caused by the cutting process.
Spine density was calculated by dividing the estimated number of total spines of thin or thick dendrites by the total length of dendritic segments of this category within the counting frames, and was expressed as the number of spines per micrometer of dendrite. Each type of spine was quantified in the same way. For the 83 brains analyzed, the combined raw data include: 7503 counting frames, 52,479 spines, and a total dendritic length of 99,037 μm (76,593 thin and 22,444 thick). Data were analyzed with the SAS statistical package (SAS Institute). The distributions of densities were tested for normality by univariate analysis (SAS Users Guide: Base, 1989) and rank transformation was applied if necessary. Densities of spines between groups of transgenic and WT mice of each age were analyzed by using a hierarchical ANOVA (GLM procedure, SAS/STAT User's Guide, version 6, 1989) and multiple comparisons between means were made using the post hoc Tukey test. The significant probability level was p ≤ 0.05.
TUNEL staining.
TUNEL staining was performed to determine whether neurodegeneration occurred in mice at 10 or 15 months of age. This assay, which detects the DNA fragmentation that occurs during neuronal death, was performed with an in situ cell death detection kit (Roche, POD). Positive controls were obtained by treatment with DNase I (3 U/ml, Roche). A second positive control for TUNEL staining (apoptotic germ cells of mouse testis) was also used to confirm the specificity of the staining. Mice (n = 4 per group) were perfused transcardially with 4% paraformaldehyde-phosphate buffer, and brains were removed and cryopreserved in 30% sucrose solution. Serial sagittal sections of hippocampus (25 μm thick) were cut using a cryostat and adjacent sections were mounted on superfrost slides and processed for TUNEL staining according to the manufacturer's instructions. Peroxidase activity was revealed with 3,3′-diaminobenzidine tetrahydrochloride (DAB) at the appropriate stage. Sections were counterstained with methylgreen. Control of the reaction was tested by omitting TdT (terminal deoxynucleotidyl transferase) from the incubation medium. Four animals per group and 10 scattered individual hippocampal slices from each animal were extensively analyzed.
Results
NMDA-R-mediated transmission, LTP and spine density are increased by expression of L286V mutation at 4–5 months of age
To determine whether expressing the L286V mutation altered synaptic functions, we performed synaptic plasticity and spine density analyses in the CA1 region of the hippocampus.
First, we analyzed two types of synaptic plasticity, paired-pulse facilitation (PPF), a form of short-term plasticity, and long-term potentiation (LTP).
The physiological substrate of information storage in the hippocampus has been proposed to involve long term potentiation (LTP), an activity-dependent, persistent increase in synaptic transmission. To study LTP, we investigated the effect of theta stimulation in the CA1 region of hippocampal slices from control and transgenic animals. At 4–5 months of age, a larger LTP response was induced in hippocampal slices from mutPS1 mice compared with slices from WT or hPS1 littermates, as previously described for other PS1 mutations (Parent et al., 1999; Barrow et al., 2000; Zaman et al., 2000; Dewachter et al., 2008) (Table 1, Fig. 1A, p < 0.05).
Table 1.
Electrophysiological recordings
| Age (months) | Genotype | NMDA-receptor I-O slope | LTP |
|---|---|---|---|
| 2–3 | WT | 0.071 ± 0.01 (n = 6; N = 4) | 1.40 ± 0.06 (n = 14; N = 8) |
| hPS1 | 0.07 ± 0.003 (n = 8; N = 4) | 1.37 ± 0.06 (n = 14; N = 7) | |
| MutPS1 | 0.07 ± 0.01 (n = 10; N = 3) | 1.39 ± 0.03 (n = 11; N = 7) | |
| 4–5 | WT | 0.08 ± 0.004 (n = 11; N = 5) | 1.35 ± 0.06 (n = 11; N = 8) |
| hPS1 | 0.07 ± 0.006 (n = 11; N = 4) | 1.41 ± 0.05 (n = 10; N = 8) | |
| MutPS1 | 0.18 ± 0.01 (n = 12; N = 5)* | 1.81 ± 0.21 (n = 13; N = 8)* | |
| 8–10 | WT | 0.08 ± 0.01 (n = 13; N = 5) | 1.47 ± 0.07 (n = 14; N = 10) |
| hPS1 | 0.17 ± 0.04 (n = 9; N = 4)* | 1.23 ± 0.03 (n = 10; N = 6)* | |
| MutPS1 | 0.19 ± 0.03 (n = 13; N = 5)* | 1.55 ± 0.07 (n = 10; N = 6) | |
| 13–14 | WT | 0.073 ± 0.002 (n = 8; N = 4) | 1.48 ± 0.07 (n = 7; N = 4) |
| HPS1 | 0.02 ± 0.003 (n = 7; N = 3)* | 1.23 ± 0.06 (n = 8; N = 3)* | |
| MutPS1 | 0.04 ± 0.006 (n = 8; N = 4)* | 1.18 ± 0.08 (n = 8; N = 4)* |
Data are expressed as mean ± SEM,
*p < 0.05. n, number of slices; N, number of animals; I-O, input-output.
Figure 1.

NMDA-R-mediated transmission, LTP, and spine density are increased by mutPS1 at 4–5 months of age. A, At 4–5 months, the LTP protocol induced a significantly larger response in mutPS1 mice than in WT mice and hPS1 mice (p < 0.05). No significant difference was observed between WT and hPS1 mice. B, PPF was significantly unchanged at 4–5 months of age. C, At 4–5 months, analysis of input-output slopes demonstrates a significant increase of NMDA receptor-mediated responses in mutPS1 compared with hPS1 and WT littermates (p < 0.05). D, No significant difference was found between the three groups of mice at any stimulus level examined for averaged input-output plots of AMPA receptor-mediated responses. E, Quantification of spine density revealed a significant increase (27%) in mutPS1 at 4–5 months of age relative to hPS1 and WT mice (*p < 0.05; Tukey's test). F, Representative segments from 4 to 5-month-old WT, hPS1, and mutPS1 mice. Segments are from the stratum radiatum layer. Scale bar, 10 μm. A Z-stack of photomicrographs was obtained with a Zeiss Axioplan microscope using 100× magnification and oil immersion, and projected on a plane using the MIP method. G, At 4–5 months of age, a significant increase in overall spine density in mutPS1 mice was the result of increases in all three categories of spines (*p < 0.05; Tukey's test).
PPF is a short lasting enhancement in synaptic strength mediated by presynaptic calcium-dependent mechanisms (Zucker and Regehr, 2002) so that the response to a second stimulation is potentiated if it is delivered within 200 ms of the first stimulus (Zucker and Regehr, 2002). Changes in synaptic release probability may cause changes in short-term plasticity of synaptic transmission. PPF was examined in 4- to 5-month-old mice using interpulse intervals of 50, 100, and 150 ms. No significant differences were observed between WT, hPS1, and mutPS1 (Fig. 1B).
To determine whether L286V mutation altered excitatory synaptic transmission, we next analyzed NMDA receptor-mediated responses by examining field potentials evoked by stimulation of the Schaeffer collateral/commissural afferent pathway, in the presence of NBQX (10 μm) to block AMPA receptors. Input-output curves were generated by plotting the slope of the field EPSP versus fiber volley amplitude (a measure of the number of presynaptic fibers activated) as stimulus intensity was increased (Fitzjohn et al., 2001). At 4–5 months of age, the magnitude of NMDA receptor-dependent responses was significantly increased in mutPS1 compared with WT and hPS1 mice (p < 0.05, Table 1, Fig. 1C).
Because CA1 pyramidal neurons have both AMPA and NMDA receptors, we also analyzed specifically the AMPA/kainate receptor-mediated synaptic transmission in the presence of d-AP5 (50 μm) to block NMDA receptors. The averaged input-output slopes of AMPA receptor-mediated transmission were not significantly different between the three groups when genotypes were compared (Fig. 1D).
To rule out a contribution of mGlu in the changes in excitatory synaptic transmission, we repeated input/output curves in the presence of d-AP5 and mGlu receptor inhibitors: MCPG ((S)-α-methyl-4-carboxyphenylglycine; group I/II) and MSOP ((RS)-α-methylserine-O-phosphate; group III). We found no differences between the three groups of mice, suggesting that the changes in fEPSPs are not caused by mGluRs, and supporting our results which implicate NMDA receptors (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material).
Because synaptic plasticity in the hippocampus is correlated with the remodeling of spines (Yuste and Bonhoeffer, 2001; Nimchinsky et al., 2002; Segal, 2005), we performed a morphological study of the hippocampal CA1 stratum radiatum using Golgi-Cox staining and optical dissector stereological procedures. We analyzed total spine density (i.e., number of spines per unit length of dendrite) and spine density by category (i.e., number of “mushroom”, “thin” or “ramified” spines per unit length of dendrite) in 4- to 5-month-old WT, hPS1, and mutPS1 mice. Analysis of the spine types revealed similar proportions of mushroom, thin and ramified spines for each age and genotype. Mushroom spine density was always higher than thin spine density, which was in turn higher than ramified spine density. Quantification revealed a significant increase (27%) in spine density in mutPS1 (p < 0.05; Tukey's test, Fig. 1E,F). This increase was observed for all categories of spines at this age (Fig. 1G).
In addition, we analyzed the level of overexpression of both human and mutant PS1, to address the concern that the demonstrated effects could be merely the result of changes in overexpression levels. We performed Western blot analysis with an anti-PS1 human C-terminal fragment (CTF) antibody on cortex extracts. In hPS1 and mutPS1 mice, the human CTF of PS1 was clearly detected and at similar levels in the two strains. In contrast, no human PS1 was observed in WT mice (Fig. 2). Because we have not detected any changes in protein levels on control hPS1 and mutPS1 mice, we can thus compare the respective effects of hPS1 and mutPS1 on synaptic functions.
Figure 2.

Similar PS1 overexpression levels in hPS1 and mutPS1 mice. Overexpression of human PS1 transgenes was examined in WT, hPS1, and mutPS1 mice (10 months of age) using Western blot of membrane protein extracts from the cerebral cortex. The human PS1 transgene is absent in WT mice and present at the same level in hPS1 and mutPS1 mice, as detected with using anti-C-terminal fragment of human PS1. Results were quantified and PS1 levels were normalized to β-actin levels (n = 3 animals per genotype), and statistical analysis (Mann–Whitney test) shows no difference between hPS1 and mutPS1 mice.
At 4–5 months of age, the PS1 L286V variant induced an increase in synaptic functions which involved NMDA receptor-mediated transmission, LTP and spine density. These effects were not observed in the hPS1 animals which rules out the overexpression effects. Analysis of synaptic plasticity and morphological effects in younger animals at 2–3 months of age showed no significant differences between the three groups, suggesting the absence of developmental effects (supplemental Figs. 2A, 3, available at www.jneurosci.org as supplemental material).
The increase of synaptic functions in mutPS1 mice is transient: PS1 overexpression effects during aging?
To determine the progression of the synaptic dysfunction and the long term effects of the L286V mutation and PS1 overexpression itself, we next examined the three genotypes at 8–10 and 13–14 months of age.
To study LTP, we examined the effect of theta stimulation at 8–10 months of age. The LTP increase observed at 4–5 months of age in mutPS1 mice did not persist at 8–10 months of age but instead LTP levels were similar to WT levels at that age (p < 0.05, Figs. 1A, 3A). LTP induction in hPS1 mice was now deficient compared with LTP in WT and mutPS1 mice when genotypes are compared (Table 1, Fig. 3A, p < 0.05). Thus, whereas levels of LTP induction were unchanged in WT animals between 4 and 5 and 8–10 months of age, the levels of LTP induced in mutPS1 and hPS1 were both smaller at 8–10 months of age compared with the ones observed in 4–5 months old animals. This result suggests that overexpression of PS1 itself decreases LTP with age. This was confirmed by analyzing even older animals. At 13–14 months of age, we found a significant decrease of LTP in both hPS1 and mutPS1 mice compared with WT animals (p < 0.05, Table 1, Fig. 4A).
Figure 3.

Overexpression effects of PS1 during aging. A, At 8–10 months of age, LTP was markedly impaired in hPS1 mice (p < 0.05). In contrast, LTP in the WT mice was normal and similar to that observed in the mutPS1 mice. B, PPF was unchanged in different genotypes analyzed. C, At 8–10 months, analysis of input-output slopes demonstrates a significant increase of basal synaptic transmission in mutPS1 and hPS1 compared with WT mice, indicating enhanced NMDA receptor-mediated synaptic transmission (p < 0.05, Kruskal–Wallis test). D, At 8–10 months of age, no statistically significant difference was detected for averaged input-output plots of AMPA receptor-mediated responses. E, The spine density increase observed at 4–5 months of age in mutPS1 mice did not persist at 8–10 months of age. No statistically significant difference in total spine density at 8–10 months of age was detected between WT, hPS1, and mutPS1 mice. F, At 8–10 months of age, hPS1 and mutPS1 mice showed a significant reduction in ramified spine density (33%) compared with WT mice (*p < 0.05; Tukey's test). If we compare ages inside each genotype for each type of spines, in mutPS1 only, the ramified spine density decreases significantly (33%) at 8–10 months of age compared with 4–5 months of age. This was confirmed by a significant interaction between age and genotype in a two way ANOVA.
Figure 4.
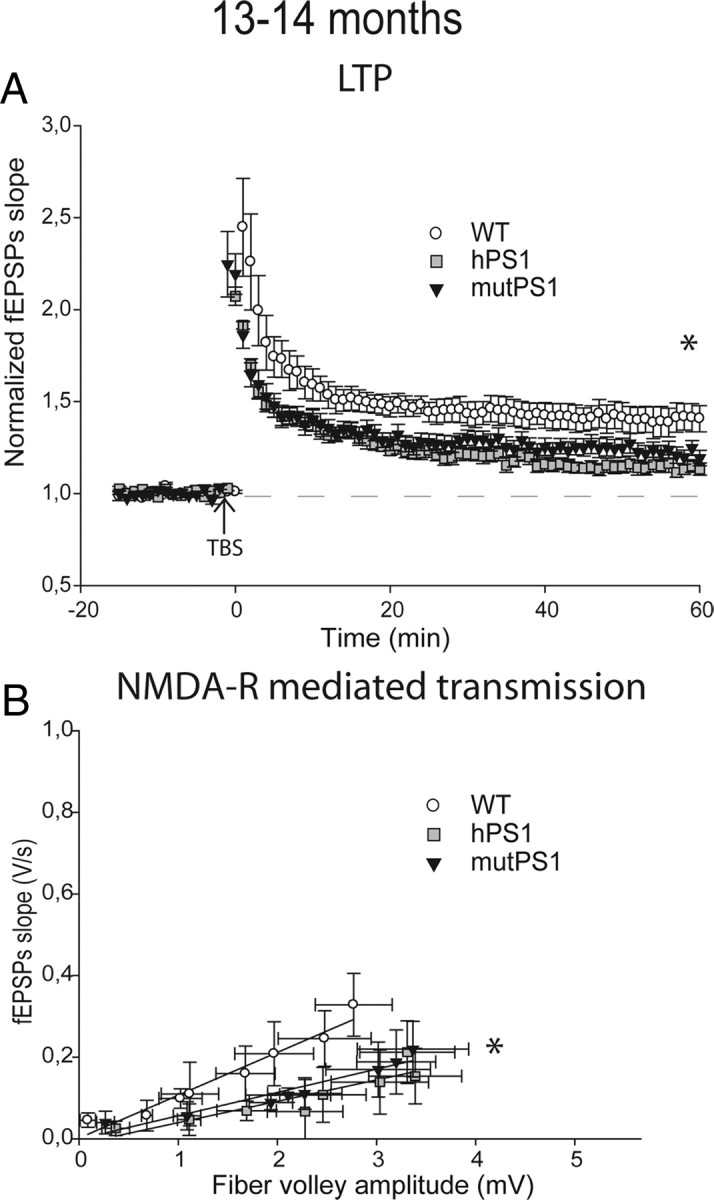
NMDA-R-mediated transmission and LTP are decreased at 13–14 months of age. A, LTP is markedly impaired in hPS1 mice and mutPS1 compared with WT animals (p < 0.05). In contrast, LTP in the WT mice was normal and similar to that observed at young ages. B, At 13–14 months, analysis of input-output slopes demonstrates a significant decrease of basal synaptic transmission in mutPS1 and hPS1 compared with WT mice, indicating an alteration of NMDA receptor-mediated synaptic transmission compared with young ages (p < 0.05, Kruskal–Wallis test).
To determine whether PS1 overexpression effects on LTP during aging was accompanied by excitatory synaptic transmission changes, we next examined NMDA-R-mediated transmission.
In mutPS1 mice, the magnitude of the NMDA-R dependent response was still increased significantly at 8–10 months of age compared with the same animal at 4–5 months of age (p < 0.05, Fig. 3C, Table 1). Surprisingly, in hPS1 mice, we found a significant increase in the magnitude of the NMDA receptor dependent response at 8–10 months of age compared with these mice at younger ages and to WT mice at 8–10 months (p < 0.05, Fig. 3C). When we further examined NMDA-R-mediated responses at 13–14 months of age, we finally found a significant decrease of NMDA-R responses in both hPS1 and mutPS1 mice compared with WT mice (p < 0.05, Table 1, Fig. 4B). Overall, these results showed a progressive decline with age of NMDA-R-mediated responses and LTP linked to PS1 overexpression.
Moreover, the analysis of PPF (Fig. 3B)-, AMPA receptor (Fig. 3D)-, or mGlu receptor-mediated transmission did not show any differences between the three groups at 8–10 months of age (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material).
To determine whether morphological parameters were affected, we also analyzed total spine density (i.e., number of spines per unit length of dendrite) and spine density by category (i.e., number of “mushroom,” “thin,” or “ramified” spines per unit length of dendrite) in 8- to 10-month-old WT, hPS1 and mutPS1 mice.
Among 8- to 10-month-old animals, no significant differences were observed in total spine density between the three genotypes (Fig. 3E), showing that the spine density increase observed at 4–5 months of age in mutPS1 mice did not persist at 8–10 months of age (Figs. 1E, 3E). When analysis was performed for each category of spines, we could detect a significant reduction in the density of ramified spines in hPS1 and mutPS1 mice (33%) compared with WT mice (p < 0.05; Tukey's test; Fig. 3F). Thus, although ramified spines only accounted for a small part (8%) of the total spines, there was a clear decrease in ramified spine density with age in mice overexpressing wild-type or mutant PS1.
To determine whether the LTP deficit observed in aging hPS1 and mutPS1 mice was linked to neuronal death in CA1 hippocampus, we performed TUNEL staining on sagittal slices at 8–10 months of age and 15 months of age by extensively analyzing 4 animals per group and 10 scattered individual hippocampal slices from each animal. TUNEL was absent in WT (Fig. 5D,D1), hPS1 (Fig. 5E,E1) and mutPS1 mice (Fig. 5F,F1) in hippocampus and particularly in the CA1 region at 8–10 (data not shown) and 15 months of age. To assess apoptosis, we also tested caspase-3 activation by immunohistochemistry on parallel sections in these three experimental conditions, and we failed to detect any “apoptotic” cell in transgenic mice (data not shown).
Figure 5.

Synaptic dysfunction are not associated with neuronal death. Dark staining indicates DNA fragmentation induced by DNase treatment of hippocampus (A) and dentate gyrus (B). Dark staining indicates apoptotic germ cells of mouse testis, a second positive control for TUNEL staining (C). Signal is absent in WT, hPS1, and mutPS1 mice in hippocampus (D–F) and particularly in the CA1 region (D1, E1, F1) at 15 months of age. Scale bars, 200 and 20 μm.
Overall, these results suggest that, during aging, PS1 overexpression itself leads to progressive decreases in LTP levels and NMDA receptor-mediated transmission. These synaptic deficits are not sustained by large changes in spine density or morphology or early neurodegeneration.
Discussion
In this study, we analyzed the pathogenic mechanisms of the L286V PS1 mutation during aging using a combination of electrophysiological and morphological analysis, and we identified a transient increase of synaptic functions that involved NMDA receptor-mediated transmission, LTP, and spine density. In addition, we demonstrated that simple wild-type PS1 overexpression, which does not lead to Aβ hypersecretion, affects progressively LTP levels and NMDA receptor-mediated transmission during aging without large changes in spine density or morphology or early neurodegeneration.
Effects of L286V PS1 mutation: a transient increase in synaptic functions
At 4–5 months of age, electrophysiological studies revealed an increase in LTP in mutPS1 mice compared with hPS1 and WT mice, in agreement with earlier publications in transgenic mice overexpressing different PS1 mutations which lead to an hypersecretion of Aβ peptides (Parent et al., 1999; Barrow et al., 2000; Zaman et al., 2000; Dewachter et al., 2008). For the first time, our study showed that the increase in LTP in mutant animals did not persist at older ages and finally decreased.
Facilitated LTP in mutPS1 mice at 4–5 months of age seems to be a consistent finding in mice overexpressing mutant PS1, but also in PS1 M146V knock-in mice without overexpression (Wang et al., 2009) suggesting that the mutation itself facilitates LTP. Recently, Puzzo et al. (2008) strongly supported a model for Aβ effects in which low concentrations play a novel positive, modulatory role on LTP, whereas high concentrations play the well-known detrimental effect (Jacobsen et al., 2006; Lacor et al., 2007; Shankar et al., 2007; Venkitaramani et al., 2007). In our mutPS1 mice, LTP follows the same pattern, being first increased and finally decreased at older ages. A full study will be necessary to clarify this issue but one possible explanation is the modulatory effect of progressive accumulation of Aβ peptides.
Another explanation often proposed by authors for the increase in LTP is that PS1 mutations cause an increase in calcium release, as the effects of PS1 mutation on endoplasmic reticulum (ER) calcium have been established by a number of groups (LaFerla, 2002; Tu et al., 2006; Nelson et al., 2007). An additional interesting hypothesis is the relationship between NMDA-R and PS1 to explain the increased LTP in mutant PS1 mice. Dewachter et al. (2008) showed that, at 4–6 months of age, PS1(A264E) mutant exhibited an increase in NMDA-R-mediated responses, in NR1/NR2B NMDA-R subunits levels and LTP. Our experiments using our mutPS1 showed also an increase in NMDA-receptor responses at 4–5 months of age. However, this issue still remains unclear as shown by recent studies. Priller et al. (2007) showed that cultured hippocampal neurons expressing a mutant PS1 exhibited an inhibition of evoked excitatory synaptic currents while the same mutation induces an increased LTP (Dewachter et al., 2008). Likewise, Wang et al. (2009) reported that PS1 mutant knock-in mice showed an increased LTP which coincides with decreased NMDA currents at the same age.
Our experiments reveal also that spine density is significantly increased in mice expressing L286V mutant PS1 at 4–5 months of age. Spines are very dynamic structures. Their size, shape, and number change throughout development and adulthood. In the adult, modulation of the number and shape of spines is associated with synaptic plasticity (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; Toni et al., 1999; Lendvai et al., 2000) and with aging (for review, see Alvarez and Sabatini, 2007). Thus, the direct analysis of spine densities is likely to provide a sensitive measure of synapse number and an early reflection of synaptic pathology. Several reports demonstrate that hippocampal CA1 neurons exhibit electrical stimulation-induced morphological plasticity and that an LTP-inducing stimulus leads to spine expansion (Nägerl et al., 2007; Yang et al., 2008). These reports suggest that LTP and spine plasticity may be functionally linked. Thus, the same mechanisms may help explain both the abnormal increase in spine density in mutPS1 and the effect of mutant PS1 on LTP although many mechanisms are involved in long term synaptic plasticity.
Long term effects of PS1 overexpression
A second finding of our study is that the overexpression of PS1 protein has its own effects which mask some of the effects of mutation during aging.
Earlier studies on young adult mice overexpressing different mutants of PS1 showed clear effects of PS1 mutation on learning and memory like increased activation of NMDA-receptors, facilitation of LTP and increased short-term cognitive performance (Parent et al., 1999; Barrow et al., 2000; Zaman et al., 2000; Dewachter et al., 2008). However, these experiments were performed in young adult mice and have never been carried on to older ages, and they never indicated that overexpression of PS1 itself has its own effects on synaptic functions.
The increase of LTP observed in mutPS1 mice at 4–5 months of age is transient and no longer present at 8–10 months. In addition, analysis of LTP at 8–10 months in hPS1 mice showed an effect during aging: diminished LTP compared with 1) the same type of animals at 4–5 months and to 2) age-matched WT and mutPS1 mice.
These effects paradoxically coincide with a transient increase in NMDA-R-mediated responses in mice overexpressing either mutPS1 or hPS1 at 8–10 months. This increase may reveal a transient compensatory mechanism to help maintain a normal LTP. This effect may also involve direct physical and functional interactions between PS1 and NMDA-Rs as previously suggested by Saura et al. (2004). Alternatively, this increase may implicate several different mechanisms since many studies have identified multiple functions for PS1 (Thinakaran and Parent, 2004). Indeed, when we further examined older mice (13–14 months of age), both hPS1 and mutPS1 transgenic mice displayed finally a decrease in both NMDA-R responses and LTP. These results strongly suggest that PS1 overexpression induces progressive decline with age of LTP and NMDA-R-mediated responses in an Aβ-independent manner, since Aβ is not overproduced in hPS1 animals.
Although no significant differences were observed in total spine density, we observed a decrease in the number of ramified spines in mice overexpressing hPS1 and mutPS1 at 8–10 months. Although the functional role of ramified spines is unknown, they may serve to modify preexisting neural circuits by forming new connections with neighboring axons, resulting in the fine control of synaptic transmission. Dhanrajan et al. (2004) showed that ramified spines increase in aged rats only after LTP. Ramified spines could disappear during normal aging in animals that do not maintain the capacity for LTP induction, such as the mice overexpressing PS1. However, this particular type of spine represents the smallest proportion of the total spines examined. This lack of massive morphological changes is unlikely to sustain the important synaptic dysfunction observed in mice overexpressing PS1.
In addition, we also used TUNEL assays to determine whether neuronal death underlies the LTP deficits observed at older ages. We found no neuronal death in the CA1 region. Thus, early neurodegeneration did not explain the synaptic plasticity dysfunction observed in older animals overexpressing PS1.
Our results show no simple correlation between the synaptic dysfunction we observed and either of spine morphology or neuronal death. Instead, previous observations have reported that PS1 is involved in the molecular mechanisms underlying cell adhesion, synapse formation, and synaptic plasticity. Indeed, PS1 is an integral protein of the γ-secretase complex, and it is involved in intramembranous proteolysis of many type I membrane proteins such as Notch, ErbB-4, CD44, N- and E-cadherins, nectin-1α, ephrinB, DCC, etc. (for review, see Thinakaran and Parent, 2004). In addition, it has been shown that PS1 interacts directly with different proteins such as Calsenilin, GSK3β, PKA, etc. (Vetrivel et al., 2006). Our results suggest that the simple overexpression of human PS1 may trigger this type of interaction leading to synaptic dysfunction at older ages, underlying multiple roles for PS1 in the adult brain.
Conclusion
Overall, this study highlights early changes in spine density which coincide with changes in synaptic plasticity in a PS1 mouse model of AD. The data also shed light on the fact that the overexpression of human PS1 triggers synaptic dysfunction at older ages. Recently, Kumar et al. (2009) reported that PS1 expression increased with age in senescence-accelerated mice, which might serve as a model for AD. It has also been shown that PS1 is highly expressed both during normal aging and in brains developing sporadic AD (Miller et al., 2008). In light of these results, the study of PS1 overexpression can be considered an interesting model of synaptic aging. Further research is needed to understand how PS1 might interfere with spine morphology and synaptic plasticity during aging.
Footnotes
This work was supported by funds from “Centre National de la Recherche Scientifique,“ Université Pierre et Marie Curie, and “l'Institut de la Longévité et du Vieillissement” (France). We thank Dr. Florence Frédéric for helping with statistical analysis, Dr. Hadi Zanjani for assistance with the Golgi-Cox experiments, and Dr. Fusheng Chen for helping with Western blots. We thank Dr. Fekrije Selimi, Dr. Ann Lohof, and Dr. Rachel Sherrard for their critical reading of this manuscript.
References
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Barrow PA, Empson RM, Gladwell SJ, Anderson CM, Killick R, Yu X, Jefferys JG, Duff K. Functional phenotype in transgenic mice expressing mutant human presenilin-1. Neurobiol Dis. 2000;7:119–126. doi: 10.1006/nbdi.1999.0276. [DOI] [PubMed] [Google Scholar]
- Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes. 2001;50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, Huerta PT. AMPA receptor downscaling at the onset of Alzheimer's disease pathology in double knockin mice. Proc Natl Acad Sci U S A. 2006;103:3410–3415. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- Dewachter I, Ris L, Croes S, Borghgraef P, Devijver H, Voets T, Nilius B, Godaux E, Van Leuven F. Modulation of synaptic plasticity and Tau phosphorylation by wild-type and mutant presenilin1. Neurobiol Aging. 2008;29:639–652. doi: 10.1016/j.neurobiolaging.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Dhanrajan TM, Lynch MA, Kelly A, Popov VI, Rusakov DA, Stewart MG. Expression of long-term potentiation in aged rats involves perforated synapses but dendritic spine branching results from high-frequency stimulation alone. Hippocampus. 2004;14:255–264. doi: 10.1002/hipo.10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Morton RA, Kuenzi F, Rosahl TW, Shearman M, Lewis H, Smith D, Reynolds DS, Davies CH, Collingridge GL, Seabrook GR. Age-related impairment of synaptic transmission but normal long-term potentiation in transgenic mice that overexpress the human APP695SWE mutant form of amyloid precursor protein. J Neurosci. 2001;21:4691–4698. doi: 10.1523/JNEUROSCI.21-13-04691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb R, Kolb B. A method for vibratome sectioning of Golgi-Cox stained whole rat brain. J Neurosci Methods. 1998;79:1–4. doi: 10.1016/s0165-0270(97)00163-5. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janus C, D'Amelio S, Amitay O, Chishti MA, Strome R, Fraser P, Carlson GA, Roder JC, St George-Hyslop P, Westaway D. Spatial learning in transgenic mice expressing human presenilin 1 (PS1) transgenes. Neurobiol Aging. 2000;21:541–549. doi: 10.1016/s0197-4580(00)00107-x. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kamino K, Sato S, Sakaki Y, Yoshiiwa A, Nishiwaki Y, Takeda M, Tanabe H, Nishimura T, Ii K, St George-Hyslop PH, Miki T, Ogihara T. Three different mutations of presenilin 1 gene in early-onset Alzheimer's disease families. Neurosci Lett. 1996;208:195–198. doi: 10.1016/0304-3940(96)12587-8. [DOI] [PubMed] [Google Scholar]
- Kumar VB, Franko M, Banks WA, Kasinadhuni P, Farr SA, Vyas K, Choudhuri V, Morley JE. Increase in presenilin 1 (PS1) levels in senescence-accelerated mice (SAMP8) may indirectly impair memory by affecting amyloid precursor protein (APP) processing. J Exp Biol. 2009;212:494–498. doi: 10.1242/jeb.022780. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Lendvai B, Stern EA, Chen B, Svoboda K. Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature. 2000;404:876–881. doi: 10.1038/35009107. [DOI] [PubMed] [Google Scholar]
- Lipton P, Aitken PG, Dudek FE, Eskessen K, Espanol MT, Ferchmin PA, Kelly JB, Kreisman NR, Landfield PW, Larkman PM. Making the best of brain slices: comparing preparative methods. J Neurosci Methods. 1995;59:151–156. doi: 10.1016/0165-0270(94)00205-u. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Oldham MC, Geschwind DH. A systems level analysis of transcriptional changes in Alzheimer's disease and normal aging. J Neurosci. 2008;28:1410–1420. doi: 10.1523/JNEUROSCI.4098-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Köstinger G, Anderson JC, Martin KA, Bonhoeffer T. Protracted synaptogenesis after activity-dependent spinogenesis in hippocampal neurons. J Neurosci. 2007;27:8149–8156. doi: 10.1523/JNEUROSCI.0511-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson O, Tu H, Lei T, Bentahir M, de Strooper B, Bezprozvanny I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest. 2007;117:1230–1239. doi: 10.1172/JCI30447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- Parent A, Linden DJ, Sisodia SS, Borchelt DR. Synaptic transmission and hippocampal long-term potentiation in transgenic mice expressing FAD-linked presenilin 1. Neurobiol Dis. 1999;6:56–62. doi: 10.1006/nbdi.1998.0207. [DOI] [PubMed] [Google Scholar]
- Priller C, Dewachter I, Vassallo N, Paluch S, Pace C, Kretzschmar HA, Van Leuven F, Herms J. Mutant presenilin 1 alters synaptic transmission in cultured hippocampal neurons. J Biol Chem. 2007;282:1119–1127. doi: 10.1074/jbc.M605066200. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, 3rd, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Parent AT. Identification of the role of presenilins beyond Alzheimer's disease. Pharmacol Res. 2004;50:411–418. doi: 10.1016/j.phrs.2003.12.026. [DOI] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D. LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature. 1999;402:421–425. doi: 10.1038/46574. [DOI] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaramani DV, Chin J, Netzer WJ, Gouras GK, Lesne S, Malinow R, Lombroso PJ. Beta-amyloid modulation of synaptic transmission and plasticity. J Neurosci. 2007;27:11832–11837. doi: 10.1523/JNEUROSCI.3478-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivel KS, Zhang YW, Xu H, Thinakaran G. Pathological and physiological functions of presenilins. Mol Neurodegener. 2006;1:4. doi: 10.1186/1750-1326-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bartheld CS. Systematic bias in an “unbiased” neuronal counting technique. Anat Rec. 1999;257:119–120. doi: 10.1002/(SICI)1097-0185(19990815)257:4<119::AID-AR2>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Wang Y, Greig NH, Yu QS, Mattson MP. Presenilin-1 mutation impairs cholinergic modulation of synaptic plasticity and suppresses NMDA currents in hippocampus slices. Neurobiol Aging. 2009;30:1061–1068. doi: 10.1016/j.neurobiolaging.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Frerking M, Zhou Q. Spine expansion and stabilization associated with long-term potentiation. J Neurosci. 2008;28:5740–5751. doi: 10.1523/JNEUROSCI.3998-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Zaman SH, Parent A, Laskey A, Lee MK, Borchelt DR, Sisodia SS, Malinow R. Enhanced synaptic potentiation in transgenic mice expressing presenilin 1 familial Alzheimer's disease mutation is normalized with a benzodiazepine. Neurobiol Dis. 2000;7:54–63. doi: 10.1006/nbdi.1999.0271. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]