

Abstract
Peroxisome proliferator-activated receptors gamma (PPARγ) are nuclear receptors with essential roles as transcriptional regulators of glucose and lipid homeostasis. PPARγ are also potent anti-inflammatory receptors, a property that contributes to the neuroprotective effects of PPARγ agonists in experimental stroke. The mechanism of these beneficial actions, however, is not fully elucidated. Therefore, we have explored further the actions of the PPARγ agonist rosiglitazone in experimental stroke induced by permanent middle cerebral artery occlusion (MCAO) in rodents. Rosiglitazone induced brain 5-lipoxygenase (5-LO) expression in ischemic rat brain, concomitantly with neuroprotection. Rosiglitazone also increased cerebral lipoxin A4 (LXA4) levels and inhibited MCAO-induced production of leukotriene B4 (LTB4). Furthermore, pharmacological inhibition and/or genetic deletion of 5-LO inhibited rosiglitazone-induced neuroprotection and downregulation of inflammatory gene expression, LXA4 synthesis and PPARγ transcriptional activity in rodents. Finally, LXA4 caused neuroprotection, which was partly inhibited by the PPARγ antagonist T0070907, and increased PPARγ transcriptional activity in isolated nuclei, showing for the first time that LXA4 has PPARγ agonistic actions. Altogether, our data illustrate that some effects of rosiglitazone are attributable to de novo synthesis of 5-LO, able to induce a switch from the synthesis of proinflammatory LTB4 to the synthesis of the proresolving LXA4. Our study suggests novel lines of study such as the interest of lipoxin-like anti-inflammatory drugs or the use of these molecules as prognostic and/or diagnostic markers for pathologies in which inflammation is involved, such as stroke.
Introduction
Inflammation is intrinsically a defensive phenomenon, with participation of both proinflammatory and anti-inflammatory responses. An unbalance resulting in excessive inflammation underlies several pathologies, including stroke. In contrast, resolution of inflammation is aimed to restitution of damaged tissues, by suppressing proinflammatory gene expression and cell trafficking, and by inducing apoptosis and phagocytosis of inflammatory cells. This process is controlled by endogenous mediators which include lipid anti-inflammatory compounds; among them, there are arachidonic acid and ω-3 polyunsaturated fatty acid derivatives, such as cyclopentenone prostaglandins and lipoxins, and resolvins and protectins, respectively (for review, see Lawrence et al., 2002; Nathan, 2002; Serhan and Savill, 2005; Serhan, 2007).
Some of these lipid mediators have been described to act as agonists of the peroxisome proliferator-activated receptor (PPAR)γ, a ligand-dependent nuclear transcription factor belonging to the nuclear hormone receptor superfamily, involved in the expression of genes related to metabolic processes, such as lipid and glucose homeostasis, but also responsible for important anti-inflammatory effects (rev. in Delerive et al., 2001; Willson et al., 2001; Berger and Moller, 2002; Clark, 2002). PPARγ is activated by several molecules that include the anti-diabetic thiazolidinediones or glitazones (Yki-Järvinen, 2004) and endogenous mediators such as the above mentioned cyclopentenone prostaglandins (Straus and Glass, 2001).
In a recent report, we found neuroprotective effects of PPARγ agonists in rats exposed to middle cerebral artery occlusion (MCAO). In this setting we also explored concomitant early transcriptional responses by using cDNA microarray analysis (Pereira et al., 2006). A surprising finding of this study was a significant and early transcriptional induction of 5-lipoxygenase (5-LO) mRNA in those rats receiving the PPARγ agonist rosiglitazone. 5-LO is a nonheme, iron-containing dioxygenase that catalyzes the oxygenation of arachidonic acid to 5-HPETE (5(S)-hydroperoxy-6-trans-8,11,14–9-eicosatetraenoic acid) and the subsequent dehydration to the unstable epoxide intermediate, leukotriene A4, the initial step in the biosynthesis of the proinflammatory leukotrienes (for reveiw, see Rådmark and Samuelsson, 2005). 5-LO is also involved in the biosynthesis of the anti-inflammatory, proresolving mediators lipoxins, such as lipoxin A4 (LXA4) (for review, see Chiang et al., 2005; Serhan, 2007). Therefore, 5-LO might theoretically exert both proinflammatory and anti-inflammatory actions, depending on the final product of its enzymatic activity.
Considering these pieces of evidence, we performed this study to elucidate the role of 5-LO in the neuroprotective effects of rosiglitazone in experimental stroke induced by MCAO, and the participation of LXA4 in this setting.
Materials and Methods
Materials.
Rosiglitazone was from Alexis, LXA4 was from Calbiochem, and T0070907 was from Cayman Chemical. Other reagents were obtained from Sigma or as indicated in the text.
Animals.
Adult male Fischer rats weighing ∼250 g were used in this study. In addition, WT controls (B6;129SF2/J) and 5-LO-deficient mice (B6;129S2-Alox5tm1Fun/J; 5-LO−/−) of 8 wks were obtained from The Jackson Laboratory. All experimental protocols adhered to the guidelines of the Animal Welfare Committee of the Universidad Complutense (following EU directives 86/609/CEE and 2003/65/CE). Animals were housed individually under standard conditions of temperature and humidity and a 12 h light/dark cycle (lights on at 8:00 A.M.) with free access to food and water.
MCAO.
All experiments were performed and quantified in a randomized manner by investigators blinded to treatment groups. Rats/mice were anesthetized with isoflurane 1.5–2% in a mixture of 70% nitrogen/30% oxygen, and body temperature was maintained at physiological levels with a heating pad during the surgical procedure and anesthesia recovery. Permanent occlusion of the middle cerebral artery was conducted as previously described for rats (De Cristóbal et al., 201) or mice (Caso et al., 2007). Rats/mice in which the MCA was exposed but not occluded served as sham-operated controls. After surgery, individual animals were returned to their cages with free access to water and food.
Experimental groups.
Several groups were used for determinations of infarct size and neurological assessment in rats: MCAO followed 10 min later by an intraperitoneal administration of vehicle (saline; n = 15) or 1 mg/kg rosiglitazone (n = 15), or 3 h later by an intraperitoneal administration of 1–5 mg/kg BWA4C (n = 9), or 10 min and 3 h after by an intraperitoneal administration of, respectively, rosiglitazone (1 mg/kg) and BWA4C (1–5 mg/kg; n = 9). Higher doses of BWA4C (10 mg/kg) caused a high mortality rate (>50%) and were not included in the study. A control group consisted of sham-operated animals 10 min before an intraperitoneal injection of saline (n = 6). For some experiments, the PPARγ antagonist T0070907 (2 mg/kg) was administered 1 h before MCAO (n = 9). Rosiglitazone is a member of the thiazolidinedione class of compounds used for its antidiabetic properties that has shown a high selectivity for PPARγ with a Kd of 40 nm. BWA4C is an acetohydroxamic acid which is a potent and selective arachidonate 5-lipoxygenase inhibitor (Darley-Usmar et al., 1989), with IC50 in the range of 0.1–0.5 μm. Injection volume was <0.4 ml/250 g body weight in all cases. For these experiments, animals were killed 5 h to 7 d after surgery and samples were collected as described below.
In a different set of experiments, some rats received an intracerebroventricular administration of LXA4 (5S,6R,15S-trihydroxy-7,9,13-trans-11-cis-eicosatetraenoic acid) after MCAO as described (Cárdenas et al., 2002; Caso et al., 2006; Pradillo et al., 2006). Injection consisted of 1 nmol (5 μl of 0.2 mm) LXA4 (n = 6) or 5 μl of 20% DMSO in phosphate buffer as control (n = 6). A subgroup of LXA4-treated animals also received an intracerebroventricular administration of the PPARγ antagonist T0070907 (10nmol, 5 μl of a 2 mm solution, n = 6) (Lee et al., 2002) before the LXA4 injection. Briefly, under anesthesia with 2% isoflurane in a mixture of 70% nitrogen/30% oxygen, an intracerebroventricular 28-gauge cannula (Alzet) was inserted into the left lateral ventricle by using a stereotaxic apparatus (David Kopf Instruments). The coordinates were as follows: anteroposterior, −0.8 mm; lateral, 1.0 mm; dorsoventral, 3.6 mm from the bregma, with the incisor bar placed at 3.3 mm below the interaural plane. Cannulas were anchored to the skull using a cyanoacrylate gel (Loctite 454, Alzet). The administration was performed immediately after the MCAO.
An additional set of experiments was designed for determination of infarct size and neurological assessment in 5-LO−/− mice (n = 6) and their wild-type littermates (n = 6) exposed to MCAO with or without an administration of 3 mg/kg rosiglitazone 10 min after the occlusion.
Experimental groups for molecular determinations were used as indicated in the text.
Animals were killed by an overdose of sodium pentobarbital for the different studies. For determination of biochemical parameters, ipsilateral cortical tissue from the peri-infarct territory was dissected. Physiological parameters (cerebral blood flow, rectal temperature, mean arterial pressure, blood glucose levels) were not significantly different between all studied groups (data not shown). No spontaneous mortality was found after MCAO with this model, and this was unaffected by the different experimental treatments, with the exception of BWA4C at 10 mg/kg.
Neurological deficit evaluation.
Before kill, 48 h after surgical procedure, neurological deficit was assessed by two independent observers, blinded to animal pharmacological treatment, using a motor behavioral test for rats (Hunter et al., 2000) as follows: score 0, no neurological deficit; score 1, failure to extend right forepaw fully; score 2, decreased grip of right forelimb while tail was pulled; score 3, spontaneous circling or walking to contralateral side; score 4, walks only when stimulated with depressed level of consciousness; score 5, unresponsive to stimulation. For mice, evaluation was performed with the grip test, in which mice are placed on a suspended wire, and latency to fall is measured (Sinz et al., 1999) or with the focal deficit test, in which seven areas are assessed and animals can be rated from 0 to 4 depending on the severity (Clark et al., 1997).
Determination of infarct size.
Two and seven d after MCAO, animals were killed by an overdose of sodium pentobarbital to assess infarct outcome. Brain was removed and cut into 1 mm (mice) or 2 mm (rats) thick coronal slices and stained with 2,3,5-triphenyltetrazolium chloride (1% TTC in 0,1 phosphate buffer). Infarct volumes were calculated sampling each side of the coronal sections with a digital camera (Nikon Coolpix 990), and the images were analyzed using ImageJ 1.33u (National Institutes of Health, Bethesda, MD).
To exclude the brain edema effects, infarct area was corrected by the ratio of the entire area of the ipsilateral hemisphere to that of the contralateral. Infarct volume was calculated as an orthogonal projection.
Western blotting.
For determination of protein levels of 5-LO and inflammatory mediators, brain tissue was collected from the peri-infarct area of rats (n = 6 for each group) killed 18 h after MCAO. Protein concentration was determined with the BCA Protein Assay (from Pierce). Equal amounts of total protein (30 μg) were resolved by SDS-PAGE and transferred to nitrocellulose membranes. Immunodetection was performed by standard procedures. The membranes were blocked with 5% nonfat milk in TBS-T (0.05% Tween 20 in TBS) and probed with mouse anti-5-LO antibody (1:250; BD Bioscience), rabbit anti-TNFα antibody (1:250; Peprotech), rabbit anti-MMP-9 (1:2000; Millipore Bioscience Research Reagents), rabbit anti-NOS2 (1:100; Santa Cruz Biotechnology) and goat anti-COX-2 (1:200; Santa Cruz Biotechnology). Mouse anti-β-actin (1:10000; Sigma) was included to ensure equal protein loading. Immunoreactive bands were visualized using the corresponding horseradish peroxidase-conjugated IgG (Santa Cruz Biotechnology) and subsequent enhanced chemiluminescence detection (PerkinElmer Life and Analytical Sciences). Specific signals were quantified using densitometry analysis software (NIH Image).
Immunohistochemistry.
Eighteen hours after MCAO, rats (n = 3 for each group) were anesthetized with sodium pentobarbital and infused through the left ventricle with phosphate buffer as a vascular rinse followed by a fixing solution containing 4% p-formaldehyde in phosphate buffer. The brains were removed and fixed for 4 h in the same solution of p-formaldehyde at room temperature. Cryoprotection was afforded by incubating the brain in a solution containing 30% sucrose for 72 h. Brains were frozen and serials of 40 μm thick coronal sections were cut with a sliding microtome (Leica SM2000R) and mounted on glass slides. Identification of the infarcted tissue in the neocortex was performed by Nissl [0.2% (w/v) cresyl violet] staining of slide-mounted coronal sections. Adjacent sections were then processed for immunohistochemistry. Briefly, sections were preincubated with 0.3% Triton X-100 and with 10% normal goat serum in PBS for 2 h at room temperature. After this time, sections were incubated at 4°C overnight with rabbit polyclonal antibody against 5-LO (1:100; Cayman Chemical) and with mouse monoclonal antibody against neuronal nuclei (NeuN Millipore Bioscience Research Reagents, 1:100), glial fibrillary acidic protein (GFAP) (Millipore Bioscience Research Reagents International; 1:1000) or OX-42 (AbD Serotec; 1:100) as the primary antibodies. Detection with secondary antibodies was performed using anti-rabbit Alexa Fluor 488 and anti-mouse Cy3TM (1:200; Invitrogen). After washing, sections were counterstained for 15 min at room temperature with 2 mm TO-PRO 3 iodide (Invitrogen Labeling and Detection) before mounting. All the immunohistochemical sections were processed together. Controls performed in parallel without primary antibodies showed very low levels of nonspecific staining. Image acquisition was performed with a laser-scanning confocal imaging system (Leica TCS SP2, and Zeiss LSM710).
Quantitative RT-PCR.
Total RNA was extracted from peri-infarct areas of rat brains collected 5 h after MCAO (n = 6 for each group) using TRIzol reagent (Invitrogen). RNA quantity was determined spectrophotometrically and the purity was confirmed by the relative absorbance at 260 nm versus 280 nm. 1 μg of RNA was reverse-transcribed with iScript cDNA Synthesis kit (Bio-Rad). Quantitative real-time PCR was performed using a Bio-Rad iQ5 Thermocycler with triplicate samples. The mRNA expression was normalized to actin and hprt1 gene expression and reported as the fold difference relative to the sham group. For lipoxygenases, denaturation at 94°C for 3 min was followed by 35 cycles of 94°C for 30 s, 57°C for 45 s, and 72°C for 45 s. For inflammatory genes, denaturation at 95°C for 2 min was followed by 40 cycles of 94°C for 10 s; 60°C for 15 s; 72°C for 20 s. Melt curve analysis was included to assure a single PCR product was formed. Specific primers for rat genes were designed using Primer Express software (Applied Biosystems) and are as follows: 5-LO (forward 5′-GTGTCTGAGGTGTTCGGTA; reverse 5′-AGTGTTGATGGCAATGGT), 12-LO (forward 5′-GGGCCACTGCAGTTCGTGA; reverse 5′-CGGCCTCTGCGCTCATC), 15-LO (forward 5′-CCGGAGACTCCAAGTACGC; reverse 5′-CGTAGCAGC-TTCCCGAGAG), iNOS (forward 5′-AACCCAAGGTCTACGTTCAAG; reverse 5′-AAAGTGGTAGCCACATCCCG), TNFα (forward 5′-GACCCTCACACTCAGATCATCTTCT; reverse 5′-TGCTACGACGTGGGCTACG); MMP9 (forward 5′-GTGCCCTGGAACTCACACAAC; reverse 5′-CCAGAAGTATTTGTCATGGCAGAA); COX-2 (forward 5′-GCATTCTTTGCCCAGCACTTCACT; reverse 5′-TTTAA GTCCACTCCATGGCCCAGT); HPRT1 (forward 5′-CAAAGCCTAAAAGACAGCGG; reverse 5′-ATGGCCACAGGACTAGAACG); actin (forward 5′-TGAGCGCAAGTACTCTGTGTGGAT; reverse 5′-TAGAAGCATTTGCGGT-GCACGATG).
Nuclear extracts.
Brain tissue from rats (n = 6) was collected from peri-infarct areas 18 h after MCAO. Nuclear extracts were prepared crushing tissue in 10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, and 0.5% Nonidet P-40. Nuclei were pelleted at 12,000 g for 1 min at 4°C and lyzed in 20 mm HEPES, pH 7.9, 15 mm MgCl2, 420 mm NaCl and 0.2 mm EDTA. After centrifugation (12,000 g for 5 min at 4°C), the pellet was discarded.
PPARγ transcriptional activity assay.
Late PPARγ activity was assessed in nuclei obtained from brains 18 h after MCAO or sham procedure using PPARγ Transcription Factor Assay kit (Cayman Chemical): briefly, nuclear extracts were incubated in a PPRE probe coated multiwell plate, and PPAR bound to PPRE probe was detected using a specific antibody against the γ isoform. Horseradish peroxidase-labeled secondary antibody was used and detected by spectrophotometry.
An assay to test the direct PPARγ activity was performed incubating isolated nuclei with LXA4 or rosiglitazone and subsequent determination of PPARγ transcriptional activity using the commercial kit described above. Briefly, rat brain isolated nuclei (final concentration: 10 mg/ml protein) were incubated for 15 min at 37°C in buffer C (20 mm HEPES, 1 mm EDTA, 1 mm EGTA, 10 mm NaCl, 1 μm DTT, 0.5 μm NaF, 10 μm NaVO4, 10 μm NaMoO4, 40 ng/ml leupeptin, 40 ng/ml TLCK, 4 ng/ml aprotinin, 1.25 μm PMSF, pH 8; final volume 30 μl) in the absence or presence of LXA4 (1–10 nm) or rosiglitazone (1–10 nm).
Determination of leukotriene A4 and LXA4 levels.
Quantitative analysis of leukotriene A4 (LTB4) and LXA4 levels in cerebral cortex dissected from the MCA territory 18 h after the occlusion was performed by ELISA (EA 35 Leukotriene B4 and EA 45 Lipoxin A4 assay kits, Oxford Biomedical Research). Samples were previously purified using C18 octadecyl mini-columns (AmprepTM Mini-Columns, GE Healthcare Bio-Sciences), and AcO2Et-eluted LXA4 containing samples were dried out using N2 stream and redissolved in assay buffer.
Gene microarray analysis.
Five hours after MCAO, ipsilateral cortices from rats (n = 6) were used immediately for total RNA extraction with the TRIzol Reagent (Invitrogen). The RNA was then cleaned (RNeasy Mini Kit, Qiagen) and analyzed in a 2100 Bioanalyzer following the instructions of the manufacturer of the array (Affymetrix). Fragmentation of biotin-labeled complementary RNA (cRNA) was performed at 94°C using 40 mm Tris-acetate, pH 8.1; 100 mm KOAc, and 30 mm MgOAc. Samples were hybridized to Affymetrix GeneChip Test3 Array and then to Affymetrix GeneChip Rat expression probe array 230A. The data were analyzed with the GeneChip Analysis Suite software.
Statistical analysis.
Results are expressed as mean ± SEM of the indicated number of experiments; statistical analysis involved one-way ANOVA (or the Kruskal–Wallis test when the data were not normally distributed) followed by individual comparisons of means (Student-Newman–Keuls, or Dunn′s method when the data were not normally distributed). p < 0.05 was considered statistically significant.
Results
The PPARγ agonist rosiglitazone increases 5-LO expression in the ischemic rat brain
At the time studied, 5-LO protein (Fig. 1A) (18 h after MCAO) and mRNA (Fig. 1B) (5 h after MCAO) expression in rat brain homogenates was not significantly altered in rats after MCAO. Treatment with rosiglitazone (1 mg/kg, administered 10 min after the surgery) induced an increase in 5-LO mRNA and protein expression in the ischemic brain (Fig. 1A,B). No effect of rosiglitazone in 5-LO expression was detected in sham brain, possibly because of the inability of this compound to cross the unaltered blood–brain barrier. The effect of rosiglitazone on 5-LO protein expression was inhibited by prior administration of the PPARγ antagonist T0070907 (2 mg/kg) (Fig. 1A). These effects were in agreement with the results obtained by cDNA microarray analysis (Pereira et al., 2006), showing that 5-lipoxygenase and not other lipoxygenases (Table 1, Fig. 1B), is upregulated 5 h after rosiglitazone administration in MCAO-exposed rat brain.
Figure 1.

Rosiglitazone increases 5-LO expression in the ischemic brain. A, Expression of 5-LO protein in sham and MCAO-exposed rat brain: Effect of rosiglitazone (RSG). Bottom shows the densitometric analysis of bands. Data are mean ± SD, n = 6, *p < 0.05 versus MCAO, #p < 0.05 versus MCAO+RSG. B, Expression of 5-, 12-, and 15-LO mRNA in sham and ischemic rat brain, untreated or treated with rosiglitazone. Expression was determined by real-time quantitative PCR assays. Actin was used as a control for RNA loading and integrity. Data are mean ± SD, n = 6, *p < 0.05 versus MCAO.
Table 1.
Effect of rosiglitazone on lipoxygenase transcripts 5 h after MCAO
| Gene title | Gene symbol | MCAO fold-change versus control (change p value) | MCAO + rosiglitazone fold-change versus MCAO (change p value) |
|---|---|---|---|
| Arachidonate 5-lipoxygenase | Alox5 | 0.3 (0.506476) n.c. | 2.27 (0.000167) |
| Arachidonate 12-lipoxygenase | Alox12 | 0.6 (0.088938) n.c. | 0.35 (0.500000) n.c. |
| Similar to arachidonate lipoxygenase | 0.19 (0.124552) n.c. | 0.13 (0.545234) n.c. |
Positive and negative fold-change values indicate increase or decrease in expression, respectively. Change p values close to 0.0 indicate likelihood for an increase in transcript expression level; values near 0.5 indicate a weak likelihood for change in any direction, whereas values close to 1.0 indicate likelihood for a decrease in transcript expression level. n.c., No change. The values for arachidonate 5-lipoxygenase were reported previously (Pereira et al., 2006).
To analyze the type of cells responsible for the increased 5-LO expression after rosiglitazone treatment in ischemia, we used double immunofluorescence staining in equivalent sections of the infarct border of neocortex corresponding to III-V layers (Fig. 2), for the different experimental groups and for each cell type. Sham animals showed some 5-LO immunoreactivity in neurons with a rounded appearance in the ipsilateral cortex (Fig. 2A), which did not significantly change after rosiglitazone administration (Fig. 2B). When compared with brain tissue of MCAO-exposed animals, treatment with rosiglitazone increased 5-LO expression mainly in neurons located in the infarct border (NeuN-positive, Fig. 2C–D). In agreement with the neuroprotective effect of rosiglitazone, we observed a more extensive transition area between core and healthy tissue in the ipsilateral neocortex of rats treated with rosiglitazone than in the untreated ischemic group; in this area, neurons were found with a triangular morphology and showing an increased expression of 5-LO (Fig. 2D).
Figure 2.

A–H, Cellular localization of 5-LO in rat brain: Effect of rosiglitazone. Double immunofluorescence of 5-LO (green) in neurons (NeuN; red) located at the infarct border in brain sections of ischemic, untreated (MCAO) (C), and ischemic, rosiglitazone-treated (MCAO+RSG) (D) animals, and in the same areas in brain sections from sham-operated animals, untreated (Sham) (A) or treated with rosiglitazone (Sham+RSG) (B). Double immunofluorescence of 5-LO (green) and cellular markers (red) for astrocytes (GFAP) and microglia (OX-42) in infarct borders of brain sections of ischemic, untreated (MCAO) (E, G), and ischemic, rosiglitazone-treated (MCAO+RSG) rats (F, H). TO-PRO-3 was used to stain the cell nuclei (blue).
In cortical areas of infarct border with high density of astrocytes and microglia (GFAP and OX42-positive cells, respectively) (Fig. 2E–H), the expression of 5-LO was not detected in these glial cells, regardless of the treatment. Interestingly, in some ischemic areas such as the one showing reactive astrocytes, we observed a high expression of 5-LO after rosiglitazone treatment in cells which presented a rounded morphology and reduced size of cell bodies and processes (Fig. 2F). Staining of cellular markers of reactive astrocytes and microglia was not evident in sham animals.
Inhibition of 5-LO reverses rosiglitazone-induced neuroprotection and PPARγ activation after MCAO in rat brain
Administration of rosiglitazone 10 min after the ischemic onset decreased both MCAO-induced infarct volume (Fig. 3A,B) and neurological deficit (neurological scores: 2.83 ± 0.18 vs 1.66 ± 0.37 in MCAO and MCAO+RSG respectively, p < 0.05, n = 9) when measured 48 h after MCAO in rats. Reduction in infarct volume was still observed 7 d after MCAO (126 ± 5 vs 78 ± 3 mm3 in MCAO and MCAO+RSG, respectively, n = 6, p < 0.05). To explore the role of 5-LO in this setting, we used the selective 5-LO inhibitor BWA4C. Its administration (BWA4C; 1–5 mg/kg) 3 h after MCAO dose-dependently reversed rosiglitazone-induced decrease in both infarct volume (Fig. 3A,B) and neurological deficit (neurological scores: 1.66 ± 0.37 vs 2.00 ± 0.40 and 2.16 ± 0.33 in MCAO+RSG, MCAO+RSG+BWA4C 1 mg/kg and MCAO+RSG+BWA4C 5 mg/kg, respectively, p < 0.05 vs MCAO+RSG, n = 9), strongly suggesting that 5-LO is involved in rosiglitazone-induced neuroprotective effect. In the absence of rosiglitazone, the administration of the 5-LO inhibitor BWA4C did not affect (1 mg/kg) or even decreased (5 mg/kg) infarct volume caused by MCAO (Fig. 3A,B).
Figure 3.

Inhibition of 5-LO abolishes rosiglitazone-induced neuroprotection and PPARγ transcriptional activity in rat brain. A, B, The 5-LO inhibitor BWA4C (1–5 mg/kg) inhibits the neuroprotective effect of rosiglitazone (RSG) on infarct volume (A) and infarct areas (B) 48 h after permanent MCAO in rat brain. Data are mean ± SEM, n = 9, *p < 0.05 versus MCAO, #p < 0.05 versus MCAO + RSG. (See Materials and Methods for details.) Photographs of brain slices from representative experiments. C, The 5-LO inhibitor BWA4C (1–5 mg/kg) inhibits PPARγ transcriptional activity induced by rosiglitazone (RSG) after MCAO in rat brain. Data are mean ± SEM, n = 9, *p < 0.05 vs MCAO, #p < 0.05 versus MCAO + RSG.
Administration of rosiglitazone was associated with PPARγ activation as shown by an increase in its transcriptional activity in nuclear extracts obtained from brain cortex 18 h after the onset of the ischemic injury (Fig. 3C). The MCAO procedure per se did not affect PPARγ transcriptional activity when compared with control animals. Interestingly, at this time point, rosiglitazone-induced PPARγ activation was dose-dependently inhibited by the 5-LO inhibitor BWA4C (Fig. 3C), suggesting the implication of 5-LO products in the effects of rosiglitazone. Rosiglitazone failed to increase PPARγ transcriptional activity in sham rat brain, in agreement with its lack of effect on 5-LO expression, again indicating its inability to cross the blood–brain barrier in control, nonischemic animals.
Inhibition of 5-LO reverses rosiglitazone-induced downregulation of the expression of inflammatory genes after ischemia
Next, we analyzed the expression of proinflammatory markers in brain homogenates from peri-infarct tissue of MCAO-injured rats. Acute expression of inflammatory mediators, such as iNOS, COX-2, MMP9 and TNF-α participates in brain damage after stroke (for review, see del Zoppo et al., 2000). Administration of rosiglitazone inhibited the expression of COX-2, iNOS, and mature forms of TNF-α and MMP9 (Fig. 4A,B). More interesting, the 5-LO inhibitor BWA4C (5 mg/kg) reversed rosiglitazone-induced inhibition of the gene expression of COX-2, iNOS and TNF-α (Fig. 4A,B). For MMP9, reversal of expression was observed only at the mRNA level, possibly because of the times selected for protein expression studies (Fig. 4A).
Figure 4.

Inhibition of 5-LO abolishes rosiglitazone-induced anti-inflammatory effects after cerebral ischemia in rat brain. A, The PPARγ agonist rosiglitazone inhibited MCAO-induced mRNA expression of COX-2, iNOS, MMP9, and TNF-α, an effect that was reversed by the 5-LO inhibitor BWA4C (5 mg/kg). Expression was determined by real-time quantitative PCR. Actin and hprt1 were used as controls for RNA loading and integrity. Data are mean ± SD, n = 6, *p < 0.05 versus MCAO. B, The LO-inhibitor BWA4C partly reversed rosiglitazone-induced inhibition of the protein expression of COX-2, iNOS, and mature forms of TNF-α caused by ischemia. Data are mean ± SD, n = 6, *p < 0.05 versus MCAO, #p < 0.05 versus MCAO+RSG.
Deletion of 5-LO gene decreases rosiglitazone-induced neuroprotection and PPARγ activation after MCAO in mouse brain
To confirm the obligatory role of 5-LO in the effects of rosiglitazone, we performed loss of function experiments using 5-LO−/− mice and their wild-type littermates. Infarct outcome was not significantly affected in 5-LO−/−mice when compared with wild-type mice (Fig. 5A–D). However, the effect of rosiglitazone on infarct size and neurological outcome was markedly decreased or abolished, respectively, in 5-LO−/− animals (Fig. 5A–D).
Figure 5.

A–E, Role of 5-LO in the actions of rosiglitazone after MCAO in mice. Effect of rosiglitazone (RSG) on infarct volume and areas (A, B), neurological outcome (C, D) and PPARγ transcriptional activity (E) in wild-type and 5-LO−/− deficient mice. Data are mean ± SEM, n = 6, *p < 0.05 versus MCAO. Photographs of brain slices from representative experiments.
Administration of rosiglitazone 10 min after MCAO induced PPARγ activation in wild-type mouse brain as shown by an increase in its transcriptional activity measured in nuclear extracts obtained from brain cortex 18 h after the onset of the ischemic injury (Fig. 5E). However, this effect was absent in mouse lacking the 5-LO gene (Fig. 5E).
Levels of the 5-LO metabolites LTB4 and LXA4 in rat brain cortex
As different by-products of lipoxygenase activity, LTB4 and LXA4 levels were measured in rat brain cortical homogenates 18 h after ischemia, a time at which 5-LO is over-expressed in rosiglitazone-treated animals. Exposure to MCAO induced an increase in rat brain LTB4 levels, which was inhibited in animals treated with rosiglitazone or with the 5-LO inhibitor BWA4C (1–5 mg/kg) (Fig. 6A).
Figure 6.

Levels of the 5-LO metabolites leukotriene B4 (LTB4) and lipoxin A4 (LXA4) in rat brain: Effect of MCAO, rosiglitazone (RSG) and the 5-LO inhibitor BWA4C. Data are mean ± SEM, n = 6, *p < 0.05 versus control, #p < 0.05 vs MCAO, &p < 0.05 versus MCAO+RSG.
Whereas ischemic animals showed no difference in LXA4 levels when compared with control animals (Fig. 6B), treatment with rosiglitazone after MCAO induced the synthesis of LXA4, an effect prevented by the 5-LO inhibitor BWA4C (Fig. 6B).
Effect of LXA4 on infarct outcome after MCAO: effect of the PPARγ antagonist T0070907
To clarify the role of LXA4 in the actions of rosiglitazone, we studied the direct effects of LXA4 on MCAO outcome. Intracerebroventricular administration of LXA4 (1 nmol) caused a decrease in both infarct volume (Fig. 5A,B) and neurological deficit (scores: 2.68 ± 0.38 vs 2.00 ± 0.00 in MCAO and MCAO+LXA4, respectively, n = 6, p < 0.05) after MCAO. Interestingly, the PPARγ antagonist T0070907 (10 nmol) inhibited the neuroprotective effect of LXA4 on infarct volume (Fig. 7A,B) and neurological deficit (scores: 2.68 ± 0.40 in MCAO+LXA4+T0070907, n = 6, p < 0.05 vs MCAO+LXA4), suggesting that at least some of the actions of this lipid are PPARγ-dependent. T0070907 did not affect per se either infarct volume (Fig. 7A,B) or neurological deficit (score: 3.00 ± 0.00 in MCAO+T0070907; n = 6; p > 0.05 vs MCAO).
Figure 7.
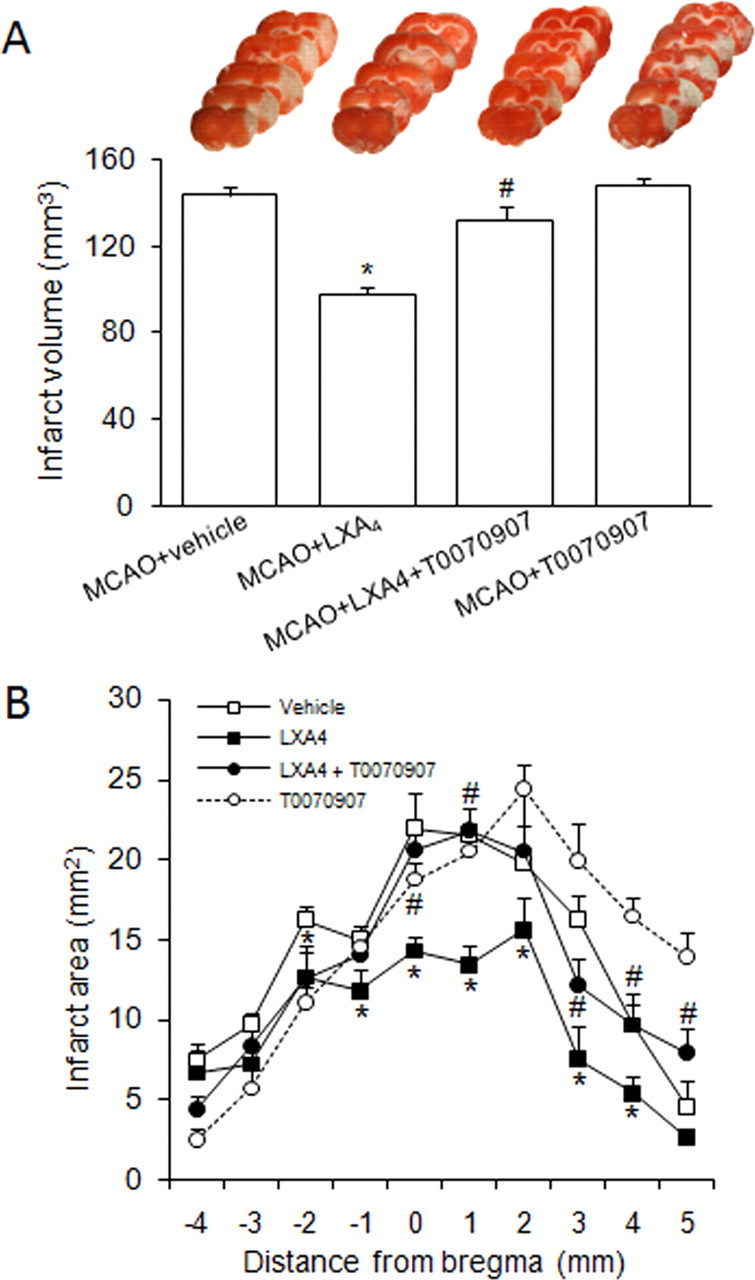
A, B, Neuroprotective effect of LXA4 in infarct volumes (A) and areas (B) after MCAO in rats. Effect of the PPARγ antagonist T070907. Data are mean ± SEM, n = 6, *p < 0.05 versus MCAO, #p < 0.05 versus MCAO + LXA4 (see Materials and Methods for details). Photographs of brain slices from representative experiments.
Effect of LXA4 on PPARγ transcriptional activity in isolated nuclei from rat brain cortex
To explore further the possible actions of LXA4 as direct PPARγ activator, isolated nuclei from rat cerebral cortex were incubated with LXA4 (1–10 nm) or rosiglitazone (1–10 nm) for 30 min. Both molecules directly induced an increase in PPARγ transcriptional activity (Fig. 8).
Figure 8.

Effect of rosiglitazone (1–10 nm) or LXA4 (1–10 nm) on PPARγ transcriptional activity in rat brain isolated nuclei. Data are mean ± SEM, n = 6, *p < 0.05 versus control (see Materials and Methods for details).
Discussion
We have explored the role of the enzyme 5-LO in the actions of the PPARγ agonist rosiglitazone in cerebral ischemia. Our results show that 1) rosiglitazone-induced 5-LO expression and subsequent lipoxin synthesis mediate most of the neuroprotective effects of rosiglitazone in experimental stroke as well as its late PPARγ agonistic effect and 2) that LXA4 is neuroprotective by acting, at least partly, as a PPARγ agonist.
Several groups have previously reported that PPARγ agonists are neuroprotective in brain ischemia (Pereira et al., 2005, 2006; Shimazu et al., 2005; Sundararajan et al., 2005; Zhao et al., 2005; Allahtavakoli et al., 2006; Chu et al., 2006; Collino et al., 2006; Lin et al., 2006; Luo et al., 2006; Ou et al., 2006; Victor et al., 2006; Y. Zhao et al., 2006; Tureyen et al., 2007), due mainly to anti-inflammatory and anti-oxidant mechanisms; anti-excitotoxic actions could also be involved, either at the presynaptic level (Romera et al., 2007) or postsynaptically (X. Zhao et al., 2006). Intriguingly, using cDNA microarray analysis, we found a robust expression of 5-LO mRNA after rosiglitazone administration, concomitant to neuroprotection (Pereira et al., 2006). Therefore, we have now sought to confirm this expression and to elucidate its role in rosiglitazone-induced neuroprotection in cerebral ischemic damage.
We have thus found that rosiglitazone increases the expression of both 5-LO mRNA and protein in rat brain after MCAO, at doses that induce neuroprotection, and that this effect is caused by PPARγ activation, because it is inhibited by the selective PPARγ antagonist T0070907, thus suggesting that 5-LO is a PPARγ target gene. Double immunostaining studies confirmed the increased expression of 5-LO after rosiglitazone administration at the infarct border of the ischemic hemisphere, and showed 5-LO immunoreactivity located in neurons, in agreement with previous studies in rat brain (Uz et al., 1998). The staining was not detected in astrocytes and microglia in this experimental setting. Interestingly, 5-LO upregulation was not evident in brains of sham rats treated with rosiglitazone. This might be attributable to the poor access of rosiglitazone through the intact blood–brain barrier described in healthy rodents (Pedersen et al., 2006), although this must not be the case when this barrier is disrupted, as in ischemia. To our knowledge, our study is the first evidence of the upregulation of 5-LO by a PPARγ agonist.
To ascertain the role of rosiglitazone-induced 5-LO upregulation, we tested the effect of a selective 5-LO inhibitor, BWA4C, on rosiglitazone-induced amelioration of infarct outcome in rats. First, this compound, depending on the dose, decreased or did not affect infarct volume on its own, suggesting a weak deleterious role of 5-LO in brain ischemia, in agreement with previous reports (Minamisawa et al., 1988; Mabe et al., 1990; Baskaya et al., 1996; Ciceri et al., 2001; Kitagawa et al., 2004; Jatana et al., 2006). Second, after administration of rosiglitazone, BWA4C inhibited the neuroprotective effect of this glitazone on infarct outcome. These findings strongly suggest that the upregulation of 5-LO induced by rosiglitazone is beneficial and mediates the neuroprotective effect of rosiglitazone after MCAO. Anti-inflammatory mechanisms have been demonstrated to mediate the neuroprotective effects of PPARγ agonists (Pereira et al., 2005, 2006; Sundararajan et al., 2005; Zhao et al., 2005; Collino et al., 2006; Luo et al., 2006; Zhao Y et al., 2006; Tureyen et al., 2007). Our present data confirm this mechanism of rosiglitazone, as shown by the inhibition of the expression of inflammatory genes with a deleterious role in cerebral ischemia, namely, COX-2, iNOS, MMP9 and TNF-α. Of note, the 5-LO inhibitor BWA4C reversed this effect of rosiglitazone, at both mRNA and protein levels, thus indicating that inhibition of inflammatory gene expression is a mechanism involved in the neuroprotective actions of 5-LO induction by rosiglitazone.
To demonstrate further this action of 5-LO after rosiglitazone administration, we performed loss of function studies using 5-LO knock-out mice. First, these experiments confirm in mice the neuroprotective effect of rosiglitazone found in rats. More important, in 5-LO knock-out mice, rosiglitazone induced a decrease in infarct volume of much lesser extent than in wild-type animals, and failed to improve neurological scores. These important results demonstrate the obligatory role of 5-LO in rosiglitazone-induced neuroprotection in experimental stroke.
We previously showed that brain PPARγ activation could be detected at early (2 h) and late times (18 h) after rosiglitazone administration (Pereira et al., 2006). Important additional mechanistical evidence which highlights the significance of 5-LO in rosiglitazone effects refers to the results showing that rosiglitazone-induced PPARγ activation found 18 h after drug administration is inhibited by the 5-LO inhibitor BWA4C, thus indicating that the late PPARγ activation by rosiglitazone depends on 5-LO activity. This suggests that the effects of rosiglitazone on PPARγ are partly indirect and mediated through 5-LO. Our finding showing that rosiglitazone fails to activate PPARγ in brains of 5-LO knock-out mice when assayed 18 h after the administration confirms this hypothesis. All these data indicate that 5-LO by-products are involved in rosiglitazone-induced activation of PPARγ.
The fact that 5-LO participates in the endogenous synthesis not only of proinflammatory mediators such as leukotrienes but also of anti-inflammatory compounds such as lipoxins (Serhan et al., 1984; Ueda et al., 1987; Serhan et al., 1987; Edenius et al., 1990; Ho and Wong, 1990; Serhan and Sheppard, 1990; Levy et al., 1993) (for review, see Nathan, 2002; Chiang et al., 2005; Serhan and Savill, 2005; Serhan, 2007) may explain the neuroprotective effect of this enzyme. Therefore, we investigated the endogenous brain levels of these mediators, specifically, the proinflammatory LTB4 and the anti-inflammatory lipoxin LXA4, in this setting. Whereas MCAO induced an increase in brain LTB4 and did not affect lipoxin synthesis, treatment with rosiglitazone after MCAO increased LXA4 levels and concomitantly blocked MCAO-induced LTB4 increase. Rosiglitazone-induced increase in LXA4 was prevented by coadministration of the 5-LO inhibitor BWA4C, confirming that 5-LO accounts for the synthesis of this proresolving mediator in this scenario. The switch from arachidonic acid-derived proinflammatory leukotrienes toward the production of proresolving lipoxins has been described as a major feature of resolution, the expected outcome of an inflammatory response that allows the tissues to return to homeostasis (rev. in Gilroy et al., 2004; Serhan, 2007). Therefore, our present results suggest that rosiglitazone acts by promoting resolution in the ischemic brain through endogenous 5-LO-dependent LXA4 synthesis. Interestingly, earlier reports showed that other drugs such as aspirin mediate some of their anti-inflammatory actions by the production of endogenous aspirin-triggered lipoxins, mainly 15-epi-LXA4 (Clària and Serhan, 1995). Now it remains to explore whether rosiglitazone may stimulate the production of other anti-inflammatory mediators, such as resolvins and protectins.
To our knowledge, there are no previous reports on the role of lipoxins in stroke. Interestingly, we found that intracerebroventricular administration of LXA4 is neuroprotective, as shown by a decrease in both infarct volume and neurological deficit after MCAO. In addition, this neuroprotective effect was partly inhibited by the PPARγ antagonist T0070907, suggesting for the first time that LXA4 is a PPARγ agonist. These results agree with our data showing that PPARγ activation found 18 h after rosiglitazone administration was abolished by 5-LO inhibition. To confirm the PPARγ agonistic properties of LXA4, we assayed PPARγ activity after adding LXA4 directly to isolated rat brain nuclei, to exclude effects from other signaling mechanisms. Our results show that LXA4 directly activates PPARγ, thus indicating that its synthesis from 5-LO accounts for, at least part, of the PPARγ-dependent, anti-inflammatory and neuroprotective effects of rosiglitazone. Several mechanisms have been reported to explain the anti-inflammatory actions of LXA4, such as its binding to a 7TM receptor termed LXA4 receptor (ALXR, activated in the nm range) (Fiore et al., 1994) (for review, see Chiang et al., 2006) as well as to the aryl hydrocarbon receptor (AhR) (which requires μm range for activation) (Schaldach et al., 1999) (for review, see Migeotte et al., 2006). On binding, these receptors have been described to initiate several signaling pathways which include inhibition of NF-κB and AP-1 (József et al., 2002), increased expression of suppressor of cytokine signaling (SOCS)-2 (Machado et al., 2006), inhibition of ERK and JNK (Svensson et al., 2007) and induction of proteasomal degradation of TRAF6 (Machado et al., 2008). Now our work supports the view that, additionally, LXA4 exerts anti-inflammatory effects by activating PPARγ receptors. To our knowledge, this is the first evidence of the PPARγ agonistic action of LXA4.
The biosynthesis of lipoxins has been described to involve several possible routes which often include more than one lipoxygenase (Serhan and Sheppard, 1990; Levy et al., 1993) (for review, see Serhan, 1997). Although we cannot discard the implication of other lipoxygenases in lipoxin synthesis in our setting, our data using a cDNA microarray analysis and quantitative RT-PCR indicate that 5-LO is the only lipoxygenase affected by rosiglitazone at the expressional level in our model.
In summary, our data show for the first time the obligatory role of 5-LO in rosiglitazone-induced PPARγ activation and neuroprotection, through the biosynthesis of the anti-inflammatory mediator LXA4. Our findings also show that LXA4 acts as a PPARγ activator (Fig. 9). Because the safety of thiazolidinediones is uncertain, our study highlights the importance of the development of novel, lipoxin-like anti-inflammatory drugs, and opens lines of study such as the elucidation of the role of lipoxins as prognostic and/or diagnostic markers of pathologies with inflammatory substrate, such as stroke.
Figure 9.

Schematic diagram of the role of 5-LO in the neuroprotective effect of rosiglitazone in experimental stroke.
Footnotes
This work was supported by Spanish Ministry of Education and Science Grants SAF2006-01753 (M.A.M.) and SAF2005-05960 (I.L.) and Spanish Ministry of Health Grant RENEVAS RD06/0026/0005 (I.L.). M.S. was funded by Local Madrid Government (MULTIMAG S-BIO-0170/2006). I.B. and D.F.L. are fellows of the Spanish Ministry of Education and Science, and J.M.P. is a postdoctoral contracted researcher funded by RENEVAS.
References
- Allahtavakoli M, Shabanzadeh AP, Sadr SS, Parviz M, Djahanguiri B. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma ligand, reduces infarction volume and neurological deficits in an embolic model of stroke. Clin Exp Pharmacol Physiol. 2006;33:1052–1058. doi: 10.1111/j.1440-1681.2006.04486.x. [DOI] [PubMed] [Google Scholar]
- Baskaya MK, Hu Y, Donaldson D, Maley M, Rao AM, Prasad MR, Dempsey RJ. Protective effect of the 5-lipoxygenase inhibitor AA-861 on cerebral edema after transient ischemia. J Neurosurg. 1996;85:112–116. doi: 10.3171/jns.1996.85.1.0112. [DOI] [PubMed] [Google Scholar]
- Berger J, Moller DE. The mechanism of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- Cárdenas A, Moro MA, Leza JC, O'Shea E, Dávalos A, Castillo J, Lorenzo P, Lizasoain I. Upregulation of TACE/ADAM17 after ischemic preconditioning is involved in brain tolerance. J Cereb Blood Flow Metab. 2002;22:1297–1302. doi: 10.1097/01.WCB.0000033968.83623.D0. [DOI] [PubMed] [Google Scholar]
- Caso JR, Lizasoain I, Lorenzo P, Moro MA, Leza JC. The role of tumor necrosis factor-alpha in stress-induced worsening of cerebral ischemia in rats. Neuroscience. 2006;142:59–69. doi: 10.1016/j.neuroscience.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- Chiang N, Arita M, Serhan CN. Anti-inflammatory circuitry: lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins Leukot Essent Fatty Acids. 2005;73:163–177. doi: 10.1016/j.plefa.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Chiang N, Serhan CN, Dahlén SE, Drazen JM, Hay DW, Rovati GE, Shimizu T, Yokomizo T, Brink C. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev. 2006;58:463–487. doi: 10.1124/pr.58.3.4. [DOI] [PubMed] [Google Scholar]
- Chu K, Lee ST, Koo JS, Jung KH, Kim EH, Sinn DI, Kim JM, Ko SY, Kim SJ, Song EC, Kim M, Roh JK. Peroxisome proliferator-activated receptor-gamma-agonist, rosiglitazone, promotes angiogenesis after focal cerebral ischemia. Brain Res. 2006;1093:208–218. doi: 10.1016/j.brainres.2006.03.114. [DOI] [PubMed] [Google Scholar]
- Ciceri P, Rabuffetti M, Monopoli A, Nicosia S. Production of leukotrienes in a model of focal cerebral ischaemia in the rat. Br J Pharmacol. 2001;133:1323–1329. doi: 10.1038/sj.bjp.0704189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clària J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell–leukocyte interactions. Proc Natl Acad Sci U S A. 1995;92:9475–9479. doi: 10.1073/pnas.92.21.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RB. The role of PPARs in inflammation and immunity. J Leukoc Biol. 2002;71:388–400. [PubMed] [Google Scholar]
- Clark WM, Lessov NS, Dixon MP, Eckenstein F. Monofilament intraluminal middle cerebral artery occlusion in the mouse. Neurol Res. 1997;19:641–648. doi: 10.1080/01616412.1997.11740874. [DOI] [PubMed] [Google Scholar]
- Collino M, Aragno M, Mastrocola R, Gallicchio M, Rosa AC, Dianzani C, Danni O, Thiemermann C, Fantozzi R. Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur J Pharmacol. 2006;530:70–80. doi: 10.1016/j.ejphar.2005.11.049. [DOI] [PubMed] [Google Scholar]
- Darley-Usmar VM, Hersey A, Garland LG. A method for the comparative assessment of antioxidants as peroxyl radical scavengers. Biochem Pharmacol. 1989;38:1465–1469. doi: 10.1016/0006-2952(89)90186-x. [DOI] [PubMed] [Google Scholar]
- De Cristóbal J, Moro MA, Dávalos A, Castillo J, Leza JC, Camarero J, Colado MI, Lorenzo P, Lizasoain I. Neuroprotective Effect of Aspirin by Inhibition of Glutamate Release After Permanent Focal Cerebral Ischaemia in Rats. J Neurochem. 2001;79:456–459. doi: 10.1046/j.1471-4159.2001.00600.x. [DOI] [PubMed] [Google Scholar]
- Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol. 2001;169:453–459. doi: 10.1677/joe.0.1690453. [DOI] [PubMed] [Google Scholar]
- del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10:95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenius C, Kumlin M, Björk T, Anggård A, Lindgren JA. Lipoxin formation in human nasal polyps and bronchial tissue. FEBS Lett. 1990;272:25–28. doi: 10.1016/0014-5793(90)80440-t. [DOI] [PubMed] [Google Scholar]
- Fiore S, Maddox JF, Perez HD, Serhan CN. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J Exp Med. 1994;180:253–260. doi: 10.1084/jem.180.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov. 2004;3:401–416. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- Ho HY, Wong PY. Transformation of 15-hydroperoxide of eicosapentaenoic acid to lipoxins and trihydroxyeicosatetraenoic acids by 5-lipoxygenase partially purified from potato tubers. Eicosanoids. 1990;3:99–104. [PubMed] [Google Scholar]
- Hunter AJ, Hatcher J, Virley D, Nelson P, Irving E, Hadingham SJ, Parsons AA. Functional assessments in mice and rats after focal stroke. Neuropharmacology. 2000;39:806–816. doi: 10.1016/s0028-3908(99)00262-2. [DOI] [PubMed] [Google Scholar]
- Jatana M, Giri S, Ansari MA, Elango C, Singh AK, Singh I, Khan M. Inhibition of NF-kappaB activation by 5-lipoxygenase inhibitors protects brain against injury in a rat model of focal cerebral ischemia. J Neuroinflammation. 2006;3:12. doi: 10.1186/1742-2094-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- József L, Zouki C, Petasis NA, Serhan CN, Filep JG. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit peroxynitrite formation, NF-kappa B and AP-1 activation, and IL-8 gene expression in human leukocytes. Proc Natl Acad Sci U S A. 2002;99:13266–13271. doi: 10.1073/pnas.202296999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Hori M. Cerebral ischemia in 5-lipoxygenase knockout mice. Brain Res. 2004;1004:198–202. doi: 10.1016/j.brainres.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Lawrence T, Willoughby DA, Gilroy DW. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat Rev Immunol. 2002;2:787–795. doi: 10.1038/nri915. [DOI] [PubMed] [Google Scholar]
- Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, Nakamura M, Miao S, Cao P, Learned RM, Chen JL, Li Y. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J Biol Chem. 2002;277:19649–19657. doi: 10.1074/jbc.M200743200. [DOI] [PubMed] [Google Scholar]
- Levy BD, Romano M, Chapman HA, Reilly JJ, Drazen J, Serhan CN. Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. J Clin Invest. 1993;92:1572–1579. doi: 10.1172/JCI116738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TN, Cheung WM, Wu JS, Chen JJ, Lin H, Chen JJ, Liou JY, Shyue SK, Wu KK. 15d-prostaglandin J2 protects brain from ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 2006;26:481–487. doi: 10.1161/01.ATV.0000201933.53964.5b. [DOI] [PubMed] [Google Scholar]
- Luo Y, Yin W, Signore AP, Zhang F, Hong Z, Wang S, Graham SH, Chen J. Neuroprotection against focal ischemic brain injury by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. J Neurochem. 2006;97:435–448. doi: 10.1111/j.1471-4159.2006.03758.x. [DOI] [PubMed] [Google Scholar]
- Mabe H, Nagai H, Suzuka T. Role of brain tissue leukotriene in brain oedema following cerebral ischaemia: effect of a 5-lipoxygenase inhibitor, AA-861. Neurol Res. 1990;12:165–168. doi: 10.1080/01616412.1990.11739937. [DOI] [PubMed] [Google Scholar]
- Machado FS, Johndrow JE, Esper L, Dias A, Bafica A, Serhan CN, Aliberti J. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat Med. 2006;12:330–334. doi: 10.1038/nm1355. [DOI] [PubMed] [Google Scholar]
- Machado FS, Esper L, Dias A, Madan R, Gu Y, Hildeman D, Serhan CN, Karp CL, Aliberti J. Native and aspirin-triggered lipoxins control innate immunity by inducing proteasomal degradation of TRAF6. J Exp Med. 2008;205:1077–1086. doi: 10.1084/jem.20072416. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006;17:501–519. doi: 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Minamisawa H, Terashi A, Katayama Y, Kanda Y, Shimizu J, Shiratori T, Inamura K, Kaseki H, Yoshino Y. Brain eicosanoid levels in spontaneously hypertensive rats after ischemia with reperfusion: leukotriene C4 as a possible cause of cerebral edema. Stroke. 1988;19:372–377. doi: 10.1161/01.str.19.3.372. [DOI] [PubMed] [Google Scholar]
- Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- Ou Z, Zhao X, Labiche LA, Strong R, Grotta JC, Herrmann O, Aronowski J. Neuronal expression of peroxisome proliferator-activated receptor-gamma (PPARgamma) and 15d-prostaglandin J2–mediated protection of brain after experimental cerebral ischemia in rat. Brain Res. 2006;1096:196–203. doi: 10.1016/j.brainres.2006.04.062. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol. 2006;199:265–273. doi: 10.1016/j.expneurol.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Pereira MP, Hurtado O, Cárdenas A, Alonso-Escolano D, Boscá L, Vivancos J, Nombela F, Leza JC, Lorenzo P, Lizasoain I, Moro MA. The nonthiazolidinedione PPARgamma agonist L-796,449 is neuroprotective in experimental stroke. J Neuropathol Exp Neurol. 2005;64:797–805. doi: 10.1097/01.jnen.0000178852.83680.3c. [DOI] [PubMed] [Google Scholar]
- Pereira MP, Hurtado O, Cárdenas A, Boscá L, Castillo J, Dávalos A, Vivancos J, Serena J, Lorenzo P, Lizasoain I, Moro MA. Rosiglitazone and 15-deoxy-delta(12,14)-prostaglandin J(2) cause potent neuroprotection after experimental stroke through noncompletely overlapping mechanisms. J Cereb Blood Flow Metab. 2006;26:218–229. doi: 10.1038/sj.jcbfm.9600182. [DOI] [PubMed] [Google Scholar]
- Pradillo JM, Hurtado O, Romera C, Cárdenas A, Fernández-Tomé P, Alonso-Escolano D, Lorenzo P, Moro MA, Lizasoain I. TNFR1 mediates increased neuronal membrane EAAT3 expression after in vivo cerebral ischemic preconditioning. Neuroscience. 2006;138:1171–1178. doi: 10.1016/j.neuroscience.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Rådmark O, Samuelsson B. Regulation of 5-lipoxygenase enzyme activity. Biochem Biophys Res Commun. 2005;338:102–110. doi: 10.1016/j.bbrc.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Romera C, Hurtado O, Mallolas J, Pereira MP, Morales JR, Romera A, Serena J, Vivancos J, Nombela F, Lorenzo P, Lizasoain I, Moro MA. Ischaemic preconditioning reveals that GLT1/EAAT2 glutamate transporter is a novel PPARγ target gene involved in neuroprotection. J Cereb Blood Flow Metab. 2007;27:1327–1338. doi: 10.1038/sj.jcbfm.9600438. [DOI] [PubMed] [Google Scholar]
- Schaldach CM, Riby J, Bjeldanes LF. Lipoxin A4: a new class of ligand for the Ah receptor. Biochemistry. 1999;38:7594–7600. doi: 10.1021/bi982861e. [DOI] [PubMed] [Google Scholar]
- Serhan CN. Lipoxins and novel aspirin-triggered 15-epi-lipoxins (ATL): a jungle of cell-cell interactions or a therapeutic opportunity? Prostaglandins. 1997;53:107–137. doi: 10.1016/s0090-6980(97)00001-4. [DOI] [PubMed] [Google Scholar]
- Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Sheppard KA. Lipoxin formation during human neutrophil-platelet interactions. Evidence for the transformation of leukotriene A4 by platelet 12-lipoxygenase in vitro. J Clin Invest. 1990;85:772–780. doi: 10.1172/JCI114503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci U S A. 1984;81:5335–5339. doi: 10.1073/pnas.81.17.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Hirsch U, Palmblad J, Samuelsson B. Formation of lipoxin A by granulocytes from eosinophilic donors. FEBS Lett. 1987;217:242–246. doi: 10.1016/0014-5793(87)80671-3. [DOI] [PubMed] [Google Scholar]
- Shimazu T, Inoue I, Araki N, Asano Y, Sawada M, Furuya D, Nagoya H, Greenberg JH. A peroxisome proliferator-activated receptor-gamma agonist reduces infarct size in transient but not in permanent ischemia. Stroke. 2005;36:353–359. doi: 10.1161/01.STR.0000152271.21943.a2. [DOI] [PubMed] [Google Scholar]
- Sinz EH, Kochanek PM, Dixon CE, Clark RS, Carcillo JA, Schiding JK, Chen M, Wisniewski SR, Carlos TM, Williams D, DeKosky ST, Watkins SC, Marion DW, Billiar TR. Inducible nitric oxide synthase is an endogenous neuroprotectant after traumatic brain injury in rats and mice. J Clin Invest. 1999;104:647–656. doi: 10.1172/JCI6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev. 2001;21:185–210. doi: 10.1002/med.1006. [DOI] [PubMed] [Google Scholar]
- Sundararajan S, Gamboa JL, Victor NA, Wanderi EW, Lust WD, Landreth GE. Peroxisome proliferator-activated receptor-γ ligands reduce inflammation and infarction size in transient focal ischemia. Neuroscience. 2005;130:685–696. doi: 10.1016/j.neuroscience.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Zattoni M, Serhan CN. Lipoxins and aspirin-triggered lipoxin inhibit inflammatory pain processing. J Exp Med. 2007;204:245–252. doi: 10.1084/jem.20061826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tureyen K, Kapadia R, Bowen KK, Satriotomo I, Liang J, Feinstein DL, Vemuganti R. Peroxisome proliferator-activated receptor-gamma agonists induce neuroprotection following transient focal ischemia in normotensive, normoglycemic as well as hypertensive and type-2 diabetic rodents. J Neurochem. 2007;101:41–56. doi: 10.1111/j.1471-4159.2006.04376.x. [DOI] [PubMed] [Google Scholar]
- Ueda N, Yamamoto S, Fitzsimmons BJ, Rokach J. Lipoxin synthesis by arachidonate 5-lipoxygenase purified from porcine leukocytes. Biochem Biophys Res Commun. 1987;144:996–1002. doi: 10.1016/s0006-291x(87)80062-1. [DOI] [PubMed] [Google Scholar]
- Uz T, Pesold C, Longone P, Manev H. Aging-associated up-regulation of neuronal 5-lipoxygenase expression: putative role in neuronal vulnerability. FASEB J. 1998;12:439–449. doi: 10.1096/fasebj.12.6.439. [DOI] [PubMed] [Google Scholar]
- Victor NA, Wanderi EW, Gamboa J, Zhao X, Aronowski J, Deininger K, Lust WD, Landreth GE, Sundararajan S. Altered PPARgamma expression and activation after transient focal ischemia in rats. Eur J Neurosci. 2006;24:1653–1663. doi: 10.1111/j.1460-9568.2006.05037.x. [DOI] [PubMed] [Google Scholar]
- Willson TM, Lambert MH, Kliewer SA. Peroxisome proliferator-activated receptor gamma and metabolic disease. Annu Rev Biochem. 2001;70:341–367. doi: 10.1146/annurev.biochem.70.1.341. [DOI] [PubMed] [Google Scholar]
- Yki-Järvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- Zhao X, Ou Z, Grotta JC, Waxham N, Aronowski J. Peroxisome-proliferator-activated receptor-gamma (PPARgamma) activation protects neurons from NMDA excitotoxicity. Brain Res. 2006;1073–1074:460–469. doi: 10.1016/j.brainres.2005.12.061. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Patzer A, Gohlke P, Herdegen T, Culman J. The intracerebral application of the PPARgamma-ligand pioglitazone confers neuroprotection against focal ischaemia in the rat brain. Eur J Neurosci. 2005;22:278–282. doi: 10.1111/j.1460-9568.2005.04200.x. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Patzer A, Herdegen T, Gohlke P, Culman J. Activation of cerebral peroxisome proliferator-activated receptors gamma promotes neuroprotection by attenuation of neuronal cyclooxygenase-2 overexpression after focal cerebral ischemia in rats. FASEB J. 2006;20:1162–1175. doi: 10.1096/fj.05-5007com. [DOI] [PubMed] [Google Scholar]