

Abstract
Pain is a complex experience involving extensive interactions between brain and spinal cord processes. Various interventions that modulate pain, such as the application of a competing noxious stimulus (counterirritation), are thought to involve cerebrospinal regulation through diffuse noxious inhibitory controls (DNICs). However, no study has yet examined the relation between brain and spinal cord activity during counterirritation analgesia in humans. This fMRI study investigates brain responses to phasic painful electrical stimulation administered to the sural nerve to evoke a spinal nociceptive response (RIII reflex) before, during and after counterirritation induced by the immersion of the left contralateral foot in cold water. Responses are compared with a control condition without counterirritation. As expected, counterirritation produced robust pain inhibition with residual analgesia persisting during the recovery period. In contrast, RIII reflex amplitude was significantly decreased by counterirritation only in a subset of subjects. Modulatory effects of counterirritation on pain perception and spinal nociception were paralleled by decreased shock-evoked activity in pain-related areas. Individual changes in shock-evoked brain activity were specifically related to analgesia in primary somatosensory cortex (SI), anterior cingulate cortex and amygdala, and to RIII modulation in supplementary motor area and orbitofrontal cortex (OFC). Moreover, sustained activation induced by the counterirritation stimulus in the OFC predicted shock-pain decrease while sustained activity in SI and the periaqueductal gray matter predicted RIII modulation. These results provide evidence for the implication of at least two partly separable neural mechanisms underlying the effects of counterirritation on pain and spinal nociception in humans.
Introduction
Pain is a complex experience involving extensive interactions between brain and spinal cord processes. Four decades ago, cerebrospinal interactions were already at the core of the leading pain theory (Melzack and Casey, 1968). Although our understanding of pain and pain modulation mechanisms has greatly advanced since then, no study has yet shown direct links between specific brain systems and spinal cord activity during interventions that modulate pain in healthy humans.
Counterirritation by the application of two competing noxious stimuli has been known for centuries as a means to induce analgesia. This effect may reflect the brain release of endogenous opioids in pain-related areas during sustained pain (Zubieta et al., 2001). Besides, human physiological studies have also shown that spinal nociceptive processes indexed by the RIII reflex, a multisynaptic flexion response, are also inhibited during and immediately following counterirritation (Willer et al., 1999). This effect is thought to rely on diffuse noxious inhibitory controls (DNIC), a spinobulbospinal loop including the caudal medulla (De Broucker et al., 1990). However, some studies found no correlation between RIII and pain modulation as counterirritation analgesia may occur without RIII reflex inhibition (Willer et al., 1979; Terkelsen et al., 2001; Bouhassira et al., 2003). This emphasizes that changes in pain and RIII reflex amplitude may be dissociated and are most likely regulated by partly distinct mechanisms.
In addition to the caudal medulla, several brainstem structures such as the periaqueductal gray matter (PAG) and the rostral ventromedial medulla (RVM) are involved in the descending modulation of nociceptive activity (Hosobuchi, 1980; Mayer, 1984; Millan, 2002). The recruitment of the PAG by different cognitive interventions (Petrovic et al., 2002; Tracey et al., 2002; Valet et al., 2004) may reflect the activation of similar pathways through top-down processes involving the anterior cingulate cortex (ACC) and the orbitofrontal cortex (OFC) (Derbyshire et al., 1998; Rainville et al., 1999; Faymonville et al., 2000; Petrovic et al., 2000; Bantick et al., 2002; Petrovic and Ingvar, 2002; Valet et al., 2004; Bingel et al., 2006). Furthermore, direct corticospinal projections from the primary somatosensory cortex (SI) (Ralston and Ralston, 1985) can inhibit nociceptive spinothalamic cells (Yezierski et al., 1983). Therefore, several brain structures potentially contribute to the effects of counterirritation on pain and RIII reflex responses.
The aim of the present study was to elucidate cerebral and cerebrospinal mechanisms of pain and RIII reflex modulation by counterirritation with combined fMRI and electrophysiological methods. Phasic painful electrical stimulation evoking a RIII reflex were administered before, during, and after counterirritation induced by the immersion of the left contralateral foot in cold water. We hypothesized that shock pain, RIII reflex amplitude, and shock-evoked brain activity would decrease as a result of the activation of cerebral and cerebrospinal inhibitory processes induced by the counterirritation stimulus. Furthermore, we investigated whether these modulatory effects reflected the sustained activation of brain structures previously involved in descending modulation of pain and RIII reflex [SI, ACC, anterior insula (aINS), OFC, amygdala (AMY), PAG, the pons, the RVM, and the caudal medulla].
Materials and Methods
Subjects.
Twelve healthy volunteers participated in the study (2 males and 10 females; 10 right handed and 2 left handed; mean age 26.7 years; SD, 4.7). The Research Ethics Board of the “Centre de recherche de l'Institut de gériatrie de Montréal” approved the study. All subjects gave written informed consent and received a compensation of C$50 for their time and commitment to the study.
Painful electrical stimulation.
Transcutaneous electrical stimulation was delivered with a Grass S48 square pulse stimulator (Astro-Med) through a constant-current stimulus-isolation unit and a radio-frequency (RF) filter, preventing artifacts in fMRI data. The stimulation consisted in a 30 ms train of 10 × 1 ms pulse, delivered on degreased skin over the retromalleolar path of the right sural nerve using a pair of surface electrodes (1 cm2, interelectrode distance 2 cm). The individual RIII reflex threshold was determined using the staircase method (Willer, 1977), immediately after positioning the subject in the scanner and before the acquisition of functional images. For the counterirritation and control scans (see below), the stimulation intensity was adjusted to 120% of the threshold (mean ± SD: 13.3 ± 4.6 mA), and remained constant through the duration of the experiment.
Experimental paradigm.
Before the scanning day, all participants were familiarized with the painful stimulations, the pain rating scale, the RIII-threshold assessment, and the counterirritation procedure in our laboratory. On the experimental day, the procedure always started with the threshold determination using the staircase method, involving several series of ascending–descending intensities administered until a stable threshold was obtained outside of the scanner (typically ∼50 stimuli). The stability of the threshold was checked again, with the subject inside the scanner, immediately before the experiment. Then, three control scans were performed with painful electrical stimulation at subthreshold and suprathreshold stimulus intensities without counterirritation, to confirm the reliability of RIII measurements obtained concurrently with the fMRI acquisition (note that only the control scan performed with electric shocks administered at 120% of the RIII threshold is reported here). The counterirritation scan was always performed last to prevent carry-over effects.
The counterirritation scan included 35 electrical stimuli administered with an interstimulus interval of 12 s (Fig. 1). The first five stimuli at the beginning of the scan (1 min before baseline) controlled for the rapid habituation effect observed occasionally on the first few trials of a series of RIII measurements. The subsequent 30 stimuli were distributed equally in three sequential conditions: painful shocks (n = 10) at baseline, painful shocks (n = 10) + foot immersion in cold water (counterirritation) and painful shocks (n = 10) after removing the foot from cold water (recovery). Counterirritation was produced by placing the subject's left contralateral foot in a MR-compatible container filled with ice and cold water for 2 min (temperature adjusted to +4°C immediately before the beginning of the scan). The foot was carefully positioned just beside the water container before the beginning of the scan. During the scan, counterirritation was visually cued to the experimenter who carefully placed the foot in cold water up to the medial and lateral malleoli and removed it after 2 min. The subjects were instructed that the foot would be moved passively into the water by the experimenter and that they should not move the rest of their body or help the movement of the leg. Subjects were covered with one or two blankets as needed to remain comfortable during the entire experiment.
Figure 1.

Counterirritation paradigm. A series of 35 electric shocks was delivered on the skin overlying the right sural nerve distributed in four conditions: (1) prebaseline, (2) baseline, (3) counterirritation, and (4) recovery. Counterirritation was applied for 2 min during the series of electric shocks by immersing the left foot in cold water (4°C). Shock pain was rated for each experimental block just after the last shock of the block. Cold-pain rating was collected at the end of the scan, just after shock-pain rating of the recovery block.
In the control scan, a similar procedure was used but no counterirritation was applied. The scan included 40 electrical stimuli administered with an interstimulus interval of 6–15 s. The first five stimuli at the beginning of the scan (1 min before baseline) controlled for the rapid habituation effect observed occasionally on the first few trials of a series of RIII measurements. The subsequent 30 stimuli were attributed to three sequential conditions to mimic the counterirritation procedure: block 1 (n = 10 painful shocks), block 2 (n = 10 painful shocks), and block 3 (n = 10 painful shocks). The last five shocks at the end of the control series were not analyzed to ensure a fair comparison with the counterirritation run which included only 35 stimuli (see fMRI data analysis below). This scan served to control for potential nonspecific temporal effects (including habituation). The data from this control scan have been used in a parallel study (M. Piché, M. Arsenault, and P. Rainville, unpublished observations).
Pain ratings.
A visual analog scale (VAS) was used to evaluate the pain induced by the electrical stimulation and the counterirritation stimulus. Participants were shown the VAS on a computer monitor backprojected onto a screen and viewed on a mirror placed on the head coil in front of the participant's eyes. The VAS was placed horizontally and included the verbal anchors “no pain” and “worst pain imaginable” at the left and right extremities, respectively (Price et al., 1994). Participants used an MRI-compatible response key to move a cursor on the VAS. They were prompted to rate shock pain immediately after each block of stimuli (condition) and were also asked to rate counterirritation pain just after shock-pain rating of the recovery condition (Fig. 1). Ratings were prompted by a phrase displayed for 1 s on the screen immediately before the pain scale appeared for 10 s. Shock-pain ratings were prompted by “Rating of shock pain” and cold pain was prompted by “Rating of cold pain on the foot.” All ratings were converted linearly to a 0–100 scale. Pain modulation by counterirritation was tested using a two-tail repeated-measures ANOVA. Analgesia during counterirritation and recovery was also calculated for each subject by subtracting pain ratings from that obtained during baseline. These values were ranked (from 1 to 11, 11 being the strongest analgesia) and served as a subject regressor coding for individual differences in analgesia in fMRI analyses (note that one subject was excluded due to excessive movement; see below). Ranks were used as a conservative measure to avoid biasing statistical estimates (e.g., correlation) due to outliers (also see RIII reflex recording and analyses). This is particularly important in small samples.
RIII reflex recording and analyses.
Electromyographic (EMG) activity of the right (ipsilateral) biceps femoris was recorded with MRI-compatible Ag–AgCl surface electrodes (Type EL-508) using Biopac MP150 system (Biopac Systems). Custom-made RF filters were used for the recording of physiological measures to prevent introducing artifact in the fMRI data. Electromyographic (EMG) activity was amplified, bandpass filtered (100–500 Hz), digitized, and sampled at 1000 Hz. EMG data were analyzed using Acqknowledge 3.8 (Biopac Systems). The raw EMG recordings were filtered off-line (120–130 Hz) and transformed using the root mean square. The resulting signal was integrated between 90 and 180 ms after the stimulus onset to quantify RIII reflex amplitude to each shock. These values were averaged for each condition (10 stimulations each) to assess RIII reflex changes during counterirritation and control scans with two-tail Friedman's ANOVAs for dependant samples. Repeated-measures ANOVAs were also performed at the subject level for counterirritation and control scans to document the significance of changes in RIII amplitude across conditions/blocks for each participant. Each trial entered the analysis as a separate observation to assess the significance of changes observed between successive blocks. For the counterirritation scan, the values were also normalized within each subject using a z-transformation and averaged for each condition to calculate an index of RIII modulation for each subject. This was done by subtracting the normalized RIII amplitude during counterirritation and recovery from that obtained during baseline. These values were ranked (from 1 to 11, 11 being the strongest RIII inhibition) and served as a subject regressor coding for individual differences in RIII-modulation in fMRI analyses (see below).
fMRI acquisition.
Imaging data were acquired at “Unité de Neuroimagerie Fonctionnelle” of the “Centre de recherche de l'Institut de gériatrie de Montréal” on a 3 T Siemens Trio scanner using a CP head coil. The head of the subject was stabilized in a comfortable position using a vacuum bag. Subjects were instructed to refrain from moving as much as possible throughout the imaging session and were given earplugs to reduce the noise from the scanner. A pelvic belt stabilizing the subject on the table of the scanner was used to limit the potential movement induced by the RIII reflex or the counterirritation procedure. Each participant underwent one high-resolution anatomical scan in addition to functional scans. The anatomical scan was a T1-weighted high-resolution scan [repetition time (TR): 13 ms; echo time (TE): 4.92 ms; flip angle: 25°; field of view: 256 mm; voxel size: 1 × 1 × 1.1 mm]. The functional scan was collected using a blood oxygen level-dependent (BOLD) protocol with a T2*-weighted gradient echo-planar imaging sequence (TR: 3.0 s with an intervolume delay of 500 ms; TE: 30 ms; flip angle: 90°; 64 × 64 matrix; 150 volume acquisitions). Electrical stimulation was always administered during the intervolume delay, thereby avoiding the potential contamination of fMRI images by shock-induced artifacts and EMG recordings by RF-pulse artifacts. The scanning planes were oriented parallel to the anterior–posterior commissure line and covered the entire brain from the vertex of the cortex to the first segments of the spinal cord (41 contiguous 5-mm-thick slices; voxel size, 3.44 × 3.44 × 5 mm).
fMRI data analyses.
Brain imaging data were analyzed using SPM2 (Wellcome Department of Cognitive Neurology, London, UK; http://www.fil.ion.ucl.ac.uk/spm/). Preprocessing included slice-time correction and realignment for movement correction. Due to excessive movement, one subject was excluded from all analyses (peak instantaneous movement = 1.25 mm; maximum instantaneous movement tolerated in all other subjects ≤0.5 mm). Anatomical and functional images were spatially normalized to a standard stereotaxic space using the Montreal Neurological Institute (MNI) template (Evans et al., 1993). Subsequently, functional images were spatially smoothed using a Gaussian kernel of twice the voxel size (FWHM: 7 × 7 × 10 mm), temporally filtered using a high-pass filter with a cutoff period of 240 s, and were corrected for serial autocorrelation using the AR(1) correction implemented in SPM.
The effects of interest were assessed using general linear models for each scan (counterirritation and control). For the counterirritation scan, individual contrasts were generated using a mixed-design model. The 2 min foot immersion was modeled as a 2 min block, while the 35 shocks were assigned to four event-related conditions: prebaseline shocks (5), baseline shocks (10), counterirritation shocks (10), and recovery shocks (10). For the control scan, the 40 shocks were assigned to five event-related conditions to mimic the counterirritation paradigm: prebaseline (5), block 1 (10), block 2 (10), block 3 (10), and shocks of no interest (5). For both scans, these vectors were convolved with a canonical hemodynamic response function. In addition, movement parameters from the realignment procedure were entered as six regressors of no interest to remove potential movement-related variance/artifacts. These two models (one for counterirritation and one for control) allowed assessing shock-evoked activity (contrast 1: baseline shocks/block 1, 10 stimulations), changes in shock-evoked activity during counterirritation/block 2 (contrast 2: counterirritation shocks/block 2 vs baseline shocks/block 1), changes in shock-evoked activity during recovery (contrast 3: recovery shocks/block 3 vs baseline shocks/block 1), and sustained activity evoked by the 2 min foot immersion (contrast 4: only for the counterirritation scan). For the group analyses, these individual contrasts were combined in random-effect models with the one-sample t test function. Parameter estimates for shock-evoked activity during baseline/block 1, counterirritation/block 2, and recovery/block 3 were also extracted from regions that were significantly modulated by counterirritation (Table 1). Repeated-measures ANOVAs were then performed to assess changes in shock-evoked activity across blocks of stimuli and between the counterirritation and the control scans (supplemental Table S1, available at www.jneurosci.org as supplemental material).
Table 1.
Brain regions in which shock-evoked activity was decreased during counterirritation (directed search)
| Brain area | BAa | Sideb | t | x, y, zc |
|---|---|---|---|---|
| Somatosensory cortex | ||||
| Postcentral gyrus (SI) | 1–3 | L | 4.73 | −7, −48, 70 |
| 1–3 | — | 4.33 | 3, −48, 75 | |
| Parietal operculum (SII) | 40 | L | 6.08 | −58, −28, 15 |
| Motor cortex | ||||
| PrCG | 4 | L | 6.35 | −7, −31, 70 |
| 4 | R | 6.00 | 14, −31, 80 | |
| SMA | 6 | L | 6.83 | −17, 0, 75 |
| 6 | R | 5.15 | 7, 3, 65 | |
| 6 | R | 5.02 | 14, 0, 75 | |
| Cingulate cortex | ||||
| ACC | 32 | — | 4.28 | 3, 17, 35 |
| 24 | R | 5.34 | 7, 3, 40 | |
| 24 | R | 5.27 | 7, 10, 35 | |
| MCC | 24 | — | 4.99 | −3, −3, 45 |
| Insula | ||||
| pINS | — | L | 7.58 | −31, −24, 10 |
| aINS | — | R | 5.63 | 45, 7, 5 |
| PHG | 34 | R | 5.93 | 17, 7, −20 |
| PFC | 9 | L | 6.65 | −31, 28, 25 |
| Thalamus | — | L | 7.92 | −14, −10, 10 |
| — | R | 4.68 | 10, −7, 10 |
Peaks of activity were thresholded at p < 0.05 corrected for multiple comparisons for the search volume, using the random field theory, minimum 3 voxels per cluster (see Materials and Methods).
aBA, putative Brodmann area.
bR, right ipsilateral side; L, left contralateral side.
cCoordinates are reported in MNI space.
The relation between brain activity and changes in pain and RIII reflex during counterirritation and recovery was further tested using multiple-regression models examining the magnitude of the modulation of shock-evoked activity during counterirritation (contrast 2) and during recovery (contrast 3). These models included subject-regressors corresponding to the magnitude of analgesia and RIII modulation (see Pain ratings and RIII reflex recording and analyses). Subsequently, to test the relation between sustained activity evoked by the foot immersion in cold water and analgesia and RIII reflex modulation, individual contrasts for sustained activity evoked by the 2 min foot immersion (contrast 4) were entered in a multiple-regression random-effect model with regressors for analgesia and RIII modulation. These analyses were performed with multiple regression models to assess brain activity that is more specifically related to analgesia or RIII modulation. Pearson's correlation coefficients were also determined for each significant peak related to analgesia or RIII modulation.
In a subsequent analysis, Pearson's correlations were performed to test the relation between sustained counterirritation activity and shock-evoked activity. Parameter estimates in brain regions activated by counterirritation and specifically related to analgesia were correlated to parameter estimates of brain regions showing decreased shock-evoked activity specifically related to analgesia. In the same way, parameter estimates of brain regions activated by counterirritation and specifically related to RIII inhibition were correlated to parameter estimates of brain regions showing decreased shock-evoked activity specifically related to RIII inhibition. These correlations were thresholded at p < 0.05 Bonferroni corrected for multiple comparisons.
Coactivation analyses were also performed to assess the brain networks involved in pain and RIII reflex modulation. Considering that sustained activity produced by counterirritation in OFC was the strongest peak related to analgesia (see Results; see also Fig. 5A and Table 3, top) and considering that OFC has been previously involved in analgesia, it was selected as the seed region. For RIII modulation, the PAG area correlated to RIII modulation (see Results; see also Fig. 5B and Table 3, bottom) and was selected as the seed region considering its well documented role in descending modulation of spinal nociceptive responses. The time courses of OFC and PAG were extracted (6 mm sphere) for the 150 volumes in each individual model. These time courses were then used as regressors in two separate models (OFC and PAG). Individual contrasts for OFC-related and PAG-related coactivations were then generated and used in group analyses (one-sample t test random-effect models). These models lead to statistical maps of networks associated with the analgesia-related peak in OFC and with the RIII-modulation peak in the PAG (see Results) (see Fig. 7 and Table 4).
Figure 5.

Sustained brain activation induced by the counterirritation stimulus associated with the magnitude of analgesia (A) and RIII inhibition (B) (see Table 3, top and bottom, respectively, for peak T values). Note one outlier in the analysis of SI activity (B, right scatterplot, data point in parenthesis). Excluding this point led to a large effect (r2 = 0.78).pACC, Pregenual anterior cingulate cortex; PPC, posterior parietal cortex.
Table 3.
Sustained activity evoked by counterirritation and specifically related to analgesia or RIII inhibition (directed search)
| Brain area | BAa | Sideb | t | r2 | x, y, zc |
|---|---|---|---|---|---|
| Regions specifically related to analgesia | |||||
| mPFC | 10 | — | 3.44 | 0.35 | 0, 65, 10 |
| OFC | 11/47 | L | 5.26* | 0.65 | −28, 31, −20 |
| Regions specifically related to RIII inhibition | |||||
| Somatosensory cortex (SI) | 1–3 | — | 3.41 | 0.31 | 0, −41, 60 |
| SMA | 6 | R | 4.26 | 0.65 | 7, −10, 75 |
| PCC | 23 | — | 3.75 | 0.37 | 0, −24, 30 |
| Midbrain (PAG) | — | — | 3.74 | 0.36 | −3, −31, −5 |
| Midbrain (lateral) | — | L | 3.78 | 0.39 | −10, −28, −15 |
Peaks of activity were thresholded at p < 0.005.
*p < 0.05 corrected for multiple comparisons for the search volume, using the random field theory, minimum 3 voxels per cluster.
aBA, Putative Brodmann area.
bR, right ipsilateral side; L, left contralateral side.
cCoordinates are reported in MNI space.
Figure 7.

Functional connectivity analysis. A, Brain regions coactivated with the analgesia-related peak in OFC. B, Brain regions coactivated with the RIII-modulation peak in the PAG. The inset is an enlargement of the RVM area. See Table 4, Regions coactivated with analgesia-related OFC and Regions coactivated with RIII-modulation peak in the PAG, respectively, for peak T values. PPC, Posterior parietal cortex; RSC, retrosplenial cortex.
Table 4.
Brain networks associated with the analgesia-related peak in OFC and the RIII-modulation peak in the PAG (directed search)
| Brain area | Regions coactivated with analgesia-related OFC |
Regions coactivated with RIII-modulation peak in the PAG |
||||||
|---|---|---|---|---|---|---|---|---|
| BAa | Sideb | t | x, y, zc | BAa | Sideb | t | x, y, zc | |
| Postcentral gyrus (SI) | — | — | — | — | 1–3 | R | 9.29 | 3, −48, 70 |
| PCL | — | — | — | — | 5 | R | 7.83 | −3, −41, 50 |
| Motor cortex | ||||||||
| SMA | — | — | — | — | 6 | — | 10.67 | 0, −10, 70 |
| Pre-SMA | — | — | — | — | 6/8 | — | 11.08 | 0, 17, 65 |
| Cingulate cortex | ||||||||
| ACC | 24/32 | L | 8.29 | −7, 10, 40 | 32 | R | 6.22 | 10, 7, 45 |
| 32 | L | 5.65 | −7, 21, 35 | 32 | — | 8.11 | −3, 38, 20 | |
| 24/32 | R | 7.81 | 10, 21, 30 | — | — | — | — | |
| 32 | — | 6.61 | 0, 31, 25 | — | — | — | — | |
| Subgenual ACC | 32 | — | 8.10 | −3, 24, −10 | — | — | — | — |
| PCC | 23 | — | 6.73 | 0, −31, 25 | 23 | L | 6.29 | −10, −24, 40 |
| — | — | — | — | 31 | R | 6.23 | −7, −48, 30 | |
| Retrosplenial cortex | — | — | — | — | 30 | — | 10.05 | −3, −45, 10 |
| aINS | — | L | 6.78 | −45, 7, −5 | — | — | — | — |
| PHG | 37 | L | 5.90 | −24, −41, −10 | 35 | R | 8.12 | 21, −17, −30 |
| 36 | R | 6.10 | 34, −10, −20 | 35 | R | 6.97 | 17, −31, −10 | |
| AMY/PHG | — | L | 6.70 | −31, −3, −20 | — | — | — | — |
| PFC | ||||||||
| Medial frontal gyrus | 10/32 | L | 9.36 | −7, 45, −15 | 9 | — | 9.23 | −3, 45, 30 |
| — | — | — | — | 9 | L | 11.23 | −10, 52, 25 | |
| — | — | — | — | 10 | R | 11.12 | 7, 62, 15 | |
| Inferior frontal gyrus | 47 | L | 8.56 | −45, 21, −15 | 9 | L | 11.80 | −55, 7, 30 |
| 47 | L | 7.86 | −41, 14, −10 | 46 | L | 7.84 | −45, 45, 10 | |
| 46 | L | 6.42 | −52, 34, 15 | 46 | R | 8.27 | 52, 41, 10 | |
| Middle frontal gyrus | 10 | L | 6.50 | −45, 45, 15 | 46 | L | 8.23 | −41, 34, 25 |
| 46 | R | 7.43 | 48, 17, 25 | 9 | R | 9.00 | 52, 7, 40 | |
| 10 | R | 6.94 | 31, 45, 25 | 9 | R | 7.40 | 31, 34, 33 | |
| 10 | R | 6.31 | 38, 48, 0 | 10 | R | 7.27 | 45, 52, 5 | |
| — | — | — | — | 10 | R | 5.89 | 41, 58, 0 | |
| Superior frontal gyrus | 9 | R | 6.61 | 38, 34, 35 | 10 | R | 10.21 | 31, 62, 20 |
| OFC | 11 | R | 10.30 | 28, 34, −15 | — | — | — | — |
| 11 | R | 9.97 | 21, 21, −15 | — | — | — | — | |
| 11 | R | 7.78 | 17, 34, −20 | — | — | — | — | |
| Thalamus | — | — | — | — | — | L | 7.06 | −3, −14, 10 |
| — | — | — | — | — | L | 6.86 | −17, −21, 15 | |
| — | — | — | — | — | R | 8.20 | 7, −14, −10 | |
| Pons | — | — | — | — | — | L | 6.91 | −7, −24, −35 |
| — | — | — | — | — | R | 6.13 | 10, −28, −40 | |
| Pons/medulla (RVM) | — | — | — | — | — | — | 8.15 | −3, −24, −45 |
Peaks of activity were thresholded at p < 0.05 corrected for multiple comparisons for the search volume, using the random-field theory, minimum 3 voxels per cluster.
aBA, Putative Brodmann area.
bR, right ipsilateral side; L, left contralateral side.
cCoordinates are reported in MNI space.
In all analysis models tested, a directed search was conducted over brain areas receiving nociceptive afferents and/or involved in the regulation of nociceptive processes (Apkarian et al., 2005). We hypothesized that structures previously shown to respond to phasic noxious stimulation, namely the thalamus, primary and second somatosensory cortices (SI-foot area and SII), the supplementary motor area (SMA), the aINS and posterior insula (pINS), the middle and anterior cingulate cortex (MCC and ACC), the amygdala and parahippocampal gyrus (PHG), the orbitofrontal cortex (OFC), and the prefrontal cortex (PFC), would show decreased shock-evoked activity during counterirritation. We also hypothesized that individual differences in analgesia and RIII reflex modulation would be associated with the activity of brain structures previously involved in modulation of nociceptive activity, including the PAG (Petrovic et al., 2002; Tracey et al., 2002; Valet et al., 2004; Bingel et al., 2006; Fairhurst et al., 2007), SI, SII, and motor cortex (Yezierski et al., 1983; Passard et al., 2007; Wise et al., 2007), cingulate cortex (Faymonville et al., 2000; Frankenstein et al., 2001; Valet et al., 2004; deCharms et al., 2005; Bingel et al., 2006,) anterior insula (Wise et al., 2007), entorhinal cortex (Ploghaus et al., 2001), orbitofrontal cortex (OFC) (Petrovic and Ingvar, 2002), and amygdala (Zubieta et al., 2001). The size of this search volume was used to account for multiple-comparison in determining the statistical threshold.
A threshold of p-corrected < 0.05 was applied to search for significant activation in target structures, corresponding to a directed-search volume estimated at 109.58 resels [resel size estimated using the effective FWHM; one-tail tests; Bonferroni-corrected for multiple comparisons based on the random field theory (Worsley et al., 1992)]. This corrected p value applied to directed searches corresponds to an uncorrected p < 0.0009. Activation maps were also examined using a more permissive criterion (p-uncorrected <0.005) to minimize the risk of type II error. This tested the activation of brain structures previously shown to be involved in pain and/or pain modulation.
Results
Pain and RIII modulation
The sustained counterirritation stimulus produced strong pain (mean ± SEM: 70.3 ± 4.2) and produced a robust analgesia on phasic pain with baseline shock-pain reduced by ∼50% during counterirritation (baseline: 43.8 ± 5.8; counterirritation: 22.4 ± 5.1, p < 0.01) (Fig. 2). Furthermore, analgesia was partly maintained during the recovery period, as shock pain remained slightly but significantly diminished relative to baseline (recovery: 37.6 ± 6.6, p < 0.05) (Fig. 2).
Figure 2.

Shock-pain ratings during baseline and counterirritation (immersion) and after counterirritation (recovery). The counterirritation stimulus produced strong analgesia that partially persisted during the recovery period. *p < 0.05; **p < 0.01.
On the other hand, the decrease in RIII reflex amplitude during counterirritation (129.2 ± 19.4 μV) and recovery (129.5 ± 24.2 μV) compared with baseline (162.9 ± 34.6 μV) did not reach statistical significance at the group level (p values >0.1) (Fig. 3A). Moreover, the changes in RIII reflex and pain produced by counterirritation were not significantly related (ρ = 0.16, p = 0.63). However, analyses of individual subjects revealed that a subgroup of 4 participants presented a robust and significant decrease in RIII reflex amplitude during counterirritation (Fig. 3B). In the other subjects, RIII reflex amplitude remained unchanged or increased slightly. Figure 3 shows one subject in whom the RIII reflex was significantly inhibited by counterirritation (Fig. 3C) and one subject showing no significant modulation (Fig. 3D). For the control scan, RIII reflex amplitude remained stable across the three blocks of electric shocks (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Moreover, individual analyses also revealed no significant modulation of RIII reflex amplitude across blocks of stimuli (all p values >0.12).
Figure 3.

RIII reflex amplitude during baseline and counterirritation (immersion) and after counterirritation (recovery). A, The counterirritation stimulus produced a decrease of RIII reflex amplitude during and after counterirritation but the effect was not significant (p > 0.1). B, Individual differences in RIII reflex modulation during counterirritation. Two subgroups are evident showing significant inhibition of RIII (gray triangles) and no inhibition (black dots). C, D, Individual examples of RIII reflex modulation: arrows indicate the beginning and the end of counterirritation. In the first subject, RIII reflex amplitude was decreased during counterirritation and slowly recovered to reach the baseline level at the end of the session (C). In the other subject, RIII reflex amplitude slightly increased during counterirritation and decreased to a stable level during recovery (D).
Changes in shock-evoked brain activity during counterirritation
Baseline electric shocks evoked robust activation in several pain-related areas (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). This event-related response was significantly decreased during counterirritation in bilateral thalamus, SI, precentral gyrus (PrCG), ACC, MCC, and SMA; left contralateral SII, pINS, and PFC; and right ipsilateral aINS and PHG (Fig. 4A, Table 1). The decrease in shock-evoked activity during counterirritation is further described by a significant decrease in parameter estimates of these regions for baseline versus counterirritation shocks (supplemental Table S1, available at www.jneurosci.org as supplemental material). Together, these results indicate that brain responses to phasic painful stimuli were consistently decreased during counterirritation. Decrease in shock-evoked activity during recovery was also significant in some pain-related regions, as shown in supplemental Figure S3A (available at www.jneurosci.org as supplemental material). This corroborates the persistent inhibition of phasic shock pain during the recovery period. In contrast, shock-evoked activity was very stable across the three blocks of electric shocks during the control scan (supplemental Table S1, available at www.jneurosci.org as supplemental material). Moreover, the changes in shock-evoked activity for counterirritation versus baseline were significantly different from changes in shock-evoked activity for block 2 versus block 1 in the control scan (supplemental Table S1, available at www.jneurosci.org as supplemental material). This clearly indicates specific effects of counterirritation on shock-evoked brain activity.
Figure 4.
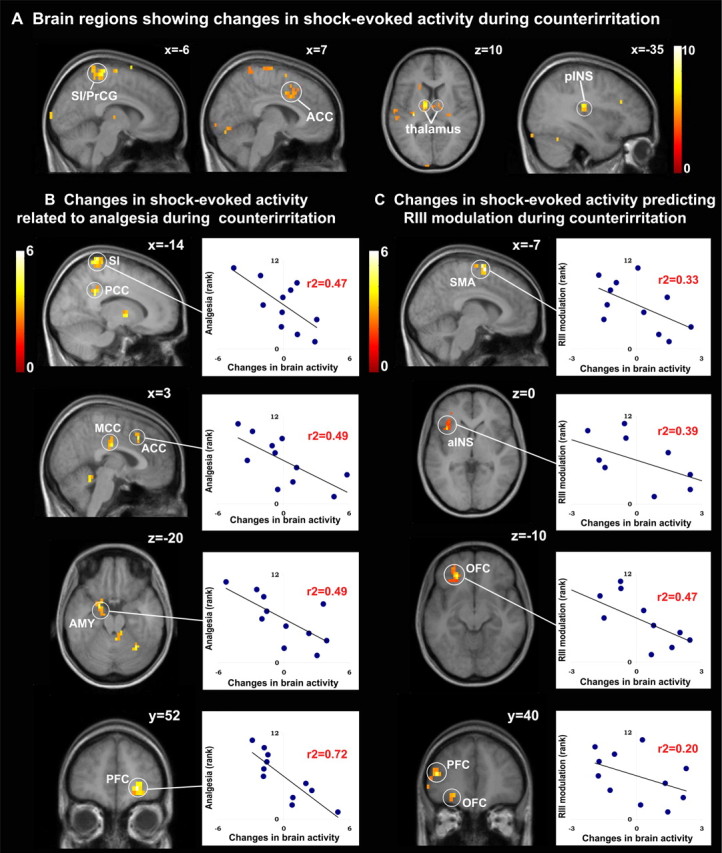
Modulation of shock-evoked brain responses during counterirritation. A, Contrast of shock-evoked activity induced during baseline versus counterirritation (see Table 1 for peak T values). B, C, Multiple regression analysis showing decreased shock-evoked activity during counterirritation proportional to analgesia (B) and RIII modulation (C). Scatter plots illustrate the individual pain and RIII modulatory effects in relation to the respective change in shock-evoked brain response (see Table 2, top and bottom, respectively, for peak T values).
Changes in shock-evoked activity during counterirritation were further examined using multiple regressions. These analyses revealed that subjects reporting stronger counterirritation analgesia displayed larger decreases in shock-evoked responses in contralateral SI, posterior cingulate cortex (PCC), and AMY; ipsilateral PFC and OFC; and bilateral ACC and MCC (Fig. 4B, Table 2, top). On the other hand, the changes in RIII amplitude were associated with the magnitude of the modulation of shock-evoked responses in contralateral SMA, aINS, PFC, and OFC and bilateral SMA (Fig. 4C, Table 2, bottom). Some of these correlations are illustrated with scatter plots in Figure 4, B and C, respectively. Changes in shock-evoked responses that were specifically related to analgesia or RIII modulation during recovery are shown in supplemental Figure S3, B and C (available at www.jneurosci.org as supplemental material).
Table 2.
Brain regions in which changes in shock-evoked responses during counterirritation were specifically related to analgesia or RIII modulation (directed search)
| Brain area | BAa | Sideb | t | r2 | x, y, zc |
|---|---|---|---|---|---|
| Specifically related to analgesia | |||||
| Postcentral gyrus (SI) | 1–3 | L | 5.08 | 0.47 | −14, −41, 80 |
| Cingulate cortex | |||||
| ACC | 32 | — | 4.39 | 0.49 | 3, 21, 40 |
| MCC | 24 | — | 4.24 | 0.59 | 3, −17, 30 |
| PCC | 31 | L | 4.09 | 0.55 | −14, −45, 35 |
| 31 | L | 5.17 | 0.49 | −7, −31, 30 | |
| PFC | |||||
| Middle frontal gyrus | 9/46 | R | 5.20 | 0.29 | 41, 41, 25 |
| 10 | R | 7.20* | 0.72 | 24, 52, −5 | |
| OFC | 11 | R | 4.94* | 0.55 | 31, 48, −10 |
| AMY | — | L | 4.74 | 0.49 | −21, 0, −20 |
| — | L | 3.51 | 0.29 | −17, −7, −15 | |
| Specifically related to RIII modulation | |||||
| SMA | 6 | L | 5.67* | 0.33 | −7, 0, 65 |
| 6 | — | 4.99 | 0.42 | 0, −14, 75 | |
| aINS | — | L | 3.83 | 0.39 | −38, 17, 0 |
| PFC | |||||
| Middle frontal gyrus | 10 | L | 4.56 | 0.20 | −45, 41, 15 |
| OFC | 11 | L | 4.40 | 0.47 | −24, 38, −10 |
Peaks of activity were thresholded at p < 0.005 uncorrected.
aBA, Putative Brodmann area.
bR, Right ipsilateral side; L, left contralateral side.
cCoordinates are reported in MNI space.
*p < 0.05 corrected for multiple comparisons for the search volume, using the random field theory, minimum 3 voxels per cluster.
Sustained brain activity evoked by the counterirritation stimulus
Counterirritation induced sustained activation in common pain-related areas (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). In addition, strong activation was found in the midbrain and the pons, consistent with the recruitment of the PAG and the reticular formation during tonic pain. Regression analyses were performed to reveal brain regions where the magnitude of cold-pain activation predicted the decrease in shock pain (i.e., counterirritation analgesia) or predicted the changes in shock-evoked RIII responses. Sustained cold-pain activity in ipsilateral OFC and bilateral medial prefrontal cortex (mPFC) was specifically related to counterirritation analgesia (Fig. 5A, Table 3, top). On the other hand, activity in bilateral SI, PCC, and midbrain (consistent with the location of the PAG) and contralateral SMA was specifically related to RIII modulation (Fig. 5B, Table 3, bottom). Interestingly, analgesia-related activity in OFC strongly predicted inhibition of shock-evoked activity in AMY (Fig. 6).
Figure 6.

Sustained activation of the OFC predicts the inhibition of shock-evoked activity in the amygdala. Counterirritation produced a robust and sustained activation of lateral OFC ipsilateral to the foot immersion that was strongly correlated to analgesia (top brain slice). Counterirritation also produced a robust inhibition of shock-evoked activity in the left amygdala, contralateral to the shock (bottom brain slice). The activity of these two structures was strongly associated, as illustrated by the scatter plot on the right.
Coactivation analyses
The analgesia-related peak in OFC was coactivated with a brain network including subregions of the cingulate cortex (PCC, ACC, and subgenual ACC), aINS, AMY, PHG, mPFC, and OFC (Fig. 7A, Table 4, Regions coactivated with analgesia-related OFC). On the other hand, structures coactivated with the RIII-modulation peak in the PAG included SI, paracentral lobule (PCL), SMA and pre-SMA, ACC, PCC, PHG, PFC, thalamus, pons, and medulla (RVM) (Fig. 7B, Table 4, Regions coactivated with RIII-modulation peak in the PAG). Notably, the OFC-related activity did not include brainstem or thalamic structures, while the PAG-related activity did not involve the OFC, suggesting at least a partial segregation between networks associated with analgesia and RIII modulation during counterirritation.
Discussion
This is the first study to examine the effects of counterirritation on pain perception and RIII reflex amplitude concurrently with BOLD measures of brain activity. As expected, counterirritation produced strong analgesia that persisted during the recovery period. On the other hand, RIII reflex amplitude was decreased by counterirritation but this effect reached statistical significance only in some subjects. The modulatory effects of counterirritation were paralleled by decreased shock-evoked activity in several pain-related brain areas. Moreover, the counterirritation stimulus produced sustained activation in brain areas involved in cortical modulation of pain and descending modulation of spinal nociception. Furthermore, coactivation analyses revealed that distinct brain networks were related to analgesia or RIII modulation. Together these results are consistent with the hypothesis that the effects of counterirritation involve both cerebral and cerebrospinal processes.
The modulation of pain by counterirritation
In the present study, shock pain to the right ankle was reduced by ∼50% during the immersion of the left foot in painfully cold water. Moreover, the analgesic effects of counterirritation persisted over the counterirritation period, indicating that analgesia to electric shocks cannot be explained solely by distraction. These results are similar to those reported in previous studies on counterirritation using ischemic or cold pain (Pertovaara et al., 1982; Jungkunz et al., 1983; Chen et al., 1985; Talbot et al., 1987). Here, specific modulation of brain responses further corroborated those modulatory effects.
The noxious shocks and the counterirritation stimulus activated a similar network of pain-related areas (see supplemental material, available at www.jneurosci.org), consistent with previous imaging studies investigating brain responses to experimental pain stimulation (Apkarian et al., 2005). Counterirritation analgesia further produced changes in shock-evoked responses in several pain-related brain areas, which were significantly greater than changes in the control condition (supplemental Table S1, available at www.jneurosci.org as supplemental material). Most notably, the phasic responses in SI, ACC, PFC, and the amygdala were decreased specifically in relation to the extent of analgesia. This further supports the role of these structures in pain-related processes (Rainville et al., 1997; Coghill et al., 1999; Hofbauer et al., 2001; Bornhövd et al., 2002; Büchel et al., 2002). Several mechanisms may have contributed to this robust and specific decrease in phasic pain-related brain responses.
The counterirritation stimulus produced sustained activation in several pain-related areas including SI, ACC, aINS, PFC, and OFC, as well as the midbrain (PAG area) and the pons. This is consistent with the recruitment of processes involved in the cortical modulation of pain (Faymonville et al., 2000; Petrovic and Ingvar, 2002; Bingel et al., 2006) as well as descending modulation of spinal nociceptive activity (Yezierski et al., 1983; Zhang et al., 1997; Jasmin et al., 2003), which potentially modulated shock-evoked brain activity. Importantly, the inhibition of phasic pain responses in ACC, PFC, and amygdala is consistent with the release of endogenous μ-opioids in those structures during sustained pain (Zubieta et al., 2001). This implies that local autoregulatory processes (e.g., opioidergic) triggered by sustained pain and affecting phasic pain responses may contribute to counterirritation analgesia. These local inhibitory effects may further be regulated by some of the brain structures activated by the counterirritation stimulus.
In line with this, sustained activity in left OFC during counterirritation was specifically related to the extent of analgesia (Fig. 5A). Moreover, sustained OFC activity covaried with the activity of PHG/amygdala (Fig. 7) and predicted the reduced amygdala response to acute shock-pain (Fig. 6). These results are consistent with the anatomical connection between OFC and amygdala (Kringelbach and Rolls, 2004), and may reflect a corticoamygdaloid regulation of endogenous opioid release during the application of the counterirritation stimulus (Zubieta et al., 2001). Activation of this emotional brain network (Goldin et al., 2008) is seldom found in pain imaging studies (Apkarian et al., 2005) but may be particularly important for the understanding of pain modulation [e.g., Petrovic and Ingvar (2002) and Petrovic et al. (2004)]. When a strongly aversive counterirritation stimulus is processed concurrently to a phasic noxious stimulus, the involvement of the OFC may reflect the assessment of the two competing stimuli, consistent with its role in the representation of appetitive/aversive value [for review, see Kringelbach and Rolls (2004) and Rolls and Grabenhorst (2008)]. Here, we suggest that OFC activation by counterirritation pain reflects the higher threat value of sustained pain, which in turn downregulates the processing of the phasic pain stimulus.
Modulation of spinal nociception by counterirritation
Despite the robust analgesic effect, the decrease in RIII reflex amplitude reached statistical significance only in a subset of participants. Furthermore the changes in pain did not correlate with changes in RIII reflex amplitude. This result indicates a dissociation between RIII reflex amplitude and pain perception and is consistent with some previous studies using segmental (Terkelsen et al., 2001) and heterosegmental (Willer et al., 1979) counterirritation. One possible explanation is that counterirritation may influence motoneuron excitability independently from its effects on ascending sensory signals, resulting in the observed uncoupling between motor-reflex responses and pain perception. However, this possibility appears unlikely because motoneuron excitability, as measured by the monosynaptic H-reflex, is not modulated by nociceptive segmental stimulation (Ge et al., 2007) nor by counterirritation (Willer et al., 1989). Therefore, the sensorimotor dissociation found in the present study most likely originates from supraspinal mechanisms affecting pain and cerebrospinal modulation separately.
Changes in shock-evoked activity were related to RIII modulation in pain-related areas, including aINS, SMA, and OFC. Modulation in the insula may reflect the changes in ascending sensory activation induced by the shock and the engagement of spinothalamocortical nociceptive pathways. In contrasts, the modulation found in the SMA and OFC may reflect sensorimotor feedback associated with the RIII response, consistent with their role in motor/behavior regulation (Picard and Strick, 1996; Fuster, 2001; Paus, 2001; Kringelbach, 2005). Thus, the magnitude of the spinal sensorimotor output evoked by noxious stimulation would predict the extent of cortical motor regulation in SMA and OFC. In line with this interpretation, the modulation of the phasic shock-evoked response in OFC is consistent with its role in the evaluation of the punishing value of stimuli. Indeed, the amplitude of the nociceptive flexion reflex provides a low-level estimate of the aversiveness of the stimulus. Changes in shock-evoked activity in OFC may therefore represent the relatively reduced punishing value of electric shocks during counterirritation, proportionally to the ongoing modulation of spinal nociceptive processes.
Interestingly, RIII reflex modulation was also related to activations partly different from those associated with counterirritation analgesia. This is consistent with the sensorimotor dissociation found in the present and previous studies (Willer et al., 1979; Terkelsen et al., 2001) and demonstrates that pain perception and nociceptive motor reflexes are regulated, at least partly, by different neural processes. Accordingly, stronger sustained activation of the PAG during counterirritation was specifically related to decrease RIII reflex amplitude. Similarly, sustained SI activation correlated with the reduction in the RIII response, an observation that points to a potential role of SI in spinal modulation. This possibility is consistent with SI projections to the dorsal horn of the spinal cord (Cheema et al., 1984; Ralston and Ralston, 1985; Casale et al., 1988) and with the inhibitory effect of this pathway on nociceptive spinothalamic cells (Yezierski et al., 1983). Furthermore, based on the coactivation analysis, several additional areas, including the motor, supracallosal cingulate, and prefrontal cortices, may contribute to this descending regulatory system.
The coactivation analysis further revealed that PAG activity covaried with RVM activity, consistent with their anatomical connections and their important role in descending modulation (Basbaum and Fields, 1984; Morgan et al., 2008). However, although the PAG can modulate counterirritation effects (Bouhassira et al., 1992b), it is not directly involved in DNIC (Bouhassira et al., 1990). Instead, DNIC is thought to mediate counterirritation effects on spinal nociceptive processes by the recruitment of the caudal medulla (De Broucker et al., 1990; Bouhassira et al., 1992a). The caudal medulla was not activated in the present study, but the absence of significant modulation of RIII in a subgroup of participants may have contributed to this negative result. Another possibility is that the fMRI methods used in this study may not have been sensitive enough to detect its activation. This last possibility seems quite likely, since the caudal medullary area (more specifically the subnucleus dorsalis reticularis or dorsal reticular nucleus) implicated in DNIC is a very small reticular region difficult to image using fMRI. Therefore, the correlations between RIII reflex modulation and brain activity in the present study should be interpreted with caution, as they may also reflect competing pronociceptive segmental or cerebrospinal processes at least in some subjects. Future studies may help clarify this ambiguity by determining how differences in the inhibition or facilitation of the reflex induced by psychological factors during counterirritation (Goffaux et al., 2007; M. Piché, M. Bouin, P. Poitras, and P. Rainville, unpublished observations) may relate to different modulatory brain processes.
Conclusion
The present study shows that modulatory effects of counterirritation on pain perception and spinal nociception are accompanied by a specific decrease in shock-evoked activity in pain-related areas and by sustained activation in brain areas involved in the cortical modulation of pain. Results suggest that the OFC is involved in the representation of the relative aversive value of competing noxious stimuli and may regulate phasic pain-related responses in the amygdala and the thalamocortical networks. In contrast, the modulation of spinal nociceptive processes was more consistently associated with activation of SI and the PAG. These results provide evidence for distinct cerebral and cerebrospinal mechanisms underlying the modulation of pain and RIII reflex by counterirritation. This experimental model further provides ground for future investigations of the cerebral and cerebrospinal mechanisms underlying cognitive and pharmacological modulation of pain-related processes in healthy volunteers and patients with chronic pain.
Footnotes
This research was supported by the Biology of Pain Young Investigator Grant from Astra/Zeneca in partnership with the Canadian Pain Society and the Canadian Institutes of Health Research (CIHR). M.P. was supported by the “Fonds de recherche en santé du Québec (FRSQ),” the CIHR, and the “Fondation de recherche chiropratique du Québec.” M.A. was supported by the FRSQ, CIHR, and the “Réseau de Formation interdisciplinaire en recherche Santé et Vieillissement.” This study was performed with the technical help of Leo Tenbokum and of Carollyn Hurst and André Cyr from the “Unité de Neuroimageie Fonctionnelle” of the “Centre de recherche de l'Institut universitaire de gériatrie de Montréal.”
References
- Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain. 2005;9:463–484. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Bantick SJ, Wise RG, Ploghaus A, Clare S, Smith SM, Tracey I. Imaging how attention modulates pain in humans using functional MRI. Brain. 2002;125:310–319. doi: 10.1093/brain/awf022. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Fields HL. Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Annu Rev Neurosci. 1984;7:309–338. doi: 10.1146/annurev.ne.07.030184.001521. [DOI] [PubMed] [Google Scholar]
- Bingel U, Lorenz J, Schoell E, Weiller C, Büchel C. Mechanisms of placebo analgesia: rACC recruitment of a subcortical antinociceptive network. Pain. 2006;120:8–15. doi: 10.1016/j.pain.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Bornhövd K, Quante M, Glauche V, Bromm B, Weiller C, Büchel C. Painful stimuli evoke different stimulus-response functions in the amygdala, prefrontal, insula and somatosensory cortex: a single-trial fMRI study. Brain. 2002;125:1326–1336. doi: 10.1093/brain/awf137. [DOI] [PubMed] [Google Scholar]
- Bouhassira D, Bing Z, Le Bars D. Studies of the brain structures involved in diffuse noxious inhibitory controls: the mesencephalon. J Neurophysiol. 1990;64:1712–1723. doi: 10.1152/jn.1990.64.6.1712. [DOI] [PubMed] [Google Scholar]
- Bouhassira D, Villanueva L, Bing Z, le Bars D. Involvement of the subnucleus reticularis dorsalis in diffuse noxious inhibitory controls in the rat. Brain Res. 1992a;595:353–357. doi: 10.1016/0006-8993(92)91071-l. [DOI] [PubMed] [Google Scholar]
- Bouhassira D, Villanueva L, Le Bars D. Effects of systemic morphine on diffuse noxious inhibitory controls: role of the periaqueductal grey. Eur J Pharmacol. 1992b;216:149–156. doi: 10.1016/0014-2999(92)90355-8. [DOI] [PubMed] [Google Scholar]
- Bouhassira D, Danziger N, Attal N, Guirimand F. Comparison of the pain suppressive effects of clinical and experimental painful conditioning stimuli. Brain. 2003;126:1068–1078. doi: 10.1093/brain/awg106. [DOI] [PubMed] [Google Scholar]
- Büchel C, Bornhovd K, Quante M, Glauche V, Bromm B, Weiller C. Dissociable neural responses related to pain intensity, stimulus intensity, and stimulus awareness within the anterior cingulate cortex: a parametric single-trial laser functional magnetic resonance imaging study. J Neurosci. 2002;22:970–976. doi: 10.1523/JNEUROSCI.22-03-00970.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casale EJ, Light AR, Rustioni A. Direct projection of the corticospinal tract to the superficial laminae of the spinal cord in the rat. J Comp Neurol. 1988;278:275–286. doi: 10.1002/cne.902780210. [DOI] [PubMed] [Google Scholar]
- Cheema SS, Rustioni A, Whitsel BL. Light and electron microscopic evidence for a direct corticospinal projection to superficial laminae of the dorsal horn in cats and monkeys. J Comp Neurol. 1984;225:276–290. doi: 10.1002/cne.902250211. [DOI] [PubMed] [Google Scholar]
- Chen AC, Treede RD, Bromm B. Tonic pain inhibits phasic pain: evoked cerebral potential correlates in man. Psychiatry Res. 1985;14:343–351. doi: 10.1016/0165-1781(85)90102-7. [DOI] [PubMed] [Google Scholar]
- Coghill RC, Sang CN, Maisog JM, Iadarola MJ. Pain intensity processing within the human brain: a bilateral, distributed mechanism. J Neurophysiol. 1999;82:1934–1943. doi: 10.1152/jn.1999.82.4.1934. [DOI] [PubMed] [Google Scholar]
- De Broucker T, Cesaro P, Willer JC, Le Bars D. Diffuse noxious inhibitory controls in man. Involvement of the spinoreticular tract. Brain. 1990;113:1223–1234. doi: 10.1093/brain/113.4.1223. [DOI] [PubMed] [Google Scholar]
- deCharms RC, Maeda F, Glover GH, Ludlow D, Pauly JM, Soneji D, Gabrieli JD, Mackey SC. Control over brain activation and pain learned by using real-time functional MRI. Proc Natl Acad Sci U S A. 2005;102:18626–18631. doi: 10.1073/pnas.0505210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire SW, Vogt BA, Jones AK. Pain and Stroop interference tasks activate separate processing modules in anterior cingulate cortex. Exp Brain Res. 1998;118:52–60. doi: 10.1007/s002210050254. [DOI] [PubMed] [Google Scholar]
- Evans A, Collins D, Mills S, Brown E, Kelly R, Peters T. 3D statistical neuroanatomical models from 305 MRI volumes. In: Klaisner L, editor. Nuclear Science Symposium and Medical Imaging Conference: 1993 IEEE Conference Record; San Francisco, CA: IEEE; 1993. pp. 1813–1817. [Google Scholar]
- Fairhurst M, Wiech K, Dunckley P, Tracey I. Anticipatory brainstem activity predicts neural processing of pain in humans. Pain. 2007;128:101–110. doi: 10.1016/j.pain.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Faymonville ME, Laureys S, Degueldre C, DelFiore G, Luxen A, Franck G, Lamy M, Maquet P. Neural mechanisms of antinociceptive effects of hypnosis. Anesthesiology. 2000;92:1257–1267. doi: 10.1097/00000542-200005000-00013. [DOI] [PubMed] [Google Scholar]
- Frankenstein UN, Richter W, McIntyre MC, Rémy F. Distraction modulates anterior cingulate gyrus activations during the cold pressor test. Neuroimage. 2001;14:827–836. doi: 10.1006/nimg.2001.0883. [DOI] [PubMed] [Google Scholar]
- Fuster JM. The prefrontal cortex—an update: time is of the essence. Neuron. 2001;30:319–333. doi: 10.1016/s0896-6273(01)00285-9. [DOI] [PubMed] [Google Scholar]
- Ge HY, Collet T, Mørch CD, Arendt-Nielsen L, Andersen OK. Depression of the human nociceptive withdrawal reflex by segmental and heterosegmental intramuscular electrical stimulation. Clin Neurophysiol. 2007;118:1626–1632. doi: 10.1016/j.clinph.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Goffaux P, Redmond WJ, Rainville P, Marchand S. Descending analgesia—when the spine echoes what the brain expects. Pain. 2007;130:137–143. doi: 10.1016/j.pain.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Goldin PR, McRae K, Ramel W, Gross JJ. The neural bases of emotion regulation: reappraisal and suppression of negative emotion. Biol Psychiatry. 2008;63:577–586. doi: 10.1016/j.biopsych.2007.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer RK, Rainville P, Duncan GH, Bushnell MC. Cortical representation of the sensory dimension of pain. J Neurophysiol. 2001;86:402–411. doi: 10.1152/jn.2001.86.1.402. [DOI] [PubMed] [Google Scholar]
- Hosobuchi Y. The current status of analgesic brain stimulation. Acta Neurochir Suppl (Wien) 1980;30:219–227. doi: 10.1007/978-3-7091-8592-6_27. [DOI] [PubMed] [Google Scholar]
- Jasmin L, Rabkin SD, Granato A, Boudah A, Ohara PT. Analgesia and hyperalgesia from GABA-mediated modulation of the cerebral cortex. Nature. 2003;424:316–320. doi: 10.1038/nature01808. [DOI] [PubMed] [Google Scholar]
- Jungkunz G, Engel RR, King UG, Kuss HJ. Endogenous opiates increase pain tolerance after stress in humans. Psychiatry Res. 1983;8:13–18. doi: 10.1016/0165-1781(83)90133-6. [DOI] [PubMed] [Google Scholar]
- Kringelbach ML. The human orbitofrontal cortex: linking reward to hedonic experience. Nat Rev Neurosci. 2005;6:691–702. doi: 10.1038/nrn1747. [DOI] [PubMed] [Google Scholar]
- Kringelbach ML, Rolls ET. The functional neuroanatomy of the human orbitofrontal cortex: evidence from neuroimaging and neuropsychology. Prog Neurobiol. 2004;72:341–372. doi: 10.1016/j.pneurobio.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Mayer DJ. Analgesia produced by electrical stimulation of the brain. Prog Neuropsychopharmacol Biol Psychiatry. 1984;8:557–564. doi: 10.1016/0278-5846(84)90015-0. [DOI] [PubMed] [Google Scholar]
- Melzack R, Casey KL. Sensory, motivational, and central control determinants of pain: a new conceptual model. In: Kenshalo D, editor. The skin senses. Springfield, IL: Thomas; 1968. pp. 423–443. [Google Scholar]
- Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Morgan MM, Whittier KL, Hegarty DM, Aicher SA. Periaqueductal gray neurons project to spinally projecting GABAergic neurons in the rostral ventromedial medulla. Pain. 2008;140:376–386. doi: 10.1016/j.pain.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passard A, Attal N, Benadhira R, Brasseur L, Saba G, Sichere P, Perrot S, Januel D, Bouhassira D. Effects of unilateral repetitive transcranial magnetic stimulation of the motor cortex on chronic widespread pain in fibromyalgia. Brain. 2007;130:2661–2670. doi: 10.1093/brain/awm189. [DOI] [PubMed] [Google Scholar]
- Paus T. Primate anterior cingulate cortex: where motor control, drive and cognition interface. Nat Rev Neurosci. 2001;2:417–424. doi: 10.1038/35077500. [DOI] [PubMed] [Google Scholar]
- Pertovaara A, Kemppainen P, Johansson G, Karonen SL. Ischemic pain nonsegmentally produces a predominant reduction of pain and thermal sensitivity in man: a selective role for endogenous opioids. Brain Res. 1982;251:83–92. doi: 10.1016/0006-8993(82)91276-8. [DOI] [PubMed] [Google Scholar]
- Petrovic P, Ingvar M. Imaging cognitive modulation of pain processing. Pain. 2002;95:1–5. doi: 10.1016/s0304-3959(01)00467-5. [DOI] [PubMed] [Google Scholar]
- Petrovic P, Petersson KM, Ghatan PH, Stone-Elander S, Ingvar M. Pain-related cerebral activation is altered by a distracting cognitive task. Pain. 2000;85:19–30. doi: 10.1016/s0304-3959(99)00232-8. [DOI] [PubMed] [Google Scholar]
- Petrovic P, Kalso E, Petersson KM, Ingvar M. Placebo and opioid analgesia—imaging a shared neuronal network. Science. 2002;295:1737–1740. doi: 10.1126/science.1067176. [DOI] [PubMed] [Google Scholar]
- Petrovic P, Carlsson K, Petersson KM, Hansson P, Ingvar M. Context-dependent deactivation of the amygdala during pain. J Cogn Neurosci. 2004;16:1289–1301. doi: 10.1162/0898929041920469. [DOI] [PubMed] [Google Scholar]
- Picard N, Strick PL. Motor areas of the medial wall: a review of their location and functional activation. Cereb Cortex. 1996;6:342–353. doi: 10.1093/cercor/6.3.342. [DOI] [PubMed] [Google Scholar]
- Ploghaus A, Narain C, Beckmann CF, Clare S, Bantick S, Wise R, Matthews PM, Rawlins JN, Tracey I. Exacerbation of pain by anxiety is associated with activity in a hippocampal network. J Neurosci. 2001;21:9896–9903. doi: 10.1523/JNEUROSCI.21-24-09896.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DD, Bush FM, Long S, Harkins SW. A comparison of pain measurement characteristics of mechanical visual analogue and simple numerical rating scales. Pain. 1994;56:217–226. doi: 10.1016/0304-3959(94)90097-3. [DOI] [PubMed] [Google Scholar]
- Rainville P, Duncan GH, Price DD, Carrier B, Bushnell MC. Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science. 1997;277:968–971. doi: 10.1126/science.277.5328.968. [DOI] [PubMed] [Google Scholar]
- Rainville P, Hofbauer RK, Paus T, Duncan GH, Bushnell MC, Price DD. Cerebral mechanisms of hypnotic induction and suggestion. J Cogn Neurosci. 1999;11:110–125. doi: 10.1162/089892999563175. [DOI] [PubMed] [Google Scholar]
- Ralston DD, Ralston HJ., 3rd The terminations of corticospinal tract axons in the macaque monkey. J Comp Neurol. 1985;242:325–337. doi: 10.1002/cne.902420303. [DOI] [PubMed] [Google Scholar]
- Rolls ET, Grabenhorst F. The orbitofrontal cortex and beyond: from affect to decision-making. Prog Neurobiol. 2008;86:216–244. doi: 10.1016/j.pneurobio.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Talbot JD, Duncan GH, Bushnell MC, Boyer M. Diffuse noxious inhibitory controls (DNICs): psychophysical evidence in man for intersegmental suppression of noxious heat perception by cold pressor pain. Pain. 1987;30:221–232. doi: 10.1016/0304-3959(87)91078-5. [DOI] [PubMed] [Google Scholar]
- Terkelsen AJ, Andersen OK, Hansen PO, Jensen TS. Effects of heterotopic- and segmental counter-stimulation on the nociceptive withdrawal reflex in humans. Acta Physiol Scand. 2001;172:211–217. doi: 10.1046/j.1365-201x.2001.00856.x. [DOI] [PubMed] [Google Scholar]
- Tracey I, Ploghaus A, Gati JS, Clare S, Smith S, Menon RS, Matthews PM. Imaging attentional modulation of pain in the periaqueductal gray in humans. J Neurosci. 2002;22:2748–2752. doi: 10.1523/JNEUROSCI.22-07-02748.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valet M, Sprenger T, Boecker H, Willoch F, Rummeny E, Conrad B, Erhard P, Tolle TR. Distraction modulates connectivity of the cingulo-frontal cortex and the midbrain during pain—an fMRI analysis. Pain. 2004;109:399–408. doi: 10.1016/j.pain.2004.02.033. [DOI] [PubMed] [Google Scholar]
- Willer JC. Comparative study of perceived pain and nociceptive flexion reflex in man. Pain. 1977;3:69–80. doi: 10.1016/0304-3959(77)90036-7. [DOI] [PubMed] [Google Scholar]
- Willer JC, Boureau F, Albe-Fessard D. Supraspinal influences on nociceptive flexion reflex and pain sensation in man. Brain Res. 1979;179:61–68. doi: 10.1016/0006-8993(79)90489-x. [DOI] [PubMed] [Google Scholar]
- Willer JC, De Broucker T, Le Bars D. Encoding of nociceptive thermal stimuli by diffuse noxious inhibitory controls in humans. J Neurophysiol. 1989;62:1028–1038. doi: 10.1152/jn.1989.62.5.1028. [DOI] [PubMed] [Google Scholar]
- Willer JC, Bouhassira D, Le Bars D. [Neurophysiological bases of the counterirritation phenomenon: diffuse control inhibitors induced by nociceptive stimulation] Neurophysiol Clin. 1999;29:379–400. doi: 10.1016/S0987-7053(00)87263-9. [DOI] [PubMed] [Google Scholar]
- Wise RG, Lujan BJ, Schweinhardt P, Peskett GD, Rogers R, Tracey I. The anxiolytic effects of midazolam during anticipation to pain revealed using fMRI. Magn Reson Imaging. 2007;25:801–810. doi: 10.1016/j.mri.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Worsley KJ, Evans AC, Marrett S, Neelin P. A three-dimensional statistical analysis for CBF activation studies in human brain. J Cereb Blood Flow Metab. 1992;12:900–918. doi: 10.1038/jcbfm.1992.127. [DOI] [PubMed] [Google Scholar]
- Yezierski RP, Gerhart KD, Schrock BJ, Willis WD. A further examination of effects of cortical stimulation on primate spinothalamic tract cells. J Neurophysiol. 1983;49:424–441. doi: 10.1152/jn.1983.49.2.424. [DOI] [PubMed] [Google Scholar]
- Zhang YQ, Tang JS, Yuan B, Jia H. Inhibitory effects of electrically evoked activation of ventrolateral orbital cortex on the tail-flick reflex are mediated by periaqueductal gray in rats. Pain. 1997;72:127–135. doi: 10.1016/s0304-3959(97)00025-0. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, Meyer CR, Koeppe RA, Stohler CS. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293:311–315. doi: 10.1126/science.1060952. [DOI] [PubMed] [Google Scholar]