

Abstract
Amyotrophic lateral sclerosis (ALS), characterized by degeneration of spinal motor neurons, consists of sporadic and familial forms. One cause of familial ALS is missense mutations in the superoxide dismutase 1 (SOD1) gene. Iron accumulation occurs in the CNS of both forms of ALS; however, its contribution to the pathogenesis of ALS is not known. We examined the role of iron in a transgenic mouse line overexpressing the human SOD1G37R mutant. We show that multiple mechanisms may underlie the iron accumulation in neurons and glia in SOD1G37R transgenic mice. These include dysregulation of proteins involved in iron influx and sensing of intracellular iron; iron accumulation in ventral motor neurons secondary to blockage of anterograde axonal transport; and increased mitochondrial iron load in neurons and glia. We also show that treatment of SOD1G37R mice with an iron chelator extends life span by 5 weeks, accompanied by increased survival of spinal motor neurons and improved locomotor function. These data suggest that iron chelator therapy might be useful for the treatment of ALS.
Keywords: ALS, iron, neurodegeneration, free radicals, spinal cord, neuron death
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by progressive paralysis of skeletal muscles and degeneration of motor neurons in the spinal cord, brainstem, and cortex. Although 90% of the cases are of the sporadic form, the rest being familial, they share similar clinical and pathological features (Boillée et al., 2006). The discovery of mutations in the free radical-scavenging protein copper/zinc superoxide dismutase (SOD1) in 20% of familial cases led to the generation of transgenic mouse models to study the disease (Rosen et al., 1993; Julien and Kriz, 2006). Mutant SOD1 causes cell death by as-yet-unknown toxic gain-of-function effects (Boillée et al., 2006; Julien and Kriz, 2006). Several mechanisms for this toxicity have been suggested, including cytoskeletal abnormalities (Williamson et al., 1998), glutamate toxicity (Rothstein, 1995), protein aggregation (Julien, 2001; Ross and Poirier, 2004), oxidative stress (Barber et al., 2006), mitochondrial dysfunction (Williamson et al., 1998), and extracellular SOD1 toxicity (Urushitani et al., 2007). All of these factors may contribute to the pathogenesis of ALS.
High levels of iron in the CNS of ALS patients have also been reported (Oba et al., 1993; Imon et al., 1995; Kasarskis et al., 1995), but its contribution to the progression of the disease is not known. Iron is essential for life, because it serves as an important cofactor for a number of enzymes involved in DNA, RNA, and protein synthesis, mitochondrial oxidation reactions, and other metabolic processes. However, its redox active nature means that it can generate free radicals and cause cell damage if not properly regulated or shielded. Mutations in the genes of some proteins involved in iron homeostasis are known to cause neurodegeneration in the CNS (Ponka, 2004; Zecca et al., 2004). There is increased oxidative damage in cases of both sporadic and familial ALS, and also in animal models of ALS (Carrí et al., 2003). Although no alteration in total copper was observed in mutant SOD1 transgenic mice (Subramaniam et al., 2002), copper, zinc, and calcium chelators have been shown to be protective (Hottinger et al., 1997; Nagano et al., 2003; Petri et al., 2007). However, there are reports of iron accumulation in the CNS of both familial and sporadic forms of ALS (Oba et al., 1993; Imon et al., 1995; Kasarskis et al., 1995). The mechanisms underlying iron accumulation, and its contribution to the progression of the disease, including effects of iron chelators, are not known. We therefore assessed iron accumulation and changes in iron homeostasis mechanisms in SOD1G37R transgenic mice, and show using an iron selective chelator that iron plays an important role in the progression of the disease.
Materials and Methods
Animals.
Heterozygous transgenic mice expressing G37R mutant SOD1 (SOD1G37R, line 29), which was generated by Wong et al. (1995), were obtained from Dr. Jean-Pierre Julien (Laval University, Quebec City, Canada). Mice were maintained on a C57BL/6 background. All procedures used were approved by the McGill University Animal Care Committee and followed the guidelines of the Canadian Council on Animal Care.
Iron histochemistry.
SOD1G37R and control mice were deeply anesthetized with ketamine/xylazine/acepromazine (50:5:1 mg/ml) and perfused with 0.1 m phosphate buffer, followed by 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.2. Modified Perl's histochemistry was performed on 14-μm-thick cryostat sections of the spinal cords to detect iron accumulation as described previously (Smith et al., 1997). Briefly, sections were incubated with 4% potassium ferrocyanide and 4% HCl, followed by a series of incubations with diaminobenzidine and hydrogen peroxide and counterstained with 0.02% methyl green (all from Sigma-Aldrich).
Ferrozine iron assay.
The ferrozine-based iron assay was used to quantify total iron levels in spinal cord homogenates of SOD1G37R and control mice. The assay was performed as described previously (Riemer et al., 2004). Briefly, the spinal cord was removed after intracardiac perfusion with PBS, the tissue was homogenized in PBS, and the protein concentration was estimated. Homogenates were then treated with acidic KMnO4 to release iron from proteins. Samples were then incubated with iron detection agent (6.5 mm ferrozine, 6.5 mm neocuproine, 2.5 m ammonium acetate, and 1 m ascorbic acid dissolved in water). A colorimetric measurement was used to calculate the amount of iron per milligram of protein by comparing the absorbance to that of a range of standard concentrations of FeCl3.
Quantitative real-time reverse transcription-PCR.
Total RNA was purified from cervical, thoracic, and lumbar segments of the spinal cord using the RNeasy Lipid Tissue kit (QIAGEN), and quantitative real-time reverse transcription (QRT)-PCR was performed using the Brilliant Probe-based QRT-PCR reagents and MX4000 (Stratagene), both following the protocols of the manufacturer. Gene-specific primers and Taqman probes were used as previously described (Jeong and David, 2006). QRT-PCR for peptidylprolyl isomerase A (PPIA) was performed for use as an internal control (Feroze-Merzoug et al., 2002). For data analysis, the sample cycle threshold values were normalized to that of PPIA and expressed as fold increase. Results are shown as the mean relative ratio (fold increase) of mRNA ± SD of the mean from three separate experiments (n = 3). The two-sample Student t test was used to determine statistical significance.
Western blotting.
Spinal cord segments from SOD1G37R and control mice were removed, and total protein was extracted as described previously (Jeong and David, 2003). Proteins were separated by SDS-PAGE, transferred to polyvinylidene fluoride membrane (Millipore), and incubated with rabbit anti-ferroportin (Fpn), rabbit anti-divalent metal transporter 1 (DMT1) (both 1:4000; Alpha Diagnostics; anti-DMT1 recognizes both forms of DMT1), rabbit anti-transferrin receptor 1 (TfR1) (1:500; Zymed), or rabbit anti-ceruloplasmin (Cp) (1:1000; Dako). Blots were washed and incubated with peroxidase-conjugated IgG (1:200,000; Jackson ImmunoResearch) and detected with enhanced chemiluminescence (PerkinElmer). Equal loading of proteins was assessed using rabbit anti-β-actin antibodies (1:400; Sigma-Aldrich).
Double immunofluorescence.
SOD1G37R and control mice were deeply anesthetized as above and perfused with 0.1 m phosphate buffer, followed by 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.2, and 14 μm cryostat sections were obtained. Double immunofluorescence labeling of tissue sections was performed as described previously (Jeong and David, 2006). Briefly, the tissue sections were incubated with PBS containing 2% normal goat serum and 1% ovalbumin to block nonspecific binding of antibodies. This was followed by an overnight incubation with rabbit anti-DMT1 or rabbit anti-Fpn antibody (1:400; both from Alpha Diagnostics) or a rabbit anti-ferritin (1:100; Dako) or a rabbit anti-mitochondrial ferritin antibody (1:100; generated by P. Arosio). After washing, the tissue sections were incubated with either monoclonal anti-GFAP for astrocytes (1:100; Sigma-Aldrich) or NeuN (for neurons; Millipore Bioscience Research Reagents) or Mac-1 (for macrophages; 1:200) as a cell-specific marker. The binding of antibodies was visualized with either fluorescein or rhodamine-conjugated secondary antibodies (1:200; Jackson ImmunoResearch). Sections were also counterstained with 100 ng/ml DAPI (4′-6-diamidino-2-phenylindole) (Vector Laboratories) and viewed with a Zeiss Axioskop 2 Plus microscope. Mitochondrial ferritin staining was quantified using the BioQuant Image analysis system by estimating the number of pixels. Identical thresholds were used to count pixels in two sections of each of the cervical, thoracic, and lumbar regions per animal (n = 4 SOD1G37R; n = 3 wild-type controls). Pixel numbers were then normalized to the area of section analyzed and reported as an arbitrary OD unit.
Immunohistochemistry.
Vectastain ABC kit (Vector Laboratories) was used to detect TfR1 expression because of its low abundance in the CNS. TfR1 was stained in the tissue samples using a monoclonal anti-TfR1 antibody (1:200; Zymed) and a goat anti-mouse biotinylated secondary antibody (1:400; Jackson ImmunoResearch). Binding of the primary antibody was visualized by using DAB (diaminobenzidine) as the chromogen.
Sciatic nerve ligation.
Three-month-old C57BL/6 mice were deeply anesthetized as above, and the left sciatic nerve was cut and ligated at the midthigh level. The right sciatic nerve was exposed and closed to serve as a sham control. After 30 d, animals were perfused with fixative as described above, and Perl's immunohistochemistry for iron was performed to detect iron in the lumbar spinal cord. The large ventral horn motor neurons were examined on both the ligated and control sides.
Gel retardation assay.
A gel retardation assay was performed to assess the interaction between iron regulatory proteins (IRPs) and iron response elements (IREs) following an established technique (Kim and Ponka, 1999; Jeong and David, 2006). Briefly, 30 μg of total protein extract of cervical, thoracic, and lumbar spinal cord from 12-month-old SOD1G37R and wild-type mice was mixed with 32P-labeled ferritin IRE RNA probe (obtained from Dr. K. Pantopoulos, McGill University, Montreal, Quebec, Canada), which was transcribed in vitro from a linearized plasmid template using T7 RNA polymerase. The samples were incubated for 10 min at room temperature with heparin (5 mg/ml) to prevent nonspecific binding. Unbound probe was degraded by incubation for 10 min with RNase T1. The formation of RNA–protein complexes was then detected by gel electrophoresis using 6% non-denaturing polyacrylamide gels. The gels were scanned with a STORM860 (PhosphorImager) and analyzed with ImageQuant for densitometry (GE Healthcare).
Mitochondrial ferritin.
Spinal cords from 12-month-old SOD1G37R transgenic and wild-type mice (n = 3) were processed immediately for mitochondrial purification using the Mitochondria Isolation Kit for Tissue (Pierce) following the manufacturer's protocol. Isolation of purified mitochondria was verified on Western blots with the anti-COX IV antibody (1:1000; Cell Signaling). The mitochondrial preparation was then solubilized in 2% SDS, and 40 μg of the protein samples were loaded onto each lane and separated on 4–12% SDS-PAGE and blotted onto polyvinylidene fluoride membranes. The membranes were incubated with a rabbit anti-mitochondrial ferritin antibody (1:500).
Iron chelator treatments and blood sample analysis.
SOD1G37R mice were treated with the iron selective chelator salicylaldehyde isonicotinoyl hydrazone (SIH) as previously described (Richardson and Ponka, 1998; Klimtova et al., 2003). Briefly, the SIH solution (50 mg/kg) was prepared fresh every time and used within 30 min. Mice (n = 12) were injected intraperitoneally starting either at 8 months of age and treated either once or twice a week. The control group of mice was treated with the vehicle solution (PBS) (n = 18). Analysis of survival was performed blind. The survival of the mice was analyzed by plotting Kaplan–Meier graphs. Two-way ANOVA was used to determine statistical significance. Blood samples from different groups of mice (n = 4 for each group) were collected via the heart at the end of experiments, and blood analysis was done by the McGill University Animal Resources Centre's Diagnostic and Research Support Service.
Histological analysis of motor neuron survival after SIH treatment.
SIH- and vehicle-treated mice were killed at 50 weeks of age (end stage for the vehicle-treated mice) by intracardiac perfusion with 4% paraformaldehyde. The spinal cord segments were removed, and frozen 14-μm-thick tissue sections were obtained. The tissue sections were stained with 0.1% cresyl violet, and the survival of the motor neurons was examined in the lumbar segment (n = 3 per group; 35 sections per animal). The two-sample Student t test was used to determine statistical significance.
Statistical analyses.
Data in all graphs except the Kaplan–Meier graph in Figure 6 are shown as mean ± SEM and assessed using Student's t test. Differences were considered significant at p < 0.05. The Kaplan–Meier survival data was analyzed using the log rank test.
Figure 6.
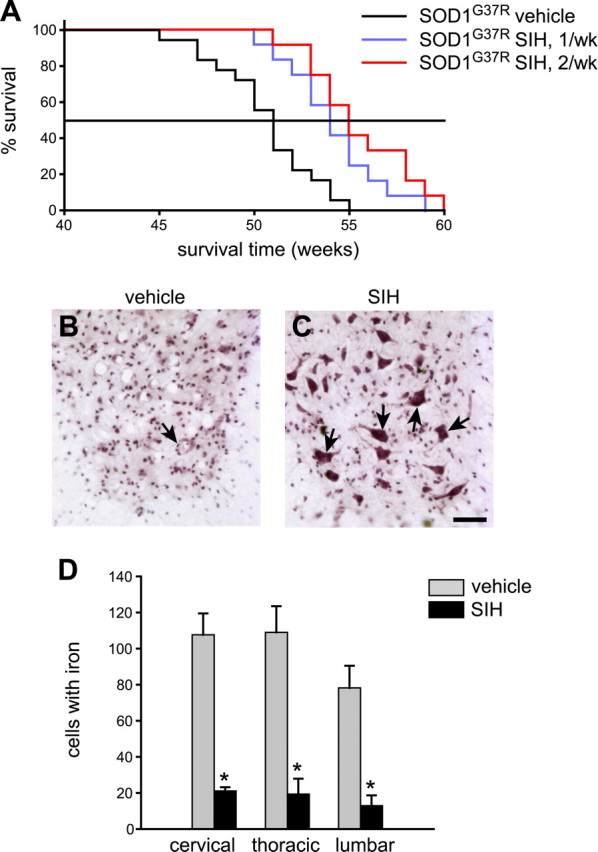
Iron chelator (SIH) treatment extends life span and increases neuronal survival in SOD1G37R mice. A, Kaplan–Meier graph shows the percentage of animals surviving with age. SOD1G37R mice were treated with SIH either once (n = 12) or twice (n = 12) a week starting at 8 months of age. Control SOD1G37R transgenic mice were injected with saline (n = 18). B, C, Representative micrographs of cresyl violet-stained lumbar spinal cord tissue sections from vehicle-treated (B) and SIH-treated (C) SOD1G37R mice at 50 weeks of age. Note the greater numbers of surviving motor neurons in the SIH-treated mouse (arrows point to some of the neurons) compared with the vehicle-treated mouse. Scale bar, 50 μm. D, Quantification of Perl's-stained sections of the spinal cord shows a marked reduction of iron-labeled cells in the ventral region in the cervical, thoracic, and lumbar cord of SIH-treated compared with vehicle-treated SOD1G37R mice (n = 3 per group; *p < 0.05). Error bars indicate SEM.
Results
Two different patterns of iron accumulation in motor neurons and glia
Iron accumulation is seen in the spinal cord of SOD1G37R transgenic mice at 12 months of age. Iron was detected in the cell bodies of the large ventral horn motor neurons (Fig. 1A–C), but not in wild-type mice of the same age (Fig. 1F–H). The iron staining in these neurons was in the form of small, round cytoplasmic inclusions (Fig. 1B,C). Some glial cells in the gray and white matter of the transgenic mice also stained positively for iron, showing a more diffuse type of labeling of the entire cell body (Fig. 1D,E), which was not seen in wild-type controls (Fig. 1I,J). Quantification of the amount of total iron in spinal cord samples taken from 12-month-old mice using a colorometric ferrozine assay showed a 56% increase in SOD1G37R mice (8.69 ± 0.5 nmol/mg) compared with age-matched wild-type controls (5.56 ± 0.3 nmol/mg; p < 0.05). We did not detect iron accumulation in the spinal cord of SOD1G93A transgenic mice, which have a shorter life span of ∼4 months. This suggests that the accumulation of iron in the CNS in this animal model is slow, which is relevant to ALS in humans with a mean age of onset at 50 years.
Figure 1.

Abnormal iron accumulation in neurons and glia of SOD1G37R mice. A, Lumbar ventral horn of 12-month-old SOD1G37R mouse shows iron accumulation in motor neurons seen as brown deposits (arrows). Note the small size of the neurons compared with wild-type mice (F) suggesting neuronal atrophy. B, C, Higher magnification of iron-containing neurons in SOD1G37R mouse (arrows). D, E, Higher magnification micrographs of iron-containing glial cells (arrows) in SOD1G37R mice. Note the homogeneous staining of the glial cells compared with neurons. F, Low-magnification micrograph of ventral horn of lumbar spinal cord of wild-type mouse at 12 months of age shows no iron accumulation. Large motor neurons are indicated by the arrows. G, H, The lack of iron in these neurons at higher magnification is shown. I, J, Higher magnification micrographs show that glia in wild-type mice do not have iron accumulation. Scale bars: A, F, 100 μm; E, J, 25 μm.
Dysregulated expression of iron homeostasis proteins
Transporters and ferroxidases
The mRNA expression of three proteins that transport iron in or out of cells (DMT1, TfR1, and iron exporter Fpn) and two ferroxidases [Cp and Heph (hephaestin)] were assessed in the cervical, thoracic, and lumbar spinal cord by QRT-PCR. This was first assessed at the end stage of the disease (i.e., 12 months of age). A striking finding is the caudal-to-rostral gradient in the mRNA levels of these proteins with the highest levels rostrally in the cervical region (Fig. 2A). DMT1 mRNA transcripts with and without iron response elements (+IRE and −IRE) are found in the CNS. In the cervical region, the increase was predominantly of DMT1 (−IRE) mRNA (5-fold), whereas TfR1, which also imports iron, showed a small increase (1.6-fold) (Fig. 2A). In contrast, in the lumbar region, the mRNA levels of DMT1 (−IRE) and TfR1 were below control levels. The increase in DMT1 (−IRE) mRNA suggests that factors other than cellular iron levels play a role in regulating its expression. However, the small but significant increase in TfR1 mRNA, which contains IREs, indicates that changes in intracellular iron levels also contribute to changes in their expression in some cell types.
Figure 2.
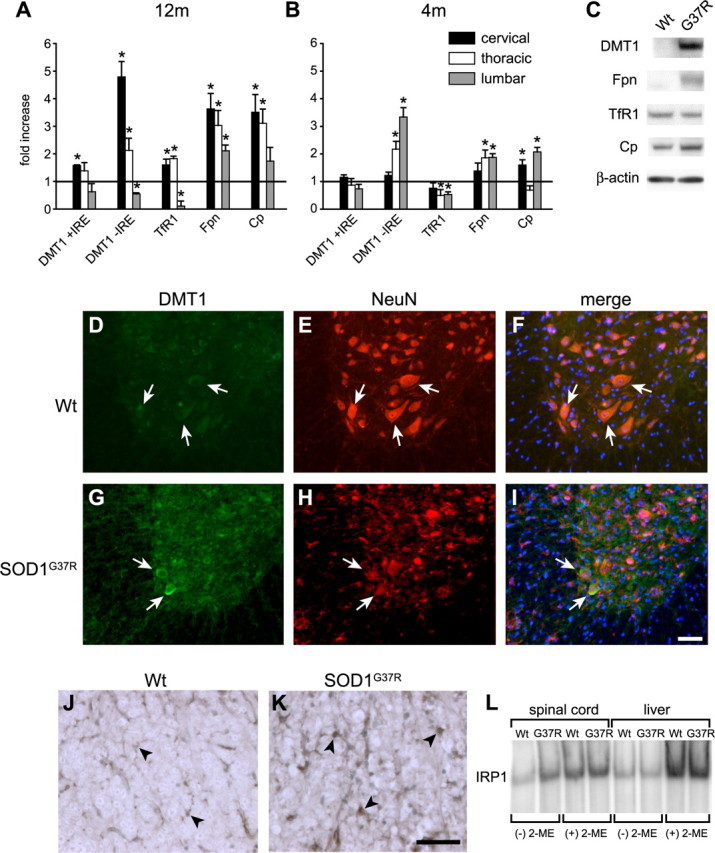
Expression of iron homeostasis proteins in SOD1G37R mice. A, QRT-PCR data showing mRNA levels at 12 months of age (end stage). Note that the increases in DMT1, Fpn, Cp, and TfR1 mRNA are much higher in the cervical cord than in the lumbar region. The increase of DMT1 mRNA is mainly of the −IRE form (*p < 0.05). B, QRT-PCR data at 4 months of age (presymptomatic stage) show that in early stages of the disease DMT1 (−IRE), Fpn, and Cp mRNA are higher in the lumbar than in the cervical region in the spinal cord of SOD1G37R mice. TfR1 expression appears to be reduced in the lumbar and thoracic regions (*p < 0.05). Error bars indicate SEM. C, Western blots of proteins from the cervical cord of 12-month-old mice show that DMT1, Fpn, and Cp are greater in the SOD1G37R than in wild-type mice. There is a greater than twofold increase in DMT1 expression in SOD1G37R mice than in Fpn and Cp. TfR1 did not show a difference in total protein expression in SOD1G37R mice compared with the wild-type mice. β-Actin was used as a loading control. D–I, Double immunofluorescence labeling shows that, in the cervical spinal cord of 12-month-old SOD1G37R transgenic mice, DMT1 expression is upregulated in surviving ventral horn neurons (G, arrows) double-labeled for the neuronal marker NeuN (H, arrows), compared with wild-type mice (D–F). Scale bar, 100 μm. J, K, TfR1 expression is markedly increased in glial cells (K, arrows) in the spinal cord white matter of SOD1G37R transgenic mouse compared with the wild-type control (J, arrows). Scale bar, 50 μm. L, Electron mobility shift assay with protein extracts of spinal cords show that there is an approximately twofold increase in IRP1 binding activity in the SOD1G37R samples (G37R) compared with wild-type (Wt) mice [(−) 2-ME samples]. This increase of IRP1 activity is only seen in the spinal cord but not in the liver samples. There was no detectable IRP2 activity in these tissues. β-Mercaptoethanol (2-ME)-treated samples [(+) 2-ME] were used as loading controls.
The iron exporter Fpn also showed a caudal-to-rostral gradient with ∼3.5-fold increase in the cervical region and a 2-fold increase in the lumbar region (Fig. 2A). In 12-month-old SOD1G37R transgenic mice, a similar caudal-to-rostral pattern was also seen with the mRNA expression of Cp, a ferroxidase that functionally partners with Fpn (Fig. 2A). No changes were seen in the expression of the ferroxidase, hephaestin (data not shown).
The caudal-to-rostral pattern of mRNA expression described above, correlates with the caudal-to-rostral progression of the disease in SOD1G37R transgenic mice. At 12 months of age, neurodegeneration is not fully underway in the cervical and thoracic spinal cord, whereas it is well advanced in the lumbar region. If this were to underlie the caudal-to-rostral pattern of expression, we reasoned that there would be a reverse gradient in mRNA expression of these iron homeostasis proteins at 4 months of age (presymptomatic stage). As expected, at 4 months of age, the mRNA expression of DMT1 (−IRE) and Fpn was highest in the lumbar region [3.5-fold for DMT1 (−IRE) and 2-fold for Fpn] (Fig. 2B) and lowest in the cervical region (both unchanged). Because the relative increase in DMT1 mRNA appears to be higher than that of Fpn, this dysregulation may contribute to an overall increase in iron uptake into cells in the spinal cord of SOD1G37R transgenic mice. At 4 months, Cp was also highest in the lumbar region (Fig. 2B).
The increased expression of DMT1, Fpn, and Cp in the cervical cord at 12 months of age was also confirmed by Western blot analysis (Fig. 2C). The increase in DMT1 was more than twofold greater than that of Fpn and Cp, indicating higher expression of iron influx proteins than efflux proteins. No difference in TfR1 protein expression was detected in SOD1G37R transgenic mice by Western blot. Double immunofluorescence labeling showed that, in 12-month-old SOD1G37R transgenic mice, the expression of DMT1 was increased in motor neurons (Fig. 2D–I) in the ventral horn of the spinal cord, and also in some GFAP+ astrocytes (data not shown), whereas TfR1 expression was increased in glia (Fig. 2J,K).
Iron storage protein ferritin
Ferritin is a sensitive indicator of cellular iron status, and an increase in ferritin reflects iron accumulation. A similar caudal-to-rostral pattern of increasing mRNA expression was also seen with ferritin heavy (FTH) and light (FTL) chains in 12-month-old SOD1G37R transgenic mice (Fig. 3A). In the cervical region, there is a fourfold increase in FTH and a ninefold increase in FTL, whereas their levels in the lumbar region were either reduced (FTH) or unchanged (FTL). Increased cytosolic ferritin expression occurs in glia, mainly microglia and some astrocytes, but not in neurons (Fig. 3B–M). The morphology of iron-positive inclusions in neurons described above, along with the lack of cytosolic ferritin staining in motor neurons suggests that the neuronal iron containing inclusions are not associated with cytosolic ferritin, but are associated with some other cytosolic compartment. These data suggest that, in SOD1G37R mice, neurons and glia might accumulate iron via different mechanisms: glia via TfR1 and ferritin and neurons via DMT1 and other mechanisms. One such mechanism that can influence the accumulation of iron in large motor neurons in SOD1G37R transgenic mice is disruption of axonal transport.
Figure 3.
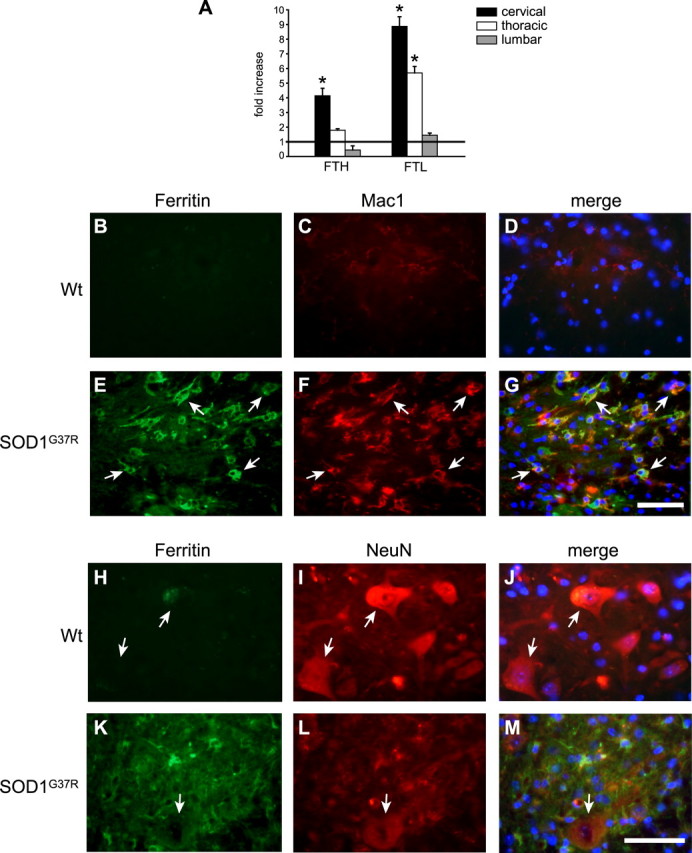
Cytosolic ferritin is increased in the macrophages but not in spinal motor neurons in SOD1G37R mice. A, QRT-PCR data show the differences in FTH and FTL mRNA in the cervical, thoracic, and lumbar cord of SOD1G37R mice at 12 months of age. The values represent fold increase over wild-type controls at the same age (horizontal line at 1). Note that the mRNA expression of the heavy and light chains of ferritin is markedly increased in the cervical than lumbar region (*p < 0.05). Error bars indicate SEM. B–G, Increased ferritin immunoreactivity is seen in Mac-1+ macrophage/microglia in SOD1G37R mice (E–G, arrows) compared with the wild-type control (B–D). No detectable ferritin or Mac1 immunoreactivity is seen in the wild-type animals. H–M, There is no increase in ferritin immunoreactivity in the large surviving motor neurons labeled with NeuN in the spinal cord of SOD1G37R mice (K–M, arrow). The same is the case with the wild-type control mouse (H–J, arrows). Scale bars: G, M, 50 μm.
Changes in SOD1G93A transgenic mice
Interestingly, increases in mRNA expression of iron homeostasis proteins was also seen in the spinal cord at the end stage (4 months) in SOD1G93A transgenic mice, particularly of DMT1 (with and without IRE), and ferritin (heavy and light chains) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This increase in ferritin mRNA suggests an increased iron load, because iron is known to regulate ferritin mRNA transcription (Hintze and Theil, 2006). As mentioned above, this short-lived transgenic mouse line does not show iron accumulation in the spinal cord by Perl's histochemistry. This is in agreement with our previous work on the aging brain, which showed that increases in ferritin mRNA are a more sensitive indicator of increased iron in the CNS than iron detectable by Perl's histochemistry.
Iron accumulation induced by disruption of anterograde axonal transport
Because alteration of axonal transport is reported in ALS and in the SOD1 transgenic mice (Sasaki and Iwata, 1996; Zhang et al., 1997; Williamson and Cleveland, 1999), we assessed whether disruption of axonal transport would result in accumulation of iron in neuronal cell bodies. To test this, we ligated the sciatic nerve unilaterally in adult wild-type mice. After 30 d, the spinal cord was assessed for iron accumulation using Perl's histochemistry. Large ventral horn motor neurons on the ligated side showed increased granular iron accumulation in the cell bodies compared with neurons in the uninjured side (Fig. 4A–C). These data suggest that blockade of axonal transport, which is known to occur in the experimental model of ALS (Zhang et al., 1997), is also likely to contribute to the iron accumulation in the motor neurons in SOD1G37R transgenic mice. This effect may be attributable to impairment of iron trafficking along the axon or attributable to retrograde neuronal changes caused by axonal damage.
Figure 4.

Blocking axonal transport causes iron accumulation in motor neurons in wild-type mice. A, B, Enhanced Perl's histochemical labeling showing granular iron deposits (arrows) in the ventral horn motor neurons on the side in which the sciatic nerve was cut and ligated. Cells were counterstained with methyl green. Scale bar, 25 μm. C, Quantification of number of iron granules in ventral horn motor neurons show that there is a marked increase in the percentage of neurons containing more than five iron granules on the ligated side compared with the uninjured side (*p < 0.05). Error bars indicate SEM.
Changes in the iron regulatory system
Another mechanism that increases iron uptake into cells is binding of IRP to IREs, which regulate the mRNA levels of several iron homeostasis proteins including TfR1. We found an increase of TfR1 mRNA in the cervical and thoracic regions in 12-month-old SOD1G37R transgenic mice. Recent in vitro studies have shown that transfection of mutant SOD1 (SOD1G93A) into a human astrocytoma cell line led to an increase in IRP1 activity and TfR1 expression (Danzeisen et al., 2006). Electromobility gel shift assays to assess IRP1 binding activity in the spinal cord of 12-month-old SOD1G37R transgenic mice revealed a twofold increase in the spinal cord of SOD1G37R transgenic mice compared with wild-type controls (Fig. 2L). This change in the IRP1 binding activity is specific for spinal cords from SOD1 transgenic mice, because no difference is seen in the liver. Of interest is that the twofold increase in IRP1 activity in the cervical and thoracic region is correlated with an expected increase in TfR1 mRNA expression. However, in the lumbar region, there is an even greater increase in IRP1 activity (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), but this is associated with a significant reduction in TfR1 mRNA levels (Fig. 2A). This dysregulation of IPR1 activity and TfR1 mRNA expression may be attributable to the effects of inflammation as discussed later.
Increase in mitochondrial iron in SOD1G37R mice
A number of lines of evidence indicate mitochondrial abnormalities in the spinal cord in ALS and in transgenic mice expressing SOD1 mutants (Wong et al., 1995; Kong and Xu, 1998; Jaarsma et al., 2000; Bendotti et al., 2001; Hervias et al., 2006). Mutant SOD1 has been shown to preferentially associate with spinal cord mitochondria via tight association or cross-linking to the outer mitochondrial membrane (Liu et al., 2004). Additionally, the SOD1G37R mutant protein was also found to be associated with the inner mitochondrial membrane (Liu et al., 2004). Such association of the mutant SOD1 protein with mitochondrial membranes is thought to lead to disruption of mitochondrial function (Liu et al., 2004). Much of the iron that normally enters cells is likely to be transported to the mitochondria, because it is the major site of generation of iron–sulfur clusters (Napier et al., 2005; Rouault and Tong, 2005; Lill et al., 2006). Iron–sulfur clusters, which serve as important cofactors, are inserted into mitochondrial proteins such as those of the oxidative chain or exported into the cytosol for incorporation into proteins and enzymes that play a variety of metabolic functions (Rouault and Tong, 2005; Lill et al., 2006). In vitro studies have shown that overexpression of SOD1G37R causes increases in mitochondrial superoxide and a shift in the redox potential (Ferri et al., 2006; Zimmerman et al., 2007). Furthermore, in vitro work has also shown that oxidation of iron–sulfur clusters by superoxide can cause release of iron (Liochev and Fridovich, 1994). Therefore, breakdown of iron–sulfur clusters in the mitochondria could lead to increased iron in mitochondria. Although mitochondrial ferritin (MtF) does not contain IREs, overexpression of MtF was shown to cause cytosolic iron deprivation and to trap iron in the mitochondria (Nie et al., 2005). We therefore assessed whether there is an increase in MtF by Western blot analysis of mitochondrial preparations purified by subcellular fractionation from the spinal cord and kidney of 12-month-old SOD1G37R and wild-type mice. There was a twofold increase in MtF in the spinal cord but not the kidney of SOD1G37R mice compared with the wild type (Fig. 5A,B). Additionally, immunofluorescence staining showed a marked increase in MtF in the ventral horn motor neurons and astrocytes in 12-month-old SOD1G37R transgenic mice but not wild-type mice of the same age (Fig. 5D–O). Quantification of this immunofluorescence labeling showed a caudal-to-rostral gradient in the increase in MtF in SOD1G37R transgenic mice (Fig. 5C). These data suggest that there is a significant increase in mitochondrial iron in motor neurons and glia in the spinal cord in this mouse model of ALS. Increased iron in the mitochondria could lead to neurodegeneration as occurs in Fredrick's ataxia, which results from mutations in the mitochondrial protein frataxin (Campuzano et al., 1996).
Figure 5.
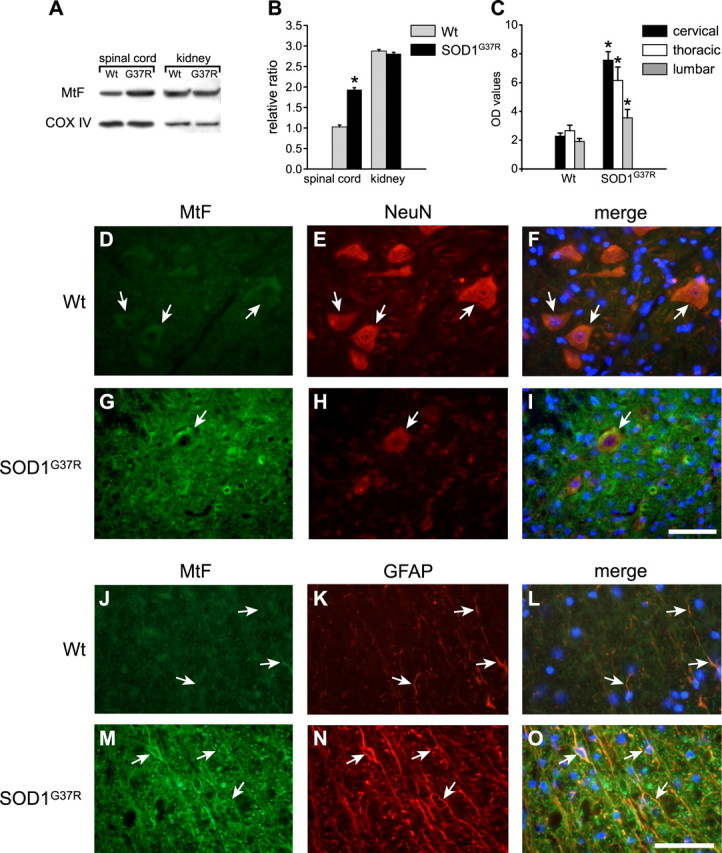
Mitochondrial ferritin is increased in motor neurons and astrocytes in SOD1G37R transgenic mice. A, Western blot for mitochondrial ferritin using purified mitochondria shows upregulation of MtF in SOD1G37R mice compared with wild-type mice (Wt). No difference in MtF is detected in mitochondrial preparations from kidney. COX IV was used for loading control. B, Quantification of the Western blot data shown in A (*p < 0.05; n = 3). C, Quantification of immunofluorescence labeling for MtF shows a caudal-to-rostral increase in MtF in cross-sections of the spinal cord of SOD1G37R transgenic mice compared with wild-type controls. Data represents OD values (mean ± SEM) (n = 4). D–I, Micrographs show that surviving motor neurons labeled with NeuN (H, arrow) in the cervical spinal cord of SOD1G37R transgenic mice show increased immunoreactivity for MtF (G, arrow) compared with wild-type control mice (D–F, arrows). J–O, Astrocytes in the transgenic mouse labeled with anti-GFAP (N) also show marked increase in MtF immunoreactivity (M–O, arrows) compared with wild-type controls (J–L). Note the increased GFAP immunoreactivity in the SOD1G37R transgenic. Scale bars: I, O, 50 μm.
Protective effects of iron chelator in vivo
Because there is disruption of iron homeostasis and iron accumulation in SOD1G37R transgenic mice, we assessed the effects of an iron chelator on the progression of the disease. The high-affinity, lipophilic iron chelator SIH (Klimtova et al., 2003; Cheah et al., 2006) administered intraperitoneally starting at 8 months of age, once a week, increased the mean life span by 3.5 weeks (Fig. 6A) (controls, 50.5 ± 2.6; SIH, 54.08 ± 2.5 weeks; log rank test; p = 0.002). Furthermore, when SIH was given twice a week starting at 8 months of age, the mean life span was increased by 5 weeks, with this group showing a lower p value of 0.00009 (Fig. 6A) (SIH, 55.5 ± 2.7; log rank test). Histological analysis showed that there were more surviving neurons (17 ± 4/105 μm2) at 50 weeks in SIH-treated SOD1G37R transgenic mice compared with vehicle-treated SOD1G37R mice (2.24 ± 1/105 μm2; p < 0.001) at the same age (Fig. 6B,C). Furthermore, SIH treatment also resulted in a fivefold to sixfold reduction in the number of iron-containing cells in Perl's stained tissue sections of the spinal cord (Fig. 6D). There was also a precipitous drop in body weight of vehicle-treated mice between 47.5 and 50 weeks of age (Fig. 7). In contrast, mice treated twice a week with SIH showed no changes in body weight during this period, indicating a delay in the onset of disease. Mice treated once a week with SIH showed body weight changes that lay in-between the controls and the twice-a-week treatment group (Fig. 7). The supplemental videos (available at www.jneurosci.org as supplemental material) show that the twice-a-week SIH-treated mice have marked improvement in locomotor function. This 4 month treatment was well tolerated, because these mice did not show hematological signs of anemia (supplemental Table 1, available at www.jneurosci.org as supplemental material). These data provide strong evidence that the disruption of iron homeostasis and increased iron accumulation in the CNS contributes to the progression of the disease in SOD1G37R transgenic mice.
Figure 7.

Changes in body weight measured twice a week up to 50 weeks of age. Note the sharp drop in body weight of vehicle-treated SOD1G37R mice beyond 47.5 weeks. SOD1G37R mice treated twice a week with SIH starting at 8 months of age do not show any loss of body weight during this period. SOD1G37R mice treated once a week with SIH show values in-between the vehicle-treated control and the twice-a-week SIH treatment groups. Values represent mean of 12 mice.
Discussion
There is evidence from the clinical literature of iron accumulation in the CNS of patients with ALS (Oba et al., 1993; Imon et al., 1995; Kasarskis et al., 1995). Although this has been known for some time, direct evidence of a role for iron in ALS has not been demonstrated thus far. Our data provide clear evidence that iron accumulation occurs in both neurons and glia but that different molecular mechanisms may contribute to iron accumulation in these two cell types. In glia, the increase in TfR1 that is likely to be mediated by the increase in IRP1 binding activity may underlie the influx of iron into these cells. Normally, IRP1 would be expected to concomitantly reduce the expression of ferritin (Pantopoulos, 2004). However, cytosolic ferritin expression goes up mainly in microglia, which could acquire iron from phagocytosis of dead and dying cells. Cytosolic ferritin mRNA expression is also known to be increased by proinflammatory cytokines or nitric oxide (Torti et al., 1988; Wei et al., 1990; Miller et al., 1991; Kwak et al., 1995; Tsuji et al., 2000; Mikhael et al., 2006). Because inflammation and increased cytokine expression have been reported in the spinal cord of SOD1 transgenic mice (Nguyen et al., 2001), this may underlie the increase of ferritin in microglia and increase their capacity to store iron. However, prolonged increases in iron could lead to loss of these cells. We have shown previously that astrocytes in Cp null mice, which lose their ability to efflux iron, markedly increase their expression of ferritin; however, they eventually die from iron-mediated toxicity (Jeong and David, 2006).
In contrast to glia, neurons in the SOD1G37R transgenic mice show increased expression of DMT1. The QRT-PCR data show that the increase in DMT1 mRNA is mainly of the −IRE transcripts, which are not regulated by changes in iron levels but by other factors such as proinflammatory cytokines that are known to be increased in the spinal cord of SOD1G37R transgenic mice (Elliott, 2001; Ghezzi and Mennini, 2001; Hensley et al., 2002, 2003). Although the iron efflux transporter Fpn is also increased in the transgenic mice, the overall level of expression of Fpn mRNA and protein appears to be twofold lower than that of DMT1. It is therefore possible that the greater increase in DMT1 might contribute, in part, to the iron accumulation in neurons. However, direct evidence for this is lacking at present. Another factor that may contribute to iron accumulation in the motor neurons in the ventral horn may be a consequence of blockage of axonal transport. A variety of structural, metabolic, and synaptic elements are transported down the axon by anterograde transport. There is some indirect evidence to suggest that iron may also be transported down the axon by anterograde transport (Wu et al., 2004; Zhang et al., 2005). We now provide evidence that ligation of the sciatic nerve in wild-type mice results in iron accumulation in the cell bodies of the ventral horn motor neurons. This may be either attributable to the blockade of anterograde transport of iron and its return and accumulation in the neuronal cell bodies or attributable to stress-related retrograde changes in the neuronal cell body as a result of nerve ligation. In either case, such mechanisms may also contribute to iron accumulation in ventral motor neurons in SOD1 transgenic mice and in ALS in which axonal aggregate formation disrupts axonal transport (Sasaki and Iwata, 1996; Zhang et al., 1997; Williamson and Cleveland, 1999). Therefore, altered axonal transport may contribute to the iron accumulation seen in neurons in ALS.
Mutant SOD1 can form nonsoluble aggregates and specifically bind to the mitochondrial membrane in the spinal cord (Liu et al., 2004). These aggregates are thought to disrupt the mitochondrial import machinery and lead to mitochondrial dysfunction that may contribute to the pathogenesis of the disease. Our current work indicates that this may also involve disruption of mitochondrial iron homeostasis leading to iron accumulation in mitochondria and iron-mediated toxicity. Mitochondria are the main sites of generation of iron–sulfur clusters, which are incorporated into a variety of proteins and serve as important cofactors for numerous enzymes (Kispal et al., 1999; Lange et al., 2000). One important Fe–S cluster-containing protein that is involved in maintaining iron homeostasis is IRP1, which is a bifunctional protein that also serves as cytosolic aconitase. When iron is low, breakdown of the Fe–S cluster in aconitase causes a conformational change that transforms it to IRP1, which binds to IREs. Binding of IRP1 to IRE causes more Fe influx into cells by stabilizing the mRNA of the iron importer TfR1. Disruption of mitochondrial Fe–S assembly or blockage of transport across the mitochondrial membrane would cause deprivation of Fe–S clusters in the cytosol and generate constitutively active IRP1 in its IRE binding form and lead to faulty iron sensing. This may underlie the iron accumulation in glia in the cervical and thoracic regions in SOD1G37R transgenic mice at 12 months of age, which is associated with an increase in IRP1 activity and increased TfR1 expression. In contrast, in the lumbar cord in these transgenic mice, the even greater IRP1 activity is associated with a significant reduction in TfR1 mRNA. It should be noted in this context that the inflammation and degenerative changes are most severe in the lumbar region and is progressively less more rostrally. It is possible therefore that dysregulation of IPR1 activity and TfR1 mRNA expression in the lumbar region may be attributable to inflammation-mediated upregulation of hepcidin (Oates and Ahmed, 2007; Wang et al., 2008), which has been shown to downregulate TfR1 mRNA independent of IRE/IRP binding activity (Martin et al., 2004).
Overexpression of SOD1G37R can also cause an increase in mitochondrial superoxide levels and shift in the redox potentials in vitro (Ferri et al., 2006; Zimmerman et al., 2007). Moreover, in vitro studies have shown that superoxide can cause oxidation of Fe–S clusters and cause release of iron (Liochev and Fridovich, 1994). Therefore, breakdown of Fe–S clusters in the mitochondria can cause accumulation of iron in mitochondria. This increased mitochondrial iron might be incorporated into mitochondrial ferritin, which we have detected is increased in the spinal cord of SOD1G37R mice. In addition, superoxide-mediated breakdown of Fe–S could also contribute to the increased IRP1 activity we have detected. This loss in the ability to sense cellular iron levels would lead to more iron uptake and iron accumulation in cells.
Treatment with the iron chelator provides strong evidence of the important role of iron-mediated toxicity in the progression of the disease. The iron chelator treatment led to increased lifespan, improved survival of ventral horn motor neurons, and reduced iron burden in the spinal cord. The enhancement of life span by 5 weeks in the slow progressing SOD1G37R transgenic line compares well with other approaches used in the SOD1G37R mice in which a single therapy was tested (Kriz et al., 2002, 2003). Our work now provides a strong rationale for the inclusion of iron chelator therapy for the treatment of ALS.
Footnotes
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR) (S.D.). S.Y.J. was supported by a studentship from the CIHR Strategic Training Program in Neuroinflammation and a McGill University Graduate Studentship; K.S. was also supported by a studentship from the CIHR Strategic Training Program in Neuroinflammation and is currently the recipient of a Multiple Sclerosis Society of Canada Studentship. We thank Hiba Kazak for technical help and Margaret Attiwell for preparing the illustrations.
References
- Barber et al., 2006.Barber SC, Mead RJ, Shaw PJ. Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim Biophys Acta. 2006;1762:1051–1067. doi: 10.1016/j.bbadis.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Bendotti et al., 2001.Bendotti C, Calvaresi N, Chiveri L, Prelle A, Moggio M, Braga M, Silani V, De Biasi S. Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J Neurol Sci. 2001;191:25–33. [Google Scholar]
- Boillée et al., 2006.Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Campuzano et al., 1996.Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- Carrí et al., 2003.Carrí MT, Ferri A, Cozzolino M, Calabrese L, Rotilio G. Neurodegeneration in amyotrophic lateral sclerosis: the role of oxidative stress and altered homeostasis of metals. Brain Res Bull. 2003;61:365–374. doi: 10.1016/s0361-9230(03)00179-5. [DOI] [PubMed] [Google Scholar]
- Cheah et al., 2006.Cheah JH, Kim SF, Hester LD, Clancy KW, Patterson SE, 3rd, Papadopoulos V, Snyder SH. NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron. 2006;51:431–440. doi: 10.1016/j.neuron.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzeisen et al., 2006.Danzeisen R, Achsel T, Bederke U, Cozzolino M, Crosio C, Ferri A, Frenzel M, Gralla EB, Huber L, Ludolph A, Nencini M, Rotilio G, Valentine JS, Carrì MT. Superoxide dismutase 1 modulates expression of transferrin receptor. J Biol Inorg Chem. 2006;11:489–498. doi: 10.1007/s00775-006-0099-4. [DOI] [PubMed] [Google Scholar]
- Elliott, 2001.Elliott JL. Cytokine upregulation in a murine model of familial amyotrophic lateral sclerosis. Brain Res Mol Brain Res. 2001;95:172–178. doi: 10.1016/s0169-328x(01)00242-x. [DOI] [PubMed] [Google Scholar]
- Feroze-Merzoug et al., 2002.Feroze-Merzoug F, Berquin IM, Dey J, Chen YQ. Peptidylprolyl isomerase A (PPIA) as a preferred internal control over GAPDH and beta-actin in quantitative RNA analyses. Biotechniques. 2002;32:776–778. 780, 782. doi: 10.2144/02324st03. [DOI] [PubMed] [Google Scholar]
- Ferri et al., 2006.Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, Gralla EB, Rotilio G, Valentine JS, Carrì MT. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci U S A. 2006;103:13860–13865. doi: 10.1073/pnas.0605814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi and Mennini, 2001.Ghezzi P, Mennini T. Tumor necrosis factor and motoneuronal degeneration: an open problem. Neuroimmunomodulation. 2001;9:178–182. doi: 10.1159/000049024. [DOI] [PubMed] [Google Scholar]
- Hensley et al., 2002.Hensley K, Floyd RA, Gordon B, Mou S, Pye QN, Stewart C, West M, Williamson K. Temporal patterns of cytokine and apoptosis-related gene expression in spinal cords of the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. J Neurochem. 2002;82:365–374. doi: 10.1046/j.1471-4159.2002.00968.x. [DOI] [PubMed] [Google Scholar]
- Hensley et al., 2003.Hensley K, Fedynyshyn J, Ferrell S, Floyd RA, Gordon B, Grammas P, Hamdheydari L, Mhatre M, Mou S, Pye QN, Stewart C, West M, West S, Williamson KS. Message and protein-level elevation of tumor necrosis factor alpha (TNF alpha) and TNF alpha-modulating cytokines in spinal cords of the G93A-SOD1 mouse model for amyotrophic lateral sclerosis. Neurobiol Dis. 2003;14:74–80. doi: 10.1016/s0969-9961(03)00087-1. [DOI] [PubMed] [Google Scholar]
- Hervias et al., 2006.Hervias I, Beal MF, Manfredi G. Mitochondrial dysfunction and amyotrophic lateral sclerosis. Muscle Nerve. 2006;33:598–608. doi: 10.1002/mus.20489. [DOI] [PubMed] [Google Scholar]
- Hintze and Theil, 2006.Hintze KJ, Theil EC. Cellular regulation and molecular interactions of the ferritins. Cell Mol Life Sci. 2006;63:591–600. doi: 10.1007/s00018-005-5285-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottinger et al., 1997.Hottinger AF, Fine EG, Gurney ME, Zurn AD, Aebischer P. The copper chelator d-penicillamine delays onset of disease and extends survival in a transgenic mouse model of familial amyotrophic lateral sclerosis. Eur J Neurosci. 1997;9:1548–1551. doi: 10.1111/j.1460-9568.1997.tb01511.x. [DOI] [PubMed] [Google Scholar]
- Imon et al., 1995.Imon Y, Yamaguchi S, Yamamura Y, Tsuji S, Kajima T, Ito K, Nakamura S. Low intensity areas observed on T2-weighted magnetic resonance imaging of the cerebral cortex in various neurological diseases. J Neurol Sci. 1995;134(Suppl):27–32. doi: 10.1016/0022-510x(95)00205-g. [DOI] [PubMed] [Google Scholar]
- Jaarsma et al., 2000.Jaarsma D, Haasdijk ED, Grashorn JA, Hawkins R, van Duijn W, Verspaget HW, London J, Holstege JC. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis. 2000;7:623–643. doi: 10.1006/nbdi.2000.0299. [DOI] [PubMed] [Google Scholar]
- Jeong and David, 2003.Jeong SY, David S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J Biol Chem. 2003;278:27144–27148. doi: 10.1074/jbc.M301988200. [DOI] [PubMed] [Google Scholar]
- Jeong and David, 2006.Jeong SY, David S. Age-related changes in iron homeostasis and cell death in the cerebellum of ceruloplasmin-deficient mice. J Neurosci. 2006;26:9810–9819. doi: 10.1523/JNEUROSCI.2922-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien, 2001.Julien JP. Amyotrophic lateral sclerosis. Unfolding the toxicity of the misfolded. Cell. 2001;104:581–591. doi: 10.1016/s0092-8674(01)00244-6. [DOI] [PubMed] [Google Scholar]
- Julien and Kriz, 2006.Julien JP, Kriz J. Transgenic mouse models of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:1013–1024. doi: 10.1016/j.bbadis.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Kasarskis et al., 1995.Kasarskis EJ, Tandon L, Lovell MA, Ehmann WD. Aluminum, calcium, and iron in the spinal cord of patients with sporadic amyotrophic lateral sclerosis using laser microprobe mass spectroscopy: a preliminary study. J Neurol Sci. 1995;130:203–208. doi: 10.1016/0022-510x(95)00037-3. [DOI] [PubMed] [Google Scholar]
- Kim and Ponka, 1999.Kim S, Ponka P. Control of transferrin receptor expression via nitric oxide-mediated modulation of iron-regulatory protein 2. J Biol Chem. 1999;274:33035–33042. doi: 10.1074/jbc.274.46.33035. [DOI] [PubMed] [Google Scholar]
- Kispal et al., 1999.Kispal G, Csere P, Prohl C, Lill R. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 1999;18:3981–3989. doi: 10.1093/emboj/18.14.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimtova et al., 2003.Klimtova I, Simunek T, Mazurova Y, Kaplanova J, Sterba M, Hrdina R, Gersl V, Adamcova M, Ponka P. A study of potential toxic effects after repeated 10-week administration of a new iron chelator—salicylaldehyde isonicotinoyl hydrazone (SIH) to rabbits. Acta Medica (Hradec Kralove) 2003;46:163–170. [PubMed] [Google Scholar]
- Kong and Xu, 1998.Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz et al., 2002.Kriz J, Nguyen MD, Julien JP. Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2002;10:268–278. doi: 10.1006/nbdi.2002.0487. [DOI] [PubMed] [Google Scholar]
- Kriz et al., 2003.Kriz J, Gowing G, Julien JP. Efficient three-drug cocktail for disease induced by mutant superoxide dismutase. Ann Neurol. 2003;53:429–436. doi: 10.1002/ana.10500. [DOI] [PubMed] [Google Scholar]
- Kwak et al., 1995.Kwak EL, Larochelle DA, Beaumont C, Torti SV, Torti FM. Role for NF-kappa B in the regulation of ferritin H by tumor necrosis factor-alpha. J Biol Chem. 1995;270:15285–15293. doi: 10.1074/jbc.270.25.15285. [DOI] [PubMed] [Google Scholar]
- Lange et al., 2000.Lange H, Kaut A, Kispal G, Lill R. A mitochondrial ferredoxin is essential for biogenesis of cellular iron-sulfur proteins. Proc Natl Acad Sci U S A. 2000;97:1050–1055. doi: 10.1073/pnas.97.3.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill et al., 2006.Lill R, Dutkiewicz R, Elsässer HP, Hausmann A, Netz DJ, Pierik AJ, Stehling O, Urzica E, Mühlenhoff U. Mechanisms of iron-sulfur protein maturation in mitochondria, cytosol and nucleus of eukaryotes. Biochim Biophys Acta. 2006;1763:652–667. doi: 10.1016/j.bbamcr.2006.05.011. [DOI] [PubMed] [Google Scholar]
-
Liochev and Fridovich, 1994.Liochev SI, Fridovich I. The role of
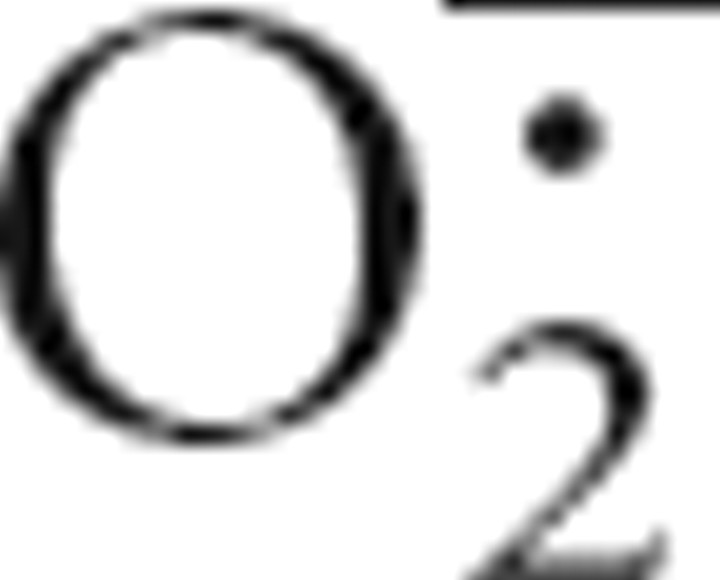 in the production of HȮ: in vitro and in vivo. Free Radic Biol Med. 1994;16:29–33. doi: 10.1016/0891-5849(94)90239-9. [DOI] [PubMed] [Google Scholar]
in the production of HȮ: in vitro and in vivo. Free Radic Biol Med. 1994;16:29–33. doi: 10.1016/0891-5849(94)90239-9. [DOI] [PubMed] [Google Scholar] - Liu et al., 2004.Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brännström T, Gredal O, Wong PC, Williams DS, Cleveland DW. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Martin et al., 2004.Martin ME, Nicolas G, Hetet G, Vaulont S, Grandchamp B, Beaumont C. Transferrin receptor 1 mRNA is downregulated in placenta of hepcidin transgenic embryos. FEBS Lett. 2004;574:187–191. doi: 10.1016/j.febslet.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Mikhael et al., 2006.Mikhael M, Kim SF, Schranzhofer M, Soe-Lin S, Lin SS, Sheftel AD, Mullner EW, Ponka P. Iron regulatory protein-independent regulation of ferritin synthesis by nitrogen monoxide. FEBS J. 2006;273:3828–3836. doi: 10.1111/j.1742-4658.2006.05390.x. [DOI] [PubMed] [Google Scholar]
- Miller et al., 1991.Miller LL, Miller SC, Torti SV, Tsuji Y, Torti FM. Iron-independent induction of ferritin H chain by tumor necrosis factor. Proc Natl Acad Sci U S A. 1991;88:4946–4950. doi: 10.1073/pnas.88.11.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano et al., 2003.Nagano S, Fujii Y, Yamamoto T, Taniyama M, Fukada K, Yanagihara T, Sakoda S. The efficacy of trientine or ascorbate alone compared to that of the combined treatment with these two agents in familial amyotrophic lateral sclerosis model mice. Exp Neurol. 2003;179:176–180. doi: 10.1016/s0014-4886(02)00014-6. [DOI] [PubMed] [Google Scholar]
- Napier et al., 2005.Napier I, Ponka P, Richardson DR. Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood. 2005;105:1867–1874. doi: 10.1182/blood-2004-10-3856. [DOI] [PubMed] [Google Scholar]
- Nguyen et al., 2001.Nguyen MD, Julien JP, Rivest S. Induction of proinflammatory molecules in mice with amyotrophic lateral sclerosis: no requirement for proapoptotic interleukin-1beta in neurodegeneration. Ann Neurol. 2001;50:630–639. doi: 10.1002/ana.1256. [DOI] [PubMed] [Google Scholar]
- Nie et al., 2005.Nie G, Sheftel AD, Kim SF, Ponka P. Overexpression of mitochondrial ferritin causes cytosolic iron depletion and changes cellular iron homeostasis. Blood. 2005;105:2161–2167. doi: 10.1182/blood-2004-07-2722. [DOI] [PubMed] [Google Scholar]
- Oates and Ahmed, 2007.Oates PS, Ahmed U. Molecular regulation of hepatic expression of iron regulatory hormone hepcidin. J Gastroenterol Hepatol. 2007;22:1378–1387. doi: 10.1111/j.1440-1746.2007.04950.x. [DOI] [PubMed] [Google Scholar]
- Oba et al., 1993.Oba H, Araki T, Ohtomo K, Monzawa S, Uchiyama G, Koizumi K, Nogata Y, Kachi K, Shiozawa Z, Kobayashi M. Amyotrophic lateral sclerosis: T2 shortening in motor cortex at MR imaging. Radiology. 1993;189:843–846. doi: 10.1148/radiology.189.3.8234713. [DOI] [PubMed] [Google Scholar]
- Pantopoulos, 2004.Pantopoulos K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann N Y Acad Sci. 2004;1012:1–13. doi: 10.1196/annals.1306.001. [DOI] [PubMed] [Google Scholar]
- Petri et al., 2007.Petri S, Calingasan NY, Alsaied OA, Wille E, Kiaei M, Friedman JE, Baranova O, Chavez JC, Beal MF. The lipophilic metal chelators DP-109 and DP-460 are neuroprotective in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem. 2007;102:991–1000. doi: 10.1111/j.1471-4159.2007.04604.x. [DOI] [PubMed] [Google Scholar]
- Ponka, 2004.Ponka P. Hereditary causes of disturbed iron homeostasis in the central nervous system. Ann N Y Acad Sci. 2004;1012:267–281. doi: 10.1196/annals.1306.022. [DOI] [PubMed] [Google Scholar]
- Richardson and Ponka, 1998.Richardson DR, Ponka P. Pyridoxal isonicotinoyl hydrazone and its analogs: potential orally effective iron-chelating agents for the treatment of iron overload disease. J Lab Clin Med. 1998;131:306–315. doi: 10.1016/s0022-2143(98)90180-9. [DOI] [PubMed] [Google Scholar]
- Riemer et al., 2004.Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Dringen R. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal Biochem. 2004;331:370–375. doi: 10.1016/j.ab.2004.03.049. [DOI] [PubMed] [Google Scholar]
- Rosen et al., 1993.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van Den Bergh R, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Ross and Poirier, 2004.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Rothstein, 1995.Rothstein JD. Excitotoxicity and neurodegeneration in amyotrophic lateral sclerosis. Clin Neurosci. 1995;3:348–359. [PubMed] [Google Scholar]
- Rouault and Tong, 2005.Rouault TA, Tong WH. Iron-sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat Rev Mol Cell Biol. 2005;6:345–351. doi: 10.1038/nrm1620. [DOI] [PubMed] [Google Scholar]
- Sasaki and Iwata, 1996.Sasaki S, Iwata M. Impairment of fast axonal transport in the proximal axons of anterior horn neurons in amyotrophic lateral sclerosis. Neurology. 1996;47:535–540. doi: 10.1212/wnl.47.2.535. [DOI] [PubMed] [Google Scholar]
- Smith et al., 1997.Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam et al., 2002.Subramaniam JR, Lyons WE, Liu J, Bartnikas TB, Rothstein J, Price DL, Cleveland DW, Gitlin JD, Wong PC. Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat Neurosci. 2002;5:301–307. doi: 10.1038/nn823. [DOI] [PubMed] [Google Scholar]
- Torti et al., 1988.Torti SV, Kwak EL, Miller SC, Miller LL, Ringold GM, Myambo KB, Young AP, Torti FM. The molecular cloning and characterization of murine ferritin heavy chain, a tumor necrosis factor-inducible gene. J Biol Chem. 1988;263:12638–12644. [PubMed] [Google Scholar]
- Tsuji et al., 2000.Tsuji Y, Ayaki H, Whitman SP, Morrow CS, Torti SV, Torti FM. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol Cell Biol. 2000;20:5818–5827. doi: 10.1128/mcb.20.16.5818-5827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urushitani et al., 2007.Urushitani M, Ezzi SA, Julien JP. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2007;104:2495–2500. doi: 10.1073/pnas.0606201104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang et al., 2008.Wang Q, Du F, Qian ZM, Ge XH, Zhu L, Yung WH, Yang L, Ke Y. Lipopolysaccharide induces a significant increase in expression of iron regulatory hormone hepcidin in the cortex and substantia nigra in rat brain. Endocrinology. 2008;149:3920–3925. doi: 10.1210/en.2007-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei et al., 1990.Wei Y, Miller SC, Tsuji Y, Torti SV, Torti FM. Interleukin 1 induces ferritin heavy chain in human muscle cells. Biochem Biophys Res Commun. 1990;169:289–296. doi: 10.1016/0006-291x(90)91466-6. [DOI] [PubMed] [Google Scholar]
- Williamson and Cleveland, 1999.Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- Williamson et al., 1998.Williamson TL, Bruijn LI, Zhu Q, Anderson KL, Anderson SD, Julien JP, Cleveland DW. Absence of neurofilaments reduces the selective vulnerability of motor neurons and slows disease caused by a familial amyotrophic lateral sclerosis-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci U S A. 1998;95:9631–9636. doi: 10.1073/pnas.95.16.9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong et al., 1995.Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Wu et al., 2004.Wu LJ, Leenders AG, Cooperman S, Meyron-Holtz E, Smith S, Land W, Tsai RY, Berger UV, Sheng ZH, Rouault TA. Expression of the iron transporter ferroportin in synaptic vesicles and the blood-brain barrier. Brain Res. 2004;1001:108–117. doi: 10.1016/j.brainres.2003.10.066. [DOI] [PubMed] [Google Scholar]
- Zecca et al., 2004.Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- Zhang et al., 1997.Zhang B, Tu P, Abtahian F, Trojanowski JQ, Lee VM. Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J Cell Biol. 1997;139:1307–1315. doi: 10.1083/jcb.139.5.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al., 2005.Zhang P, Land W, Lee S, Juliani J, Lefman J, Smith SR, Germain D, Kessel M, Leapman R, Rouault TA, Subramaniam S. Electron tomography of degenerating neurons in mice with abnormal regulation of iron metabolism. J Struct Biol. 2005;150:144–153. doi: 10.1016/j.jsb.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman et al., 2007.Zimmerman MC, Oberley LW, Flanagan SW. Mutant SOD1-induced neuronal toxicity is mediated by increased mitochondrial superoxide levels. J Neurochem. 2007;102:609–618. doi: 10.1111/j.1471-4159.2007.04502.x. [DOI] [PubMed] [Google Scholar]