

Abstract
The discovery of the molecular mechanisms regulating the abundance of synaptic NMDA receptors is essential for understanding how synaptic plasticity, as well as excitotoxic events, are regulated. However, a complete understanding of the precise molecular mechanisms regulating the composition of the NMDA receptor complex at hippocampal synapse is still missing. Here, we show that 2 h of CaMKII inhibition leads to a specific reduction of synaptic NR2B-containing NMDA receptors without affecting localization of the NR2A subunit; this molecular event is accompanied by a dramatic reduction in the induction of long-term potentiation (LTP), while long-term depression induction is unaffected. The same molecular and functional results were obtained by disrupting NR2B/PSD-95 complex with NR2B C-tail cell permeable peptide (TAT-2B). These data indicate that NR2B redistribution between synaptic and extrasynaptic membranes represents an important molecular disturbance of the glutamatergic synapse and affects the correct induction of LTP.
Keywords: NMDA receptor, synaptic plasticity, trafficking, CaMKII, MAGUK, postsynaptic density
Introduction
Understanding the molecular components guiding the correct assembly of the glutamatergic synapse is a challenge in our comprehension of synaptic strength in physiological and pathological plasticity. Indeed, several studies indicate that different ensembles of proteins in the postsynaptic densities (PSD) perform different synaptic functions (Lau and Zukin, 2007). In particular, the localization of ionotropic glutamate receptors in the PSD is key in the modulation of the response of the postsynaptic neuron to different stimuli, both in activity-dependent synaptic plasticity and in cell death (Gardoni, 2008).
In the PSD, it has been demonstrated that NMDA-type receptors are bound to scaffolding and signaling proteins, which regulate the strength of synaptic transmission. The C-tails of the NR2A/B subunits of NMDA receptor bind to Calcium/Calmodulin-dependent Protein Kinase II (CaMKII) and to members of the MAGUK family, such as PSD-95 (Gardoni et al., 1998; Strack and Colbran, 1998). Even if the molecular details of these interactions have been addressed, the physiological function of these interactions still remains to be clarified. Interestingly, recent data have put forward new roles for NMDA receptor-CaMKII complex in synaptic plasticity (Barria and Malinow, 2005; Zhou et al., 2007).
The role of CaMKII in synaptic plasticity has been examined both at cellular and behavioral level using both pharmacological and genetic approaches (Malinow et al., 1989; Silva et al., 1992; Hinds et al., 1998). Of relevance, recent works show that the binding of CaMKII to the NMDA channel is necessary for long-term potentiation (LTP) induction (Barria and Malinow, 2005).
Recent observation has shown that CaMKII in the PSD acts not only as a signaling element but also as a structural protein that bundles F-actin through a specific interaction; interestingly, such capability is required for the maintenance of the postsynaptic architecture of the dendritic spine (Okamoto et al., 2007). Notably, it has been proposed that clustered CaMKII works as a seed for PSD protein assembly (Hudmon et al., 2005), gathering various CaMKII-binding PSD proteins.
Despite these evidences on the structural-molecular and functional roles of active CaMKII in the PSD, less information are available on the effects of changes in CaMKII activity on the molecular composition of the PSD as well as on the effects of these events on synaptic plasticity.
Thus, considering also that αCaMKII is by far the highest abundant protein in the adult forebrain PSD (Dosemeci et al., 2006; Sheng and Hoogenraad, 2007), and that the active form of αCaMKII represents a main interacting partner for NMDA receptor in the PSD, it is of great interest to understand how modulation of CaMKII activity can affect NMDA receptor composition at synapses as well as NMDA-dependent synaptic plasticity.
To address this issue, we used a cell-permeable inhibitor peptide (Ant-AIP-2) strategy to induce CaMKII-inhibition. Here, we show that this treatment leads to a reduction of NR2B-containing NMDARs at synapses and to a significant reduction of LTP but not long-term depression (LTD) induction. Furthermore, mimicking the decreased NR2B-containing NMDARs at synapses by disrupting NR2B/PSD-95 complex induces the same effect on synaptic plasticity events.
Materials and Methods
Neuronal cultures and acute hippocampal slices.
Low-density or high-density hippocampal neuronal cultures were prepared from embryonic day 18 (E18)–E19 rat hippocampi (Brewer et al., 1993; Sala et al., 2001). Acute hippocampal slices were prepared as described previously (Gardoni et al., 2001). Slices were quickly frozen at −80°C after pharmacological treatments.
Subcellular fractionation.
As described previously, Triton insoluble fractions (TIFs) were isolated from cultured hippocampal neurons harvested at 14 d in vitro (DIV14) or acute hippocampal slices (Gardoni et al., 2003). Subcellular fractionation of hippocampal tissue was performed as previously reported with minor modifications (Gardoni et al., 2001). Primary hippocampal cultures or slices were homogenized in ice-cold sucrose 0.32 m containing 1 mm Hepes,1 mm MgCl2, 1 mm EDTA, 1 mm NaHCO3, 0.1 mm PMSF, at pH 7.4. The homogenized tissue was centrifuged at 1000 × g for 5 min. The resulting supernatant (S1) was centrifuged at 13,000 × g for 15 min to obtain a crude membrane fraction (P2 fraction). The pellet was resuspended in buffer containing 75 mm KCl and 1% Triton X-100 and centrifuged at 100,000 × g for 1 h. The supernatant was stored and referred as Triton soluble fraction (TSF). The final pellet was homogenized in a glass-glass potter in 20 mm Hepes. Then, an equal volume of glycerol was added and this fraction, referred as TIF, was stored at −80°C until processing. TIF was used instead of the classical PSD, because the amount of the starting material was very limited. All purifications were performed in presence of a complete set of protease inhibitors (Complete, Roche) and of both Ser/Thr- and Tyr-phosphatase inhibitor cocktails (Sigma-Aldrich).
Immunoprecipitation.
Neuronal lysates (50 μg proteins) or crude membrane fractions (P2, 50 μg proteins) were incubated overnight at 4°C in a RIA buffer containing: 200 mm NaCl, 10 mm EDTA, 10 mm Na2HPO4, 0.5% NP-40, 0.1% SDS, NaF 10 mm, with antibody against NR2B (antibody dilution 1:200). Samples were at first solubilized in RIA buffer in the presence of 1% SDS, and only subsequently diluted 10 times in RIA buffer to obtain a final 0.1% SDS concentration. Protein A-agarose beads (Santa Cruz), washed in the same buffer, were added, and incubation continued for 2 h. The beads were collected by centrifugation and washed five times, sample buffer for SDS-PAGE was added, and the mixture was boiled for 5 min. Beads were pelleted by centrifugation, and supernatants were applied to 6% SDS-PAGE.
Immunofluorescence labeling.
Hippocampal neurons were fixed in 100% methanol at −20°C for 15 min. Primary (1:100) and secondary (1:200) antibodies were applied in a buffer containing 30 mm phosphate buffer, pH 7.4, 0.2% gelatin, 0.5% Triton X-100, and 0.8 m NaCl. Fluorescence images were acquired using Bio-Rad Radiance 2100 confocal microscope.
Image acquisition, quantification, and statistical analysis.
Confocal images were obtained using a Nikon 60× objective with sequential acquisition setting at 1024 × 1024 pixels resolution. Each image was a z-series projection of images taken at 0.5 μm depth intervals. Pharmacologically treated neurons were chosen randomly for quantification from three to five independent experiments by taking from each experiment from two to five coverslips. Quantification of confocal experiments was performed using Bio-Rad Laserpix software (Bio-Rad). Image acquisition, quantification of the fluorescence signal and colocalization analysis were performed by investigators who were “blind” to the experimental conditions. Quantification of Western blot analysis was performed by means of computer-assisted imaging (Quantity-OneR System; Bio-Rad), and statistical evaluations were performed according to one-way ANOVA followed by Bonferroni as post hoc comparison test. Paired Student's t test was used if the experiment included only two experimental conditions. All data are presented as mean ± SEM and, if not indicated otherwise, as percentage of control deriving from three to six independent experiments.
Surface expression assays.
For proteolysis experiments, cells were incubated and lysed as described previously (Hall and Soderling, 1997); in brief, cells were incubated with 1 mg/ml chymotrypsine (Pierce) in D-PBS or only D-PBS (after a quick wash incubation with D-PBS) for 10 min with agitation at 37°C. After having aspirated the D-PBS, plates were washed three times with ice-cold lysis buffer: PBS containing PMSF 1 mm, ethanolamine 50 mm, EDTA 1 mm, and a complete set of protease inhibitors (Complete) and thereafter lysed in harvest buffer.
Cell-ELISA.
Quantification of total and surface-expressed NR2B receptor in neuronal cultures was determined using a modification of the ELISA-based assay (cell-ELISA) described previously (Pickard et al., 2000). Briefly, hippocampal cells were grown on 6-well plates. Cultures were incubated with blocking solution in the presence and absence of 1% Triton X-100 and then with primary antibody against NR2B in blocking solution for 1 h at room temperature. The addition of 1% Triton X-100 was omitted for detection of surface-expressed proteins. Cells were washed three times with blocking solution, incubated with peroxidase-conjugated secondary antibody in blocking solution for 1 h at RT, and washed four times in PBS. Samples in each well were then incubated with K-Blue substrate (Neogen) for 10 min. After the colorimetric reaction, each well was washed in PBS and solubilized. To determine total NR2B, neurons were permeabilized with Triton X-100 after fixation. Protein concentration was determined by Bradford assay (Bio-Rad). Each OD450 value of the ELISA reaction was normalized to protein levels of that particular well. For each experimental condition three parallel samples were used. Surface NR2B levels were calculated as percentage of total NR2B determined in the Triton X-100 permeabilized samples. Control experiments (plates) without hippocampal cells, were included routinely to determine background value, which was subtracted from the OD450 readings.
CaMKII activity.
CaMKII activity was measured by means of SignaTECT CaMKII assay system (Promega) according to the manufacturer's instructions.
Recording of evoked field potentials.
Rats were anesthetized with alotane before decapitation. The brain was quickly removed and immersed for 2–3 min in ice-cold artificial CSF (ACSF) containing (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 10 glucose, and 25 NaHCO3, continuously bubbled with 95% O2 and 5% CO2, pH 7.4. The hippocampus was extracted and cut in ice-cold ACSF with a vibratome (Pelco 1000 plus) into 400-μm-thick transverse slices, which were allowed to recover in oxygenated ACSF at 30°C for 30 min, and then at room temperature for an additional 1–2 h before experimental recordings. Some slices were then maintained in ACSF with Ant-AIP-2 10 μm, TAT-2B 1 μm or TAT-2B(-SDV) 1 μm at room temperature (23°–26°C) 2 h before the electrophysiological recordings. All the experiments were conducted in conformity with the European Communities Council Directive of November 1986 (86/609/ECC).
A slice was transferred into the recording chamber and submerged with ACSF at a constant rate of 2.5 ml/min at a temperature of 30°C. Recording electrodes were made from borosilicate glass capillaries (GC150F-10; Harvard Apparatus) and filled with 2 m NaCl (resistance, 10–15 mΩ).
Under visual control, the stimulating electrode was inserted into the Schaffer collateral fibers, and the recording electrode was inserted into CA1 region. Testing stimuli of 0.1 Hz, 10 μs duration and 20–30 V amplitude evoked field EPSPs that were 50–70% of maximum slope. fEPSPs were filtered at 3 kHz, digitized at 10 kHz, and stored on PC. An Axoclamp 2B amplifier (Axon Instruments) was used for extracellular recordings. After recording a stable baseline for 20 min, LTD was induced with 900 pulses at 1 Hz low-frequency stimulation (LFS), at baseline stimulation intensity. LTP was induced by high-frequency stimulation (HFS) consisting of one train of one second at 100 Hz at baseline stimulation intensity. The initial slope of the response was used to assess changes in synaptic strength.
For all of the experiments, data are presented as mean ± SEM (n is a number of slices). ANOVA and Bonferroni's post hoc test were used for statistical analysis. The significance level was established at p < 0.001.
Antibodies.
The following antibodies were used: monoclonal αCaMKII and polyclonal GluR1 antibodies were purchased from Millipore; monoclonal PSD-95 antibody was purchased from Affinity BioReagents; monoclonal anti-NR2A and polyclonal anti-NR2B (N-terminal) were purchased from Zymed; monoclonal GluR2 and NR2B were purchased from NeuroMab. Polyclonal anti-NR2A, anti-NR2B (C-terminal) and AlexaFluor 488, 555, 568 secondary antibodies were purchased from Invitrogen.
Results
CaMKII inhibition modulates NR2B localization in the postsynaptic fraction
The key role of active CaMKII in driving synaptic plasticity events has been widely described previously (Colbran and Brown, 2004; Griffith, 2004); less is known about the structural role of CaMKII in defining the composition of the excitatory PSD (Okamoto et al., 2007).
To address this issue, at first we analyzed the molecular effects on NMDA receptor complex composition induced by CaMKII inhibition. To this, we treated DIV14 primary hippocampal neurons with the CaMKII autoinhibitory peptide (AIP-2) fused to the antennapedia peptide (Ant) for 2 h (Ant-AIP-2, 10 μm, 2 h). At first, we checked that the inhibitor was still effective during this long period of time measuring in vitro CaMKII activity in the homogenized samples. The long treatment still results in effective CaMKII inhibition (−46.2 ± 17.2%, p < 0.05 Ant-AIP-2 vs control).
Western blot analysis performed on the homogenate and postsynaptic TIF reveals that, as expected (Shen et al., 2000), CaMKII inhibition causes a significant decrease of αCaMKII in the TIF (Fig. 1) (αCaMKII TIF, −23.2 ± 5.1%, p < 0.05, Ant-AIP-2 vs control), confirming that CaMKII activation modulates the amount of PSD-associated kinase (Shen et al., 2000). Moreover, 2 h CaMKII inhibition affects also the localization of the NMDA receptor NR2B subunit (Fig. 1) (NR2B TIF, −50.6 ± 24.2%, p < 0.001, Ant-AIP-2 vs control). Interestingly, this treatment does not significantly alter NR2A TIF levels (−4.3 ± 13.2%, p = 0.782, Ant-AIP-2 vs control), or of PSD-95 (+7.1 ± 12.4%, p = 0.525, Ant-AIP-2 vs control), the other main PSD constituent together with CaMKII (Sheng and Hoogenraad, 2007). Total levels of the same proteins analyzed in homogenate fraction were unchanged (Fig. 1).
Figure 1.

CaMKII inhibition with Ant-AIP-2 modulates NR2B localization in the postsynaptic fraction in cultured neurons. Western blot analysis of the homogenate (Homo) and postsynaptic TIF obtained from control and Ant-AIP-2 (10 μm, 2 h)-treated DIV14 primary hippocampal cultures. The same amount of proteins were loaded in each lane. Ant-AIP-2 treatment leads to a decreased NR2B localization in the TIF leaving the total amount of NR2B unaltered. NR2A and PSD-95 immunostaining in the TIF were not affected. Notably, αCaMKII levels in the TIF were also decreased (NR2B TIF, p < 0.001, Ant-AIP-2 vs Control; αCaMKII TIF, p < 0.05, Ant-AIP-2 vs Control).
Reduced NR2B localization to synaptic sites after 2 h of CaMKII inhibition
To strengthen the evidence that 2 h CaMKII inhibition can influence the subcellular localization of NR2B, we used confocal microscopy. We treated DIV14 cultured hippocampal neurons with Ant-AIP-2, and then we evaluated the subcellular distribution of NR2A and NR2B subunits by confocal imaging. Staining with PSD-95 was used as marker of the postsynaptic compartment. In untreated cultures (Fig. 2), both NR2A and NR2B display a high synaptic staining, measured by a very good colocalization with the postsynaptic marker PSD-95 (Fig. 2A,C) (NR2B/PSD-95 colocalization control: 46.9 ± 16.0%; NR2A/PSD-95 colocalization control: 48.8 ± 15.7%). Treating neurons with Ant-AIP-2 brings to a more diffuse staining of NR2B leading to a decreased colocalization with PSD-95 (Fig. 2B) (NR2B/PSD-95 colocalization Ant-AIP-2: 11.4 ±6.6%, p < 0.00005, Ant-AIP-2 vs control). NR2A synaptic immunostaining was not altered after CaMKII inhibition as showed by PSD-95 colocalization (Fig. 2C) (NR2A/PSD-95 colocalization Ant-AIP-2: 39.5 ± 22.0%, Ant-AIP-2, p = 0.23, AIP-2 vs control). Ant-AIP-2 treatment, however, did not alter the total number of PSD-95 positive clusters (5.52 ± 0.24/10 μm control; 5.31 ± 0.28/10 μm Ant-AIP-2, p = 0.46).
Figure 2.
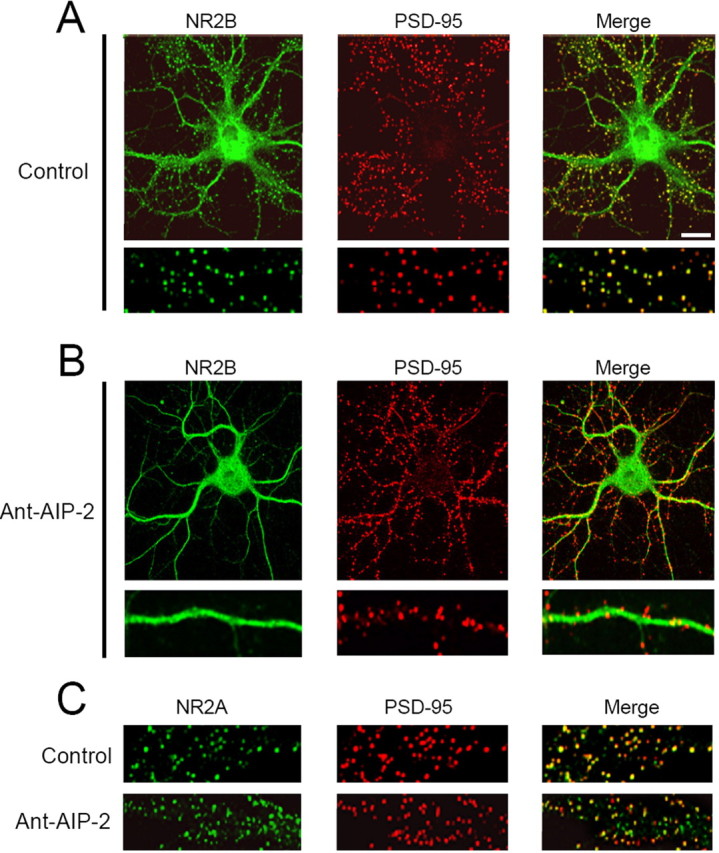
Altered NR2B subcellular localization in cultured neurons after long-lasting CaMKII inhibition. Hippocampal neurons were either left untreated (control) or treated with Ant-AIP-2 (10 μm) fixed, and immunolabeled for NR2B (A, B) or NR2A (C) and PSD-95 (A–C) as postsynaptic marker. Scale bar, 10 μm. CaMKII inhibition results in a significant decrease of NR2B/PSD-95 colocalization without altering NR2A/PSD-95 colocalization. A, B, Bottom, Magnification of the top ones.
NR2B dispersal from the Triton-insoluble synaptic fraction and less associated to PSD-95 clusters could indicate an internalization or a lateral movement of this NMDA receptor subunit to extrasynaptic sites (Groc et al., 2006). To address this issue, we used the membrane-impermeable protease chymotrypsin as surface expression assay (Mauceri et al., 2004). Neuronal cultures were treated with Ant-AIP-2 (2 h) and then exposed or not to chymotrypsin to cleave and consequently measure the amount of surface NR2B (Fig. 3A). NR2B intracellular content (Fig. 3, see arrowhead corresponding to the uncleaved NR2B band in presence of chymotrypsin) did not differ in the hippocampal cultures treated with Ant-AIP-2 (Fig. 3A) (−0.77 ± 11.7%, p = 0.951, Ant-AIP-2 vs control). Similarly, no modification of GluR1 intracellular content was observed after 2 h of Ant-AIP-2 treatment (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These results suggest that long-lasting CaMKII inhibition results in a lateral movement of the NR2B subunit that still remains on neuronal surface. To better address this key point, we tested the surface expression of NR2B after long-time CaMKII inhibition by means of cell-ELISA. We measured the levels of surface-expressed and total immunoreactivity of NR2B using intact versus Triton X-100 permeabilized, paraformaldehyde-fixed neurons by means of N-terminal NR2B-antibody. Ant-AIP-2 treatment does not change the percentage of surface NR2B (control 61.58 ± 2.35%; Ant-AIP-2, 62.1 ± 2.21%, p = 0.914). Using an antibody specific to the intracellular C-terminal tail of NR2B allowed us to test the integrity of the plasma membrane after fixation; this antibody was unable to interact with NR2B in neurons without Triton X-100 permeabilization (data not shown).
Figure 3.

NR2B subunit is still expressed on the cell surface after removal from synaptic sites. A, Western blot of NR2B from control and Ant-AIP-2-treated hippocampal cultures exposed or not exposed to the protease chymotrypsin. B, Ant-AIP-2, promoting NR2B extrasynaptic localization, causes the dephosphorylation of CREB. Lysates from control and Ant-AIP-2-treated hippocampal cultures were subjected to Western blot analysis for pCREB. Afterward, membranes were stripped and reprobed with total CREB.
To further confirm these data, we used a different strategy, measuring the activation of downstream targets associated to extrasynaptic or synaptic NR2B. In particular, it was previously demonstrated that NR2B-containing NMDA extrasynaptic receptors trigger CREB dephosphorylation, while synaptic ones promote the phosphorylated state of CREB (Hardingham et al., 2002). After 2 h of Ant-AIP-2 treatment, we measured the phosphorylation state of CREB using a phospho-specific antibody. Two hours of CaMKII inhibition effectively lowers CREB phosphorylation levels by removing NR2B from synaptic contacts (Fig. 3B) (−63.4 ± 18.1%, p < 0.005, Ant-AIP-2 vs control) without affecting total CREB.
CaMKII inhibition with Ant-AIP-2 modulates NR2B interaction with αCaMKII
To better address the functional impact of these events, the same experimental treatment with Ant-AIP-2 was repeated in acute hippocampal slices. Also under these experimental settings, 2 h Ant-AIP-2 treatment was able to affect CaMKII activity in the homogenate (−43.4 ± 6.2%, p < 0.001, Ant-AIP-2 vs control). As shown in Figure 4A, Ant-AIP-2 treatment in acute slices induced a decreasing αCaMKII (Fig. 4A) (−28.6 ± 6.2%, p < 0.05, Ant-AIP-2 vs control) and NR2B (Fig. 4A) (−54.7 ± 29.9%, p < 0.05, Ant-AIP-2 vs control) levels in the TIF. No significant changes of NR2A and PSD-95 content in the TIF were observed (NR2A, p = 0.621, Ant-AIP-2 vs control). Furthermore, no modification of both GluR1 and GluR2 subunits of the AMPA receptor was found in the TIF (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 4.

Treatment with Ant-AIP-2 modulates NR2B localization in the postsynaptic fraction in acute hippocampal slices. A, Western blot analysis of the homogenate or TIF obtained from control and Ant-AIP-2 (10 μm, 2 h)-treated hippocampal slices. Same amount of proteins was loaded in each lane. Ant-AIP-2 treatment leads to a decreased NR2B localization in the TIF leaving the total amount of NR2B unaltered. NR2A and PSD-95 immunostaining in the TIF were not affected. Notably, αCaMKII levels in the TIF were also decreased. The graph displays the results of Western blot analysis expressed as control percentage (NR2B, *p < 0.05; αCaMKII, *p < 0.05, Ant-AIP-2 vs Control). B, Crude membrane fractions (P2) and TSF from control and Ant-AIP-2 (10 μm, 2 h)-treated hippocampal slices were analyzed by Western blot analysis with NR2B and αCaMKII antibodies. The graph displays the results of Western blot analysis expressed as control percentage (NR2B, #p < 0.01; αCaMKII, *p < 0.05, Ant-AIP-2 vs Control).
To study in more detail the subcellular distribution of NR2B and αCaMKII in Ant-AIP-2-treated slices, Western blotting analysis was performed also in the P2 crude membrane fraction and in the TSF (see Materials and Methods). No alteration of both NR2B and αCaMKII levels was found in the P2 crude membrane fraction after 2 h of Ant-AIP-2 treatment (Fig. 4B). Notably, analysis of NR2B and αCaMKII levels in the TSF showed a significant higher level of both proteins in this soluble fraction in Ant-AIP-2-treated slices (Fig. 4B) (+37.5 ± 5.6%, p < 0.01, NR2B Ant-AIP-2 vs control; +23.3 ± 7.1%, p < 0.05, αCaMKII Ant-AIP-2 vs control). These results suggest a redistribution of NR2B from insoluble to soluble membrane fractions after 2 h of CaMKII inhibition, most likely from synaptic to extrasynaptic sites.
Several possible mechanisms could be responsible for NR2B dispersal from synaptic sites after 2 h of CaMKII inhibition. A major hypothesis is that reduced NR2B levels in the Triton insoluble postsynaptic fraction could be the result of a decreased direct interaction between αCaMKII and NR2B after Ant-AIP-2 treatment. This effect could be induced basically by two main mechanisms: (1) a direct competition of Ant-AIP-2 with αCaMKII binding to NR2B (Strack et al., 2000), and (2) a decreased affinity of αCaMKII for NR2B as direct consequence of CaMKII inhibition (Strack et al., 1998; Gardoni et al., 2006b).
The Ant-AIP-2 inhibitor peptide used in our study is highly homologous to the autocamtide-2 inhibitor peptide that has been previously shown to compete in a direct manner with αCaMKII binding to NR2B (Strack et al., 2000), thus suggesting that also Ant-AIP-2 could be able to interfere directly with the αCaMKII/NR2B complex. To test this hypothesis, coimmunoprecipitation (co-i.p.) experiments were performed in the P2 crude membrane fractions purified from control and Ant-AIP-2-treated slices (Gardoni et al., 2006b). In particular, NR2B was immunoprecipitated to reveal the coprecipitation of αCaMKII after 30 min and 2 h Ant-AIP-2 treatment. As shown in Figure 5A–C, 30 min treatment with Ant-AIP-2 induced a significant decrease of αCaMKII coprecipitation with NR2B (−44.3 ± 5.44, #p < 0.005, Ant-AIP-2 30 min vs control) without a concomitant significant decrease of NR2B levels in the TIF at this time point (Fig. 5D). This result indicates that the disruption of αCaMKII/NR2B complex by Ant-AIP-2 represents an early process that comes first the dispersal of NR2B from synaptic fraction. Alteration of αCaMKII/NR2B interaction was still significant after 2 h Ant-AIP-2 treatment (−28.9 ± 6.2%, *p < 0.05, Ant-AIP-2 2 h vs control) indicating a prolonged competition effect of Ant-AIP-2. As above described for the 2 h Ant-AIP-2 treatment (Fig. 4B), no modification of αCaMKII and NR2B protein levels in the P2 fraction was observed after 30 min Ant-AIP-2 treatment (Fig. 5A,B); thus, a modification of αCaMKII/NR2B complex was not ascribed to alteration of the abundance of the two proteins in the P2 fraction used for the co-i.p. assay.
Figure 5.
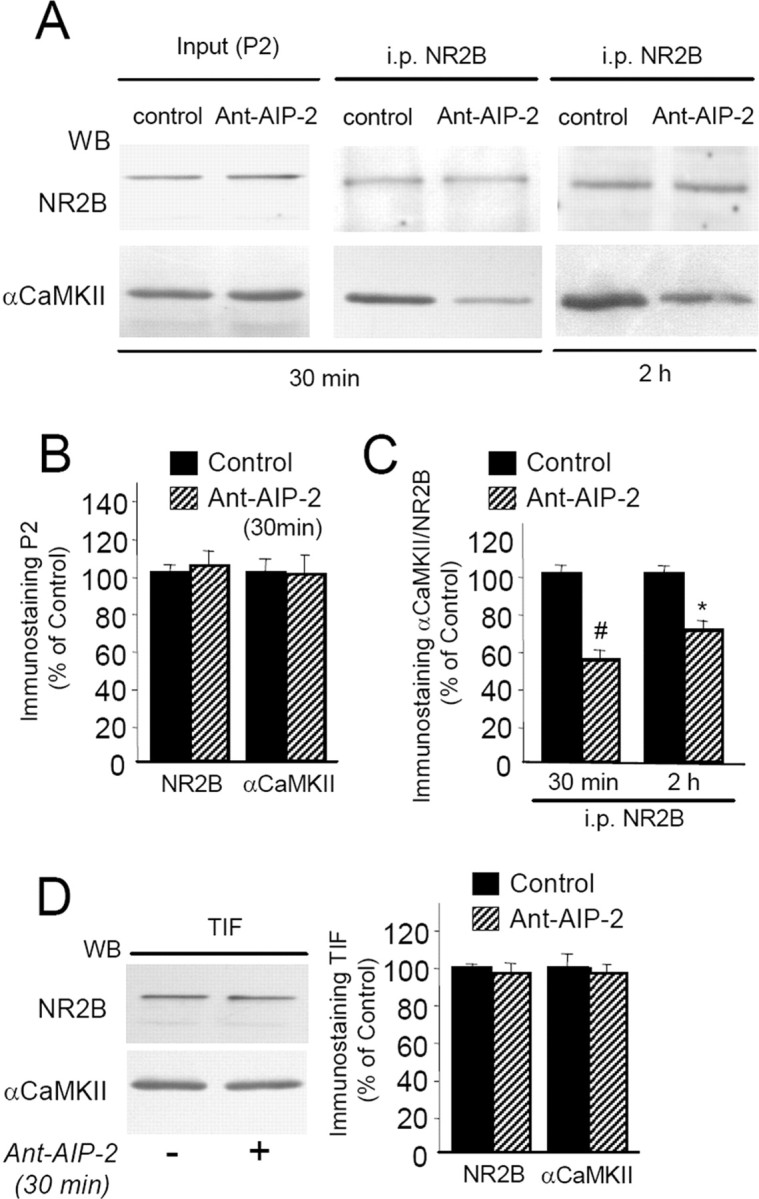
Ant-AIP-2 treatment modulates αCaMKII interaction with NR2B in acute hippocampal slices. A, Proteins from P2 crude membrane fraction purified from control and Ant-AIP-2-treated slices were immunoprecipitated with a polyclonal antibody raised against NR2B subunit of NMDA receptor; Western blot analysis was performed in the immunoprecipitated material with anti-NR2B and anti-αCaMKII monoclonal antibodies. B, Diagram illustrating statistical analysis of Western blotting experiments performed in the P2 fraction obtained from control and Ant-AIP-2 (10 μm, 30 min)-treated hippocampal slices. Same amount of proteins was loaded in each lane. Thirty-minute-Ant-AIP-2 treatment does not affect NR2B and αCaMKII localization in the P2 fraction. C, Diagram illustrating statistical analysis of Western blotting experiments performed on NR2B-immunoprecipitated material. Data are expressed as the ratio αCaMKII/NR2B (#p < 0.005 Ant-AIP-2 30 min vs control; *p < 0.05 Ant-AIP-2 2 h vs control). D, Western blot analysis of the TIF obtained from control and Ant-AIP-2 (10 μm, 30 min)-treated hippocampal slices. Same amount of proteins were loaded in each lane. Thirty-minute-Ant-AIP-2 treatment does not affect NR2B and αCaMKII localization in the TIF.
Decreased NR2B subunit synaptic levels induced by CaMKII-inhibition impair LTP but not LTD induction
To test the effects of 2 h CaMKII inhibition on synaptic plasticity, hippocampal slices were incubated for 2 h with the Ant-AIP-2 inhibitor before recording field EPSPs and inducing LTP or LTD. CaMKII inhibitor was removed before induction of LTP or LTD, and CaMKII activity was monitored before induction in parallel sample to check that in absence of the inhibitor kinase activity was back to control levels (data not shown); this was necessary to be sure that abnormalities in LTP/LTD induction could be ascribed to an altered composition of the NMDA receptor complex and not to a reduction of CaMKII activity. As shown in Figure 6A, we found that exposure of hippocampal slices for prolonged periods to CaMKII inhibitor (Ant-AIP-2) had no detectable effect on the ability to elicit robust LTD when compared with untreated control slices (p = 1.0; n = 10). However, the same treatment was sufficient to almost completely abolish the induction of LTP by HFS compared with untreated slices (Fig. 6B) p < 0.0001; n = 10).
Figure 6.

Treatment with Ant-AIP-2 cell-permeable peptide affects LTP but not LTD. A, The plots show that both in Ant-AIP-2-treated slices and in control slices, an LFS stimulating protocol (arrow) produces an LTD of fEPSP. The reported examples of fEPSPs before the LFS application and 60 min post-LFS reveal a similar LTD in control conditions (top traces) as well as in an Ant-AIP-2-treated slice (bottom traces). B, Time course of the normalized field potential (fEPSP) slope recorded in Ant-AIP-2-treated slices respect to nontreated (control) slices. The HFS stimulating protocol (arrow) produces an LTP of the fEPSP in control slices, whereas in Ant-AIP-2-treated slices, only a short-term potentiation can be observed. Note that the group treated with Ant-AIP-2 displays a reduced LTP in respect to control (p < 0.0001). Example traces of fEPSPs acquired before the application of the HFS protocol and 60 min post-HFS, in control conditions (top traces) and in an Ant-AIP-2-treated slice (bottom traces).
Overall, the above described results show that 2 h reduction of CaMKII activity in hippocampal neurons leads to an unbalanced composition of the NMDA receptor at synapse with a decreased synaptic content of NR2B-containing NMDA receptors. From a functional point of view, these molecular events are associated with a severe impairment in induction of LTP. Thus, it was of major relevance to understand whether NR2B reduction at synaptic sites was sufficient per se to induce a modification of LTP independently of CaMKII localization/activity.
Decreased NR2B subunit synaptic levels induced by TAT-2B peptide impair LTP but not LTD induction
It is well established that interactions of NR2B subunit C-terminal domain with the PDZ domains of PSD-MAGUK proteins, above all with PSD-95, play an important role in the regulation of NMDA receptors trafficking/clustering at synaptic sites (Prybylowski and Wenthold, 2004; Lau and Zukin, 2007). Previous reports showed that modulation of NR2B interaction with PSD-95 by using TAT-2B cell permeable peptide, corresponding to the last 9aa of the receptor subunits fused to the TAT sequence, was sufficient to induce a decrease in the synaptic levels of NR2B-containing NMDA receptors that accumulate at extrasynaptic sites (Aarts et al., 2002; Gardoni et al., 2006a); however, the consequences of the disruption of NR2B-PSD-95 complexes on physiological events such as synaptic plasticity are still not completely understood (Lim et al., 2003; Prybylowski et al., 2005). Thus, treatment of hippocampal slices with the TAT-2B peptide could represent the ideal approach to obtain a specific delocalization of the NR2B subunit (not affecting αCaMKII), to confirm the role of synaptic versus extrasynaptic NR2B-containing NMDA receptors in the induction of synaptic plasticity processes and to show that LTP reduction was not due to a decrease of αCaMKII synaptic levels.
Based on these observations, we performed a long-time disturbance of NR2B interaction with PSD-95 in hippocampal slices using TAT-2B (1 μm, 2 h) to test the effects of a reduced interaction with PSD-95 on synaptic NR2B localization and on the induction of synaptic plasticity. Treatments with TAT-2B(-SDV) peptide (lacking the last C-terminal three amino acids of NR2B) were performed as control.
At first, we verified whether treatment with the TAT-2B peptide (1 μm, 2 h) could effectively perturb NR2B/PSD-95 protein complexes by coimmunoprecipitation experiments performed on homogenate fraction from hippocampal slices. As expected (Aarts et al., 2002; Gardoni et al., 2006a), TAT-2B treatment reduces NR2B interaction with PSD-95 (Fig. 7A) [−34.1 ± 4.8.%, p < 0.01, TAT-2B vs TAT-2B(-SDV)] inducing a concomitant decrease of NR2B but not NR2A localization in the Triton insoluble postsynaptic compartment (TIF) (Fig. 7B,C) (NR2B −38.1 ± 13.3%, p < 0.05). No effect of TAT-2B treatment was detected in Western blotting performed on the whole homogenate indicating that the peptide did not affect NR2B expression (Fig. 7B). Of relevance, no modifications of PSD-95 and αCaMKII localization in the TIF were observed after TAT-2B treatment.
Figure 7.
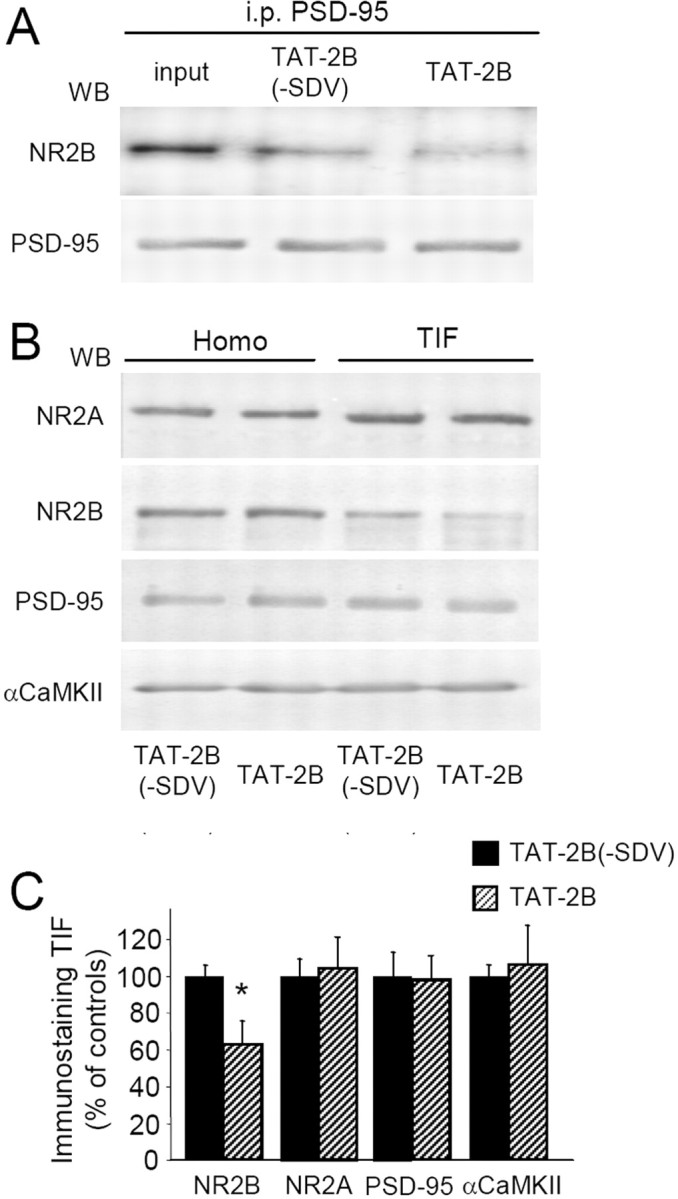
Decreased NR2B subunit synaptic levels induced by TAT-2B. A, Acute hippocampal slices were treated with TAT-2B active peptide (1 μm) or with TAT-2B(-SDV) control peptide (1 μm) for 2 h. Coimmunoprecipitation experiments were performed on the homogenized samples. TAT-2B peptide results in a significant decrease of NR2B coprecipitation with PSD-95 as shown by Western blot analysis. B, Western blot analysis of the homogenate or TIF obtained from TAT-2B active peptide (1 μm) or with TAT-2B(-SDV) control peptide (1 μm)-treated hippocampal slices. Same amount of proteins was loaded in each lane. TAT-2B treatment leads to a decreased NR2B localization in the TIF leaving the total amount of NR2B unaltered. NR2A and PSD-95 immunostaining in the TIF were not affected. The graph displays the results of Western blot analysis expressed as control percentage (NR2B, *p < 0.05).
We then tested whether synaptic plasticity was affected by a long-time dissociation of the PSD-95/NR2B interaction with the TAT-2B peptide leading to NR2B reduced levels at synapse. As for the molecular experiments, treatments with TAT-2B(-SDV) peptide were always performed as control (Gardoni et al., 2006a). Hippocampal slices were incubated for 2 h with the control or active peptide before recording field EPSPs and inducing LTP or LTD.
We found that exposure of hippocampal slices for prolonged periods to TAT-2B had no detectable effect on the ability to elicit robust LTD (Fig. 8A) when compared with slices treated with TAT-2B(-SDV) control peptide (p = 0.22; n = 10). However, treatment with TAT-2B was sufficient to almost completely abolish the induction of LTP by HFS (Fig. 8B) (p < 0.0001; n = 10).
Figure 8.

TAT-2B-cell-permeable peptide affects LTP but not LTD. A, Time course of the normalized fEPSP slope recorded in slices treated with TAT2B and TAT2B(-SDV) peptides. In both groups, an LFS protocol (arrow) produces a robust LTD. Traces of fEPSP acquired before LFS and 60 min post-LFS indicating a similar fEPSP reduction in TAT-2B(-SDV)-treated slices (bottom traces) and in TAT2B-treated slices (top traces). B, Time course of the normalized fEPSP slope recorded in slices treated with the TAT-2B peptide and in slices treated with the TAT-2B(-SDV) peptide. Notably, in TAT2B-treated slices, the HFS protocol (arrow) fails to induce the LTP, whereas in TAT2B(-SDV)-treated slices, this form of synaptic plasticity is expressed (p < 0.0001). The reported example shows that in a slice treated with the TAT2B peptide, the fEPSP slope acquired before HFS remains unaltered 60 min after HFS application (top traces), whereas it is significantly increased in a TAT2B(-SDV)-treated slice (bottom traces).
Discussion
The main finding of this work is that a significant reduction of NR2B-containing NMDA receptors at synapses leads to an alteration of synaptic plasticity processes with an impairment of LTP but not LTD induction.
A high number of reports in the last five years have indicated that NR2A- and NR2B- containing receptors may have different roles in the regulation of the induction of synaptic plasticity events in the hippocampus (Bartlett et al., 2007; Bellone and Nicoll, 2007; Morishita et al., 2007). In a recent study (Morishita et al., 2007), three independent groups have shown that normal LTD can be generated in the CA1 region of the hippocampus in the presence of selective antagonists of NR2B-containing NMDA receptors (Morishita et al., 2007). These studies have provided strong evidence that NR2B-containing NMDA receptors are not required for the triggering of LTD and therefore that NR2A-containing NMDA receptors are in fact sufficient to generate this form of plasticity (Morishita et al., 2007). However, a role for NR2B receptors in LTP has been shown in some but not all previous studies. A recent observation (Bartlett et al., 2007) showed that a selective NR2B antagonist (Ro 25–6981) significantly reduced LTP but not LTD. In addition, antisense knockdown of NR2B abolished LTP in 2-month-old rats (Clayton et al., 2002). Conversely, Liu et al. (2004) showed that NR2A receptors are required for hippocampal LTP, but not LTD, while NR2B receptors are required for LTD, but not LTP.
Together, all these studies try to address a specific role for NMDA receptors regulatory subunits in synaptic plasticity events; major differences on the technical approaches used may explain some conflicting points that are still highly debated. For example, information are still lacking for understanding in minute details the precise role of NR2B-containing NMDA receptors in LTP.
Here, we show that a proper synaptic localization of NR2B-containing receptor is key for a physiological induction of LTP; a reduction of ∼40–50% of the NR2B subunit in the Triton insoluble postsynaptic compartment is sufficient to induce a dramatic impairment of LTP induction.
In particular, in the present study, we decided to use a different experimental approach to study NMDAR subunits role in synaptic plasticity in hippocampus; we have induced a reduction of NR2B localization at synaptic sites, either through inhibition of CaMKII (leading to a reduction of αCaMKII/NR2B interaction) or through a reduction of NR2B/PSD-95 interaction.
At first, our data support the idea that maintenance of the PDZ interaction is a critical element in keeping NMDA receptors at the synapse and that disrupting this interaction may be the first step in the removal of the receptor. From a functional point of view, a report from Lim et al. (2003) showed that pretreating hippocampal slices with the membrane-permeable NR2B C-tail peptide blocked NR2B interaction with PSD-95 but not LTP. However, they performed only a short time window treatment (30 min) using a very low concentration of the peptide (50 nm). In the present study, we show that an higher concentration of TAT-2B (1 μm) is able to interfere with NR2B but not NR2A localization at synapse; furthermore, we show that this molecular event is associated with a decreased LTP induction after 2 h pretreatment, confirming the existence of a previously suggested long-time effect of NR2B C-terminal peptides (Aarts et al., 2002; Gardoni et al., 2006a).
Second, we identified a new role for CaMKII in the modulation of NMDA receptor synaptic localization and function. Recent studies have described the functional role of CaMKII/NMDA receptor complex at synapses. It has been shown that binding of CaMKII to the NR2B subunit is necessary for LTP induction (Barria and Malinow, 2005). More recently, inducible and reversible disruption of NR2B/CaMKII interactions in transgenic mice has been shown to be involved in downregulation of αCaMKII autophosphorylation and reduction of LTP in the hippocampus (Zhou et al., 2007). Our data are in agreement with all these previous observations: here, we show that CaMKII inhibition by means of Ant-AIP-2 leads to a disruption of αCaMKII/NR2B complex and to a subsequent reduction of αCaMKII and NR2B protein levels at synapses without affecting NR2A subunit localization, and that this molecular event is associated with a decreased LTP induction.
Thirty-minute-Ant-AIP-2 treatment is sufficient to detect a significant dissociation of αCaMKII/NR2B complex (Strack et al., 2000), while still no reduction of NR2B synaptic levels is visible at this time point. Furthermore, both αCaMKII/NR2B interaction and NR2B synaptic levels are impaired after 2 h Ant-AIP-2 treatment. These data indicate that, as expected (Strack et al., 2000), Ant-AIP-2 is capable of interfering with αCaMKII/NR2B complex and that this molecular event can represent an early step of the dispersal of the NR2B subunit outside the synapse. Notably, these results indicate that a direct anchoring to αCaMKII represents a key mechanism to keep NR2B anchored to the synapse, thus suggesting that NR2B subunit interaction with both αCaMKII and PSD-95 is a needed mechanism for NR2B-containing NMDA receptor stabilization at synaptic sites. Reduction of either αCaMKII/NR2B or PSD-95/NR2B association is sufficient to decrease NR2B synaptic content in hippocampal neurons.
Interestingly, we show here that reduction of synaptic NR2B leads to a shut off of the CREB signal and not to a decreased level of NR2B inserted in the membrane thus indicating a lateral movement of this subunit to extrasynaptic sites after disruption of αCaMKII/NR2B complex.
Finally, the present study indicates that synaptic localization of NMDA receptor NR2B subunit, already described to be crucial in the pathogenesis of CNS disorders (Gardoni et al., 2006a), is also essential for a correct induction of physiological neuronal processes such as LTP.
Footnotes
This work was supported by Ministero Istruzione Università e Ricerca 2006 (MIUR2006) to F.C., and by Fondo per gli Investimenti della Ricerca di Base 2003 (FIRB2003) and Fondazione Cassa di Risparmio di Perugia to P.C. We thank Elisa Zianni and Cristiano Spaccatini for their excellent technical assistance.
References
- Aarts et al., 2002.Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- Barria and Malinow, 2005.Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Bartlett et al., 2007.Bartlett TE, Bannister NJ, Collett VJ, Dargan SL, Massey PV, Bortolotto ZA, Fitzjohn SM, Bashir ZI, Collingridge GL, Lodge D. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology. 2007;52:60–70. doi: 10.1016/j.neuropharm.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Bellone and Nicoll, 2007.Bellone C, Nicoll RA. Rapid bidirectional switching of synaptic NMDA receptors. Neuron. 2007;55:779–785. doi: 10.1016/j.neuron.2007.07.035. [DOI] [PubMed] [Google Scholar]
- Brewer et al., 1993.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Clayton et al., 2002.Clayton DA, Mesches MH, Alvarez E, Bickford PC, Browning MD. A hippocampal NR2B deficit can mimic age-related changes in long-term potentiation and spatial learning in the Fischer 344 rat. J Neurosci. 2002;22:3628–3637. doi: 10.1523/JNEUROSCI.22-09-03628.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbran and Brown, 2004.Colbran RJ, Brown AM. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol. 2004;14:318–327. doi: 10.1016/j.conb.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Dosemeci et al., 2006.Dosemeci A, Tao-Cheng JH, Vinade L, Jaffe H. Preparation of postsynaptic density fraction from hippocampal slices and proteomic analysis. Biochem Biophys Res Commun. 2006;339:687–694. doi: 10.1016/j.bbrc.2005.11.069. [DOI] [PubMed] [Google Scholar]
- Gardoni, 2008.Gardoni F. MAGUK proteins: New targets for pharmacological intervention in the glutamatergic synapse. Eur J Pharmacol. 2008;585:147–152. doi: 10.1016/j.ejphar.2008.01.048. [DOI] [PubMed] [Google Scholar]
- Gardoni et al., 1998.Gardoni F, Caputi A, Cimino M, Pastorino L, Cattabeni F, Di Luca M. Calcium/calmodulin-dependent protein kinase II is associated with NR2A/B subunits of NMDA receptor in postsynaptic densities. J Neurochem. 1998;71:1733–1741. doi: 10.1046/j.1471-4159.1998.71041733.x. [DOI] [PubMed] [Google Scholar]
- Gardoni et al., 2001.Gardoni F, Schrama LH, Kamal A, Gispen WH, Cattabeni F, Di Luca M. Hippocampal synaptic plasticity involves competition between Ca2+/calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor. J Neurosci. 2001;21:1501–1509. doi: 10.1523/JNEUROSCI.21-05-01501.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni et al., 2003.Gardoni F, Mauceri D, Fiorentini C, Bellone C, Missale C, Cattabeni F, Di Luca M. CaMKII-dependent phosphorylation regulates SAP97/NR2A interaction. J Biol Chem. 2003;278:44745–44752. doi: 10.1074/jbc.M303576200. [DOI] [PubMed] [Google Scholar]
- Gardoni et al., 2006a.Gardoni F, Picconi B, Ghiglieri V, Polli F, Bagetta V, Bernardi G, Cattabeni F, Di Luca M, Calabresi P. A critical interaction between NR2B and MAGUK in l-DOPA induced dyskinesia. J Neurosci. 2006a;26:2914–2922. doi: 10.1523/JNEUROSCI.5326-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni et al., 2006b.Gardoni F, Polli F, Cattabeni F, Di Luca M. Calcium-calmodulin-dependent protein kinase II phosphorylation modulates PSD-95 binding to NMDA receptors. Eur J Neurosci. 2006b;24:2694–2704. doi: 10.1111/j.1460-9568.2006.05140.x. [DOI] [PubMed] [Google Scholar]
- Griffith, 2004.Griffith LC. Calcium/calmodulin-dependent protein kinase II: an unforgettable kinase. J Neurosci. 2004;24:8391–8393. doi: 10.1523/JNEUROSCI.2888-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc et al., 2006.Groc L, Heine M, Cousins SL, Stephenson FA, Lounis B, Cognet L, Choquet D. NMDA receptor surface mobility depends on NR2A–2B subunits. Proc Natl Acad Sci U S A. 2006;103:18769–18774. doi: 10.1073/pnas.0605238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall and Soderling, 1997.Hall RA, Soderling TR. Quantitation of AMPA receptor surface expression in cultured hippocampal neurons. Neuroscience. 1997;78:361–371. doi: 10.1016/s0306-4522(96)00525-8. [DOI] [PubMed] [Google Scholar]
- Hardingham et al., 2002.Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hinds et al., 1998.Hinds HL, Tonegawa S, Malinow R. CA1 long-term potentiation is diminished but present in hippocampal slices from alpha-CaMKII mutant mice. Learn Mem. 1998;5:344–354. [PMC free article] [PubMed] [Google Scholar]
- Hudmon et al., 2005.Hudmon A, Lebel E, Roy H, Sik A, Schulman H, Waxham MN, De Koninck P. A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. J Neurosci. 2005;25:6971–6983. doi: 10.1523/JNEUROSCI.4698-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau and Zukin, 2007.Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Lim et al., 2003.Lim IA, Merrill MA, Chen Y, Hell JW. Disruption of the NMDA receptor-PSD-95 interaction in hippocampal neurons with no obvious physiological short-term effect. Neuropharmacology. 2003;45:738–754. doi: 10.1016/s0028-3908(03)00276-4. [DOI] [PubMed] [Google Scholar]
- Liu et al., 2004.Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Malinow et al., 1989.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Mauceri et al., 2004.Mauceri D, Cattabeni F, Di Luca M, Gardoni F. Calcium/calmodulin-dependent protein kinase II phosphorylation drives synapse-associated protein 97 into spines. J Biol Chem. 2004;279:23813–23821. doi: 10.1074/jbc.M402796200. [DOI] [PubMed] [Google Scholar]
- Morishita et al., 2007.Morishita W, Lu W, Smith GB, Nicoll RA, Bear MF, Malenka RC. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology. 2007;52:71–76. doi: 10.1016/j.neuropharm.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Okamoto et al., 2007.Okamoto K, Narayanan R, Lee SH, Murata K, Hayashi Y. The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc Natl Acad Sci U S A. 2007;104:6418–6423. doi: 10.1073/pnas.0701656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard et al., 2000.Pickard L, Noël J, Henley JM, Collingridge GL, Molnar E. Developmental changes in synaptic AMPA and NMDA receptor distribution and AMPA receptor subunit composition in living hippocampal neurons. J Neurosci. 2000;20:7922–7931. doi: 10.1523/JNEUROSCI.20-21-07922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski and Wenthold, 2004.Prybylowski K, Wenthold RJ. N-Methyl-D-aspartate receptors: subunit assembly and trafficking to the synapse. J Biol Chem. 2004;279:9673–9676. doi: 10.1074/jbc.R300029200. [DOI] [PubMed] [Google Scholar]
- Prybylowski et al., 2005.Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala et al., 2001.Sala C, Piëch V, Wilson NR, Passafaro M, Liu G, Sheng M. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31:115–130. doi: 10.1016/s0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
- Shen et al., 2000.Shen K, Teruel MN, Connor JH, Shenolikar S, Meyer T. Molecular memory by reversible translocation of calcium/calmodulin-dependent protein kinase II. Nat Neurosci. 2000;3:881–886. doi: 10.1038/78783. [DOI] [PubMed] [Google Scholar]
- Sheng and Hoogenraad, 2007.Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Silva et al., 1992.Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Strack and Colbran, 1998.Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/ calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl- D-aspartate receptor. J Biol Chem. 1998;273:20689–20692. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- Strack et al., 2000.Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2000;275:23798–23806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- Zhou et al., 2007.Zhou Y, Takahashi E, Li W, Halt A, Wiltgen B, Ehninger D, Li GD, Hell JW, Kennedy MB, Silva AJ. Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. J Neurosci. 2007;27:13843–13853. doi: 10.1523/JNEUROSCI.4486-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]