

Abstract
Axon degeneration contributes widely to neurodegenerative disease but its regulation is poorly understood. The Wallerian degeneration slow (WldS) protein protects axons dose-dependently in many circumstances but is paradoxically abundant in nuclei. To test the hypothesis that WldS acts within nuclei in vivo, we redistributed it from nucleus to cytoplasm in transgenic mice. Surprisingly, instead of weakening the phenotype as expected, extranuclear WldS significantly enhanced structural and functional preservation of transected distal axons and their synapses. In contrast to native WldS mutants, distal axon stumps remained continuous and ultrastructurally intact up to 7 weeks after injury and motor nerve terminals were robustly preserved even in older mice, remaining functional for 6 d. Moreover, we detect extranuclear WldS for the first time in vivo, and higher axoplasmic levels in transgenic mice with WldS redistribution. Cytoplasmic WldS fractionated predominantly with mitochondria and microsomes. We conclude that WldS can act in one or more non-nuclear compartments to protect axons and synapses, and that molecular changes can enhance its therapeutic potential.
Keywords: axon degeneration, Wallerian degeneration, neurodegeneration, slow Wallerian degeneration gene, neuroprotection, neuromuscular junction
Introduction
Axon degeneration occurs in many neurodegenerative diseases and often precedes neuronal cell body death (Raff et al., 2002; Coleman, 2005; Saxena and Caroni, 2007). Wallerian degeneration, a classical experimental model for axon degeneration, is a rapid sequence of events in distal axons after a period of separation from the cell body (Waller, 1850; Beirowski et al., 2005). It is substantially delayed by the WldS gene in mice (Mack et al., 2001), rats (Adalbert et al., 2005), and Drosophila (Hoopfer et al., 2006; MacDonald et al., 2006). WldS is an in-frame fusion protein arising from an 85-kb tandem triplication that does not alter expression of the two parent proteins (Coleman et al., 1998; Conforti et al., 2000; Mack et al., 2001). It comprises the N-terminal 70 aa of multiubiquitination factor Ube4b (N70-Ube4b), the complete sequence of Nmnat1, a key enzyme of nicotinamide adenine dinucleotide (NAD+) biosynthesis, and a short joining sequence of 18 aa with no known function.
The WldS gene delays axon degeneration in mouse models of several neurodegenerative disorders, suggesting a molecular similarity between processes regulating Wallerian degeneration and degeneration in some axonopathies (Coleman, 2005; Beirowski et al., 2009). This further indicates the need to dissect the molecular mechanisms of WldS.
Previous studies addressing which WldS domains are responsible for neuroprotection illustrate the importance of in vivo experiments. Whereas NAD+ overproduction by heavily overexpressed Nmnat isoforms confers axon protection in neuronal explant cultures and, to some extent, in Drosophila (Araki et al., 2004; Wang et al., 2005; MacDonald et al., 2006; Sasaki et al., 2006), overexpression of Nmnat1 in transgenic mice at levels similar to WldS provides no detectable axon protection (Conforti et al., 2007). Thus, WldS and NAD+ overproduction by Nmnat1 are not interchangeable, and the full axoprotective effect in vivo and in vitro requires more N-terminal sequences of WldS.
A key question about the WldS mechanism surrounds the subcellular site of its action (Fainzilber and Twiss, 2006). In vivo studies have consistently detected WldS only in the nucleus (Mack et al., 2001; Samsam et al., 2003; Sajadi et al., 2004; Wilbrey et al., 2008), suggesting that WldS confers its axonal effect indirectly by putative nuclear mechanisms (Araki et al., 2004; Gillingwater et al., 2006; Simonin et al., 2007a). However, the complete absence of WldS in other cellular compartments could not be proven experimentally owing to detection limits and there are precedents for proteins acting in a subcellular compartment where they are barely detectable (Hamilton et al., 2001). Interestingly, some indirect, in vitro evidence suggests bioenergetic or other axonal roles for WldS and its putative mediators (Wang et al., 2005; Yang et al., 2007b) compatible with cytoplasmic and axonal WldS presence.
To elucidate the subcellular locus of WldS action in vivo, we generated transgenic mouse lines with reduced nuclear targeting and cytoplasmic redistribution of WldS. These mice showed surprisingly strong protection of axons and synapses suggesting cytoplasmic WldS protected them more effectively for equivalent expression levels. We also show WldS exists at low concentrations in extranuclear compartments, and higher concentrations in the ΔNLS WldS variant, consistent with a direct axonal role for WldS.
Materials and Methods
In vitro/in vivo expression of WldS variants
To reduce WldS nuclear targeting, arginines 213 and 215 were mutated to alanine using the Stratagene QuikChange Site-Directed Mutagenesis Kit and the WldS transgene construct (Mack et al., 2001) as template. The two primers used were exactly reverse complementary, the forward primer having the following sequence (R→A encoding mutations underlined): 5′-GCACTGGAAAAGCCTGGGGCGAAGGCGAAGTGGGCTGATCAAAAG-3′.
The sequence contained in pBluescript CK+ (Stratagene) was verified and subcloned into pHβ-Apr1 containing β-actin promoter (Mack et al., 2001) or into pcDNA3 vector (Invitrogen). For in vitro expression of variant WldS EGFP fusion proteins, the ΔNLSR213A,R215A WldS cDNA was amplified by high-fidelity PCR from the above construct using BamHI- and HindIII-tagged primers and subcloned in-frame to the EGFP sequence into pEGFP-N1 vector (BD Bioscience).
For generation of ΔNLS WldS transgenic mice, the ΔNLSR213AR215A WldS cDNA including β-actin promoter from above pHβ-Apr1 vector was linearized using EcoRI and NdeI restriction enzymes and pronuclear injection of the fragment into an F1 C57BL/CBA strain was performed by the in-house Gene Targeting Facility of the Babraham Institute. Eleven founder mice for the ΔNLS WldS strain and their transgene-positive offspring were identified by Southern blotting of BamHI plus HindIII double-digested genomic tail DNA hybridized with a 32P-labeled WldS cDNA probe and by PCR using appropriate primers. Founders with medium to high copy number integrations were selected for further study (to generate transgenic lines 1–8) and crossed to homozygous YFP-H mice (The Jackson Laboratory) to breed mice hemizygous for the ΔNLSR213AR215A WldS and YFP transgene. Subsequently, these mice were intercrossed to obtain mice homozygous for the ΔNLSR213AR215A WldS and positive for the YFP transgene. Mice positive for the YFP transgene were identified by Southern blotting using a 32P-labeled YFP cDNA probe. Furthermore, we used double heterozygous native WldS/YFP-H mice, triple heterozygous tg-WldS/WldS/YFP-H mice, homozygous natural WldS mice, and homozygous WldS transgenic rats from line 79 (Adalbert et al., 2005) for this study. Triple heterozygous tg-WldS/WldS/YFP-H mice express levels of WldS protein similar to those of homozygous natural mutant WldS mice and display a similarly retarded time course of axon degeneration (Beirowski et al., 2005).
Cell culture
Culture and transfection of PC12 and HeLa cells using Lipofectamine 2000 (Invitrogen) was performed as described previously (Wilbrey et al., 2008). Dissociated hippocampal and DRG neuron cultures were prepared from embryonic day 14.5 (E14.5)–E16.5 mouse embryos and transfected using Lipofectamine LTX with PLUS reagent (Invitrogen) as described previously (Conforti et al., 2007; Wilbrey et al., 2008). Cells were plated on 35 mm Petri dishes (μ-Dish, ibidi) for subsequent labeling experiments and high-resolution confocal imaging. For mitochondrial colocalization studies, transfected neurons were treated with 40 nm Mitotracker Red CMXRos (Invitrogen) for 30 min according to the manufacturer's instructions for live cell staining. Additionally, the pDsRed2-Mito vector (Clontech, PT3633-5) was used for mitochondrial labeling.
For SCG explant cultures SCGs were dissected from 1- or 2-d-old mouse pups using sterile technique. Explants were placed into L15 (Leibovitz) medium (Invitrogen) containing 0.01% fetal bovine serum for removal of other tissue, and three explants were then placed in the center of 3.5 cm tissue culture dishes precoated with poly-l-lysine (20 μg/ml for 2 h; Sigma) and laminin (20 μg/ml for 2 h; Sigma). Explants were cultured in DMEM containing 4500 mg/L glucose and 110 mg/L sodium pyruvate (Sigma), 2 mm glutamine, 1% penicillin/streptomycin, and 100 ng/ml 7S NGF (all from Invitrogen), 20 μm uridine and fluorodeoxyuridine (both Sigma) to block proliferation of non-neuronal cells, and 10% fetal bovine serum (Sigma). After 7 d in culture, explants and their radial neurite networks from three dishes were separated by transection and collected separately for lysis directly in Laemmli sample buffer. Explant fractions contain the cell bodies and proximal neurites while the distal neurite fraction is almost exclusively neurites.
For dissociated cultures, explants were treated as described previously (Whitfield et al., 2004). Approximately 10,000 dissociated neurons were plated in a 1 cm2 area of a laminin-coated 35 mm Petri dish (μ-Dish, ibidi) and maintained under the same conditions as above.
Biochemical assessment of variant WldS protein levels
Brains, lumbar spinal cord, and sciatic nerve segments were homogenized in RIPA buffer and prepared for Western blotting as previously described (Mack et al., 2001; Conforti et al., 2007).
Subcellular fractionation was as previously published with modifications (Spencer et al., 2000; Okado-Matsumoto and Fridovich, 2001; Liu et al., 2004; Fang et al., 2005). In initial experiments for generation of crude nuclear, cytoplasmic, and cytosolic fractions (see Fig. 2A) mouse brains were snap-frozen by immersion in liquid nitrogen and stored on dry ice. The nuclear transcription factor SP1 and β-actin served as nuclear and cytoplasmic markers respectively and as loading controls for these respective fractions (Sau et al., 2007).
Figure 2.
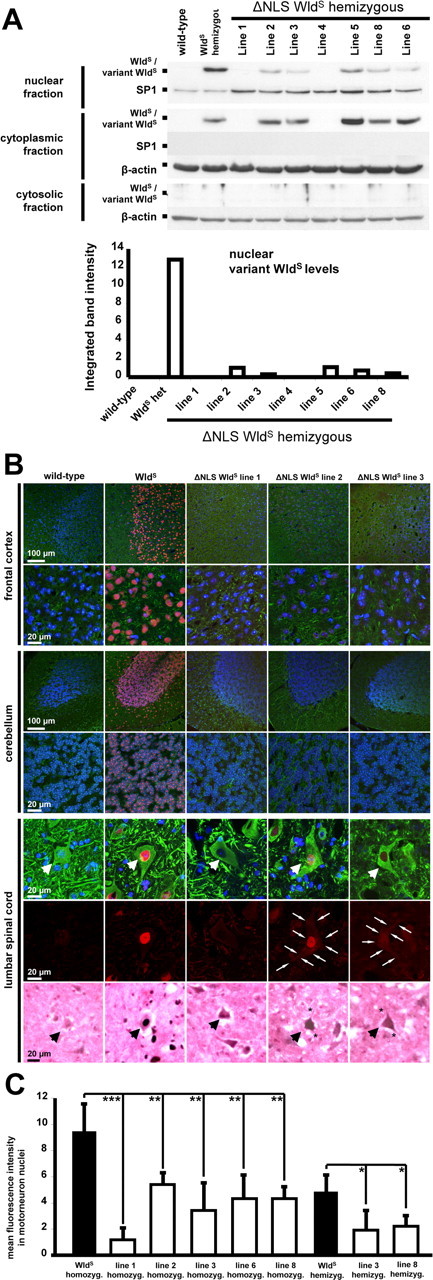
Reduced nuclear targeting of ΔNLS WldS in various neuronal subtypes in vivo. A, Western blots of nuclear, cytoplasmic, and cytosolic brain fractions demonstrating dramatically reduced nuclear targeting in individual mice from different ΔNLS WldS lines compared with spontaneous WldS mutant. The bottom graph demonstrates corresponding relative optic densities of nuclear WldS bands normalized to SP1 for individual mice. These Western blots are representative of three individual experiments. B, Rows 1–6, Confocal images showing (variant) WldS protein immunolabeling (red) with β-III tubulin (green) and Hoechst 33258 (blue) counterstaining on frozen sections from homozygous mice. Transgenics shown represent lines without detectable variant WldS protein expression in Western blotting (line 1) and with strong (line 2) and medium (line 3) variant WldS protein expression levels. Row 7, Light microscopic pictures from lumbar motorneurons (arrows) showing WldS immunolabeling using 3,3′-diaminobenzidine. C, Graph showing normalized fluorescence intensities for nuclear variant WldS protein signals in lumbar motoneurons. For each genotype group, three mice were assessed (10 motorneurons each). *p < 0.05; **p < 0.005; ***p < 0.001; one-way ANOVA.
Each brain was homogenized using a Teflon-glass pestle (no. B15541, Thomas; 10 strokes, 700 rpm) at 1:5 (w/v) ratio in ice-cold homogenization buffer containing 10 mm HEPES, 6 mm MgCl2, 1 mm EDTA, 10% sucrose, pH 7.2, and protease inhibitor cocktail (Roche Diagnostics). Unbroken cells and connective tissue were removed by filtration using a cell strainer (40 μm nylon, BD Falcon). Homogenized brain tissue was centrifuged at 2000 × g for 5 min. Further centrifugation of the resulting pellet A and supernatant B at different speeds yielded the following fractions.
Nuclear fraction.
The pellet A was washed repeatedly and finally resuspended in 0.25 ml of homogenization buffer containing additionally 0.5 m NaCl. The suspension was then incubated for 1 h in an ice bath with frequent vortexing. After incubation the suspension was centrifuged at 8000 × g for 10 min, and the final supernatant was regarded as nuclear fraction.
Cytoplasmic fraction.
The supernatant B was further centrifuged at 8000 × g, and the final supernatant was used as cytoplasmic fraction.
Cytosolic fraction.
The supernatant B was centrifuged at 100,000 × g for 30 min, and the final supernatant was regarded as cytosolic fraction.
In follow-up subcellular fractionation experiments for generation of enriched mitochondrial and microsomal preparations (see Fig. 9A), mouse and rat brains were homogenized as described above in buffer containing 50 mm Tris, 6 mm MgCl2, 1 mm EDTA, 10% sucrose, pH 7.2, and protease inhibitor cocktail (Roche Diagnostics). After 5 min of centrifugation at 2000 × g, the resulting pellet C and supernatant D were used for generation of the following fractions.
Figure 9.
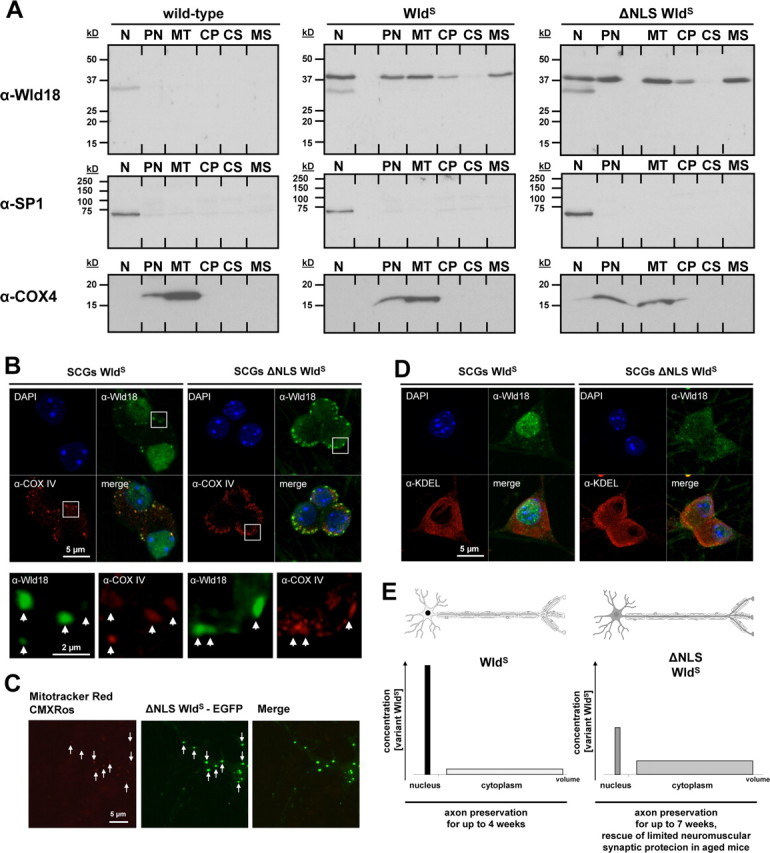
WldS and variant ΔNLS WldS proteins associate with mitochondria and microsomes. A, Western blot of WldS variants in subcellular fractions from mouse brain: N, nuclear; PN, postnuclear; MT, mitochondrial; CP, cytoplasmic; CS, cytosolic; MS, microsomal. Nuclear SP-1 and mitochondrial marker COX-IV were used to assess the fractionation. B, Confocal images of SCG neurons labeled with Wld18 (green; Alexa488-tyramide signal amplification) and COXIV (red) antibodies. White squares represent higher magnification insets shown in lower panel. Arrows indicate partial colocalization of Wld18 and COXIV. C, Confocal live cell imaging of hippocampal neuron expressing variant ΔNLS WldS fused to EGFP (green). The preparation was labeled with Mitotracker Red CMXRos (red). Arrows indicate partial colocalization of ΔNLS WldS-EGFP and mitochondria. D, Confocal images of SCG neurons labeled with Wld18 (green; Alexa488-tyramide signal amplification) and KDEL (red; as ER marker) antibodies. Note many extranuclear variant WldS foci overlap with the KDEL endoplasmic reticulum marker signals. E, Schematic illustration summarizing non-nuclear-mediated delay of Wallerian degeneration: altered ratio of nuclear versus cytoplasmic WldS content results in more robust neuroprotection in ΔNLS WldS transgenics. The graphs demonstrate distribution of WldS (left) with high concentration in the smaller nuclear compartment and low concentration in the much larger cytoplasmic compartment. In contrast, ΔNLS WldS (right) is altered in favor of the large cytoplasmic compartment resulting in easier detectability and in more robust axon and synapse protection.
Nuclear and postnuclear fraction.
The pellet C was resuspended in homogenization buffer plus 0.5 m NaCl for 1 h on ice and the suspension centrifuged at 8000 × g for 10 min. The resulting pellet was again resuspended in homogenization buffer and used as nuclear fraction. The supernatant D was centrifuged at 8000 × g for 10 min to obtain the postnuclear fraction (supernatant).
Cytoplasmic and mitochondria-enriched fraction.
The postnuclear fraction was centrifuged at 21,000 × g for 20 min to obtain mitochondria-enriched pellet and cytoplasmic fraction (supernatant). The mitochondria-enriched pellet was washed in homogenization buffer and resuspended in buffer containing 150 mm NaCl, 50 mm Tris/HCl, and 10% SDS, pH 8.0. This final suspension was regarded as mitochondria-enriched fraction.
Microsome-enriched and cytosolic fraction.
The cytoplasmic fraction was centrifuged at 135,000 × g for 1 h to obtain the microsome-enriched fraction (pellet) and cytosolic fraction (supernatant). The microsome-enriched pellet was processed in the same way as the mitochondria-enriched pellet to obtain the final microsome-enriched fraction.
All described centrifugation steps were performed at 4°C using a Sanyo MSE Harrier 18/80 and Beckman MAX Ultracentrifuge. Subcellular fractions were stored at −80°C and subsequently used for Western blot analysis.
For Western blotting SDS-PAGE and standard wet protein transfer (Bio-Rad) to PVDF membranes were performed as previously described with modifications (Mack et al., 2001; Conforti et al., 2007). Variant WldS was detected using rabbit polyclonal antiserum Wld18 (1:2000) with mouse monoclonal anti-β-actin (1:3000, Abcam, ab8226) or mouse monoclonal anti-neuronal class βIII-tubulin (1:2000, Covance, MMS-435P) as loading controls. In fractionation experiments the following antibodies were used as loading controls: mouse monoclonal anti-SP1 (1:1000, Santa Cruz Biotechnology, PEP2 sc-59); mouse monoclonal anti-histones H1 (1:500, Chemicon International, MAB052); and mouse monoclonal anti-COXIV (1:5000, Abcam, ab14744).
For quantification, integrated optical density (OD) of bands from three blots per experimental group was determined by NIH ImageJ software, normalized to the loading controls and expressed as a percentage of reference WldS expression level.
Nmnat enzyme activities from sagittally divided half mouse brains were determined as described previously (Conforti et al., 2007).
Assessment of axon preservation
All experiments were performed in accordance with the Animals (Scientific Procedures) Act, 1986, under Project License PPL 80/1778. Mice were anesthetized by intraperitoneal injection of Ketanest (5 mg/kg; Parke Davis) and Rompun (100 mg/kg; Bayer). Right sciatic nerves were transected or crushed close to the “foramen intrapiriforme” with contralateral side as control. A 5 mm segment was removed to prevent regeneration from complicating analysis of degeneration at longer lesion durations (14–49 d). The lesion site was also inspected upon nerve removal to be sure the proximal and distal stumps had been fully separated and remained separate. After 6 h (for crush experiments) and 3, 5, 14, 21, 28, 35, and 49 d (for transection experiments), the mice were humanely killed and sciatic/tibial nerve segments were dissected.
We tested the structural preservation of axons in the sciatic and tibial nerve following unilateral sciatic nerve transection by confocal microscopy of a YFP-labeled axonal subset (longitudinal imaging) and by light/electron microscopy using highly established methods for evaluation of axon integrity (Mack et al., 2001; Beirowski et al., 2004, 2005; Adalbert et al., 2005; Conforti et al., 2007).
For analysis of axonal preservation in YFP-labeled nerves, ∼1.5-cm-long nerve stumps containing the sciatic and tibial nerve segments were dissected from humanely killed mice following sciatic nerve transection, processed as previously described (Beirowski et al., 2004, 2005), and mounted on conventional glass slides in Vectashield mounting medium for subsequent analysis on a Zeiss LSM 510 Meta Confocal system. Confocal z-stack series from both longitudinally embedded sciatic and tibial nerve were taken using a 20× magnification objective, and z-projections were electronically generated for final presentation. Criteria for survival were unfragmented YFP-positive fibers in the analyzed segment.
For semithin light microscopy and electron microscopy, sciatic and tibial nerve segments were fixed for at least 3 d in 0.1 m phosphate buffer containing 4% paraformaldehyde and 2.5% glutardialdehyde, embedded in Durcupan resin (Fluka), and processed as described previously (Conforti et al., 2007). Morphological criteria for intact axons were normal myelin sheaths, uniform axoplasm, and intact mitochondria. The percentage of surviving axons in nerve segments was calculated in relation to unlesioned preparations.
Electrophysiology and vital labeling of neuromuscular junctions
Mice were killed by cervical dislocation. Previous section of the sciatic nerve was verified by reexposing the wound in the thigh. Tibial nerve–flexor digitorum brevis (FDB) preparations were dissected and pinned to a Sylgard-lined dish and bathed in oxygenated mammalian physiological saline (137 mm Na+, 4 mm K+, 2 mm Ca2+, 1 mm Mg2+, 147 mm Cl−, 5 mm glucose, and 5 mm HEPES, pH 7.2–7.4, equilibrated with 100% oxygen). The nerve was stimulated using a suction electrode connected to a Powerlab 4/20T and stimulated using 50–200 μs pulses, 0.1–1 mA intensity at 1–20 Hz. Muscle contractions were recorded as short (10–35 s) movies through a Wild M5A dissecting microscope using a Nikon Coolpix 4500 digital cameral fitted with an ocular adapter. The three distal tendons of FDB were then tied together using Ethicon 7/0 suture and connected to a Grass force transducer and the Powerlab bridge amplifier. Twitch contractions to single stimuli delivered at 1–5 Hz and fused contractions evoked by 20 Hz stimulation were recorded using Chart v4.1.1 and Scope v3.6.8 software on an Apple Macintosh Powerbook computer. The preparation was then dismounted from the force transducer and a pair of fine electromyographic recording needles, insulated to within 0.2 mm of their tips with a coating of nail varnish, was inserted into the belly of the FDB muscle. The bathing solution was earthed (grounded) using an Ag/AgCl wire and all three electrodes were connected to the Bioamp input of the Powerlab unit. Stimuli to the tibial nerve were delivered either from the Powerlab using parameters indicated above, or using a A-M Systems 210 isolated pulse stimulator with variable 1–10 V pulses, 200 μs in duration and at frequencies of 1–40 Hz. EMG signals were low-pass (2 kHz), high-pass (10 Hz), 50 Hz notch, and mains filtered and averaged using Scope software. In some cases, a simpler preparation was used for EMG recording. Isolated hindlimbs were stripped of their skin and pinned to a Sylgard-lined dish in oxygenated mammalian saline as above. The tibial nerve was dissected free to the ankle, the foot was amputated at the tibia, and EMG needles were inserted immediately into the FDB/interosseous muscles. The tibial nerve was stimulated with a suction electrode and the EMG response recorded and averaged using the Powerlab Scope software. Sample records were extracted from the Chart and Scope raw data files and pasted into Powerpoint.
For morphological quantification of functionally preserved neuromuscular junctions (NMJs) in tibial nerve–FDB preparations recycled synaptic vesicles of motor nerve terminals were stained using AM1-43 (Nerve Terminal Staining Kit II, Biotium) with 20 Hz nerve stimulation. AM1-43 is a fixable form of styryl dye FM1-43, widely used for vital labeling of NMJs, where it indicates functional synaptic transmission (Betz et al., 1992; Barry and Ribchester, 1995; Mack et al., 2001). Unspecific background fluorescence of AM1-43 was quenched with ADVASEP-7 (Kay et al., 1999). Acetylcholine receptors were subsequently stained with tetramethylrhodamine isothiocyanate conjugates of α-bungarotoxin (TRITC-α-BTX) (Biotium). FDB muscles were fixed in 0.1 m PBS containing 4% paraformaldehyde for 30 min, cleaned of connective tissue and mounted on conventional glass slides in Vectashield mounting medium for analysis. For quantification of endplate occupancy following sciatic nerve lesion occupied and vacant bungarotoxin-labeled NMJs in the three subcompartments of the FDB muscle (see supplemental movies, available at www.jneurosci.org as supplemental material) were counted using an IX81 Olympus fluorescence microscope. Fifty to one hundred endplates were assessed per FDB preparation and compared with the contralateral unlesioned preparation from each mouse.
Immunocytochemistry and immunohistochemistry
For conventional indirect immunofluorescence detection of WldS variant expression in primary and secondary cell culture, cells were fixed in 4% paraformaldehyde (PFA) 48 h after transfection, permeabilized with 1% Triton X-100 for 10 min, blocked [5% NGS (Sigma) in PBS, 1 h], and immunostained using rabbit polyclonal Wld18 antibody (1:500) and secondary Alexa568-goat anti-rabbit antibody (1:200) diluted in 5% NGS/PBS. Nuclear counterstaining was performed with DAPI and cells were mounted in Vectashield (Vector Laboratories).
For immunofluorescence detection of variant WldS protein on brain, lumbar spinal cord, DRG, and sciatic nerve obtained from perfusion fixed mice, 20 μm cryostat sections on poly-l-lysine-coated glass slides (VWR SuperFrost Plus) were incubated overnight in citrate buffer, pH 6.0, at 50°C or for 45 min in 0.05% citraconic anhydride solution, pH 7.4, at 98°C (Namimatsu et al., 2005) and subsequently permeabilized with 0.1% Triton X-100 plus 0.05 m NH4Cl in 0.05 m TBS for 10 min. The sections were then rinsed in fresh TBS, immunoblocked with 5% bovine serum albumin (Sigma) in TBS for 1 h, and incubated overnight at 4°C in primary antibody solution (Wld18 antibody, 1:500 in 0.8% bovine serum albumin in TBS). After extensive washes, the secondary antibody solution (Alexa568-goat anti-rabbit, 1:200 in TBS) was applied for 1 h at room temperature and slices were rinsed in TBS and dH2O. The samples were counterstained with primary mouse monoclonal anti-neuronal class βIII-tubulin (1:500, Covance, MMS-435P), mouse monoclonal anti-neurofilament 200 (1:500, Sigma, clone 52, N0142), or mouse monoclonal anti-APP (1:200, Chemicon, MAB348) and secondary Alexa488-goat anti-mouse (1:200) antibodies as above. Nuclear staining was performed with DAPI or Hoechst 33258. Samples were mounted in Vectashield mounting medium (Vector Laboratories).
For high-sensitivity detection of low-abundance WldS protein variants on dissociated SCG and DRG neuronal cultures exploiting catalyzed reported deposition of tyramide-Alexa 488 or tyramide-Alexa 568 conjugate (Molecular Probes TSA detection kit, Invitrogen), cells were fixed for 15 min with 4% PFA in 0.1 m PBS, rinsed, and permeabilized with 1% Triton X-100 for 10 min. After quenching endogenous peroxidase activity with 3% hydrogen peroxide in PBS for 1 h, cells were treated with 1% TSA blocking buffer (Molecular Probes, Invitrogen) for 1 h, and primary antibody Wld18 (1:500 in 1% TSA blocking buffer) was applied overnight at 4°C. Cells were extensively washed in PBS and incubated with HRP-coupled goat anti-rabbit secondary antibody (1:100 in 1% TSA blocking buffer) for 1 h. The tyramide-fluorophore conjugate solution was applied for 10 min according to the manufacturers instructions. Cells were extensively washed and conventionally counterstained with the following primary antibodies: mouse monoclonal anti-neurofilament 200 (1:500, Sigma, clone 52, N0142); mouse monoclonal anti-COXIV (1:100, Abcam, ab14744); mouse monoclonal anti-KDEL (1:100, MAC 256, gift from Geoff Butcher, Babraham Institute, Cambridge, UK); mouse monoclonal anti-LAMP-2 (1:10, ABL-93, gift from Aviva Tolkovsky, University of Cambridge, Cambridge, UK). Secondary Alexa488-goat anti-mouse (1:200) antibodies were applied, and cells were washed in PBS and dH2O and mounted in Vectashield mounting medium.
For high-sensitivity detection of variant WldS on cryostat sections of peripheral and central nerves, ∼5 mm sciatic and optic nerve segments were dissected and postfixed for 2 h in 4% PFA in 0.1 m PBS. After extensive PBS washes, the nerves were incubated overnight in 20% sucrose, embedded in OCT embedding medium (Shandon), and frozen in a −80°C freezer. Serial 20 μm cryostat sections were cut and mounted onto poly-l-lysine-coated slides. Sections were incubated at 98°C with 0.05% citraconic anhydride solution, pH 7.4, for 45 min and permeabilized with 0.5 m NH4Cl + 0.25% Triton in 0.1 m PBS, and low-abundance WldS protein was detected using HRP-catalyzed tyramide-fluorophore conjugate deposition as described above. In some experiments, the sections were counterstained with Hoechst 33258 and finally mounted in Vectashield mounting medium.
For nonfluorescence immunohistochemical detection of WldS variants, frozen sections were pretreated by incubating overnight at 50°C in citrate buffer, pH 6.0, for antigen retrieval. The Vectastain Elite ABC Kit (PK-6100, Vector Laboratories) was applied according to the manufacturer's instructions, and the peroxidase reaction was visualized with 3,3′-diaminobenzidine (DAB) plus nickel enhancement (ABC method). The WldS18 primary antibody was used at a dilution of 1:500. DAB-labeled sections were counterstained with nuclear fast red (Vector Laboratories).
Nonfluorescent immunohistochemically stained tissue sections were imaged with a Leitz DM RB microscope coupled to a Leica DC camera system and Leica DC Twain Software.
Confocal imaging and fluorescence intensity quantification
Immunostained tissue sections and vital dye-labeled muscle preparations were imaged on a Zeiss LSM 510 Meta Confocal system, and z-series were merged using algorithms from Zeiss LSM Software Release 3.2. Colocalization analysis used Multi-track configuration mode to avoid cross talk between individual fluorophores and bleed-through. Object based colocalization analysis was performed by assessment of overlap between linear fluorescence intensity profiles in individual focal planes. Nuclear variant WldS expression in lumbar spinal cord motoneurons was measured in the ventral horn (10 motoneurons per mouse). Confocal z-series stacks of Hoechst 33258-labeled entire nuclear profiles were recorded and mean variant WldS fluorescence intensities were measured on each single optical section using algorithms from Zeiss LSM Software Release 3.2. The measured mean intensities were normalized to reference signal derived from endothelial nuclear profiles without variant WldS expression from the same section (nuclei of endothelial cells).
Statistical analysis
Data are presented as mean ± SD. One-way ANOVA was used for group comparisons and statistical significance or high significance was considered if p < 0.05 or p < 0.005, respectively.
Results
Two point mutations redistribute WldS from nucleus to cytoplasm in vitro
To reduce targeting of full-length WldS protein from nuclei we introduced two point mutations (R213A, R215A) within the NLS of the Nmnat1 domain. We confirmed nuclear WldS reduction by transiently transfecting primary hippocampal and dorsal root ganglion (DRG) cultures and HeLa and PC12 cells. Immunofluorescence with Wld18 antibody, directed against the unique linker region in WldS (Samsam et al., 2003), showed almost complete exclusion from nuclei in HeLa and PC12 cells and substantially reduced nuclear targeting in primary culture (Fig. 1A and data not shown). Thus, the previously identified Nmnat1 NLS (Sasaki et al., 2006) is also the main determinant of nuclear localization of full-length WldS protein. We additionally identified a weak predicted NLS (PSORTII and NucPred predictions) within the N-terminal 16 aa of WldS but chose not to mutate this region because its influence on WldS biochemistry (Laser et al., 2006) could affect phenotype in other ways.
Figure 1.
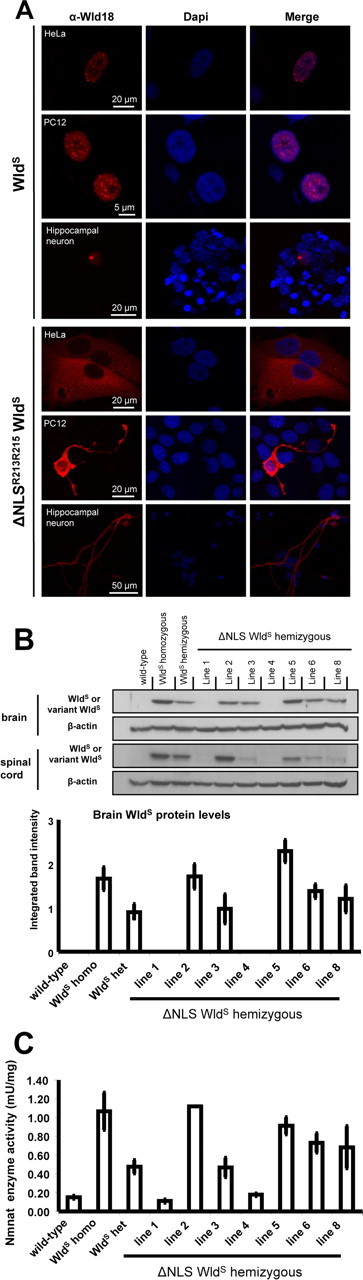
Subcellular localization of ΔNLS WldS in different cell types in vitro and initial characterization of ΔNLS WldS transgenic mice. A, Immunofluorescence using Wld18 primary and Alexa dye-coupled secondary antibodies showing ΔNLS WldS cytoplasmic translocation in transfected HeLa and PC12 cells and hippocampal neurons. B, Top, Representative Westerns blots from brain and lumbar spinal cord crude extracts probed with Wld18 antibody and β-actin loading control. Bottom, Intensities of individual variant WldS protein Western blot bands from brain were densitometrically quantified and normalized to β-actin loading control (N = 3 mice for each group tested). C, Normalized brain Nmnat enzyme activities in individual ΔNLS WldS transgenic lines, wild-type controls, and WldS (N = 2–8 mice for each group tested).
Interestingly, in neurons we detected a strong signal in neuritic trees, especially prominent in neurites >500 μm (Fig. 1A and data not shown). This suggests that the ΔNLS WldS molecule either diffuses or is transported into neurites in vitro. Neuritic immunosignal was not observed in cells expressing native WldS using this method.
Generation of transgenic mice overexpressing ΔNLS WldS
After testing in vitro we coupled the ΔNLSR213A,R215A WldS cDNA to a β-actin promoter, a system that consistently confers a WldS phenotype in mice and rats using native WldS (Mack et al., 2001; Adalbert et al., 2005). Lines were established from seven of the eight founders (lines 1–6, 8) by breeding to YFP-H mice (Feng et al., 2000) for convenient assay of axon degeneration (Beirowski et al., 2004). Line 3, 6, and 8 hemizygotes showed very similar total brain expression levels to that of WldS in heterozygous WldS mice, whereas variant WldS levels were higher in lines 2 and 5 (Fig. 1B). Lumbar spinal cord levels were lower relative to WldS heterozygotes in lines 3, 6, and 8. This difference between brain and spinal cord may reflect the increased presence of variant WldS in the axon (see below), most of which lies outside the spinal cord. As expected, breeding to homozygosity elevated ΔNLS WldS protein expression in brain approximately twofold (data not shown). Variant WldS was not detectable in Western blots from lines 1 and 4, even in homozygous mice, probably reflecting insertional silencing. Enzyme assays showed increased Nmnat activity in brains of highly expressing lines broadly in line with these protein concentrations (Fig. 1C), also confirming that mutation of the NLS did not impair NAD+ synthesis efficacy, which is critical for the neuroprotective phenotype (Araki et al., 2004) (L. Conforti, A. Wilbrey, G. Morreale, L. Janeckova, B. Beirowski, R. Adalbert, F. Mazzola, M. Di Stefano, R. Hartley, E. Babetto, T. Smith, J. Gilley, R. Billington, A. Genazzani, R. Ribchester, G. Magni, and M. Coleman, unpublished work).
NLS mutation reduces nuclear targeting of WldS in vivo
To assess subcellular redistribution of the ΔNLS WldS protein in vivo, we performed Western blots of nuclear, cytoplasmic, and cytosolic fractions (Fig. 2A). Nuclear targeting was reduced by >90% in brain tissue in all expressing lines relative to native WldS (Fig. 2A), although curiously cytoplasmic levels were consistently increased only in transgenic line 5. This could reflect the enormous dilution upon redistribution from nucleus to the much larger cytoplasmic compartment, especially in projection neurons. We were surprised to find significant quantities of native WldS in cytoplasm, but this may have been missed previously due to the use of paraffin-embedded sections, sometimes a less specific antibody (Mack et al., 2001), and Western blotting of just one brain region (cortex) (Fang et al., 2005). In accordance with earlier results (Fang et al., 2005), however, we did not detect WldS in cytosol.
Immunofluorescence of brain and lumbar spinal cord frozen sections also showed strongly reduced nuclear targeting (Fig. 2B). In some cortical and cerebellar neurons, confocal imaging showed only very faint nuclear WldS staining (Fig. 2B, rows 1–4). Faint signals in motor neuron nuclei (Fig. 2B, rows 5–7) were highly significantly reduced in multiple lines relative to spontaneous WldS mice (Fig. 2C). Reflecting variant WldS redistribution, a faint outline of individual cell bodies was readily visible in homozygotes of lines 2 and 3 (Fig. 2B, row 6, arrows). Similar redistribution with reductions in nuclear targeting was seen in DRG neurons (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Using 3,3′-diaminobenzidine immunostaining on frozen sections, we found clear WldS cytoplasmic signals in ΔNLS WldS spinal cord motorneurons with distinct labeling of proximal neuronal processes (Fig. 2B, row 7, asterisks). Interestingly, we also could detect weak cytoplasmic variant WldS signals in some motorneurons from ΔNLS line 1 this way, suggesting the transgene is expressed in this line but at a very low level.
Thus, ΔNLS WldS transgenic lines express the variant WldS protein with full Nmnat enzyme activity and considerably reduced nuclear targeting and relative cytoplasmic redistribution in various neuronal subtypes.
ΔNLS WldS delays Wallerian degeneration more robustly than native WldS
We and others have previously shown that delay of Wallerian degeneration and related axon pathologies by WldS is strongly dose dependent (Perry et al., 1990; Mack et al., 2001; Samsam et al., 2003; Adalbert et al., 2005). In particular, the strength of axon protection in transgenic mice correlates closely with WldS protein level (Mack et al., 2001) (and unpublished observations) (note: the lower expressing lines are no longer available for comparison). Thus, if WldS works through an intranuclear mechanism, reducing nuclear targeting should weaken the protective phenotype.
Surprisingly, however, axonal continuity was substantially better preserved 3, 5, and 14 d after nerve lesion in ΔNLS WldS transgenic lines 2, 3, 5, 6, and 8 than in native WldS mice (Fig. 3A and data not shown). Fragmentation in wild type and partial fragmentation in WldS heterozygotes (Fig. 3A, asterisks) was not seen in hemizygous ΔNLS WldS transgenics. Similar results were obtained in transverse semithin sections 3–14 d after nerve lesion (Fig. 3B; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Quantification showed significantly to highly significantly more intact axons in line 2, 3, 5, 6, and 8 hemizygotes than in WldS heterozygotes (p < 0.05 and p < 0.005, one-way ANOVA). For example, after 14 d, 24.2 ± 2.2% intact axons were intact in WldS (N = 3) compared with 67.4 ± 2.8% in line 3 (N = 3).
Figure 3.

Time course of Wallerian degeneration in ΔNLS WldS/YFP-H transgenics at 3, 5, and 14 d following sciatic nerve transection. A, Representative confocal z-series stacks of sciatic and tibial nerves from wild type, heterozygous WldS (“WldS het”), and ΔNLS WldS transgenics (“ΔNLS WldS het”) additionally expressing YFP in a representative axonal subset (YFP-H transgene) to facilitate longitudinal imaging of axons 3, 5, and 14 d after transection. No interruptions are detectable in hemizygous ΔNLS WldS nerves, whereas axons in nerves from WldS heterozygotes increasingly fragment at the indicated time points (asterisks). Note that we observed proximo-distal gradients of axonal fragmentation along the sciatic/tibial nerve consistent with earlier data (Beirowski et al., 2005). B, Quantification of axonal preservation following nerve lesion in semithin sections showing percentage of intact myelinated sciatic nerve axons (percentage of contralateral unlesioned nerve) at 3, 5, and 14 d following axotomy (see supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Intriguingly, lines 1 and 4 showed axon preservation up to 14 d (supplemental Fig. 3, available at www.jneurosci.org as supplemental material, and data not shown) despite the lack of detectable variant WldS on Western blots (Fig. 1B). In contrast, we previously reported that a low but detectable level of native WldS was insufficient to confer any detectable axon protection (WldS transgenic line 4839 hemizygotes) (Mack et al., 2001). Thus, the amount of protein expressed in transgenic WldS line 4839 should be at least as great, while axon protection is far weaker than in ΔNLS WldS lines 1 and 4. Unfortunately the previously generated WldS line 4839 is no longer available for direct comparison, but this result also suggests WldS has greater efficacy after a reduction in nuclear targeting.
Reduction of WldS nuclear targeting extends the maximum preservation of axon continuity and ultrastructure
We then asked whether the maximum time for which axon continuity and ultrastructure can be preserved increases when WldS nuclear targeting is reduced. For this we used lines 3 and 8 with expression levels similar to native WldS mice. In WldS heterozygotes all axons degenerated by 21 d, whereas in line 3 ΔNLS WldS transgenics some axons remained continuous for at least 35 d (Fig. 4A). In homozygotes the maximum preservation of continuous axons was extended from <35 to >49 d (Fig. 4B and data not shown). It is also interesting to note that YFP has a sufficiently long half-life in these axons to give a strong signal for 7 weeks after being isolated from any further synthesis in the cell body. To confirm that the loss of signal in WldS is not simply due to degradation of YFP, and to study the underlying axon ultrastructure, we then performed electron microscopy at these time points (Fig. 4C). Many ultrastructurally normal axons were preserved up to 49 d in ΔNLS WldS lines 3 and 8, with uniformly distributed axoplasm and unswollen mitochondria, whereas all axonal profiles were completely degraded in spontaneous WldS mice (Fig. 4C).
Figure 4.

Long-term axon survival after sciatic nerve lesion in ΔNLS WldS transgenics. A, Representative confocal images of lesioned sciatic/tibial nerves from heterozygous WldS (“WldS het”) and hemizygous ΔNLS WldS transgenics from line 3 crossed to YFP-H. While sciatic and tibial nerves from WldS heterozygotes show pronounced degradation of YFP-positive axon fragments from 21 d, ΔNLS WldS line 3 hemizygotes have uninterrupted axons up to 35 d (arrows in inset 1 and 2). B, Representative confocal images of lesioned sciatic/tibial nerves from WldS (“WldS homo”) and ΔNLS WldS line 3 homozygotes. While WldS homozygotes show pronounced degradation of YFP-positive axon fragments from 35 d, ΔNLS WldS line 3 mice display uninterrupted axons up to 49 d (arrows in inset 3 and 4). C, Transmission electron microscopy of distal sciatic and tibial nerve from WldS homozygotes (left) and line 3 (middle) and line 8 (right) ΔNLS WldS transgenics 35 and 49 d following nerve section. In contrast to WldS, some ΔNLS WldS axons 49 d after axotomy are ultrastructurally preserved, showing intact myelin sheaths, uniform, regularly spaced cytoskeleton, and normal-appearing mitochondria. Scale bars, 2 μm.
Denervated ΔNLS WldS axons and neuromuscular junctions remain functional in young and aged transgenics
To test whether NMJs were also preserved after axotomy, we recorded muscle contractions, electromyography, and vital labeling of synaptic terminals in FDB preparations. Functional protection of axotomized motor nerve terminals and NMJs in spontaneous and transgenic WldS mice is highly age dependent, with innervation 3 d after a nerve lesion falling from >80% at 1–2 months to <20% at 12 month (Gillingwater et al., 2002). As expected, young WldS homozygotes (∼2 months) showed robust conduction of action potentials and synaptic transmission 3 and 6 d after axotomy, but this protective phenotype was almost completely lost in older mice (aged >7 months).
Three days after sciatic nerve transection, 2-month-old wild-type FDB muscles showed no activity, while age-matched FDB from ΔNLS WldS line 3 or spontaneous WldS mutants showed robust contractile and electromyographic responses to nerve stimulation indicating functional preservation of both motor axons and NMJs (supplemental Fig. 4A–F, Movies 1, 2, available at www.jneurosci.org as supplemental material). Quantitative, functional labeling using the activity-dependent nerve terminal dye AM1-43 showed almost 100% innervated NMJs in FDB preparations from both ΔNLS WldS and spontaneous WldS mutants (supplemental Fig. 4G, available at www.jneurosci.org as supplemental material). Six days after sciatic nerve transection the proportion of occupied NMJs decreased to ∼50% in young WldS mutants, whereas young ΔNLS WldS transgenics still showed almost 100% intact NMJs, indicating stronger protection of synaptic terminals (Fig. 5E; supplemental Fig. 4G, available at www.jneurosci.org as supplemental material). After 10 d, <10% of intact NMJs remained in ΔNLS WldS FDB (supplemental Fig. 4G, available at www.jneurosci.org as supplemental material).
Figure 5.
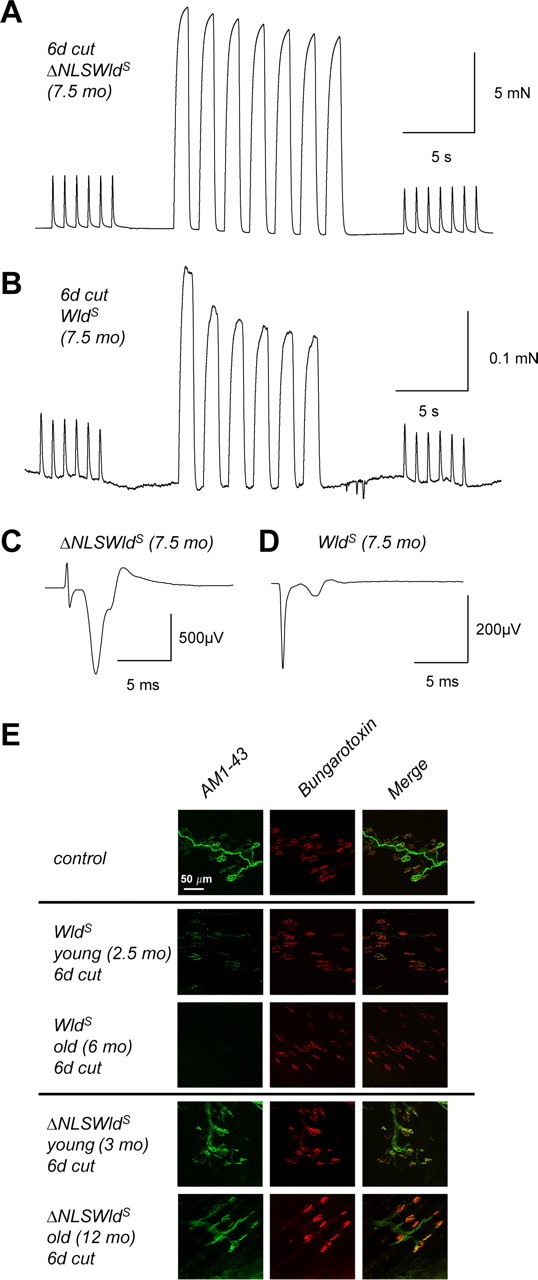
Maintained preservation of neuromuscular function in aged ΔNLS WldS transgenic mice. A, B, Recordings of FDB repetitive 1 Hz muscle twitch contractions and 20 Hz fused contractile responses, 6 d after sciatic nerve lesion from 7.5-month-old ΔNLS WldS transgenic (A) and age-matched native WldS mutant (B). Note the amplitude of isometric force generated by nerve stimulation in ΔNLS WldS is ∼50 times that of the native WldS. The fluctuating baseline discernible in B is due partly to the increased amplifier gain required to register the muscle response and partly to distributed asynchronous spontaneous contractions typically observed in axotomized WldS muscle. C, D, Averaged extracellular EMG recordings using nerve/muscle preparations from A and B. The initial spike is the stimulus artifact and the slower waves are produced by muscle fiber action potentials. Note the ∼10-fold greater amplitude of the evoked EMG response in ΔNLS WldS (C) compared with WldS (D). E, Representative confocal z-series projections of paraformaldehyde-fixed FDB nerve muscle preparations double labeled presynaptically with the activity-dependent nerve terminal dye AM1-43 and postsynaptically by the acetylcholine receptor marker TRITC-α-bungarotoxin. Colocalization of the green and red signals thus indicates functionally preserved NMJs.
We then studied mice aged 6–12 months old (Fig. 5), when native WldS mice almost completely lose neuromuscular synaptic protection. As expected, 7.5-month-old WldS muscles showed only weak or no contraction upon stimulation of the axotomized distal nerve stump (supplemental Movie 4, available at www.jneurosci.org as supplemental material; Fig. 5B). Electromyographic responses were also very weak (Fig. 5D). AM1-43 functional labeling in a 6-month-old WldS mutant confirmed almost complete absence of occupied NMJs (Fig. 5E). In marked contrast, axotomized FDB from 7.5-month-old ΔNLS WldS mice showed robust contractions after nerve stimulation, with contractile forces and electromyographic responses indistinguishable from nonaxotomized muscles (N = 2) (supplemental Movie 3, available at www.jneurosci.org as supplemental material; Fig. 5A,C). In accord with these results, quantitative AM1-43 functional labeling in a 12-month-old ΔNLS WldS mouse 6 d after lesion revealed 95% endplate occupancy (Fig. 5E). Together, these results suggest that the decline of synaptic protection with age in native WldS mice is significantly reduced by extranuclear targeting of variant WldS. Thus, functional survival of motor axons and their nerve terminals increases and becomes independent of age if WldS is partially translocated from the nucleus to the cytoplasm in vivo.
Detection of WldS protein variants in axons
These findings raise the possibility that even native WldS protein may function within axons. Western blotting with Wld18 at a dilution at which it is completely specific for WldS revealed a faint 43 kDa band in WldS mouse and transgenic WldS rat sciatic nerves that was absent in wild type (Fig. 6A,B). Consistent with a local protective action in axons, ΔNLS WldS transgenic mice showed substantially higher amounts in nerve extracts from lines 3 and 8 (Fig. 6A). Nevertheless, we could see no WldS-specific conventional immunofluorescence staining on sections or whole-mount preparations from paraformaldehyde fixed PNS and CNS tissue (data not shown). Interestingly, antigen retrieval using citraconic anhydride (Namimatsu et al., 2005) revealed a marked glial signal in sciatic and optic nerve sections from native WldS mice and rats and ΔNLS WldS transgenics (Fig. 6C and data not shown). This signal increased near the lesion site in injured nerves (Fig. 6D), but its significance for axon protection is unknown since neuronal expression is sufficient for WldS neuroprotection and delay of Wallerian degeneration can be observed in vitro in the virtual absence of glia (Glass et al., 1993; MacDonald et al., 2006; Conforti et al., 2007). Using this method and high-power confocal imaging we also observed weak axonal staining in ΔNLS WldS but not in native WldS and wild-type sciatic nerve sections (Fig. 6E; supplemental Fig. 5, available at www.jneurosci.org as supplemental material; and not shown). Axoplasmic ΔNLS WldS protein was distributed throughout nodes and internodes of individual sciatic nerve axons in a fine granular staining pattern (Fig. 6E). To test whether variant WldS protein remains detectable after axotomy we conducted Western blotting and immunofluorescence on distal sciatic nerve stumps from ΔNLS WldS transgenic mice 1 week following lesion (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). The protein was still present at this time, consistent with a direct axonal role, and no changes in staining pattern could be observed (supplemental Fig. 6B, available at www.jneurosci.org as supplemental material).
Figure 6.
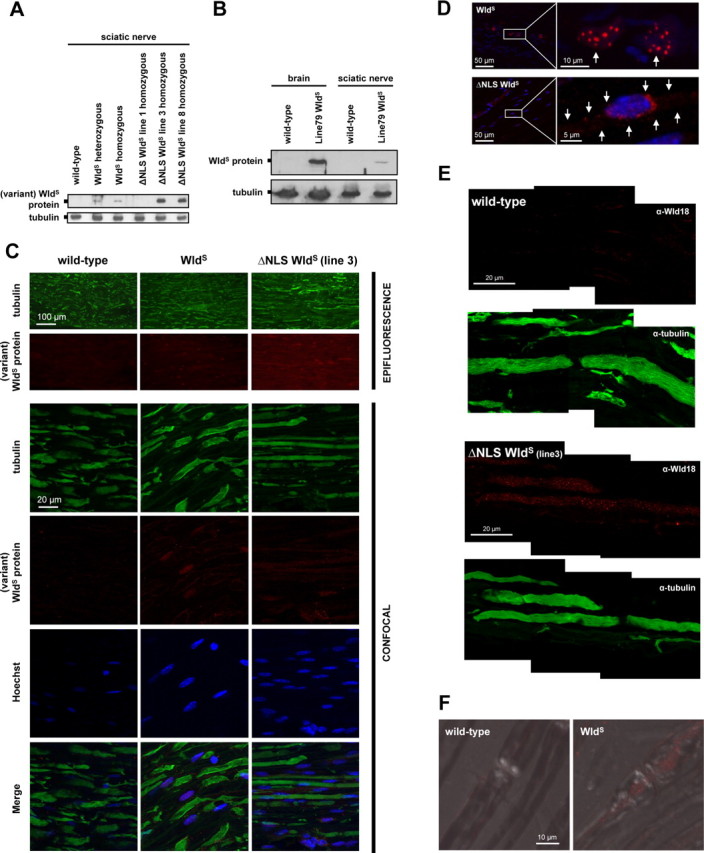
Detection of WldS and variant ΔNLS WldS proteins in nerves in vivo. A, Western blot showing presence of variant WldS proteins in sciatic nerve extracts from both spontaneous WldS mice and ΔNLS WldS transgenics. Note markedly higher levels of variant WldS protein in extracts from ΔNLS WldS lines 3 and 8 compared with WldS (heterozygous and homozygous). B, Western blot showing presence of WldS protein in sciatic nerve extracts from transgenic WldS rat (line 79, homozygous). For comparison, detection of WldS in similar total protein amount from brain homogenate from the same rat is also shown. C, Fluorescence immunostaining on wild-type, WldS (homozygous), and ΔNLS WldS (line 3, homozygous) longitudinal sciatic nerve sections using tubulin (green) and Wld18 (red) antibodies. Conventional epifluorescence microscopy (rows 1 and 2) demonstrates more intense Wld18 labeling in sciatic nerve from ΔNLS WldS mouse than from WldS. Higher-resolution confocal images (rows 3–6) from the same sections show variant WldS protein signals in glial cells whose nuclei were counterstained with Hoechst 33258 (blue). Note more prominent Wld18 staining in sample from ΔNLS WldS sciatic nerve. D, Confocal images showing induction of variant WldS expression (red) in glial cells 6 h after sciatic nerve injury located distally from the lesion site. Note nuclear WldS foci in activated glial cells (arrows, upper row) from native WldS mutant, whereas induced ΔNLS WldS protein expression shows a more cytoplasmic staining pattern demarcating the cell body (arrows, lower row). Blue, Hoechst 33258 counterstain. E, High-power confocal composite (z-series projection) shows presence and homogenous distribution of variant WldS protein (red) in the nodal and internodal axoplasm of ΔNLS WldS sciatic nerve (line 3) which is counterstained with tubulin (green). Note the fine granular staining pattern of Wld18 antibody within the axoplasm. Variant WldS protein immunoreactivity is absent from wild-type sciatic axons. F, Confocal images (z-series projection + DIC merge) showing localization of WldS protein (red) in nodal and perinodal axoplasm of native WldS axon in contrast to wild-type fiber where WldS signal is absent (Wld18 antibody plus Alexa568-tyramide signal amplification).
The presence of variant WldS protein in the axoplasm raises the possibility that it could be transported anterogradely and/or retrogradely. To test this and to further enhance the signal, we performed focal sciatic nerve crush injury and immunostained cryosections with Wld18 as above and additionally with APP antibody (Fig. 7). As previously shown (Cavalli et al., 2005), sciatic nerve crush caused a focal block of axonal transport, and APP accumulated primarily at the proximal side close to the injury point 6 h following lesion in mice of each genotype (Fig. 7A,B). A small increase in WldS signal was seen at crush sites of native WldS nerves, although this may reflect glial signal induced by the lesion (see above). An altogether more striking and clearly axonal signal was evident on both sides of the crush in ΔNLS WldS axons. This suggests that at least the variant ΔNLS WldS protein is transported both anterogradely and retrogradely in fast axonal transport, to account for this build up within 6 h, although more direct evidence will be needed to confirm this.
Figure 7.

Accumulation of variant WldS at the site of nerve constriction. A, B, Fluorescence double immunostaining on longitudinal sciatic nerve sections proximal and distal to crush site (6 h p.o.) using Wld18 (red) and APP (green) antibodies. Confocal projections demonstrate no Wld18 signal in wild type (A, left), weak Wld18 signal in nerve portions from native WldS (A, right) and substantial accumulation of variant WldS protein signal proximal and distal to the crush site in ΔNLS WldS sciatic nerves (B, lines 2 and 3 homozygous). APP immunosignal is enriched in proximal portions close to the crush site in all samples at this time point. Confocal survey composites in bottom panels show the position of surgical nerve constriction (arrows) in relation to adjacent nerve segments. C, Control double immunostaining on unlesioned longitudinal sciatic nerve segments from wild-type, native WldS, and ΔNLS WldS mice for comparison with A and B.
To assess whether presence of WldS can be demonstrated in native WldS axons by increasing detection sensitivity we exploited catalyzed reporter deposition (CARD) of tyramide derivatives (Van Heusden et al., 1997). Similar to the results in nerves from ΔNLS WldS transgenics, this method revealed also axoplasmic WldS in granular staining pattern at native WldS nodes and internodes (Fig. 6F), with lack of signal in wild type indicating specificity.
For further corroboration we then studied axonal WldS in superior cervical ganglion (SCG) explants (Fig. 8A) to exclude glia, reduce the dilution of WldS into the large axonal volume and to isolate neurites in the virtual absence of cell bodies. A robust WldS phenotype was present in explants from both WldS and ΔNLS WldS mice (data not shown). Neuritic extracts from WldS and ΔNLS WldS explants showed a clear Western blotting signal that was markedly stronger in the latter (N = 4 experiments) (Fig. 8A). CARD immunostaining confirmed protein redistribution showing decreased nuclear staining and distinct labeling of neurites in ΔNLS WldS SCGs and DRGs (Fig. 8B and data not shown). Interestingly, cell bodies and occasionally axons of many neurons showed discrete variant WldS foci of varying size (Figs. 8C,D, 9B,D). Longer tyramide deposition reaction times revealed cytoplasmic foci also for native WldS but not for wild-type controls. Variant WldS foci were also present in hippocampal cultures transfected with a ΔNLS WldS construct fused to EGFP (Fig. 8E). However, such foci were never observed in situ in brain or spinal cord neurons from ΔNLS WldS transgenics or spontaneous WldS mice, presumably due to lower concentrations of the variant WldS proteins in vivo and/or antigen masking through fixation and embedding procedures.
Figure 8.

Visualization of WldS and variant ΔNLS WldS distribution in primary neuronal culture. A, Left, Western blot from SCG cell body/proximal neurite (“cell bodies”) and distal neurite fractions (“neurites”) showing WldS and variant WldS in neurites. Note the reduced level of variant ΔNLS WldS in cell bodies and significantly increased levels in neurites. To rule out nuclear contamination derived from glial and other cells in neurite fractions, Western blots were probed with the nuclear marker Histones H1. Right, Densitometric quantification of variant WldS protein (normalized to β-actin). Data from two independent experiments are presented as mean ± SD. B, Left, Confocal images showing cell bodies (and proximal neurites) from dissociated SCG preparations labeled with Wld18 antibody (green; Alexa488-tyramide signal amplification), neurofilament antibody (red) and DAPI (blue). Note cytoplasmic redistribution of the ΔNLS WldS protein variant relative to WldS. Right, Higher magnification confocal images demonstrating variant WldS in SCG neurites (green; Alexa488-tyramide signal amplification). C, D, High-power confocal projections demonstrating peri-nuclear variant WldS foci (green) in SCG preparation from ΔNLS WldS transgenic mouse (C) and occasional foci in proximal neurites (D, arrows). E, Confocal projection showing transfected hippocampal neuron expressing ΔNLS WldS-EGFP fusion protein. Note cytoplasmic ΔNLS WldS-EGFP foci in cell body and neurites.
Together, these results suggest presence of extranuclear WldS in axons with higher axoplasmic levels in ΔNLS WldS transgenic mice and the possibility of axonal transport of WldS variants.
WldS and variant ΔNLS WldS proteins associate with mitochondria and intracellular membranes
Finally, we studied the association of WldS and variant WldS with organelles in subcellular fractionation of brain tissue and in cell culture. WldS and variant WldS were detectable both in enriched mitochondrial (MT) and intracellular membrane (microsome) fractions (MS) but absent in wild type (Fig. 9A). Similar data were obtained from transgenic WldS rats (data not shown). In CARD analysis, ∼85% of extranuclear native or variant WldS foci partially colocalized with mitochondria, although many mitochondria were not colocalized (Fig. 9B). Again, wild-type controls lacked these foci (supplemental Fig. 7A, available at www.jneurosci.org as supplemental material). In transfected hippocampal primary cultured neurons and PC12 cells, ∼90% and 60% of ΔNLS WldS-EGFP foci partially colocalized with Mitotracker Red CMXRos (Fig. 9C) or DsRed2-Mito, respectively, and linear fluorescence intensity profiles confirmed partial colocalization (supplemental Fig. 7B, available at www.jneurosci.org as supplemental material). Many other WldS foci were adjacent to mitochondria, but conversely most mitochondria in PC12 cells were not detectably associated with WldS, suggesting association with a subset of mitochondria. Additionally, we observed partial association between extranuclear WldS variants and the endoplasmic reticulum marker KDEL (anti-MAC256 antibody) in SCG neurons (Fig. 9D). Analysis of SCG neurons costained with LAMP-2 antibodies (anti-ABL-93) did not indicate any association with lysosomes (data not shown).
Discussion
These data show that the effectiveness of WldS mediated neuroprotection in vivo is highly dependent on non-nuclear levels of the mutant protein, indicating a cytoplasmic or even direct axonal role for WldS (Fig. 9E). We detect native WldS protein outside the nucleus and in axons for the first time in vivo and report novel subcellular localization to mitochondria and microsome fractions. In addition to these insights into the protective mechanism, the increased efficacy will improve assessment of which neurodegenerative disorders involve Wallerian-like degeneration and points to more optimal therapeutic strategies based around WldS.
WldS protection has been shown to be strongly dose-dependent by the weaker phenotype of C57BL/WldS heterozygotes in both injury and disease (Perry et al., 1992; Mack et al., 2001; Samsam et al., 2003; Mi et al., 2005) and by strong correlation between expression level and phenotype strength in WldS transgenic lines (Mack et al., 2001; Adalbert et al., 2005). Surprisingly, we found that reduced nuclear targeting of WldS without altering total expression level strengthens the protective phenotype, rather than weakening it as a nuclear action would predict. The maximum axon survival following sciatic nerve transection was extended from 4 to 7 weeks and very weakly expressing lines were able to confer a robust WldS phenotype. Intriguingly, the increased efficacy of the ΔNLS WldS variant is particularly striking at motor nerve terminals from older mice, where native WldS is far less effective than in the axon trunk (Mack et al., 2001; Gillingwater et al., 2002).
The reason for the substantial weakening of functional NMJ protection in older WldS mice is unknown but appears not to involve any decrease in WldS expression (Gillingwater et al., 2002). In contrast to spontaneous and transgenic WldS mice, ΔNLS WldS dramatically retained its ability to preserve neuromuscular synapses in older mice as well as increasing the maximum survival of axotomized NMJs in younger mice. We previously reported enhanced NMJ protection after sciatic nerve lesion in transgenic WldS rats and speculated that the longer distal axon stump relative to mice might be responsible (Adalbert et al., 2005). In WldS mice lengthening the distal stump delays degeneration of NMJs by 1–2 d/cm (Ribchester et al., 1995). One model to explain this could be that WldS neuromuscular synapses require continuous supply of a neuroprotective factor for their survival. We hypothesize now that this putative factor could be axonally transported WldS itself. As rate of axonal transport declines with age (Cross et al., 2008) the weakening of protective phenotype in old WldS mutants could be explained with this model. Axonal WldS protein may fall below an efficacy threshold for synaptic maintenance as mice age. This might not occur in old ΔNLS WldS transgenics due to the overall higher variant WldS levels in axons (as shown for sciatic nerves). Although we provide preliminary data suggesting fast axonal transport of at least variant ΔNLS WldS protein in sciatic nerve, further experiments addressing the relative levels of WldS variants being transported and at synapses will be needed to test this hypothesis.
Several hypotheses for WldS action now need to be reexamined to ask how each fits with these new data. For instance, mechanisms involving action of WldS exclusively in nuclei now appear unlikely, although we cannot rule out a simultaneous action in both cytoplasm and nuclei. Reports of gene expression changes in WldS mice (Gillingwater et al., 2006; Simonin et al., 2007b) may be less linked to the high nuclear WldS concentration than expected and feedback mechanisms from cytoplasmic WldS could be one other explanation for the gene expression data. Previous data based on strong lentiviral overexpression of Nmnat1 in DRG neurons suggested efficacy was independent of subcellular targeting (Sasaki et al., 2006) although it is not clear whether this reflects the axon protection mechanism in vivo (Conforti et al., 2007). Altered gene regulation driven by the NAD+ dependent deacetylase sirtuin 1 (Sirt1), a nuclear enzyme (Araki et al., 2004) now appears unlikely and a role for Sirt1 was already hard to reconcile with axon protection in Sirt1 null neurons (Wang et al., 2005) (M. Avery, S. Sheehan, K. Kerr, J. Wang, and M. Freeman, unpublished work). A non-nuclear site of action for WldS now casts further doubt on a mechanism involving nuclear Sirt1. Instead, our data are consistent with local axonal protection mechanisms, proposed previously based on in vitro data (Wang et al., 2005). Although we demonstrate localization of WldS variants to axons in vivo additional targeting studies will be needed to address whether the critical site is within the axon itself or in the cell body cytoplasm.
Once WldS is targeted to a specific non-nuclear site, the associated, essential Nmnat activity (Araki et al., 2004) is likely to produce a high local NAD+ concentration. The existence of multiple pathways for NAD+ catabolism (Berger et al., 2004) may help explain why this increase has not been detected more generally (Mack et al., 2001; Araki et al., 2004), as once generated NAD+ will be rapidly and locally degraded. While it persists, however, this tightly localized NAD+ may be used to influence downstream calcium signaling (Berger et al., 2004), bioenergetics (Di Lisa and Ziegler, 2001; Wang et al., 2005), protein modification (Berger et al., 2004) or other functions near its site of production (Berger et al., 2007). The presence of WldS in the mitochondrial fraction and the partial colocalization with a subset of mitochondria in vitro, as observed for other proteins (Kang et al., 2008), are consistent with a bioenergetics or signaling role, as mitochondria require NAD+ to synthesize ATP and to regulate signaling pathways necessary for cell viability (Di Lisa and Ziegler, 2001; Yang et al., 2007a). However, although WldS axons maintain both NAD+ and ATP levels after axon lesion better than wild type (Ikegami and Koike, 2003; Wang et al., 2005), a causal role for bioenergetic metabolism in WldS mechanism is unproven. A recent in vitro study suggests an alternative mechanism based on Nmnat blocking production of reactive oxygen species (ROS) from mitochondria (Press and Milbrandt, 2008).
Other data regarding the molecular mechanism of WldS mediated axon protection suggest involvement of valosin-containing protein (VCP) that binds the N-terminal 16 aa of WldS through a VCP binding motif (Laser et al., 2006). This region is necessary but not sufficient for axon protection in mice (Conforti et al., 2007) (Conforti, Wilbrey, Morreale, Janeckova, Beirowski, Adalbert, Mazzola, Di Stefano, Hartley, Babetto, Smith, Gilley, Billington, Genazzani, Ribchester, Magni, and Coleman, unpublished work) and is necessary for full strength phenotype in Drosophila (Avery, Sheehan, Kerr, Wang, and Freeman, unpublished work). Another recent study showed that VCP binding is required for WldS to localize to discrete subnuclear foci (Wilbrey et al., 2008). These nuclear foci were not required for the protective phenotype, but may reflect a more general fine localization mechanism that is relevant also to WldS in other compartments. VCP is a ubiquitous cellular protein with high concentrations in the neuronal cytoplasm (Wang et al., 2004; Laser et al., 2006). The high cytoplasmic VCP content is likely to drive WldS-VCP binding, particularly at sites where VCP is most abundant. One such site is the endoplasmic reticulum (ER), where protein binding interactions with Hrd1, gp78, and Derlin-1 recruit VCP (Schulze et al., 2005). The presence of WldS in microsome fraction where detection of VCP has been also reported (Madeo et al., 1998) is consistent with an ER localization, although the incomplete colocalization with ER suggests either that WldS is restricted to ER subdomains and/or that it is restricted to a different microsome component.
ΔNLS WldS transgenic mice should considerably enhance axon and synapse protection also in neurodegeneration models. Thus far, WldS showed significant axon protection in several disorders such as progressive motor neuronopathy (pmn) (Ferri et al., 2003), peripheral neuropathy (Samsam et al., 2003) and chronic or induced glaucoma (Howell et al., 2007; Beirowski et al., 2009), but was less effective in others, most notably in mouse models of familial ALS (Vande Velde et al., 2004; Fischer et al., 2005). The modest or even undetectable neuroprotective effect in WldS/SOD1 mutants was unexpected, given that these mice have axonal transport deficiencies (Vande Velde et al., 2004) and that axonal transport impairment appears to underlie the pmn phenotype. In fact, WldS does confer significant protection of motor nerve terminals in SOD1 (G93A) mutant mice at least up to 80 d of age but not thereafter (Fischer et al., 2005), suggesting that the age-dependent weakening of synaptic protection in WldS mice could explain its inability to alter the SOD1 phenotype strongly. It will be important to test now whether the ΔNLS WldS variant could overcome this limit of neuroprotection.
We conclude that WldS is able to exert extensive neuroprotective effects on axons and synapses through a non-nuclear action, indicating an urgent need to address the roles of NAD+ synthesis and other WldS functions such as VCP binding in these locations. We show that the efficacy of WldS can be increased by appropriate subcellular targeting, so future studies can now address whether the critical location is axonal or cytoplasmic, whether mitochondria or one of the microsome compartments are involved, and whether the enhanced protection offered by the ΔNLS WldS variant can be developed into more effective therapy for axonopathies.
Footnotes
This work was supported by the Biotechnology and Biological Sciences Research Council and the German Federal Ministry of Education and Research (BMBF-LPD 9901/8-128). R.R.R. held a Royal Society of Edinburgh/Scottish Executive Support Research Fellowship during the conduct of this study. We are grateful to the staff of the Babraham Institute animal facilities for excellent technical assistance with mouse breeding and to Dr. Simon Walker (Babraham Institute) for assistance with confocal microscopy.
References
- Adalbert et al., 2005.Adalbert R, Gillingwater TH, Haley JE, Bridge K, Beirowski B, Berek L, Wagner D, Grumme D, Thomson D, Celik A, Addicks K, Ribchester RR, Coleman MP. A rat model of slow Wallerian degeneration (WldS) with improved preservation of neuromuscular synapses. Eur J Neurosci. 2005;21:271–277. doi: 10.1111/j.1460-9568.2004.03833.x. [DOI] [PubMed] [Google Scholar]
- Araki et al., 2004.Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Barry and Ribchester, 1995.Barry JA, Ribchester RR. Persistent polyneuronal innervation in partially denervated rat muscle after reinnervation and recovery from prolonged nerve conduction block. J Neurosci. 1995;15:6327–6339. doi: 10.1523/JNEUROSCI.15-10-06327.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski et al., 2004.Beirowski B, Berek L, Adalbert R, Wagner D, Grumme DS, Addicks K, Ribchester RR, Coleman MP. Quantitative and qualitative analysis of Wallerian degeneration using restricted axonal labelling in YFP-H mice. J Neurosci Methods. 2004;134:23–35. doi: 10.1016/j.jneumeth.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Beirowski et al., 2005.Beirowski B, Adalbert R, Wagner D, Grumme DS, Addicks K, Ribchester RR, Coleman MP. The progressive nature of Wallerian degeneration in wild-type and slow Wallerian degeneration (WldS) nerves. BMC Neurosci. 2005;28:1166–1179. doi: 10.1186/1471-2202-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski et al., 2009.Beirowski B, Babetto E, Coleman PM, Martin RM. The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur J Neurosci. 2009 doi: 10.1111/j.1460-9568.2008.06426.x. in press. [DOI] [PubMed] [Google Scholar]
- Berger et al., 2004.Berger F, Ramírez-Hernández MH, Ziegler M. The new life of a centenarian: signalling functions of NAD(P) Trends Biochem Sci. 2004;29:111–118. doi: 10.1016/j.tibs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Berger et al., 2007.Berger F, Lau C, Ziegler M. Regulation of poly(ADP-ribose) polymerase 1 activity by the phosphorylation state of the nuclear NAD biosynthetic enzyme NMN adenylyl transferase 1. Proc Natl Acad Sci U S A. 2007;104:3765–3770. doi: 10.1073/pnas.0609211104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz et al., 1992.Betz WJ, Mao F, Bewick GS. Activity-dependent fluorescent staining and destaining of living vertebrate motor nerve terminals. J Neurosci. 1992;12:363–375. doi: 10.1523/JNEUROSCI.12-02-00363.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli et al., 2005.Cavalli V, Kujala P, Klumperman J, Goldstein LS. Sunday Driver links axonal transport to damage signaling. J Cell Biol. 2005;168:775–787. doi: 10.1083/jcb.200410136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman, 2005.Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Coleman et al., 1998.Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH. An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc Natl Acad Sci U S A. 1998;95:9985–9990. doi: 10.1073/pnas.95.17.9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti et al., 2000.Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc Natl Acad Sci U S A. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti et al., 2007.Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, Magni G, Glass JD, Coleman MP. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ. 2007;14:116–127. doi: 10.1038/sj.cdd.4401944. [DOI] [PubMed] [Google Scholar]
- Cross et al., 2008.Cross DJ, Flexman JA, Anzai Y, Maravilla KR, Minoshima S. Age-related decrease in axonal transport measured by MR imaging in vivo. Neuroimage. 2008;39:915–926. doi: 10.1016/j.neuroimage.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Di Lisa and Ziegler, 2001.Di Lisa F, Ziegler M. Pathophysiological relevance of mitochondria in NAD(+) metabolism. FEBS Lett. 2001;492:4–8. doi: 10.1016/s0014-5793(01)02198-6. [DOI] [PubMed] [Google Scholar]
- Fainzilber and Twiss, 2006.Fainzilber M, Twiss JL. Tracking in the Wlds—the hunting of the SIRT and the luring of the Draper. Neuron. 2006;50:819–821. doi: 10.1016/j.neuron.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Fang et al., 2005.Fang C, Bernardes-Silva M, Coleman MP, Perry VH. The cellular distribution of the Wld s chimeric protein and its constituent proteins in the CNS. Neuroscience. 2005;135:1107–1118. doi: 10.1016/j.neuroscience.2005.06.078. [DOI] [PubMed] [Google Scholar]
- Feng et al., 2000.Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Ferri et al., 2003.Ferri A, Sanes JR, Coleman MP, Cunningham JM, Kato AC. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol. 2003;13:669–673. doi: 10.1016/s0960-9822(03)00206-9. [DOI] [PubMed] [Google Scholar]
- Fischer et al., 2005.Fischer LR, Culver DG, Davis AA, Tennant P, Wang M, Coleman M, Asress S, Adalbert R, Alexander GM, Glass JD. The Wld(S) gene modestly prolongs survival in the SOD1(G93A) fALS mouse. Neurobiol Dis. 2005;19:293–300. doi: 10.1016/j.nbd.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Gillingwater et al., 2002.Gillingwater TH, Thomson D, Mack TG, Soffin EM, Mattison RJ, Coleman MP, Ribchester RR. Age-dependent synapse withdrawal at axotomised neuromuscular junctions in Wld(s) mutant and Ube4b/Nmnat transgenic mice. J Physiol. 2002;543:739–755. doi: 10.1113/jphysiol.2002.022343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater et al., 2006.Gillingwater TH, Wishart TM, Chen PE, Haley JE, Robertson K, MacDonald SH, Middleton S, Wawrowski K, Shipston MJ, Melmed S, Wyllie DJ, Skehel PA, Coleman MP, Ribchester RR. The neuroprotective WldS gene regulates expression of PTTG1 and erythroid differentiation regulator 1-like gene in mice and human cells. Hum Mol Genet. 2006;15:625–635. doi: 10.1093/hmg/ddi478. [DOI] [PubMed] [Google Scholar]
- Glass et al., 1993.Glass JD, Brushart TM, George EB, Griffin JW. Prolonged survival of transected nerve fibres in C57BL/Ola mice is an intrinsic characteristic of the axon. J Neurocytol. 1993;22:311–321. doi: 10.1007/BF01195555. [DOI] [PubMed] [Google Scholar]
- Hamilton et al., 2001.Hamilton MH, Tcherepanova I, Huibregtse JM, McDonnell DP. Nuclear import/export of hRPF1/Nedd4 regulates the ubiquitin-dependent degradation of its nuclear substrates. J Biol Chem. 2001;276:26324–26331. doi: 10.1074/jbc.M101205200. [DOI] [PubMed] [Google Scholar]
- Hoopfer et al., 2006.Hoopfer ED, McLaughlin T, Watts RJ, Schuldiner O, O'Leary DD, Luo L. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Howell et al., 2007.Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, Whitmore AV, Masland RH, John SW. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007;179:1523–1537. doi: 10.1083/jcb.200706181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami and Koike, 2003.Ikegami K, Koike T. Non-apoptotic neurite degeneration in apoptotic neuronal death: pivotal role of mitochondrial function in neurites. Neuroscience. 2003;122:617–626. doi: 10.1016/j.neuroscience.2003.08.057. [DOI] [PubMed] [Google Scholar]
- Kang et al., 2008.Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–148. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay et al., 1999.Kay AR, Alfonso A, Alford S, Cline HT, Holgado AM, Sakmann B, Snitsarev VA, Stricker TP, Takahashi M, Wu LG. Imaging synaptic activity in intact brain and slices with FM1–43 in C. elegans, lamprey, and rat. Neuron. 1999;24:809–817. doi: 10.1016/s0896-6273(00)81029-6. [DOI] [PubMed] [Google Scholar]
- Laser et al., 2006.Laser H, Conforti L, Morreale G, Mack TG, Heyer M, Haley JE, Wishart TM, Beirowski B, Walker SA, Haase G, Celik A, Adalbert R, Wagner D, Grumme D, Ribchester RR, Plomann M, Coleman MP. The slow Wallerian degeneration protein, WldS, binds directly to VCP/p97 and partially redistributes it within the nucleus. Mol Biol Cell. 2006;17:1075–1084. doi: 10.1091/mbc.E05-04-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu et al., 2004.Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brännström T, Gredal O, Wong PC, Williams DS, Cleveland DW. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- MacDonald et al., 2006.MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Mack et al., 2001.Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Madeo et al., 1998.Madeo F, Schlauer J, Zischka H, Mecke D, Fröhlich KU. Tyrosine phosphorylation regulates cell cycle-dependent nuclear localization of Cdc48p. Mol Biol Cell. 1998;9:131–141. doi: 10.1091/mbc.9.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi et al., 2005.Mi W, Beirowski B, Gillingwater TH, Adalbert R, Wagner D, Grumme D, Osaka H, Conforti L, Arnhold S, Addicks K, Wada K, Ribchester RR, Coleman MP. The slow Wallerian degeneration gene, WldS, inhibits axonal spheroid pathology in gracile axonal dystrophy mice. Brain. 2005;128:405–416. doi: 10.1093/brain/awh368. [DOI] [PubMed] [Google Scholar]
- Namimatsu et al., 2005.Namimatsu S, Ghazizadeh M, Sugisaki Y. Reversing the effects of formalin fixation with citraconic anhydride and heat: a universal antigen retrieval method. J Histochem Cytochem. 2005;53:3–11. doi: 10.1177/002215540505300102. [DOI] [PubMed] [Google Scholar]
- Okado-Matsumoto and Fridovich, 2001.Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- Perry et al., 1990.Perry VH, Lunn ER, Brown MC, Cahusac S, Gordon S. Evidence that the rate of Wallerian degeneration is controlled by a single autosomal dominant gene. Eur J Neurosci. 1990;2:408–413. doi: 10.1111/j.1460-9568.1990.tb00433.x. [DOI] [PubMed] [Google Scholar]
- Perry et al., 1992.Perry VH, Brown MC, Tsao JW. The effectiveness of the gene which slows the rate of Wallerian degeneration in C57BL/Ola mice declines with age. Eur J Neurosci. 1992;4:1000–1002. doi: 10.1111/j.1460-9568.1992.tb00126.x. [DOI] [PubMed] [Google Scholar]
- Press and Milbrandt, 2008.Press C, Milbrandt J. Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J Neurosci. 2008;28:4861–4871. doi: 10.1523/JNEUROSCI.0525-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff et al., 2002.Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Ribchester et al., 1995.Ribchester RR, Tsao JW, Barry JA, Asgari-Jirhandeh N, Perry VH, Brown MC. Persistence of neuromuscular junctions after axotomy in mice with slow Wallerian degeneration (C57BL/WldS) Eur J Neurosci. 1995;7:1641–1650. doi: 10.1111/j.1460-9568.1995.tb01159.x. [DOI] [PubMed] [Google Scholar]
- Sajadi et al., 2004.Sajadi A, Schneider BL, Aebischer P. Wld(s)-mediated protection of dopaminergic fibers in an animal model of Parkinson disease. Curr Biol. 2004;14:326–330. doi: 10.1016/j.cub.2004.01.053. [DOI] [PubMed] [Google Scholar]
- Samsam et al., 2003.Samsam M, Mi W, Wessig C, Zielasek J, Toyka KV, Coleman MP, Martini R. The Wlds mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy. J Neurosci. 2003;23:2833–2839. doi: 10.1523/JNEUROSCI.23-07-02833.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki et al., 2006.Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sau et al., 2007.Sau D, De Biasi S, Vitellaro-Zuccarello L, Riso P, Guarnieri S, Porrini M, Simeoni S, Crippa V, Onesto E, Palazzolo I, Rusmini P, Bolzoni E, Bendotti C, Poletti A. Mutation of SOD1 in ALS: a gain of a loss of function. Hum Mol Genet. 2007;16:1604–1618. doi: 10.1093/hmg/ddm110. [DOI] [PubMed] [Google Scholar]
- Saxena and Caroni, 2007.Saxena S, Caroni P. Mechanisms of axon degeneration: from development to disease. Prog Neurobiol. 2007;83:174–191. doi: 10.1016/j.pneurobio.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Schulze et al., 2005.Schulze A, Standera S, Buerger E, Kikkert M, van Voorden S, Wiertz E, Koning F, Kloetzel PM, Seeger M. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J Mol Biol. 2005;354:1021–1027. doi: 10.1016/j.jmb.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Simonin et al., 2007a.Simonin Y, Perrin FE, Kato AC. Axonal involvement in the Wlds neuroprotective effect: analysis of pure motoneurons in a mouse model protected from motor neuron disease at a pre-symptomatic age. J Neurochem. 2007a;101:530–542. doi: 10.1111/j.1471-4159.2006.04366.x. [DOI] [PubMed] [Google Scholar]
- Simonin et al., 2007b.Simonin Y, Ferrer-Alcon M, Ferri A, Kato AC. The neuroprotective effects of the Wld s gene are correlated with proteasome expression rather than apoptosis. Eur J Neurosci. 2007b;25:2269–2274. doi: 10.1111/j.1460-9568.2007.05501.x. [DOI] [PubMed] [Google Scholar]
- Spencer et al., 2000.Spencer RL, Kalman BA, Cotter CS, Deak T. Discrimination between changes in glucocorticoid receptor expression and activation in rat brain using Western blot analysis. Brain Res. 2000;868:275–286. doi: 10.1016/s0006-8993(00)02341-6. [DOI] [PubMed] [Google Scholar]
- Vande Velde et al., 2004.Vande Velde C, Garcia ML, Yin X, Trapp BD, Cleveland DW. The neuroprotective factor Wlds does not attenuate mutant SOD1-mediated motor neuron disease. Neuromolecular Med. 2004;5:193–203. doi: 10.1385/NMM:5:3:193. [DOI] [PubMed] [Google Scholar]
- Van Heusden et al., 1997.Van Heusden J, de Jong P, Ramaekers F, Bruwiere H, Borgers M, Smets G. Fluorescein-labeled tyramide strongly enhances the detection of low bromodeoxyuridine incorporation levels. J Histochem Cytochem. 1997;45:315–319. doi: 10.1177/002215549704500216. [DOI] [PubMed] [Google Scholar]
- Waller, 1850.Waller A. Experiments on the section of glossopharyngeal and hypoglossal nerves of the frog and observations of the alterations produced thereby in the structure of their primitive fibres. Philos Trans R Soc Lond B Biol Sci. 1850;140:423–429. [Google Scholar]
- Wang et al., 2005.Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang et al., 2004.Wang Q, Song C, Li CC. Molecular perspectives on p97-VCP: progress in understanding its structure and diverse biological functions. J Struct Biol. 2004;146:44–57. doi: 10.1016/j.jsb.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Whitfield et al., 2004.Whitfield J, Neame SJ, Ham J. Methods for culturing primary sympathetic neurons and for determining neuronal viability. Methods Mol Biol. 2004;282:157–168. doi: 10.1385/1-59259-812-9:157. [DOI] [PubMed] [Google Scholar]
- Wilbrey et al., 2008.Wilbrey AL, Haley JE, Wishart TM, Conforti L, Morreale G, Beirowski B, Babetto E, Adalbert R, Gillingwater TH, Smith T, Wyllie DJ, Ribchester RR, Coleman MP. VCP binding influences intracellular distribution of the slow Wallerian degeneration protein, Wld(S) Mol Cell Neurosci. 2008;38:325–340. doi: 10.1016/j.mcn.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Yang et al., 2007a.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, de Cabo R, Sauve AA, Sinclair DA. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007a;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang et al., 2007b.Yang Y, Kawataki T, Fukui K, Koike T. Cellular Zn(2+) chelators cause “dying-back” neurite degeneration associated with energy impairment. J Neurosci Res. 2007b;85:2844–2855. doi: 10.1002/jnr.21411. [DOI] [PubMed] [Google Scholar]