

Abstract
To understand the intrinsic operation of spinal networks generating locomotion, we need to not only characterize the constituent neurons and their connectivity, but also determine the role of intrinsic modulation in shaping the final motor output. We have focused on the effects of nitric oxide (NO) on the locomotor frequency and the underlying synaptic mechanisms in the lamprey spinal cord. To identify the source of NO, we used NADPH-diaphorase histochemistry and nNOS immunocytochemistry. Gray matter and sensory neurons were positively labeled using both methods. Preparations preincubated with NO synthase inhibitors displayed slower locomotor frequency that increased upon washout of the inhibitors, suggesting that NO is an endogenous neuromodulator in the spinal cord. Application of NO donors increased the locomotor frequency that was blocked by an NO scavenger and partially reduced by an inhibitor of sGC. To analyze the synaptic modulation underlying the NO-induced increase of the locomotor frequency we performed intracellular recordings from motoneurons and interneurons. The NO-induced increase in locomotor frequency was associated with a decrease in the midcycle inhibition and an increase in on-cycle excitation. To determine the site of action of NO, we examined the effect of NO donors on miniature PSCs. NO increased both the frequency and amplitude of mEPSCs while it only decreased the frequency of mIPSCs, suggesting the increased excitation is mediated by both presynaptic and postsynaptic mechanisms, while the decrease in inhibition involves only presynaptic mechanisms. Our results demonstrate a significant role of NO in adult vertebrate motor control which, via modulation of both excitatory and inhibitory transmission, increases the locomotor burst frequency.
Introduction
Nitric oxide (NO) is a phylogenetically conserved, widely distributed and important biological signaling molecule which participates in a huge diversity of physiological systems and pathological conditions (Prast and Philippu, 2001). NO synthase (NOS), the synthetic enzyme for NO, is also widely distributed (Vincent, 1995). It has been suggested that no neuron in the mammalian brain is >25 μm from a source of NO (Garthwaite, 2008). NO plays a role in many brain functions including, for example, hippocampal LTP, cerebellar LTD, and olfactory processing (Prast and Philippu, 2001; Garthwaite, 2008). In each of these systems synaptic plasticity is important and the contribution of NO may be linked to its ability to act in a retrograde manner to alter synaptic strengths. The synaptic activation of neurons can trigger an influx of calcium (e.g., through NMDA channels) and the resulting NOS activation produces NO that can influence neighboring cells, including presynaptic terminals, thereby providing a link between presynaptic and postsynaptic activity. In addition, NO signaling can interact with endocannabinoids to modify transmitter release (Kyriakatos and El Manira, 2007; Makara et al., 2007). However, the sources and mechanisms of NO action in the nervous system are not completely understood.
NO has recently been implicated in the regulation of motor behavior including locomotory and respiratory activity (McLean and Sillar, 2000; Del Bel et al., 2005; Hedrick et al., 2005). NO modulates locomotor activity via actions at multiple levels along the neuroaxis from the cortex (Del Bel et al., 2005), to the brainstem and the spinal cord (McLean and Sillar, 2002, 2004). In Xenopus tadpoles, NOS-positive neurons are restricted to three prominent clusters located in the brainstem (McLean and Sillar, 2000, 2001). NO exerts a potent inhibitory influence on swimming activity mediated in part via presynaptic facilitation of GABAergic and glycinergic transmission (McLean and Sillar, 2002, 2004) and in part via direct actions on spinal motorneurons. However, the role of NO in the modulation of an adult vertebrate locomotor network has not been studied previously. The aim of this study, therefore, was to examine the origin, effects and mechanisms of action of NO in the swimming system of the lamprey. The present study was partly prompted by the recent demonstration that NO is involved in the potentiation of the locomotor frequency following mGluR1 receptor activation (Kyriakatos and El Manira, 2007).
We report that in adult lampreys nNOS labeling is found in particular subsets of spinal cord neurons including gray matter and sensory neurons. NO is endogenously released and increases the locomotor burst frequency, accompanied by a decrease in the amplitude of the midcycle inhibition and an increase in the on-cycle excitation. Analysis of miniature synaptic currents shows that these differential effects of NO on excitation and inhibition in the network are mediated presynaptically via effects on the transmitter release machinery, with an additional postsynaptic locus for the NO modulation of excitation. The results have not been published previously except in abstract form (Kyriakatos et al., 2008; Sillar et al., 2008).
Materials and Methods
Animals/preparation.
All experiments were performed on in vitro preparations of the isolated, intact spinal cord from adult lampreys (Lampetra fluviatilis and Petromyzon marinus). All protocols were approved by the Animal Research Ethical Committee, Stockholm. Lampreys were anesthetized with MS 222 (100 mg/L; Sigma) and eviscerated, and the lateral muscle walls were removed. The spinal cord and notochord were dissected and pinned in a cooled (8–12°C) Sylgard-lined experimental chamber continuously perfused with physiological solution. The control solution was composed of (in mm) 138 NaCl, 2.1 KCl, 1.8 CaCl2, 1.2 MgCl2, 4 glucose, and 2 HEPES, bubbled with O2 and pH adjusted to 7.4. Fictive swimming activity was induced by adding 50–100 μm NMDA (Grillner et al., 1981) to the physiological solution. Alternating locomotor burst activity was recorded by en passant glass suction electrodes placed on two opposing ventral roots at their exits from the spinal cord. After perfusion of NMDA, the locomotor burst frequency increased gradually over time and reached a stable level after 3–4 h. Pharmacological reagents were normally added only after the burst frequency had been stable for at least 1 h. In preincubation experiments, the spinal cord was perfused with the NOS inhibitor l-NAME, l-NNA, ODQ, or carboxy-PTIO for 1–2 h before application of NMDA. Two minute recordings of ventral root activity were sampled every 5 min. The cycle duration was measured and averaged from 100 to 200 consecutive cycles. The cycle duration was defined as the time interval between the onsets of two consecutive bursts and the burst frequency was calculated as the inverse of the averaged cycle duration.
To examine the changes in midcycle reciprocal inhibition during locomotion, intracellular recordings were made from motoneurons (MNs) and rhythmically active unidentified interneurons with 3 m potassium acetate-filled thin-walled glass microelectrodes with a resistance of 15–30 MΩ. Motoneurons were identified by recording their axonal action potentials in a one-to-one manner from the corresponding ventral root through the extracellular suction electrode. All neurons recorded during fictive locomotion received phasic excitation alternating with phasic inhibition. The concurrent recordings of the locomotor frequency and of the activity of motorneurons and network interneurons allowed us to correlate changes in the synaptic amplitude with changes in locomotor frequency. Two minutes recordings of intracellular and extracellular activity recordings were sampled every 5 min. The spikes were removed using a low-pass filter and the synaptic input was averaged from 100 to 200 cycles and their duration was normalized. The peak to trough amplitude of the locomotor-related synaptic input was measured and monitored in control, in DEA/NO, and after washout.
To examine the effects of NO donors on ipsilateral excitatory synaptic transmission the recording chamber was divided into two pools by an agar barrier (Dale, 1986). Locomotor activity was induced in the rostral pool by NMDA (100 μm) while the spinal cord in the caudal pool was perfused with physiological solution containing strychnine (5 μm) to block inhibitory synaptic transmission. After 30 min in strychnine, intracellular recordings were made from neurons 2–3 segments away from the agar barrier. In the presence of strychnine, excitatory synaptic input occurred in phase with the activity of the ipsilateral ventral root in the rostral pool. Two minute recordings of intracellular and extracellular activities were sampled every 4 min and the amplitude the excitatory input was monitored in control, in NO donor, and after washout.
Intracellular recordings were made in bridge mode current-clamp mode with an Axoclamp 2B amplifier (Molecular Devices). pClamp software (Molecular Devices) was used for data acquisition and analysis on a personal computer equipped with an analog–digital interface (Digidata 1300). Recordings were also acquired and stored digitally using pClamp. Data analysis of burst frequency and synaptic transmission was done off line with the program Spike 2 (CED). We used MiniAnalysis 6 (Synaptosoft) to detect mPSC and create cumulative amplitude histograms of the pooled data.
Spontaneous mPSCs were recorded using whole-cell patch-clamp recordings in voltage-clamp mode from the isolated spinal cord from Petromyzon marinus. For this the spinal cord was dissected free of the notochord, the meningeal layers were removed and a thin slice above the gray matter was cut using a vibratome to allow access of the recording electrode (Cochilla and Alford, 1998; Nanou et al., 2008). The intracellular solution contained (in mm) 102 KCH3SO3, 1.2 MgCl2, 1 CaCl2, 10 EGTA, 10 glucose, 10 HEPES, 1 Na-GTP, 3 Mg-ATP, and 5 Na2-phosphocreatine, pH 7.4. In experiments in which mIPSCs were examined, the intracellular KCH3SO3 was substituted with KCl. The preparation was continuously perfused with normal physiological solution containing TTX while strychnine or AP5 and GYKI 52466 was added to recorded mEPSPs or mIPSPs, respectively. Spontaneous mPSC were recorded with a MultiClamp 700B amplifier, acquired and stored digitally using pClamp. We used MiniAnalysis 6 (Synaptosoft) to detect mPSP events, create cumulative amplitude histograms of the pooled data. The following drugs were stored as stock solutions at −20°C and were dissolved to their final concentrations in physiological solution before their application: AP5, GYKI 52466, l-NAME, l-NNA, NMDA, ODQ, carboxy-PTIO and strychnine (Tocris Bioscience), DEA/NO (A.G. Scientific), SNAP (Chemistry Department, University of St. Andrews), and TTX (Sigma-Aldrich). All drugs were diluted in water except for GYKI 52466, ODQ, and SNAP, which were dissolved in DMSO. The same concentration of the solvent DMSO had no effect on the locomotor frequency and was always added to control solutions throughout the experiments. We used a range of concentrations of NOS inhibitors l-NAME (100–300 μm) and l-NNA (100–200 μm) as well as NO donors DEA/NO (20–200 μm) and SNAP (50–250 μm). Since there were no concentration-dependent effects, the data were pooled.
Statistical significance was assessed with paired or unpaired Student's t test and differences were considered significant if p < 0.05. Kolmogorov–Smirnov test was used for comparison of cumulative probability distributions of mPSC.
NADPH-d histochemistry.
The NADPH-diaphorase (NADPH-d) histochemical method is a simple and reliable staining method for revealing the location of NOS (Hope et al., 1991). This technique relies upon the fact that NOS uses NADPH as an electron source and is the only “diaphorase” enzyme that is resistant to paraformaldehyde fixation. Nitroblue tetrazolium acts are an electron acceptor to generate a blue reaction product. The NADPH-d histochemical method was applied as described previously for Xenopus (McLean and Sillar, 2000; McLean et al., 2001). The isolated lamprey spinal cord was fixed in 4% paraformaldehyde (pH 7.4, 4°C) for 2 h on a rocking agitator. It was then washed in phosphate buffer (PB; 3 × 5 min), transferred to 30% sucrose in 0.1 m PB and stored in the refrigerator until they sank. The spinal cord was then immersed in 5 ml of NADPH-d staining solution consisting of 5 mg of NADPH (Sigma-Aldrich), 4.95 ml of 0.3% PB-TX, and 50 μl of Nitroblue tetrazolium salt (NBT; Sigma-Aldrich) made up from 5 mg of NBT dissolved in 0.5 ml of PB-TX and incubated at 37°C for 2 h. The spinal cord was then washed in 0.1 m PB (3 × 5 min), dehydrated in acetone/alcohol series, and cleared in xylene before being mounted with DPX in slide, sealed, and topped with a coverslip, ready for viewing.
nNOS immunofluorescence.
Neuronal NOS (nNOS) immunocytochemistry was performed using protocols developed previously for Xenopus (Ramanathan et al., 2006). The isolated lamprey spinal cord was fixed in 4% paraformaldehyde, pH 7.4, overnight at room temperature, washed in 0.1 m PB (3 × 10 min). Fixed preparations were cryoprotected in 30% sucrose and mounted in OCT compound for cryostat sectioning. Horizontal slices (20 μm) were incubated in blocking serum and primary antibody. This solution consisted of 1% bovine serum albumin (Sigma-Aldrich) in PBS-TX (pH 7.4; 0.01 m PBS plus 0.3% Triton X-100); and primary rabbit polyclonal antibody (1:50 v/v, NOS, Santa Cruz Biotechnology). Following incubation for 48 h, the specimen were washed in 0.01 m PBS (3 × 10 min), then incubated with a secondary antibody (pH 7.4; Rhodamine Red, conjugated to donkey anti-rabbit, Jackson ImmunoResearch) at 1:200 v/v for 24 h. They were then washed in 0.01 m PBS (3 × 10 min), mounted with DPX and topped with a coverslip. Control experiments consisted of using a blocking peptide or only the secondary antibody. We also used a sheep nNOS antibody (1:1000) (Kishimoto et al., 1993) on 15-μm-thick transverse spinal cord sections. Similar results were obtained using both antibodies.
Results
NADPH-d labeling in the spinal cord of the lamprey
Segments of lamprey spinal cord (n = 5) were processed for NADPH-d histochemistry in whole mount (Fig. 1) and in two preparations cross sections of the spinal cord were also made to supplement the whole-mount data (data not shown). Staining was observed in 3 of 5 preparations with a variety of cells consistently appearing (Fig. 1). In segments of whole-mount spinal cord, the stretch-sensitive edge cells, in the lateral column were found to stain reliably and were distributed irregularly along the length of the spinal cord segment (Fig. 1A,D). In addition cutaneous mechanosensory dorsal cells as well as motoneurons and interneurons in the gray matter also labeled (Fig. 1A–C). Furthermore, less intense NADPH-d labeling was also found in cells lining the central canal (Fig. 1A,C).
Figure 1.
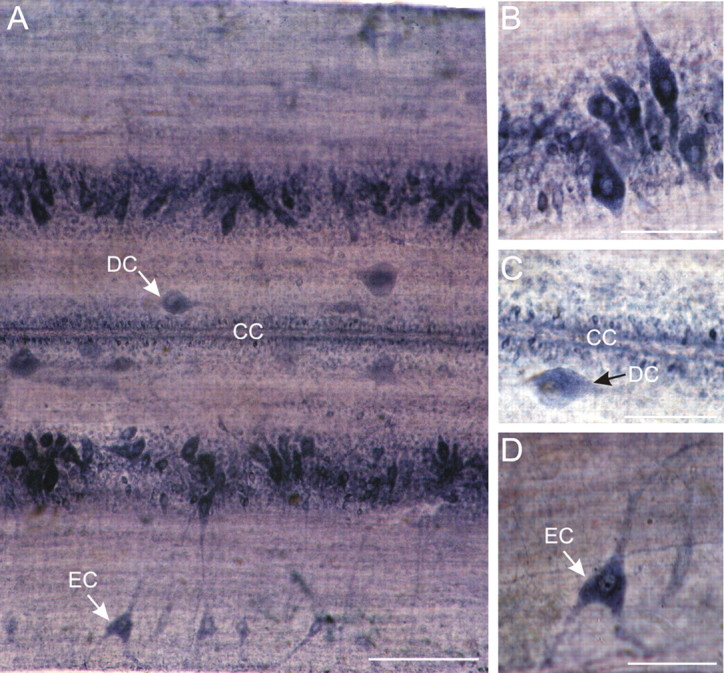
NADPH-d labeling in the spinal cord of the lamprey. A, Whole-mount segment of spinal cord viewed dorsally. NADPH-d staining can be observed in different types of cell. Edge cells (EC) are located in the lateral columns and have long dendrites projecting toward edge of cord. Dorsal cells (DC) are large, rounded, and irregularly distributed along the medial columns. Ependymal cells surrounding the central canal (CC) are marked. Scale bar, 200 μm. B, Detail of a part of the gray column, which includes motoneurons and interneurons. C, Detail showing a dorsal cell (DC) medial to the central canal (CC). Note that the ependymal cells surrounding the central canal show NADPH-d reactivity. D, Enlargement of edge cell with its lateral dendrites. Scale bars: B–D, 100 μm.
nNOS immunohistochemistry
NADPH-d histochemistry is generally a reliable stain for the presence of NOS; however, due to the possibility that it can yield false positives (for example in cases in which tissue is overfixed or incubated for an excessive period of time), nNOS immunohistochemistry was used to corroborate initial findings. Segments (n = 2) ∼4 mm in length or horizontal sections (n = 2) were incubated with antibodies against nNOS. Confocal microscopy revealed a pattern of immunofluorescence very similar to the pattern of NADPH-d labeling just described (Fig. 2; compare with Fig. 1). Mechanosensory edge cells (Fig. 2A) and the cutaneous sensory neurons (dorsal cells) (Fig. 2B) were stained very reliably. Cells in the gray matter labeled brightly (Fig. 2C,D), and appeared similar in shape and size to the cells labeling for NADPH-d. The nNOS-ir cells (arrows) and dendrites (open arrowheads) could be clearly distinguished from negatively labeled cells (filled arrowheads). No signal was detected when specimen were incubated with the blocking peptide or by omission of the primary antibody (data not shown). Overall, the pattern of labeling observed for nNOS immunohistochemistry was found to closely resemble that of NADPH-d labeling in the spinal cord.
Figure 2.

nNOS immunoreactivity in the spinal cord of the lamprey. A, A stacked series of optical sections of lateral margin of spinal cord segment, showing edge cells (arrows). Dendrites of edge cells are clearer with nNOS immunohistochemistry than with NADPH-d labeling. B, Dorsal cells (DC) in the medial column label for nNOS (arrows). There were also DC negative for nNOS (arrowheads). C–D, Detail of the gray matter column showing presumed motor neurons and interneurons positive for nNOS (arrows) and others negative for nNOS (arrowhead). Dendritic processes were also labeled for nNOS (open arrowheads). Note that the shape and size of labeled cells closely correlates with those of labeled cells in spinal cord processed for NADPH-d histochemistry.
Intrinsic activation of NO synthesis increases the locomotor frequency
Since lamprey spinal cord neurons contain the necessary machinery for NO production and since NOS is a calcium- and therefore potentially an activity-dependent enzyme, we next asked whether the lamprey spinal cord synthesizes NO during fictive locomotion and whether it contributes to setting the baseline frequency. If NO is synthesized in the active spinal locomotor network then blocking its synthesis should result in a change in the locomotor frequency. To test this idea, we preincubated preparations with the NOS inhibitor l-NAME (100–300 μm) or l-NNA (100–200 μm) for 1 h before the induction of swimming with NMDA (100 μm), and after the frequency had been stable for 1 h (Fig. 3A), we washed out the inhibitor (Fig. 3B). Upon washout of l-NAME, the locomotor frequency increased significantly by 23.6 ± 2.4% (Fig. 3C) (p < 0.0001, n = 6), while the increase was by 20.6 ± 2.4% after washout of l-NNA (Fig. 3D) (p < 0.0001, n = 4). These results indicate that there is an intrinsic NOS-dependent NO modulatory tone during locomotion.
Figure 3.

Release of endogenous NO during fictive locomotion. A, Locomotor activity was induced in NMDA (100 μm) in the presence of the NOS inhibitor l-NAME (100 μm). B, Washing out l-NAME resulted in an increase in the frequency of the locomotor rhythm, suggesting a continuous endogenous release of NO. C, Averaged data from all the experiments showing a significant increase in the locomotor frequency upon washout of l-NAME (p < 0.0001, n = 6). D, Averaged data from all the experiments showing a significant increase in the locomotor frequency upon washout of l-NNA (p < 0.0001, n = 4).
Nitric oxide donors potentiate the locomotor frequency
To confirm the excitatory actions of NO on the locomotor frequency, the effects of NO donors on the fictive locomotor rhythm were tested. NMDA (100 μm) was applied and the locomotor rhythm was monitored by recording two opposing ventral roots with extracellular suction electrodes. After the locomotor frequency had been stable for a period of at least 1 h, we bath applied either DEA/NO or SNAP, two different NO donors. Applications of DEA/NO (20–200 μm, 20–40 min) increased the locomotor frequency by 69.0 ± 12.1% (Fig. 4A–C) (p < 0.0001, n = 18). Similarly, applications of SNAP (50–250 μm, 30–60 min) resulted in an increase of the locomotor frequency by 34.3 ± 13.3% (Fig. 4D) (p < 0.0001, n = 8). Both the onset and recovery of the effects of SNAP were slower than DEA/NO. In addition, in some preparations the SNAP-induced increase of the locomotor frequency was preceded by a short-term reduction in the locomotor frequency. Since in all preparations the effects of DEA/NO were faster and more robust than those of SNAP, the experiments that follow were performed using DEA/NO as an NO donor. To check that the effects of DEA/NO were due to its release of NO, the NO scavenger carboxy-PTIO was preapplied before application of the donor. In the presence of equimolar concentration of carboxy-PTIO (150 μm), the ability of DEA/NO to increase the locomotor frequency was occluded (Fig. 5A–C) (98.7 ± 0.8%, p > 0.05, n = 4). Thus, NO is released endogenously within the locomotor networks and increases the locomotor frequency.
Figure 4.

NO donors increase the locomotor frequency. A, Locomotor activity was induced by NMDA (100 μm) and monitored by recording ventral roots on opposite sides in the same segment. B, Subsequent application of the NO donor DEA/NO (150 μm) increases the locomotor frequency. C, Averaged data from all the experiments showing the potentiating effect of DEA/NO on the locomotor frequency (p < 0.0001, n = 18). D, Averaged data from all the experiments showing the increase in the frequency induced by SNAP (p < 0.0001, n = 8).
Figure 5.

The effects of NO are blocked by the NO scavenger carboxy-PTIO. A, Locomotor activity was induced in NMDA (100 μm) in the presence of the NO scavenger carboxy-PTIO (150 μm). B, Equimolar application of DEA/NO failed to increase the locomotor frequency. C, Averaged data from all the experiments showing the failure of DEA/NO to increase the locomotor frequency in the presence of carboxy-PTIO (p > 0.05, n = 4). D, Inhibition of soluble guanylate cyclase with ODQ blocked part of the increase of the DEA/NO-induced increase in the locomotor frequency. The graph represents averaged data from all the experiments and shows a significant block of DEA/NO effect by ODQ (p < 0.02, n = 12).
sGC is a target of NO to mediate the increase in locomotor frequency
NO can exert its effects via direct modulation of ion channels and direct nitrosylation of target proteins, but the primary target of NO is the enzyme, soluble guanylate cyclase (sGC); activation of sGC leads to the elevation of cGMP levels and a subsequent increase in protein kinase G (PKG) levels which in turn target downstream effector proteins. To test whether the NO-mediated increase of the locomotor rhythm involves activation of sGC, we preincubated preparations in the sGC inhibitor ODQ (10–20 μm) for 1 h before DEA/NO (20–150 μm) application. Thereafter, ODQ was washed and DEA/NO applied for a second time. ODQ blocked part of the DEA/NO-induced increase in the locomotor frequency by 13.1 ± 3.1% (Fig. 5D) (p < 0.02, n = 12). The partial block of ODQ could be due to the fact that inadequate inhibition of sGC was achieved with the concentrations used. However, if it is assumed that 10–20 μm ODQ is a saturating concentration producing a full block of sGC, then the remaining increase in locomotor frequency must involve other, as yet unknown mechanisms of NO action, such as S-nitrosylation of target proteins.
NO depresses midcycle inhibition during locomotion via a presynaptic mechanism
To evaluate the mechanisms that underlie the NO-mediated increase of the locomotor frequency we performed intracellular recordings from motoneurons during fictive locomotion before, during, and after DEA/NO application. In control conditions (NMDA alone), all recorded motoneurons and interneurons (n = 8) received phasic excitation and fired action potentials in phase with the ipsilateral ventral root discharge alternating with inhibition when the contralateral ventral root was active (Fig. 6A,C). Applications of DEA/NO increased the locomotor frequency, as also shown above, by 53.5 ± 17.7% (Fig. 6B,D) (p < 0. 001, n = 8), and this increase was reversible (99.7 ± 4.4% compared with control, p > 0.05, n = 8). In parallel with the increase of the frequency there was an associated decrease in the amplitude of the midcycle inhibition by 38.8 ± 5.6% (Fig. 6C,D) (p < 0.0001, n = 8) that in 3 of 8 preparations partially recovered to 89.6 ± 11.8% (p < 0.0001) and in 5 of 8 recovered fully to baseline (p < 0.0001). The time course of the decrease of the midcycle inhibition was a mirror image of the frequency potentiation (Fig. 6D).
Figure 6.

NO donor DEA/NO increases the locomotor frequency via depression of midcycle inhibition. A, Intracellular recording from a MN that received phasic on-cycle excitation with the ipsilateral ventral root (vr-i) and midcycle inhibition in phase with the contralateral ventral root (vr-c). B, Application of DEA/NO (100–200 μm) increased the locomotor frequency in parallel with a depression of the midcycle inhibition. C, Averaged synaptic inputs received by the MN. The action potentials were filtered out using a low pass filter. DEA/NO decreased the amplitude of the midcycle inhibition, while the on-phase excitation was not decreased. D, Averaged data from all experiments showing the time course of the NO-mediated increase of the locomotor frequency (p < 0.001, n = 8) is in parallel with a decrease in the midcycle inhibition (p < 0.0001, n = 8).
To explore in more detail the locus of the NO modulation on the inhibitory transmission we performed an analysis of miniature IPSCs. Patch-clamp recordings were made using an intracellular solution with a high intracellular chloride concentration in the presence of TTX (0.5 μm) to block action potentials, GYKI 52466 (30 μm) to block AMPA receptors and AP5 (50 μm) to block NMDA receptors. Under these conditions, we recorded spontaneous mIPSCs that were reversed in sign at a holding potential of −60 mV. Upon application of DEA/NO (100 μm), the frequency of these events significantly decreased (Fig. 7A,C) (interevent interval increased from 821 ± 338 ms to 1204 ± 443 ms; p < 0.05; n = 5), an effect that reversed upon washout of DEA/NO (Fig. 7A). In contrast, DEA/NO had no detectable effect on the amplitude of the miniature IPSCs (Fig. 7B,D) (control: 20.8 ± 3.5 pA and DEA/NO: 21.1 ± 4.5 pA; p > 0.05; n = 5). DEA/NO did not induce any change in the membrane potential or the holding current of the recorded neurons. These experiments suggest that the NO-mediated modulation of glycinergic transmission involves a presynaptic inhibition of the vesicular release machinery, with no detectable parallel postsynaptic effect.
Figure 7.

NO acts via presynaptic mechanisms to depress the inhibitory transmission. A, mIPSCs from lamprey gray matter neurons recorded with a high intracellular chloride electrode in the presence TTX (0.5 μm), GYKI 52466 (30 μm), and AP5 (50 μm). The membrane potential was clamped at −60 mV. Application of DEA/NO (100 μm) decreased the frequency of mIPSCs. B, The amplitude of the mIPSCs was not affected by DEA/NO. C, Frequency distribution of mIPSCs in the presence of DEA/NO shifted toward lower values because of the increased in the interval between mIPSCs compared with control (p < 0.05, n = 5). D, Amplitude distribution of mIPSCs in the presence of DEA/NO remained unchanged compared with control (p > 0.05, n = 5).
NO increases the on-cycle excitatory drive during locomotion via presynaptic and postsynaptic mechanisms
In addition to depressing the midcycle inhibition, we also examined whether NO modulated the excitatory synaptic transmission during fictive locomotion. To do this we used a split-bath recording chamber in which two pools were separated by agar (Fig. 8A) and locomotor activity was induced in the rostral pool with NMDA (100 μm). In the caudal pool no NMDA was added and glycinergic synaptic transmission was blocked with strychnine (5 μm). In this configuration, the locomotor-related rhythmic excitatory synaptic drive that is recorded arises from impulses in interneurons in the rostral pool whose axons project ipsilaterally across the agar barrier. Applications of DEA/NO (100 μm) in the caudal pool reversibly increased the amplitude of the on-cycle excitation by 24.8 ± 4.3% (Fig. 8B–E) (p < 0.0001, n = 8), indicating that NO facilitates excitatory transmission during fictive locomotion to increase the frequency.
Figure 8.
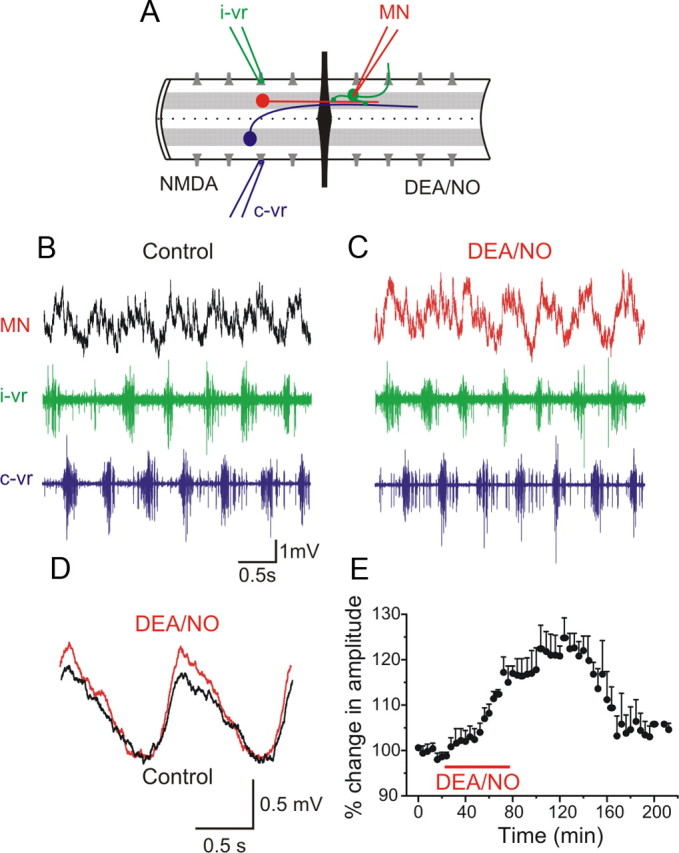
NO enhances the on-cycle excitation. A, Experimental arrangement with the recording chamber divided into two pools with an agar barrier. In the rostral part of the spinal cord locomotor activity was induced by NMDA while the caudal part was perfused with normal solution containing strychnine to block inhibition and intracellular recording was made from an MN. The red neuron represents ipsilateral excitatory interneurons and the blue neuron represents a commissural inhibitory interneuron. B, In control, the MN received excitation in phase with the ipsilateral ventral root (i-vr) burst. C, Application of DEA/NO (100 μm) in the caudal pool increased the amplitude of the excitatory synaptic input received by the MN. D, Averaged excitatory synaptic input received by the MN showing an increase in amplitude induced by DEA/NO. E, Plot of averaged data from a different experiment showing the time course of the change in the amplitude of the excitation before, during and after washout of DEA/NO (p < 0.0001, n = 8).
To examine further the NO modulation of excitatory transmission we performed an analysis of miniature EPSCs by making patch-clamp recordings in the presence of TTX (0.5 μm) and strychnine (5 μm). DEA/NO increased the frequency of mEPSCs (Fig. 9A,C) (interevent interval decreased from 446 ± 100 ms to 253 ± 34.5 ms; p < 0.05; n = 5). The amplitude of mEPSCs was also increased from 6.6 ± 1.5 pA to 8.5 ± 1.6 pA (Fig. 9B,D) (p < 0.05, n = 5). These results show a simultaneous presynaptic and postsynaptic modulation of the transmitter release machinery by NO at these excitatory synapses. In summary, the NO-mediated increase in the locomotor frequency is associated with two parallel and differential effects on synaptic transmission: a depression of inhibition and a facilitation of excitation.
Figure 9.

NO acts via presynaptic and postsynaptic mechanisms to facilitate the excitatory transmission. A, mEPSCs from lamprey gray matter neurons recorded in TTX (0.5 μm) in the presence of strychnine (5 μm) and the membrane potential was clamped at −60 mV. B, Application of DEA/NO (100 μm) increased the amplitude and the frequency of mEPSCs. C, Frequency distribution of mEPSCs in the presence of DEA/NO shifted toward higher values because the interval between the events increased compared with control. D, Amplitude distribution of mEPSCs in the presence of DEA/NO shifted toward higher values compared with control. A significant increase in the frequency and amplitude of mEPSCs was seen in all neurons tested (p < 0.05, n = 5).
Discussion
Lamprey spinal cord neurons contain the NO synthesis machinery
We have provided evidence, using NADPH-d histochemistry and nNOS immunohistochemistry, that several classes of neurons in the lamprey spinal cord express NOS and are therefore potential intrinsic sources of NO. At least some of the positively labeled neurons are known to be involved in motor control and sensorimotor integration such as gray matter neurons (motoneurons and interneurons) and proprioceptive edge cells (Grillner et al., 1984; Vinay et al., 1996). Edge cells are depolarized and fire upon stretching of the lateral margin of the spinal cord, which occurs when the curvature of the spinal cord changes as the lamprey's body bends during swimming or turning. Via their synaptic outputs edge cells they are able to entrain the locomotor activity to the animal's own movements by releasing both excitatory and inhibitory transmitters onto the locomotor network a role in (Di Prisco et al., 1990). It is conceivable that NO production from edge cells is triggered when the lamprey begins to swim with the resulting excitatory effects of NO serving to facilitate locomotor activity. Dorsal cells in the medial columns (El Manira et al., 1997), on either side of the central canal also label for NADPH-d/nNOS. These primary mechanosensory neurons are similar to the Rohon-Beard cells of Xenopus tadpoles (Clarke et al., 1984), which do not label for nNOS (McLean and Sillar, 2000). Also labeled were cells lining the central canal.
Although NOS is expressed in these different spinal neuron populations the effects of exogenous NO that we describe in the isolated spinal cord could reflect actions of NO deriving from neurons in other parts of the CNS, such as the brainstem, which have been removed during the dissection. Indeed NADPH-d staining has been reported previously in lamprey reticulospinal neurons including Mauthner and Muller cells (Schober et al., 1994). Thus, some of the excitatory actions of NO could be linked to the activation of spinal locomotor networks by descending commands, as well to CPG operation and its modulation by proprioceptive feedback from edge cells during movement.
NO increases the locomotor frequency
Applications of two different NO donors, SNAP and DEA/NO, produced a similar effect on fictive swimming: namely, an increase in rhythm frequency. The effect of the two drugs differed in that the SNAP effect on frequency was smaller, took longer to become established and was more difficult to wash off. The reasons for these differences are unknown but the faster time course of the DEA/NO effects made this the drug of choice for a more detailed analysis of NO modulation. However, the effects of DEA/NO were occluded by carboxy-PTIO, an NO scavenger, providing strong evidence to support the conclusion that it is indeed the NO released by these donors that increases the locomotor frequency. One hypothesis based on these results is that NO production during swimming produces a positive feedback effect on the network to maintain the rhythm at a high frequency. Indeed, locomotor activity induced in the presence of the NOS inhibitors l-NAME or l-NNA had a lower frequency, which increased following washout of these inhibitors, suggesting that NO contributes to setting the baseline frequency.
A similar finding has been reported in the Xenopus tadpole (McLean et al., 2000), where nNOS inhibition by l-NNA exerts the opposite effect on swimming to NO donors. However, the effects of NO on tadpole swimming are predominantly inhibitory, in contrast to the excitatory effects we describe here in the lamprey. These differences may be due both to the immature stage of development in the tadpole and the specific sites of modulatory action of NO in this system. Around the time of hatching, Xenopus tadpoles spend much of their time motionless, a behavioral strategy that may be suited to predator avoidance early in development. NO promotes this immobility by selectively enhancing GABAergic descending inhibition from midhindbrain reticulospinal interneurons and by enhancing glycinergic inhibition during swimming (McLean and Sillar, 2002). This latter effect involves metamodulation of the effects of noradrenaline (McLean and Sillar, 2004). Therefore, compared with the lamprey, the predominant effects of NO are (1) inhibitory and (2) mediated in the brainstem. In the present study on the isolated lamprey spinal cord, the dissection removes influences mediated from brainstem sources, but differences in the spinal actions of NO are also apparent compared with the tadpole. First, in the lamprey, NO does not exert a direct depolarizing effect on spinal locomotor neurons as it does in the tadpole (McLean and Sillar, 2002), and second, NO increases transmitter release from excitatory interneurons, an effect not described for the tadpole. It is interesting to note that in Rana temporaria tadpoles, in contrast to Xenopus at the equivalent stage of development, NO has an excitatory effect, triggering a slow nonrhythmic motor pattern that may be associated with hatching (McLean et al., 2001). These species- and stage-specific differences could be accounted for by the differential distribution of the targets of NO in different neuronal classes.
The precise sources of NO that increase locomotor frequency in vivo in the lamprey is not known, and as mentioned above, it seems likely that more than one source contributes to the overall effect of NO in the intact lamprey. In the mammalian hippocampus, it has been shown recently that LTP requires the summation of a tonic NO signal from the vascular endothelium and a phasic NO signal from a neuronal sources (Hopper and Garthwaite, 2006). This type of arrangement is not possible in the lamprey, because there is no intraspinal vasculature. However, it remains conceivable that two or more different neuronal sources of NO act cooperatively to increase locomotor frequency and, in the intact lamprey, these sources could include not only spinal cord classes but conceivably also those in more rostral centers.
Mechanisms of NO-mediated modulation
The mechanisms by which NO modulates lamprey locomotor activity are complex and include two parallel effects on synaptic transmission: a reduction in midcycle inhibition and a facilitation of on-cycle excitation. Voltage-clamp analysis of miniature synaptic currents suggests that these effects result from a purely presynaptic modulation of the probability of glycine release together with a simultaneous presynaptic increase of the probability of excitatory transmitter release and a postsynaptic modulation of the amplitude of the mEPSCs. The extent to which these mechanisms explain the effects of DEA/NO on evoked transmitter release is not known but we found no evidence to suggest that NO mediates significant effects upon the electrical properties of spinal neurons. For example, there was no change in the holding current following applications of DEA/NO under voltage clamp. In other preparations it has been shown that NO can lead to membrane potential depolarization and an associated increase in neuronal input resistance (McLean and Sillar, 2002), but if such a phenomenon occurs in lamprey neurons it was not detected and so presumably does not contribute significantly to the effects of NO we have described on fictive locomotion.
In addition to its direct effect on synaptic transmission, NO seems to act in a synergistic manner together with endocannabinoids to mediate the long-term potentiation of the locomotor frequency induced by activation of mGluR1 (Kyriakatos and El Manira, 2007). Although a long-lasting effect of NO donors on the locomotor frequency was seen in some preparations, this was not always the case. It could be that the mGluR-mediated long-term potentiation requires both NO and other modulators, such as endocannabinoids (Kettunen et al., 2005). The shift in balance between inhibition and excitation during locomotion induced by NO is reminiscent of that induced by mGluR1 and by endocannabinoids, suggesting that similar synaptic targets are modulated by different modulatory pathways (El Manira et al., 2002; LeBeau et al., 2005; El Manira et al., 2008).
In summary, we have illuminated some of the synaptic mechanism by which NO modulates the lamprey motor pattern and also described the potential sources of NO that might engage these mechanisms in the intact animal. However, it will be important in future experiments to marry these two sets of observations and in so doing determine whether there may be other sources of NO that are also important such as from the descending reticulospinal systems that have previously been shown to be NADPHd positive (Schober et al., 1994). Also important will be a continued effort to decipher the interactions between NO and endocannabinoids in the mGluR1 receptor-mediated potentiation of the locomotor frequency.
Footnotes
This work was supported by the Swedish Research Council, Söderberg Foundation, the European Commission (Health-F2-2007-201144), and the Karolinska Institutet. Support was also provided in part by a Royal Society International Joint Project to K.T.S. and A.E.M. We thank Dr. Russell Hill for comments on this manuscript and Dr. Jon Lundberg for discussions.
References
- Clarke JD, Hayes BP, Hunt SP, Roberts A. Sensory physiology, anatomy and immunohistochemistry of Rohon-Beard neurones in embryos of Xenopus laevis. J Physiol. 1984;348:511–525. doi: 10.1113/jphysiol.1984.sp015122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochilla AJ, Alford S. Metabotropic glutamate receptor-mediated control of neurotransmitter release. Neuron. 1998;20:1007–1016. doi: 10.1016/s0896-6273(00)80481-x. [DOI] [PubMed] [Google Scholar]
- Dale N. Excitatory synaptic drive for swimming mediated by amino acid receptors in the lamprey. J Neurosci. 1986;6:2662–2675. doi: 10.1523/JNEUROSCI.06-09-02662.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bel EA, Guimarães FS, Bermúdez-Echeverry M, Gomes MZ, Schiaveto-de-souza A, Padovan-Neto FE, Tumas V, Barion-Cavalcanti AP, Lazzarini M, Nucci-da-Silva LP, de Paula-Souza D. Role of nitric oxide on motor behavior. Cell Mol Neurobiol. 2005;25:371–392. doi: 10.1007/s10571-005-3065-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Prisco GV, Wallén P, Grillner S. Synaptic effects of intraspinal stretch receptor neurons mediating movement-related feedback during locomotion. Brain Res. 1990;530:161–166. doi: 10.1016/0006-8993(90)90675-2. [DOI] [PubMed] [Google Scholar]
- El Manira A, Tegnér J, Grillner S. Locomotor-related presynaptic modulation of primary afferents in the lamprey. Eur J Neurosci. 1997;9:696–705. doi: 10.1111/j.1460-9568.1997.tb01418.x. [DOI] [PubMed] [Google Scholar]
- El Manira A, Kettunen P, Hess D, Krieger P. Metabotropic glutamate receptors provide intrinsic modulation of the lamprey locomotor network. Brain Res Brain Res Rev. 2002;40:9–18. doi: 10.1016/s0165-0173(02)00184-4. [DOI] [PubMed] [Google Scholar]
- El Manira A, Kyriakatos A, Nanou E, Mahmood R. Endocannabinoid signaling in the spinal locomotor circuitry. Brain Res Rev. 2008;57:29–36. doi: 10.1016/j.brainresrev.2007.06.020. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillner S, McClellan A, Sigvardt K, Wallén P, Wilén M. Activation of NMDA-receptors elicits “fictive locomotion” in lamprey spinal cord in vitro. Acta Physiol Scand. 1981;113:549–551. doi: 10.1111/j.1748-1716.1981.tb06937.x. [DOI] [PubMed] [Google Scholar]
- Grillner S, Williams T, Lagerbäck PA. The edge cell, a possible intraspinal mechanoreceptor. Science. 1984;223:500–503. doi: 10.1126/science.6691161. [DOI] [PubMed] [Google Scholar]
- Hedrick MS, Chen AK, Jessop KL. Nitric oxide changes its role as a modulator of respiratory motor activity during development in the bullfrog (Rana catesbeiana) Comp Biochem Physiol A Mol Integr Physiol. 2005;142:231–240. doi: 10.1016/j.cbpb.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Hope BT, Michael GJ, Knigge KM, Vincent SR. Neuronal NADPH diaphorase is a nitric oxide synthase. Proc Natl Acad Sci U S A. 1991;88:2811–2814. doi: 10.1073/pnas.88.7.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper RA, Garthwaite J. Tonic and phasic nitric oxide signals in hippocampal long-term potentiation. J Neurosci. 2006;26:11513–11521. doi: 10.1523/JNEUROSCI.2259-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettunen P, Kyriakatos A, Hallén K, El Manira A. Neuromodulation via conditional release of endocannabinoids in the spinal locomotor network. Neuron. 2005;45:95–104. doi: 10.1016/j.neuron.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Kishimoto J, Keverne EB, Hardwick J, Emson PC. Localization of nitric oxide synthase in the mouse olfactory and vomeronasal system: a histochemical, immunological and in situ hybridization study. Eur J Neurosci. 1993;5:1684–1694. doi: 10.1111/j.1460-9568.1993.tb00236.x. [DOI] [PubMed] [Google Scholar]
- Kyriakatos A, El Manira A. Long-term plasticity of the spinal locomotor circuitry mediated by endocannabinoid and nitric oxide signaling. J Neurosci. 2007;27:12664–12674. doi: 10.1523/JNEUROSCI.3174-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakatos A, Molinari M, Grillner S, Sillar KT, El Manira A. Source and modulatory actions of nitric oxide in the lamprey spinal locomotor network. Soc Neurosci Abstr. 2008;34:575–5. [Google Scholar]
- LeBeau FE, El Manira A, Griller S. Tuning the network: modulation of neuronal microcircuits in the spinal cord and hippocampus. Trends Neurosci. 2005;28:552–561. doi: 10.1016/j.tins.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Makara JK, Katona I, Nyíri G, Németh B, Ledent C, Watanabe M, de Vente J, Freund TF, Hájos N. Involvement of nitric oxide in depolarization-induced suppression of inhibition in hippocampal pyramidal cells during activation of cholinergic receptors. J Neurosci. 2007;27:10211–10222. doi: 10.1523/JNEUROSCI.2104-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean DL, Sillar KT. The distribution of NADPH-diaphorase-labelled interneurons and the role of nitric oxide in the swimming system of Xenopus laevis larvae. J Exp Biol. 2000;203:705–713. doi: 10.1242/jeb.203.4.705. [DOI] [PubMed] [Google Scholar]
- McLean DL, Sillar KT. Spatiotemporal pattern of nicotinamide adenine dinucleotide phosphate-diaphorase reactivity in the developing central nervous system of premetamorphic Xenopus laevis tadpoles. J Comp Neurol. 2001;437:350–362. doi: 10.1002/cne.1288. [DOI] [PubMed] [Google Scholar]
- McLean DL, Sillar KT. Nitric oxide selectively tunes inhibitory synapses to modulate vertebrate locomotion. J Neurosci. 2002;22:4175–4184. doi: 10.1523/JNEUROSCI.22-10-04175.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean DL, Sillar KT. Metamodulation of a spinal locomotor network by nitric oxide. J Neurosci. 2004;24:9561–9571. doi: 10.1523/JNEUROSCI.1817-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean DL, Merrywest SD, Sillar KT. The development of neuromodulatory systems and the maturation of motor patterns in amphibian tadpoles. Brain Res Bull. 2000;53:595–603. doi: 10.1016/s0361-9230(00)00393-2. [DOI] [PubMed] [Google Scholar]
- McLean DL, McDearmid JR, Sillar KT. Induction of a non-rhythmic motor pattern by nitric oxide in hatchling Rana temporaria embryos. J Exp Biol. 2001;204:1307–1317. doi: 10.1242/jeb.204.7.1307. [DOI] [PubMed] [Google Scholar]
- Nanou E, Kyriakatos A, Bhattacharjee A, Kaczmarek LK, Paratcha G, El Manira A. Na+-mediated coupling between AMPA receptors and KNa channels shapes synaptic transmission. Proc Natl Acad Sci U S A. 2008;105:20941–20946. doi: 10.1073/pnas.0806403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prast H, Philippu A. Nitric oxide as modulator of neuronal function. Prog Neurobiol. 2001;64:51–68. doi: 10.1016/s0301-0082(00)00044-7. [DOI] [PubMed] [Google Scholar]
- Ramanathan S, Combes D, Molinari M, Simmers J, Sillar KT. Developmental and regional expression of NADPH-diaphorase/nitric oxide synthase in spinal cord neurons correlates with the emergence of limb motor networks in metamorphosing Xenopus laevis. Eur J Neurosci. 2006;24:1907–1922. doi: 10.1111/j.1460-9568.2006.05057.x. [DOI] [PubMed] [Google Scholar]
- Schober A, Malz CR, Schober W, Meyer DL. NADPH-diaphorase in the central nervous system of the larval lamprey (Lampetra planeri) J Comp Neurol. 1994;345:94–104. doi: 10.1002/cne.903450107. [DOI] [PubMed] [Google Scholar]
- Sillar KT, Kyriakatos A, Molinari M, Grillner S, El Manira A. Nitric oxide potentiation of lamprey locomotor network activity. FENS Abstr. 2008;4 doi: 10.1523/JNEUROSCI.3069-09.2009. 021.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinay L, Barthe JY, Grillner S. Central modulation of stretch receptor neurons during fictive locomotion in lamprey. J Neurophysiol. 1996;76:1224–1235. doi: 10.1152/jn.1996.76.2.1224. [DOI] [PubMed] [Google Scholar]
- Vincent S. R. Localization of nitric oxide neurons in the central nervous system. In: Jenner P, editor. Nitric oxide in the nervous system. New York: Academic; 1995. pp. 83–102. [Google Scholar]