

Abstract
Coordinated functions of the actin cytoskeleton and microtubules, which require careful control in time and space, are indispensable for the drastic alterations of neuronal morphology during neuromorphogenesis and neuronal network formation. Actin filament formation driven by the Arp2/3 complex and its activator neural Wiskott-Aldrich syndrome protein (N-WASP) is important for proper axon development. The underlying molecular mechanisms for targeting to and specific activation of N-WASP at the neuronal plasma membrane, however, have thus far remained elusive. We show that syndapin I is critical for proper neuromorphogenesis and hereby uses N-WASP as a cytoskeletal effector. Upon N-WASP binding, syndapins release N-WASP autoinhibition. Syndapins hereby cooperate with Cdc42 and phosphatidyl-inositol-(4,5)-bisphosphate. Syndapins furthermore specifically bind to phosphatidylserine-containing membranes via their extended F-BAR domain. Dissecting the syndapin functions actin nucleation and direct membrane binding in vivo, we demonstrate that both functions are physiologically relevant and required. Constitutive plasma membrane-targeting experiments in vivo indicate that specifically actin nucleation at the cell cortex is triggered by syndapins. Consistent with syndapins steering N-WASP as downstream effector for cortical actin nucleation, syndapin-induced neuronal arborization is N-WASP and Cdc42 dependent. The functions of syndapin–N-WASP complexes in neuromorphogenesis were revealed by loss-of-function studies. Knockdown of syndapin I leads to impaired axon development and especially phenocopies the aberrant axon branching observed upon N-WASP and Arp2/3 complex deficiency. In contrast, proper length control involves another N-WASP-binding protein, Abp1. Our data thus reveal that syndapin I is crucial for neuromorphogenesis and that different N-WASP activators ensure fine control of N-WASP activity and have distinct functions during neuronal network formation.
Introduction
The topology of the plasma membrane of eukaryotic cells is dictated by underlying actin cytoskeletal structures. Extensive changes of cell morphology are required during neuromorphogenesis. The induction and elaboration of axonal and dendritic morphologies are fundamental aspects of neuronal network formation and information processing in the CNS. Actin filament formation at defined sites at the cell cortex is a source for the forces required for the induction of the different protrusive structures formed by neurons, such as neurites, branches within the neuritic arbor, and dendritic spines (Cingolani and Goda, 2008; Witte and Bradke, 2008). Actin polymerization, which also plays an important role in growth cone and spine dynamics and plasticity, depends on the help of specialized actin nucleation promoting factors, such as the Arp2/3 complex, which is activated by members of the Wiskott-Aldrich syndrome protein (WASP) family. To control and fine-tune actin nucleation, the neural Wiskott-Aldrich syndrome protein (N-WASP) is kept in an autoinhibited state (Pollard, 2007). N-WASP reduction results in defects in proper axon development (Kakimoto et al., 2006; Pinyol et al., 2007) and in reduction of postsynaptic spines and synapses (Wegner et al., 2008). N-WASP overexpression in neurons leads to excessive formation of neurites and neuritic branches (Pinyol et al., 2007).
Surprisingly, the C terminus of N-WASP, which strongly activates the Arp2/3 complex but is unable to mediate interactions with further N-WASP-binding proteins, causes neuronal phenotypes similar to Arp2/3 complex loss of function (Strasser et al., 2004; Pinyol et al., 2007; Wegner et al., 2008). These observations suggest that a recruitment of N-WASP to the cell cortex is of utmost importance for N-WASP/Arp2/3 complex-dependent processes during neuromorphogenesis. Brain-enriched N-WASP-binding proteins that could mediate such plasma membrane recruitment include syndapin I (Qualmann et al., 1999). Syndapin I (also called PACSIN1) belongs to the F-BAR family within the BAR domain superfamily (Itoh et al., 2005)—a large and diverse protein family that can discern the curvature of a membrane by offering curved, crescent-shaped membrane interaction surfaces (Peter et al., 2004). Members of the F-BAR family are marked by the presence of a Fes/Cip4-homology (FCH) domain (Aspenström et al., 2006; Dawson et al., 2006; Chitu and Stanley, 2007). For syndapin I, the FCH domain has been shown to represent a self-association interface within a larger module, the extended F-BAR domain (eF-BAR), which is responsible for self-association in vivo (Kessels and Qualmann, 2006). The diversity of BAR proteins might reflect different types of membrane interfaces and biological functions.
Here we demonstrate that syndapins bind to membranes directly via their eF-BAR domain and are capable of releasing N-WASP autoinhibition. Our studies clearly demonstrate that SH3 domain-mediated N-WASP activation along with membrane binding are important aspects of syndapin function in vivo. Gain-of-function and loss-of-function studies furthermore show that proper neuromorphogenesis relies on syndapin-mediated N-WASP- and Arp2/3 complex-dependent actin nucleation at the plasma membrane and that loss of syndapin I, similar to effects caused by loss of N-WASP and Arp3, results in improper axon development.
Materials and Methods
DNA constructs and recombinant proteins.
Plasmids encoding for Xpress-tagged syndapin I full-length and ΔSH3 were described by Qualmann et al. (1999) and Qualmann and Kelly (2000), GFP-SH3 was described by Kessels and Qualmann (2006), and Flag-EHD1 G429R was described by Braun et al. (2005). Further syndapin constructs were generated by subcloning or by PCR, cloned into the appropriate expression vectors, and analyzed by DNA sequencing. The SH3 domain of human amphiphysin I corresponding to the amino acids 625-695 of the transcript variant I (gi:21536405) was amplified via PCR and cloned into pEGFP-C2. A plasmid encoding for dominant-negative Eps15 (Eps15 Δaa95-295) was kindly provided by A. Benmerah (Université Paris Descartes, Paris, France) (Benmerah et al., 1999).
Plasma membrane-attached syndapin I and II full-length and deletion and point mutants thereof were generated by subcloning into a derivative of the pCMV-Tag2 mammalian expression vector (Clontech), in which the sequence encoding for the N-terminal 20 aa of growth-associated protein 43 (GAP-43) encompassing a palmitoylation motif and GFP were inserted by PCR.
To generate RNAi constructs against rat syndapin I, the following oligonucleotides were annealed and integrated into the pRNAT H1.1-GFP vector (GenScript). Syndapin I RNAi sequence #2: 5′-GATCCGCTCGTTCAGTAGGCTGTTCTTGATATCCGGAACAGCCTACTGAACGAGTTTTTTA-3′ and 5′-AGCTTAAAAAACTCGTTCAGTAGGCTGTTCCGGATATCAAGAACCAGCCTACTGAACGAGG-3′; syndapin I RNAi sequence #3: 5-GATCCGCTTCTGCCAGTTCTTGACTTTGATATCCGAGTCAAGAACTGGCAGAAGTTTTTTA-3′ and 5′-AGCTTAAAAAACTTCTGCCAGTTCTTGACTCGGGATATCAAAGTCAAGAACTGGCAGAAGC-3′.
To generate an RNAi-resistant syndapin I mutant, several silent mutations were introduced by PCR into the syndapin I cDNA site targeted by RNAi sequence #2. Residues mutated for generation of an RNAi-resistant syndapin I are underlined in the above sequence. The PCR product was then cloned into pCMV-Tag2 (Clontech) and verified by DNA sequencing. RNAi tools against N-WASP, Arp3, Abp1, and the nonsilencing RNAi control were described by Pinyol et al. (2007).
GST-fusion proteins of syndapins were expressed and purified as described previously (Qualmann et al., 1999; Qualmann and Kelly, 2000). For actin polymerization assays and for liposome binding assays, GST-fusion proteins were liberated from the GST-tag using thrombin (Sigma) and purified by gel filtration.
Antibodies.
Rabbit anti-syndapin I, anti-syndapin II, and anti-GST antibodies were raised and affinity purified as described previously (Qualmann et al., 1999; Qualmann and Kelly, 2000). Rabbit anti-mAbp1 antibodies were described by Haeckel et al. (2008), and guinea pig anti-N-WASP antibodies and guinea pig anti-GST antibodies were described by Kessels and Qualmann (2002).
Monoclonal anti-Flag (M2), anti-tubulin, and anti-MAP2 antibodies were from Sigma. Polyclonal rabbit anti-MAP2 and anti-GFP (290) antibodies were from Abcam. Monoclonal anti-Xpress antibodies were from Invitrogen. Rabbit anti-Flag antibodies were from Zymed. Monoclonal anti-GFP antibodies (B34) were purchased from Babco.
Secondary antibodies used in this study included goat anti-mouse-peroxidase, donkey anti-rabbit-peroxidase, goat anti-rabbit Cy5, and donkey anti-mouse Cy5 antibodies (Dianova). Alexa Fluor 568 goat anti-mouse, Alexa Fluor 350, 488, and 568 goat anti-rabbit, and Alexa Fluor 350, 488, 568, and 680 goat anti-mouse antibodies were from Invitrogen. DyLight 800-conjugated goat anti-rabbit antibodies were from Pierce. Donkey anti-guinea pig antibodies coupled to IRDye680 and IRDye800CW, respectively, were from LI-COR Bioscience.
Preparation of liposomes and liposome binding assays.
Liposomes were prepared from phosphatidylethanolamine (PE) and additional lipids, such as phosphatidylcholine (PC), phosphatidylserine (PS) (Avanti polar lipids, Sigma), and phosphatidyl-inositol-(4,5)-bisphosphate (PIP2) (Alexis), by a respective reduction of PE percentage. Lipids were mixed and dried to thin films. Lipid films were then rehydrated in 20 mm HEPES buffer, pH 7.4, containing 0.3 m sucrose with occasional low-speed vortexing.
For visualization, liposomes were dotted with 5% of Rhodamine B-conjugated PE (Avanti polar lipids, Sigma). Floatation assays with liposomes were conducted according to Bigjay et al. (2005) with slight modifications. Protein (1 μm) was mixed with 1 mm liposomes and incubated at room temperature for 10 min. Then the samples were brought to 30% sucrose concentration (final volume 250 μl) and overlaid with 200 μl of 25% sucrose and 0% sucrose solution each in the above HEPES buffer. The samples were then centrifuged at 240,000 × g for 30 min at 4°C. Fractions of 100 μl were collected from top to bottom and subjected to immunoblotting with anti-GST antibodies.
Subcellular fractionation and floatation assays.
COS-7 cells were transfected with epitope-tagged syndapins, harvested, and lysed mechanically in ice-cold 10 mm HEPES buffer, pH 7.4, with 150 mm NaCl, 1 mm EGTA, and 0.1 mm MgCl2 48 h later. Lysate (525 μl) was supplemented with 175 μl of 1 m sucrose in PBS and centrifuged at 1000 × g (3 min at 4°C). The resulting supernatant was subjected to floatation assays according to Kretzschmar et al. (1996). Two hundred microliters of the supernatant were diluted to 1 ml by adding sucrose (70% final), placed under two cushions of 65% (7 ml) and 10% sucrose (3.5 ml), and centrifuged overnight at 4°C at 100,000 × g. Fractions (1 ml) were collected from top to bottom. Proteins were precipitated by TCA, solubilized in SDS-PAGE sample buffer, and analyzed by immunoblotting.
Reconstitution and analyses of F-actin polymerization in vitro.
Actin polymerizations on beads and pyrene-actin polymerizations in fluorimeter cuvettes were performed and analyzed essentially as described by Pinyol et al. (2007) and Ahuja et al. (2007). For the latter, exclusively syndapin fusion proteins liberated from the GST-tag were used. For the examinations of lipid effects, proteins were preincubated with 50 μm liposomes (30 min, 25°C).
Cell culture and examinations of the actin cytoskeleton in HeLa cells.
Culturing of COS-7 and HeLa cells and evaluations of F-actin phenotypes induced by overexpression of syndapins and mutants thereof were done according to Kessels and Qualmann (2006).
Preparation of brain homogenates and relative and quantitative determination of protein contents.
Whole rat brains of developmental stages E18, P0, and P4 (n = 2 each; Rattus norvegicus, Wistar strain) were collected and stored at −80°C until usage. Subsequent steps were performed on ice or at 4°C. Brains were homogenized in homogenization buffer (10 mm HEPES-NaOH, pH 7.4, 10 mm NaCl, 1 mm EGTA, and 0.1 mm MgCl2) using an Ultraturrax T8-S8N5G (Ika) as previously described (Braun et al., 2005). Lysis was completed by incubation with Triton X-100 (1% final) for 1 h. Afterward, SDS-PAGE sample buffer and urea (1 m final) were added and the samples were incubated at 70°C for 10 min. Fifty micrograms of total protein of whole-brain homogenates of the different developmental stages were subjected to Western blot analyses.
Immunosignals of syndapin I and Abp1, respectively, were quantified using a LI-COR Odyssey detection system (LiCOR Bioscience). Different amounts of GST (between 0.25 and 40 ng) were loaded on the same blots used for detection of syndapin I and Abp1, respectively, and served to calibrate signal intensities for anti-Abp1 and anti-syndapin I immunoreactivity using LI-COR Odyssey V3.0 software (settings: reciprocal fit).
For relative analyses of protein expression during different stages of development, tubulin was used for normalization. Antibody/tubulin ratios at E18 were set to 100%.
Preparations of primary hippocampal neurons and morphometric analyses.
Primary hippocampal cultures were prepared, processed for immunofluorescence, and morphometrically analyzed as described by Pinyol et al. (2007). Transfections of neurons were performed with Lipofectamine 2000 (Invitrogen) at days 2 and 4 in vitro, respectively. Neurons were fixed in 4% PFA in PBS, pH 7.4, for 8 min at room temperature 24 and 48 h after transfection, respectively.
Images were recorded using a Zeiss Axioplan 2 microscope or a Zeiss Axio-Imager.D1, both equipped with a Zeiss Plan-Apochromat 63×/1.4 and a CCD camera 2.1.1 from Diagnostic Instruments, and processed in MetaVue or SpotSoftware and Adobe Photoshop.
Morphometric measurements and knockdown intensity measurements of transiently transfected hippocampal neurons were performed with ImageJ. Each experiment was repeated 2–10 times with independent neuronal preparations. Neurons were identified by anti-MAP2 staining and sampled completely based on anti-MAP2 immunosignals to identify neurites and to exclude other types of cellular protrusions.
The number of dendrites, the number of dendritic branching points, the summarized dendritic arborization, and/or axon branching were determined after 6 d in culture from 31–199 neurons for each condition.
After 3 d in culture, axon length and branching and the length of the summarized axonal arbor as well as dendrite number and branching and summarized dendritic points of arborization were measured in 24–143 neurons each. Axons of neurons were defined as the longest neurite at that time, as dendritic and axonal markers are not yet showing a polarized distribution in immature neurons.
All data were normalized to the corresponding controls run in parallel in each individual experiment and neuronal preparation. Statistical analysis was performed using the two-tailed Student's t test.
Results
Syndapin I mediates N-WASP-dependent cell morphology changes in developing neurons
Actin filament organization and dynamics is crucial for neuronal development by critically influencing structure, polarity, and functionality of neuronal cells. Syndapin I, which is predominantly expressed in neurons (Qualmann et al., 1999), can interface with the Arp2/3 complex actin polymerization machinery via an interaction with the Arp2/3 complex activator N-WASP (Qualmann and Kelly, 2000). We therefore hypothesized that syndapin I may be important for establishment and plastic changes of neuronal morphology—processes that are important for proper neuronal network formation and function.
Morphologies of hippocampal neurons transfected with syndapin I at DIV 4 and processed for morphometric analyses 2 d later indeed strongly differed from control neurons (Fig. 1A,B). Both neurite number and dendrite branching was highly increased in syndapin I-transfected cells (Fig. 1B). Quantitative evaluations confirmed these observations (Fig. 1G–I). Dendrite number, dendritic branching, and the summarized dendritic arborization were increased by ∼50%.
Figure 1.
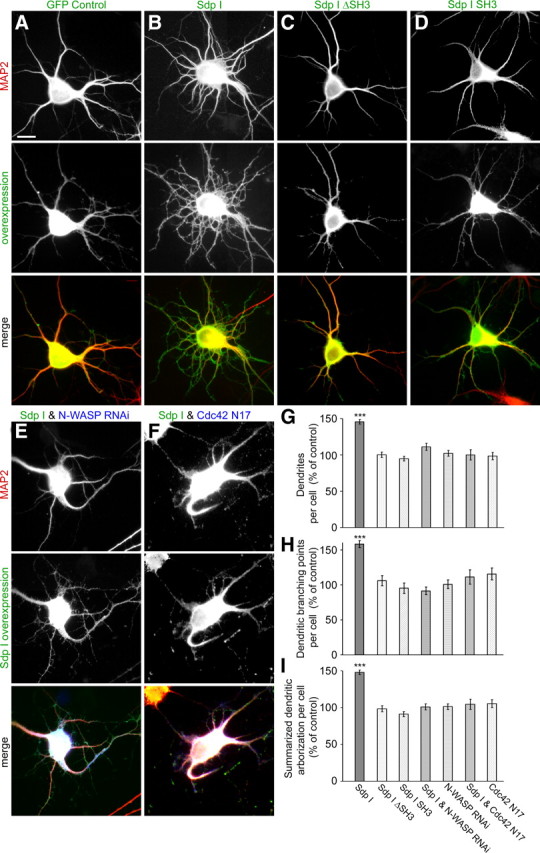
Syndapin I induces dendrite formation and branching. Syndapin I expression in primary hippocampal neurons (B) resulted in a strong increase in the number of dendrites and dendritic branching points when compared with control cells (A). The cells were transfected as indicated at DIV4 and processed for immunofluorescence microscopy 2 d later. Upper images show the anti-MAP2 staining used to evaluate neuronal morphology. Middle panels show overexpressed proteins, and lower panels show the merged images. In E and F, the merged images include a third channel (in blue) showing the expression of N-WASP RNAi (E) and Cdc42 N17 (F), respectively. Quantitative evaluations demonstrate a highly significant increase of the numbers of dendrites (G) and dendritic branching points (H) as well as of the summarized dendritic arborization (I) of syndapin I-transfected neurons that is not observed with syndapin I deletions lacking the SH3 domain (Sdp I ΔSH3; C) or lacking the N-terminal part (Sdp I SH3; D). The syndapin I-induced phenotype leading to increased dendritic arborization is completely abolished upon RNAi-based knockdown of N-WASP expression (E) or coexpression of Cdc42 N17 (F). Morphological data are represented as mean ± SEM (***p < 0.0001). Scale bar, 10 μm.
Although the SH3 domain of syndapin I is necessary and sufficient for the association with N-WASP (Qualmann and Kelly, 2000), the SH3 domain was proven to be critical but not sufficient for the induction of dendrites and enhanced branching. Deletion analyses showed that the syndapin I-induced phenotype was dependent on both the SH3 domain and the N-terminal part, as neurons transfected with mutants lacking these domains did not differ from control cells (Fig. 1A,C,D,G–I).
The syndapin I effect on neuronal morphology clearly depends on N-WASP, because both the number of dendrites and branching points in syndapin I-expressing cells were reduced to control levels upon simultaneous knockdown of N-WASP expression, whereas N-WASP RNAi alone had no effect on the dendritic compartment (Fig. 1E,G–I). In line with N-WASP being used as a cytoskeletal effector by syndapin I, a dominant-negative mutant of the N-WASP-activating GTPase Cdc42 (Cdc42 N17) (Allen et al., 1997) also suppressed the syndapin I-induced phenotype completely (Fig. 1F–I).
Syndapin I-induced changes in neuronal morphology are not based on impaired endocytosis or endosomal recycling
Syndapins have been identified as dynamin-interacting proteins and play a role in endocytosis and endosomal receptor recycling (Qualmann et al., 1999; Qualmann and Kelly, 2000; Braun et al., 2005; Andersson et al., 2008). Both functions also involve N-WASP (Kessels and Qualmann, 2002; Benesch et al., 2005; Parsons et al., 2005). In addition to interference with the function of syndapin complexes (Braun et al., 2005), at least overexpression of GFP-syndapin I was reported to negatively influence clathrin-mediated endocytosis of transferrin receptors (Modregger et al., 2000). We therefore analyzed whether the syndapin-induced change in neuromorphology correlated with a disruption of any of these membrane trafficking functions. Two independent, well established molecular tools were used to block receptor-mediated endocytosis; however, neither overexpression of amphiphysin I SH3 domain (Wigge et al., 1997) nor overexpression of a dominant-negative mutant of Eps15 (Eps15 Δaa95-295; Benmerah et al., 1999) led to effects similar to those induced by syndapin I (Fig. 2A–C). Quantitative analyses furthermore demonstrated that also overexpression of EHD1 G429R, an EHD1 mutant, which has been shown to block endosomal receptor recycling (Lin et al., 2001), failed to cause any syndapin I-related phenotypes (Fig. 2A–C).
Figure 2.
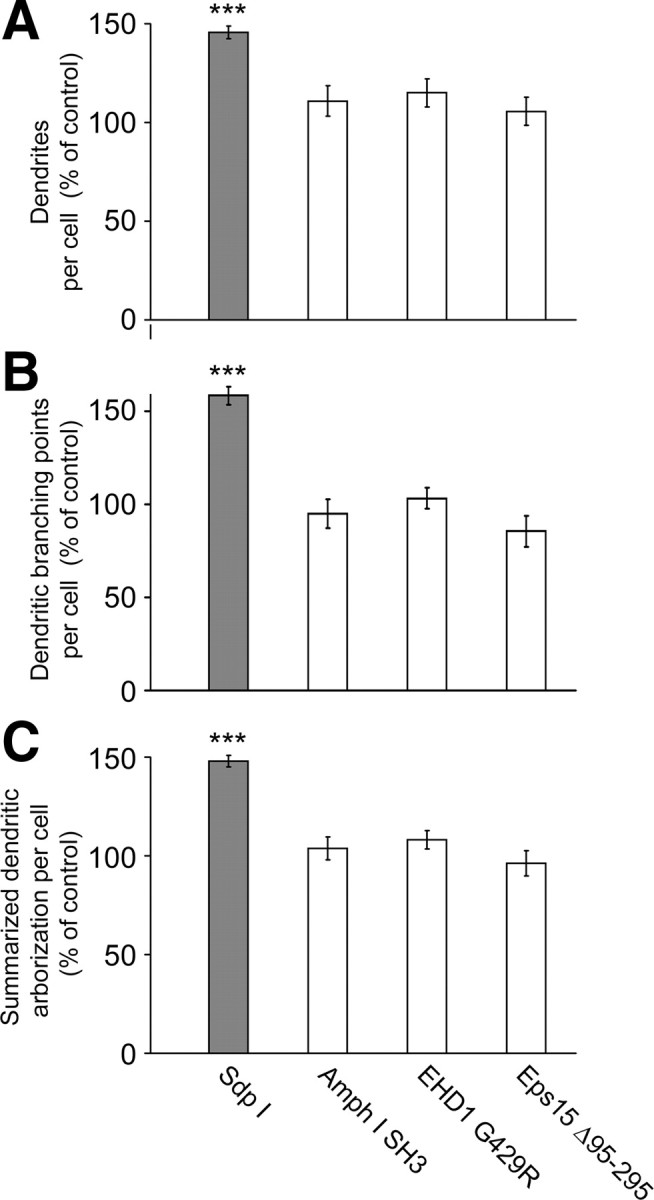
Syndapin I-induced excessive arborization of the dendritic compartment is not caused by impairments of endocytosis and endosomal recycling. Primary hippocampal neurons were transfected at DIV4, processed for immunofluorescence microscopy 2 d later, and their morphology was quantitatively evaluated. Neither inhibition of endocytosis via overexpression of the amphiphysin I SH3 domain and of Eps15 Δaa95-295 nor block of endosomal recycling via overexpression of EHD1 G429R resulted in syndapin I-like phenotypes in dendrite number (A), dendritic branching points (B), or the summarized dendritic arborization of the developing neurons (C). The morphological data are represented as mean ± SEM (***p < 0.0001).
Syndapins release the N-WASP autoinhibition
The modulation of neuronal morphology by syndapin I (Fig. 1), the lack of influences of membrane trafficking aspects (Fig. 2), and the observation that both N-WASP and Cdc42 are required for syndapin-mediated changes of neuronal morphology (Fig. 1) strongly suggested that syndapins interface with the Arp2/3 complex actin nucleation machinery and may influence its activity. For the requirement of the syndapin N-terminal part in addition to the N-WASP-binding C-terminal SH3 domain (Fig. 1), two mechanistic explanations seemed plausible. First, the full-length context might be important for targeting the actin polymerization machinery to the cell cortex. The N terminus including the F-BAR domain of syndapins may hereby mediate the membrane association, and the SH3 domain, which we also found to be crucial for phenotype induction (Fig. 1C,G–I), in turn recruits an interaction partner, such as N-WASP, which alters cellular morphology by reorganizing the cortical actin cytoskeleton. Second, it was also possible that the ΔSH3 domain part of syndapins is required for the activation of the N-WASP/Arp2/3 complex actin polymerization machinery.
The observation of F-actin halos around syndapin-coated beads incubated with brain extracts supplemented with fluorescently labeled G-actin indeed demonstrated the ability of syndapins to trigger actin filament formation (Fig. 3A–F). Since the SH3 domain of both syndapin I (Fig. 3A) and syndapin II (Fig. 3C) was as efficient for halo induction as full-length syndapin I (Fig. 3E), the syndapin SH3 domains are sufficient for triggering F-actin formation. Consistently, beads coated with mutant SH3 domains did not assemble fluorescent F-actin halos but remained as dark as controls (Fig. 3B,D,F).
Figure 3.

Syndapins stimulate N-WASP-mediated Arp2/3 complex-dependent actin polymerization. A–F, Different GST-syndapin I and II fusion proteins comprising a functional SH3 domain that were immobilized on beads elicited fluorescent F-actin halos in brain extracts endowed with Alexa Fluor 568-labeled G-actin, whereas mutated SH3 domains and the GST control did not. Scale bar, 50 μm. G–I, In vitro reconstitutions of actin nucleation and polymerization analyzed by the fluorescence increase of pyrene-labeled actin plotted against time. Concentrations were as follows: actin, 4 mm; pyrene-actin, 0.4 mm; Arp2/3 complex, 10 nm; Flag-tagged N-WASP, 80 nm; and syndapin, 100 nm, unless otherwise stated. Polymerization induced by Arp2/3 complex and N-WASP WA (80 nm) is shown for comparison. All samples contain actin and Arp2/3 complex; other components added are indicated. G, Dose-dependent activation of N-WASP/Arp2/3 complex-dependent actin polymerization by increasing amounts of the syndapin I SH3 domain. H, I, Addition of full-length syndapin I (H) and syndapin II-l (I) to N-WASP activated with GTPγS-loaded Cdc42 under saturation conditions (400 nm Cdc42; 80 nm N-WASP) leads to a further activation of N-WASP.
The syndapin I gain-of-function phenotype on neuronal morphology was N-WASP dependent (Fig. 1). Since syndapins bind to N-WASP (Qualmann et al., 1999), we hypothesized that syndapins can interface with the Arp2/3 complex by modulating the activity of N-WASP. To test this hypothesis, we reconstituted actin polymerization reactions with purified actin, Arp2/3 complex, N-WASP, and syndapin in a fluorimeter and followed actin polymerization by the resulting fluorescence increase of pyrene-labeled actin. Whereas actin and Arp2/3 complex had very little effects (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) and also the addition of N-WASP did not dramatically accelerate filament formation (Fig. 3G–I), addition of syndapin to N-WASP, Arp2/3 complex, and actin promoted actin nucleation significantly (Fig. 3G–I). The effects of syndapin I and II were very similar (Fig. 3, compare H, I).
In line with our reconstitutions with complex brain extracts (Fig. 3A–F), the syndapin SH3 domain was sufficient and yielded a dose-dependent increase of N-WASP activation (Fig. 3G). Control reactions without N-WASP did not lead to any significant generation of actin filaments (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), demonstrating that syndapins do not interface with actin and/or the Arp2/3 complex but function via activation of N-WASP.
The ability of syndapin to activate N-WASP was approximately comparable to the N-WASP activation by the established N-WASP activator Cdc42 [compare data in Fig. 3H,I [with an excess of Cdc42 (400 nm)] with the syndapin effects at saturation conditions in Fig. 3G (300 nm)]. We therefore asked whether syndapins and Cdc42 compete for N-WASP activation or work together. Putative syndapin effects on Cdc42-activated N-WASP molecules can only be proven under Cdc42 saturation conditions. We thus used a fivefold excess of GTPγS-loaded Cdc42 to activate N-WASP (Fig. 3H,I). Interestingly, addition of syndapin I or II led to a further activation of Cdc42-activated N-WASP. The effects were about additive (Fig. 3H,I).
Syndapins associate with phosphatidylserine-containing membranes via their eF-BAR domains
Our experiments did not support the working hypothesis that the N terminus of syndapins is required for triggering N-WASP-mediated Arp2/3 complex-dependent actin filament formation. To address the role of the syndapin N terminus, we therefore focused on the alternative hypothesis, a crucial role in membrane recruitment.
Syndapins are involved in endocytosis and can reorganize the cortical actin cytoskeleton. Both functions require action at the cell cortex. Since syndapins exhibit similarities to BAR domain proteins (Fig. 4A), we tested for direct association of syndapin I and II with membranes. Using liposomes with defined compositions we also asked whether syndapins would show any preferences for membrane compositions preferred by their binding partners dynamin, N-WASP, and EHD proteins, i.e., PS and/or PIP2 (Praefcke and McMahon, 2004; Daumke et al., 2007; Takenawa and Suetsugu, 2007). In line with a report, which also shows that the F-BAR protein FBP17 associates with PS and used a liposome precipitation assay (Itoh et al., 2005), we observed that syndapin I bound to liposomes tight enough to withstand floatation through a sucrose gradient and efficiently floated with PE/PC-based liposomes accumulating in density gradient fraction 2 (for liposome detection, see Fig. 4H) when 10% PS was included (Fig. 4B). Likewise, syndapin II binding to liposomes was promoted by PS (Fig. 4C). Syndapin II lipid binding appeared somewhat stronger than that of syndapin I. The observed binding of both syndapin isoforms to PS-containing liposomes suggests that PS-binding is a general property of syndapins.
Figure 4.
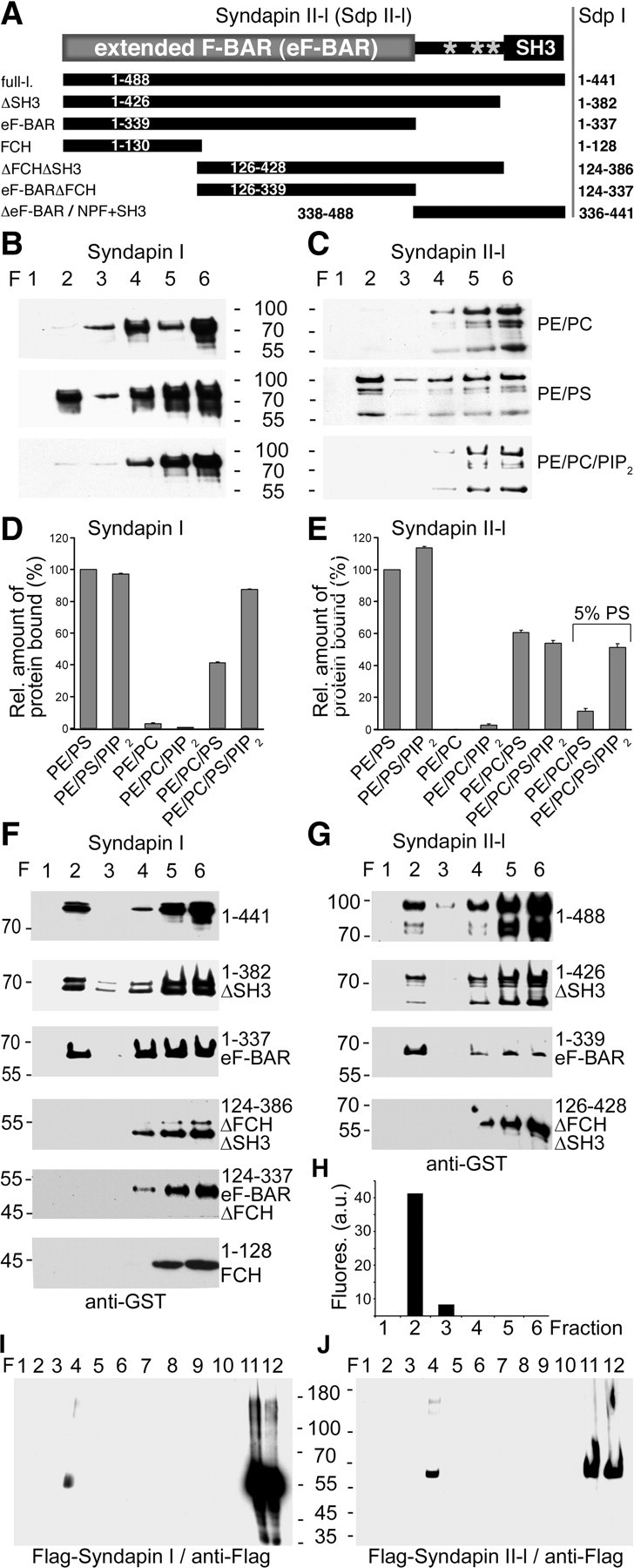
Syndapins associate with PS-containing membranes via eF-BAR domains. A, Syndapin II domain structure (gray stars, NPFs) and schematic representation of different syndapin II constructs and the corresponding syndapin I proteins (right column). B, C, Syndapin I and II-l directly bind to PS-containing liposomes. Shown are liposome floatation analyses of GST-syndapin I (B) and II-l (C) with liposomes composed of (1) 80% PE and 20% PC (PE/PC); (2) 90% PE and 10% PS (PE/PS); and (3) 70% PE, 20% PC, and 10% PIP2 (PE/PC/PIP2). Density gradient fractions were immunoblotted with anti-GST antibodies. Liposomes floated into fraction 2, as proven by incorporation of fluorescent lipids and fluorescence measurements (compare with H). Note that both syndapin I as well as syndapin II-l protein material specifically associates with PS-containing liposomes tightly enough to withstand floating through the sucrose gradients and can therefore be detected together with the liposomes in fraction 2. D, E, Quantitative analyses of binding of GST-syndapin I (D) and II-l (E) to liposomes. F, G, Analyses of different syndapin I and II-l deletion mutants for floatation with 60% PE/20% PC/10% PIP2/10% PS liposomes. H, Distribution of incorporated fluorescent lipids demonstrates that liposomes float to fraction 2 of the density gradients. I, J, Lysates of COS-7 cells overexpressing Flag-syndapin I (I) and Flag-syndapin II-l (J) subjected to density gradient separations according to Kretzschmar et al. (1996). Immunoblotting of the fractions with anti-Flag antibodies shows that a syndapin I and II-l subpool associates with plasma membrane material enriched in fraction 4.
Neither syndapin I nor syndapin II bound to PIP2 (Fig. 4B,C). Quantitative analyses confirmed that there was no stable association with PIP2-containing PE/PC-liposomes (Fig. 4D,E). Further experiments showed that PIP2 is able to promote syndapin binding to PS-containing PE/PC-liposomes but not to PS-containing PE-liposomes (Fig. 4D). This effect is reminiscent of the behavior of FBP17 (Itoh et al., 2005). In the case of syndapin II, this promotion of PS-binding by PIP2 could only be visualized under reduced amount of PS (Fig. 4E) (5% PS).
Experiments with deletion mutants showed that syndapin I and II-l mutants lacking the C-terminal SH3 domains (ΔSH3) as well as syndapin mutants comprising only the N-terminal eF-BAR domains floated with PS-containing liposomes (Fig. 4F,G). Thus, the eF-BAR domains are sufficient for lipid association.
Removal of the FCH domain completely abolished the liposome associations (Fig. 4F,G). For syndapin I, this was corroborated by an alternative mutant additionally lacking the NPF region. Experiments with the syndapin I FCH domain alone demonstrated that, in contrast to its role in syndapin self-association (Kessels and Qualmann, 2006), the FCH domain and the C-terminal half of the eF-BAR domain are not acting as independent modules but are both needed for lipid association (Fig. 4F). This is in line with the recently published crystal structure of syndapin I showing a curved, tightly packed bundle of α-helices of the F-BAR domains of two syndapin molecules (Wang et al., 2009).
Both crude biochemical fractionations (our unpublished data) and density gradient analyses revealed that a subpool of syndapins in cellular extracts is associated with membranes. Both syndapin I and II-l accumulated in fraction 4 (Fig. 4I,J)—a fraction that corresponds to floating plasma membrane material (Kretzschmar et al., 1996) (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Thus, syndapin–lipid interactions are not restricted to in vitro reconstitution experiments, such as those shown in Figure 4B–H, but can also be observed in cell extracts (Fig. 4I,J).
Syndapins activate N-WASP-mediated Arp2/3 complex-dependent actin polymerization in cooperation with PIP2
If the in vivo function of syndapin indeed was to recruit and activate N-WASP specifically at the plasma membrane, it is required to carefully analyze the effects of both syndapin- and N-WASP-binding lipids on syndapin-mediated release of N-WASP autoinhibition. Since PIP2 binding to N-WASP has also been described to overcome the N-WASP autoinhibition (for review, see Takenawa and Suetsugu, 2007), we analyzed a putative coactivation of N-WASP via syndapins and PIP2. Addition of a high molar excess of PIP2-containing liposomes indeed led to a relatively strong activation of N-WASP, which was only slightly weaker than that induced by syndapin II (Fig. 5A). When syndapin II-l and PIP2-containing liposomes were added, a significant enhancement of the performance of PIP2-activated N-WASP was detected. As observed in our experiments showing that syndapin and the N-WASP activator Cdc42 work together, the effects of syndapin II-l and PIP2 on N-WASP activation were about additive (Fig. 5A).
Figure 5.

Stimulation of N-WASP-mediated Arp2/3 complex-dependent actin polymerization by syndapins and lipid association is additive. A, B, In vitro reconstitutions of actin nucleation and polymerization analyzed by the fluorescence increase of pyrene-labeled actin plotted against time. Concentrations were as follows: actin, 4 mm; pyrene-actin, 0.4 mm; Arp2/3 complex, 10 nm; Flag-tagged N-WASP, 80 nm; syndapin, 100 nm; and lipid concentration of liposomes, 50 μm. Polymerization induced by Arp2/3 complex and N-WASP WA (80 nm) is shown for comparison. All samples contain actin and Arp2/3 complex; other components added are indicated. A, Syndapin- and PIP2-mediated activation of N-WASP is additive. B, In contrast, neither N-WASP basal activity nor the syndapin II-mediated activation of N-WASP is significantly increased by PS-containing liposomes.
In contrast, liposomes containing the syndapin-binding lipid PS did not result in any significant activation of N-WASP. Also, PS-liposome addition did not lead to a strong increase of syndapin-mediated N-WASP activation (Fig. 5B), at the same time, the ability of PS-liposome-bound syndapins to activate N-WASP was not diminished either. These data are in line with the syndapin SH3 domain being fully accessible in the full-length context of the protein and with the syndapin SH3 domain being sufficient for N-WASP activation (Fig. 3).
Syndapins modulate the cortical actin cytoskeleton and the topology of the plasma membrane by a mechanism that depends on membrane targeting and SH3 domain interactions
Experiments with the ubiquitously expressed syndapin II isoform in HeLa cells demonstrate that the requirement of the lipid-binding extended F-BAR domain (Fig. 4) in addition to the N-WASP activating SH3 domain (Fig. 3) is a generalized feature of this subfamily of F-BAR proteins in organization of the cortical actin cytoskeleton and cell morphology control (Fig. 6). Overexpression of syndapin II led to a strong induction of F-actin-rich, kinky lamellipodia decorated with filopodial structures (Fig. 6A,I). In contrast, cells transfected with the isolated SH3 domain, the ΔSH3 part, and with syndapin II-l ΔeF-BAR mutants (NPF+SH3) were indistinguishable from control cells (Fig. 6B,C,I). This demonstrated that both the SH3 domain and the eF-BAR domain are required and that neither the SH3 nor the eF-BAR domain is sufficient. With the ability of syndapins to associate with membranes (Fig. 4) we have unraveled a potential molecular explanation for this necessity of the eF-BAR domain in cortical cytoskeleton and membrane topology organization.
Figure 6.
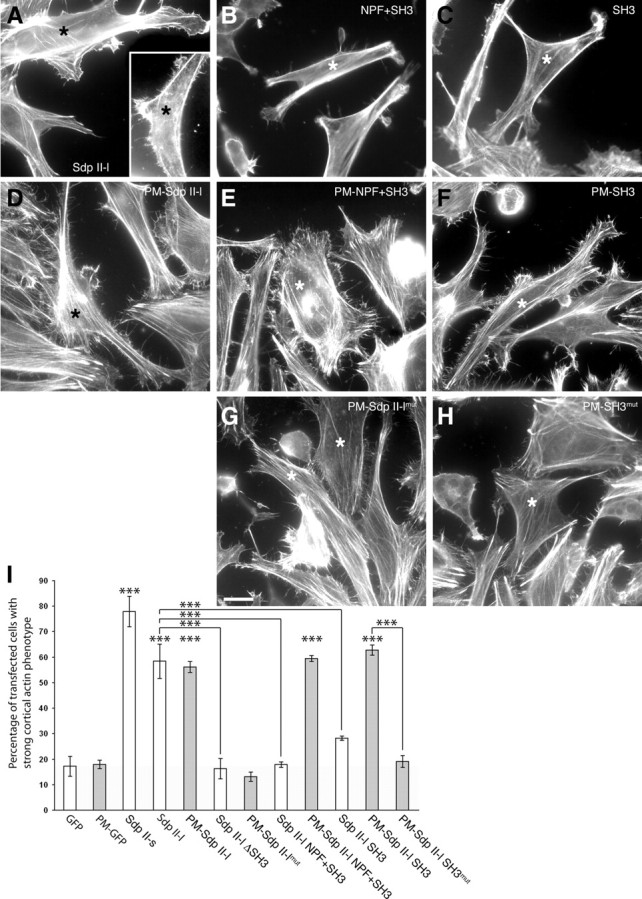
The syndapin eF-BAR is required for syndapin-induced cortical actin rearrangements and exercises its function by targeting the syndapin SH3 domain to the plasma membrane. A–H, Phalloidin staining of the actin cytoskeleton of HeLa cells transiently transfected (stars) with GFP-tagged syndapin II-l and deletion mutants thereof as well as with plasma membrane-anchored (PM) syndapin II-l mutants. Note that only syndapin II-l full-length (A) and PM-anchored proteins with an intact SH3 domain (D–F) exhibited drastic cortical actin rearrangements manifesting in F-actin-rich, kinky, and filopodia-decorated lamellipodia. Scale bar, 20 μm. I, Quantitative examination of the effects of indicated syndapin constructs. A total of 134–528 transfected cells from several independent assays were scored. Background effects of GFP are shaded in gray. Error bars represent SDs (***p < 0.001).
Such a putative importance of the eF-BAR domain for membrane association in vivo and in turn cell morphology control was addressed experimentally in vivo by fusing syndapin II-l and parts thereof with palmitoylation consensus sequences. Characterization of the constructs by density gradient centrifugation and analysis by confocal microscopy (supplemental Fig. S2, available at www.jneurosci.org as supplemental material) confirmed that syndapin proteins carrying a palmitoylation consensus sequence (PM-syndapins) are indeed very efficiently targeted to the plasma membrane and anchored there.
Phalloidin staining revealed that GFP-tagged PM-syndapin II-l led to a phenotype similar to that caused by the nonmodified full-length protein (Fig. 6A,D,I). Strikingly, it was possible to induce the syndapin overexpression phenotype with the C-terminal ΔeF-BAR part or even with merely the SH3 domain fused to the plasma membrane targeting sequence (Fig. 6E,F,I). As corresponding constructs carrying a mutated SH3 domain (P480L) incapable of binding to proline-rich motifs failed to elicit the cortical phenotype (Fig. 6G–I), only constructs that have the ability to interact with membranes [i.e., that contain the eF-BAR domain or alternatively carry a palmitoyl anchor (or both)] and additionally comprise a functional SH3 domain modulate plasma membrane topology and cell shape.
Syndapin-mediated cell morphology changes in developing neurons depend on plasma membrane association
Deletion analyses showed that the syndapin-induced changes in neuronal morphology are dependent on both the SH3 domain and the N-terminal part mediating the lipid interaction (Fig. 1). Since in contrast, the SH3 domain of syndapin I is sufficient for the association with N-WASP and the induction of actin polymerization in vitro (Fig. 3), we hypothesized that the full-length protein context might be important for targeting the actin polymerization machinery to the cell cortex. Our observations with the syndapin II isoform in non-neuronal cells are in line with such a hypothesis (Fig. 6). If this hypothesis was valid and of general importance for all syndapin isoforms, constitutive targeting of the neuronal form of syndapins, i.e., of syndapin I, or of its SH3 domain to the plasma membrane should cause excessive dendritic arborization similar to overexpression of the unmodified full-length protein (Fig. 1). Indeed, overexpression of PM-syndapin I SH3 led to increases in the number of dendrites and in dendritic branching points when compared with PM-GFP controls that were indistinguishable from those induced by full-length syndapin I and by PM-syndapin I, respectively (Fig. 7).
Figure 7.
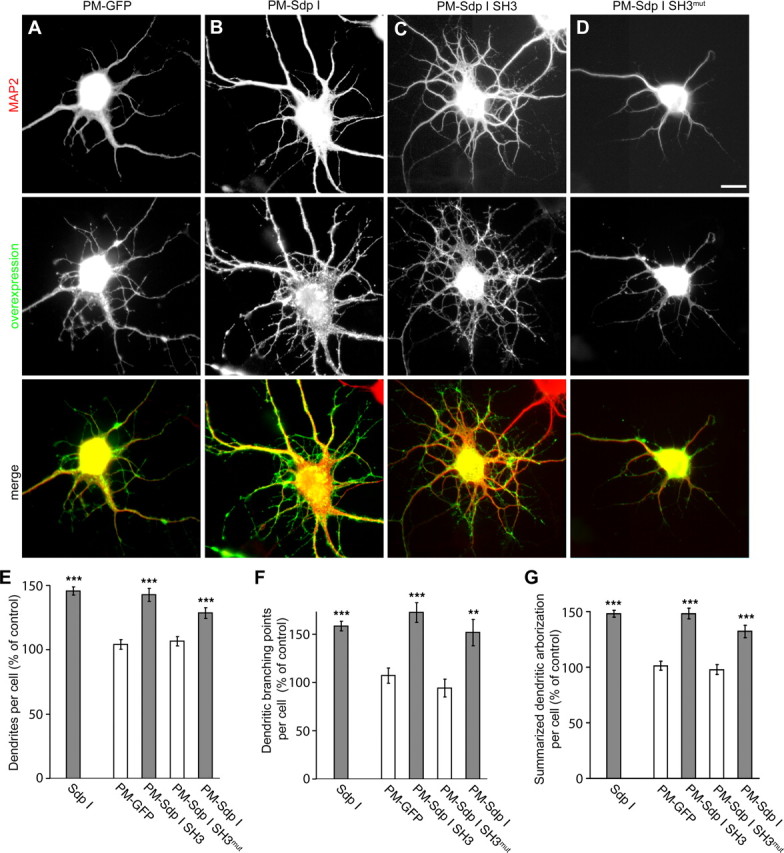
Syndapin I-induced dendrite formation and branching relies on plasma membrane targeting. Primary hippocampal neurons were transfected as indicated at DIV4 and processed for immunofluorescence microscopy 2 d later. Upper images show the anti-MAP2 staining used to evaluate neuronal morphology. Middle images show the overexpressed protein and lower images the merged images. The highly significant increase in dendrites and dendritic branching points in syndapin I-transfected neurons (compare with Fig. 1) can be mimicked by fusing a plasma membrane targeting sequence to either full-length syndapin I (B) or the isolated SH3 domain (C), whereas a plasma membrane-associated mutant version of the SH3 domain (P434L) (D) and PM-GFP (A) did not induce such effects. E–G, Quantitative examination of the effects of indicated constructs confirm these observations on changes in neuronal morphology. Morphological data are represented as mean ± SEM (**p < 0.01; ***p < 0.001). Scale bar, 10 μm.
Since silencing the syndapin I SH3 domain by introduction of a P434L mutation into the plasma membrane-targeted syndapin I abolished the phenotype induction completely (Fig. 7D–G), it can be concluded that a combination of efficient membrane targeting and SH3 domain-mediated N-WASP interaction is indispensable for the role of syndapin I in neuromorphogenesis control.
Syndapin expression increases during development
If syndapin I together with N-WASP indeed played a role in the development of the brain, it is required that both proteins are expressed during early development. Therefore, we subjected brain homogenates of embryonic and new-born rats to Western blot analyses and quantitatively determined the protein content with fluorescently labeled antibodies. At E18, the time point at which hippocampal primary cultures were prepared for our morphometric analyses, both N-WASP (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material) and syndapin I were detected (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material). Normalizing the sample loadings to tubulin showed that the expression of syndapin I strongly increases from E18 to P0 and P4, whereas the relative levels of N-WASP remained relatively constant (supplemental Fig. S3C, available at www.jneurosci.org as supplemental material). This suggests that the influence of the N-WASP activator syndapin I is steeply increasing from E18 to P4. Using purified recombinant protein as standards (data not shown), we were able to quantitatively determine the syndapin I protein content in brains of different developmental stages. The syndapin protein content increases from 1.2 ng of syndapin I per 50 μg of protein of brain homogenates at E18 to 3.4 ng of syndapin I per 50 μg of protein of brain homogenates of P4 animals (supplemental Fig. S3D, available at www.jneurosci.org as supplemental material).
It has to be noted, however, that syndapin I is the neurospecific isoform of the syndapin family (Qualmann et al., 1999; Qualmann and Kelly, 2000). The syndapin content in neurons may thus be as high or higher than the value obtained for the brain homogenates.
Syndapin I knockdown phenocopies effects of N-WASP deficiency on proper axon development
Syndapin I knockdown should unravel whether syndapin I is not only involved in but is also crucial for proper neuromorphogenesis. Furthermore, a comparison of phenotypes should highlight functional relations to N-WASP- and Arp2/3 complex-mediated actin nucleation. Different RNAi constructs were generated, analyzed in quantitative immunofluorescence experiments (supplemental Fig. S4, available at www.jneurosci.org as supplemental material) and were then used in morphometric analyses (Fig. 8). Since colocalization studies of overexpressed as well as endogenous syndapin I and N-WASP with the dendritic marker MAP2 suggested that both syndapin I and N-WASP are not restricted to the dendritic or axonal compartment (supplemental Fig. S5, available at www.jneurosci.org as supplemental material), we decided to carefully examine both compartments. Whereas the dendritic compartment of syndapin I RNAi-transfected neurons did not show any striking differences in dendrite number and dendritic branching compared with control cells (supplemental Fig. S6, available at www.jneurosci.org as supplemental material), the axonal compartment of developing neurons was strongly altered in syndapin I RNAi-transfected cells and showed excessive growth (Fig. 8C).
Figure 8.

Syndapin I knockdown results in increased axon length and branching. A–H, Fluorescence of GFP used as cell filler highlighting the morphology of primary hippocampal neurons transfected with the RNAi vector pRNAT (A), nonsilencing RNAi (B), with a pRNAT-driven syndapin I RNAi construct (sequence #2; C), cotransfected with syndapin I RNAi sequence #2 and an RNAi-insensitive syndapin I construct (Sdp Ires; D), N-WASP RNAi (E), Arp3 RNAi (F), N-WASP CA (G), and Abp1 RNAi (H), respectively. Cells were fixed and processed for immunofluorescence microscopy at DIV3. I–K, Quantitative evaluations based on MAP2 staining highlight that neurons expressing the pRNAT-driven syndapin I RNAi sequence #2 exhibit a statistically significant increase in the summarized length of the axonal arbor (I). Interestingly, this effect is not mainly reflecting an elongated axon length (J), as observed upon Abp1 RNAi, but a strongly increased axonal branching of syndapin I-deficient neurons (K). In contrast to the distinct syndapin and Abp1 loss-of-function defects, lack of functional N-WASP and Arp2/3 complex, respectively, consistently leads to a combination of both phenotypes. The various controls (pRNAT, nonsilencing RNAi, nonfunctional syndapin I RNAi sequence #3) and the rescue of the syndapin I RNAi effects with RNAi-insensitive syndapin I demonstrate the specificity of the syndapin RNAi effects. Morphological data were normalized to control (pRNAT) and are represented as mean (in percentage) ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001). Scale bar, 10 μm.
Quantitative analyses showed that the summarized length of the axonal arbor increased to 140 ± 11% of control in syndapin I-deficient neurons (Fig. 8I). This effect was specific, because cells transfected with the RNAi vector pRNAT, with nonsilencing RNAi or with syndapin I RNAi sequence #3, which did not result in a significant syndapin I knockdown (supplemental Fig. S4, available at www.jneurosci.org as supplemental material), all did not show any significant increase of axonal arborization (Fig. 8A,B,I). Furthermore, cotransfection of syndapin I RNAi-transfected cells with an RNAi-resistant syndapin I mutant largely abolished the phenotype (Fig. 8D). Quantitative examinations showed that, in such cells, neuronal morphology was almost restored to the wild-type situation (Fig. 8I).
Strikingly, the syndapin I loss-of-function phenotype observed mirrors the effects seen for N-WASP deficiency, Arp3 deficiency, and Arp2/3 complex inhibition by overexpression of the C terminus of N-WASP, which binds the Arp2/3 complex but fails to activate it (Strasser et al., 2004). All of these manipulations lead to excessive growth of the axonal compartment (Fig. 8E–G,I). Our data therefore demonstrate that syndapin I is crucial for proper neuromorphogenesis and suggest that it exerts its function in close collaboration with the Arp2/3 complex activator N-WASP.
Deficiency for the F-actin and N-WASP-binding protein Abp1 has also been demonstrated to mirror N-WASP and Arp2/3 complex deficiency as well as Arp2/3 complex inhibition, as it led to significant axon length increase (Pinyol et al., 2007)—one of the parameters changed upon N-WASP RNAi (Kakimoto et al., 2006). The other parameter changed upon N-WASP RNAi was axonal branching (Kakimoto et al., 2006). We therefore analyzed the changes of the axonal compartment in more detail. Interestingly, syndapin RNAi only led to some minor increase of axon length but strikingly and highly significantly increased axonal branching to 207 ± 11% of control (Fig. 8J,K). Again, this phenotype was specifically caused by syndapin I deficiency, as similar to the increased length of axonal arbor, also axonal branching of syndapin I RNAi-transfected neurons was restored to wild-type levels by coexpression of an RNAi-resistant syndapin I mutant (Fig. 8K).
Also later in development, syndapin I RNAi led to excessive axon branching. The effect was even more pronounced than that observed in early neuronal development and amounted to ∼300% of control (supplemental Fig. S7, available at www.jneurosci.org as supplemental material) compared with ∼200% upon transfections at DIV2 (Fig. 8K). Syndapin I functions are thus not restricted to early developmental processes but consistent with its rising expression during development (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) also play an important role in later phases of neuronal network formation.
Consistent with the effect of syndapin I loss of function, overexpression of syndapin I caused an opposite effect and decreased axonal branching below control to 60.5 ± 8.1% of control. The higher numbers of transfected cells obtainable in transfections after 4 d in culture allowed the syndapin I overexpression effect to become statistically significant despite the already low level of wild-type branching (supplemental Fig. S7, available at www.jneurosci.org as supplemental material).
In contrast to syndapin I RNAi, Abp1 RNAi did not show any statistically significant increase in axonal branching but exclusively affected axonal length, whereas Arp3 and N-WASP deficiency causes both phenotypes (Fig. 8H–K). These data show that the two N-WASP-binding and activating proteins syndapin I and Abp1 fine-tune N-WASP functions in neuromorphogenesis and have distinct physiological functions.
Discussion
The ability of the actin cytoskeleton to reorganize rapidly in response to external stimuli and internal cues is indispensable for changes in cell shape and motility and also for a variety of intracellular functions. Our data provide clear evidence that syndapins, members of the diverse F-BAR protein superfamily, are critically involved in Arp2/3 complex-mediated actin polymerization controlled by N-WASP. In vitro reconstitutions of actin polymerization with purified components revealed that syndapins activate the Arp2/3 complex via relieving the autoinhibition of its catalytic activator N-WASP.
The physiological importance of the ability of syndapins to interface with N-WASP-driven actin nucleation, which was demonstrated by our in vitro reconstitution and overexpression experiments, for neuronal morphogenesis is underscored by two findings. First, protein complexes of endogenous syndapin I and N-WASP were indeed detected in brain (Qualmann et al., 1999). Second, the increased axonal branching observed upon syndapin I RNAi is consistent with similar aberrant axon branching upon Arp3 and N-WASP knockdown. The comparative data we collected for N-WASP RNAi are hereby in line with a previous report (Kakimoto et al., 2006). Also in the study of Kakimoto et al. (2006), N-WASP deficiency was found to impair proper axon development. Our loss-of-function data show that likewise syndapins play a crucial role in axon development. This finding is well in line with our findings that syndapin binds to N-WASP, releases its autoinhibition, and controls the activity of the Arp2/3 complex at the cell cortex.
In apparent contrast to our findings, a recent study in Drosophila did not report any obvious N-WASP/Arp2/3 complex-related developmental defects in syndapin knock-out animals (Kumar et al., 2009). It has to be noted, however, that the single fly syndapin resembles more the muscle-enriched syndapin III isoform in mammals. Consistently, fly syndapin was exclusively found on the muscle side of neuromuscular junctions (Kumar et al., 2009). In contrast, mammalian syndapin I is almost exclusively expressed in the brain (Qualmann et al., 1999). It may thus specifically support functions of more complex vertebrate brains.
The observation that syndapin I-induced morphology changes critically depend on N-WASP demonstrates that—in clear analogy to in vitro reconstitutions—syndapins act upstream of N-WASP in vivo. Consistently, in addition to similar defects in neuromorphogenesis in loss-of-function experiments, also the syndapin I overexpression phenotype mirrors that of N-WASP overexpression, excessive neurite induction and arborization (Pinyol et al., 2007). The observed phenotypes for syndapin I and N-WASP overexpression are furthermore consistent with the increased neuritogenesis and branching observed upon reduction of PICK1-mediated Arp2/3 complex inhibition (Rocca et al., 2008). Increased N-WASP levels detected in Alzheimer's disease brains were suggested to relate to the aberrant neuronal sprouting observed as a pathological characteristic of Alzheimer's disease (Kitamura et al., 2003).
Strikingly, both syndapin I (this study) and N-WASP gain-of-function phenotypes (Pinyol et al., 2007) depend on Cdc42, which we found to cooperate with syndapins in N-WASP activation. Comparisons with the activation brought about by saturation concentrations of established N-WASP activators, such as Cdc42 and PIP2 (Rohatgi et al., 2000), show that syndapin-induced N-WASP activation is a powerful mechanism for autoinhibition release. The in vitro reconstitutions also demonstrated that syndapin-mediated N-WASP activation does not compete with the activating mechanisms of Cdc42 and PIP2 but that syndapins work together with these N-WASP activators and give rise to additive effects. This shows that N-WASP integrates several molecular stimuli leading to Arp2/3 complex-mediated actin nucleation. The cooperative action of several components upstream of N-WASP may provide a molecular solution to the need for spatial and temporal fine control of N-WASP function during endocytosis, neuronal development, and neuronal network formation—cellular functions impaired by N-WASP loss of function (Kessels and Qualmann, 2002; Benesch et al., 2005; Kakimoto et al., 2006; Pinyol et al., 2007; Wegner et al., 2008).
In vitro, the N-WASP-binding syndapin SH3 domain was both critical and sufficient for N-WASP/Arp2/3 complex-mediated actin polymerization. In vivo, however, when actin nucleation was analyzed via the resulting morphology changes and thus specifically needed to occur at the cell cortex, the eF-BAR domain was required in addition. Reconstitution experiments showed that the eF-BAR domain associates with PS-containing liposomes. In vivo, it was possible to functionally replace the eF-BAR by a palmitoylation consensus sequence, which effectively and constitutively assures plasma membrane anchoring. These data clearly show not only that the syndapin eF-BAR domain is a dimerization motif (Kessels and Qualmann, 2006) but also that it is important for membrane association and for syndapin function in cell morphology control in vivo.
Our biochemical examinations and our functional studies in neurons suggest that the molecular mechanisms by which syndapins control the functions of their downstream effector N-WASP are (1) release of N-WASP autoinhibition upon SH3 domain binding and (2) mediation of membrane association of syndapin–N-WASP complexes. That syndapin–N-WASP interactions are strong enough to recruit N-WASP to syndapin-enriched membranes in vivo has been demonstrated (Kessels and Qualmann, 2002). Biochemical fractionations show that N-WASP is mostly found in the cytosolic fraction and that membrane-associated N-WASP accounts only for some percentage of the intracellular pool (Kessels and Qualmann, 2002). Nevertheless, recruitment of N-WASP to membranes seems to be of utmost importance in N-WASP/Arp2/3 complex-dependent actin filament formation. Overexpression of the C terminus of N-WASP, which strongly activates the Arp2/3 complex but lacks the more N-terminal binding sites for proteins allowing for membrane recruitment, such as syndapin, severely inhibits cellular processes that depend on cortical, Arp2/3 complex-mediated actin nucleation, such as lamellipodia formation (Machesky and Insall, 1998; Rogers et al., 2003; Steffen et al., 2006), synaptogenesis (Wegner et al., 2008), and proper axon development (Kakimoto et al., 2006; Pinyol et al., 2007). For all of these processes, N-WASP and the Arp2/3 complex need to be recruited to and activated at the plasma membrane. Syndapins can promote association with the cell cortex by their preferential association with PS, as this lipid is mainly enriched in the inner leaflet of the plasma membrane (Yamaji-Hasegawa and Tsujimoto, 2006). Therefore, PS and PIP2 together may represent important spatial cues for N-WASP/Arp2/3 functions in neuromorphogenesis. Our detailed morphometric studies of the axonal defects caused by syndapin I, N-WASP, and Arp3 knockdown suggest that syndapin/N-WASP/Arp2/3 complex-mediated cortical actin nucleation may serve to ensure the establishment and maintenance of cortical F-actin structures, which prevent aberrant branching of axons by the action of Arp2/3 complex-unrelated actin nucleation-promoting factors, such as formins or the newly discovered WH2 domain-containing nucleators (Pollard, 2007; Qualmann and Kessels, 2009). Since on the other side overexpression of syndapin I suppresses branching of the axon but promotes growth of the dendritic arbor by cortical Arp2/3 complex activation, it seems that dendritic and axonal actin organization is mediated by very distinct mechanisms.
In addition to syndapin I, also the F-actin-binding protein Abp1 (Kessels et al., 2000) cooperates with N-WASP in morphogenesis of hippocampal neurons (Pinyol et al., 2007; Haeckel et al., 2008). Similar to syndapins, Abp1 activates N-WASP in concert with Cdc42 in vitro and in vivo. Both Abp1 and syndapin interact via their C-terminal SH3 domain with the central N-WASP PRD (Kessels and Qualmann, 2002; Pinyol et al., 2007) and may thus form distinct N-WASP protein complexes. We found that both syndapin I and Abp1 are relatively abundant in brains of developing rats and that the absolute amounts of both N-WASP-binding proteins are in the same order of magnitude (compare supplemental Figs. S3, S8, available at www.jneurosci.org as supplemental material). Interestingly, Abp1 loss of function leads to excessive axon growth, a phenotype also observed upon N-WASP deficiency (Pinyol et al., 2007) but only weakly observed upon syndapin I deficiency. On the other hand the axon branching phenotype observed upon N-WASP deficiency that syndapin I deficiency mirrors very well is very weak upon Abp1 deficiency. Thus, syndapin and Abp1 have tight functional relationships with N-WASP but are very specific modulators of N-WASP functions in the two different aspects of axon development, which are dependent on N-WASP/Arp2/3 complex-mediated actin nucleation, i.e., control of axonal branching and length.
The close physiological relationship with N-WASP in neuromorphogenesis significantly distinguishes Abp1 and syndapin I from proteins suggested to have syndapin-related functions, as they also bind to lipids and to N-WASP, such as FBP17/Toca-1 (Itoh et al., 2005; Tsujita et al., 2006). FBP17/Toca-1 has been shown to interact with N-WASP and to elicit N-WASP autoinhibition release in vitro (Ho et al., 2004). Surprisingly, however, knockdown of FBP17/Toca-1 failed to phenocopy N-WASP deficiency in hippocampal neurons (Kakimoto et al., 2006). Instead, recent reports revealed that FBP17/Toca-1 is required in N-WASP-induced filopodia formation in N1E115 neuroblastoma cells (Bu et al., 2009) and for Shigella-induced, N-WASP-dependent actin filament formation in HeLa cells (Leung et al., 2008). It therefore seems that FBP17/Toca-1-N-WASP, Abp1-N-WASP, and syndapin I-N-WASP complexes—albeit all mediated by SH3 domain interactions with the central PRD of N-WASP—have very distinct and specific functions in vivo.
Together, our results experimentally demonstrate that syndapins act as important integration platforms, which allow cells to shape membranes in a highly controlled spatial and temporal manner. Syndapins execute their functions in neuromorphogenesis by associating with the plasma membrane and by triggering activity of their downstream effectors N-WASP and Arp2/3 complex at the cell cortex—functions crucial for proper axonal development.
Footnotes
This work was supported by grants from the Deutsche Forschungsgemeinschaft to B.Q., to M.M.K., and to both (Qu116/2-1, 2-3, 4-1, 5-1; KE685/2-2, 2-3; and Qu116/3-1, 3-2, respectively), as well as by a grant from the Schram Foundation (T287/16245/2006). We thank Kathrin Hartung, Sabine Opitz, Annett Kreusch, Dirk Schlobinski, Anett Ritter, and Katja Hille for their assistance and Alexandre Benmerah for the Eps15 Δaa95-295-expressing plasmid.
References
- Ahuja R, Pinyol R, Reichenbach N, Custer L, Klingensmith J, Kessels MM, Qualmann B. Cordon-bleu is an actin nucleation factor and controls neuronal morphology. Cell. 2007;131:337–350. doi: 10.1016/j.cell.2007.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen WE, Jones GE, Pollard JW, Ridley AJ. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. J Cell Sci. 1997;110:707–720. doi: 10.1242/jcs.110.6.707. [DOI] [PubMed] [Google Scholar]
- Andersson F, Jakobsson J, Löw P, Shupliakov O, Brodin L. Perturbation of syndapin/PACSIN impairs synaptic vesicle recycling evoked by intense stimulation. J Neurosci. 2008;28:3925–3933. doi: 10.1523/JNEUROSCI.1754-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspenström P, Fransson A, Richnau N. Pombe Cdc15 homology proteins: regulators of membrane dynamics and the actin cytoskeleton. Trends Biochem Sci. 2006;31:670–679. doi: 10.1016/j.tibs.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Benesch S, Polo S, Lai FP, Anderson KI, Stradal TE, Wehland J, Rottner K. N-WASP deficiency impairs EGF internalization and actin assembly at clathrin-coated pits. J Cell Sci. 2005;118:3103–3115. doi: 10.1242/jcs.02444. [DOI] [PubMed] [Google Scholar]
- Benmerah A, Bayrou M, Cerf-Bensussan N, Dautry-Varsat A. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J Cell Sci. 1999;112:1303–1311. doi: 10.1242/jcs.112.9.1303. [DOI] [PubMed] [Google Scholar]
- Bigay J, Casella JF, Drin G, Mesmin B, Antonny B. ArfGAP1 responds to membrane curvature through the folding of a lipid packing sensor motif. EMBO J. 2005;24:2244–2253. doi: 10.1038/sj.emboj.7600714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun A, Pinyol R, Dahlhaus R, Koch D, Fonarev P, Grant BD, Kessels MM, Qualmann B. EHD proteins associate with syndapin I and II and such interactions play a crucial role in endosomal recycling. Mol Biol Cell. 2005;16:3642–3658. doi: 10.1091/mbc.E05-01-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu W, Chou AM, Lim KB, Sudhaharan T, Ahmed S. The Toca-1-N-WASP complex links filopodia formation to endocytosis. J Biol Chem. 2009;284:11622–11636. doi: 10.1074/jbc.M805940200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Stanley ER. Pombe Cdc15 homology (PCH) proteins: coordinators of membrane-cytoskeletal interactions. Trends Cell Biol. 2007;17:145–156. doi: 10.1016/j.tcb.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Daumke O, Lundmark R, Vallis Y, Martens S, Butler PJ, McMahon HT. Architectural and mechanistic insights into an EHD ATPase involved in membrane remodelling. Nature. 2007;449:923–927. doi: 10.1038/nature06173. [DOI] [PubMed] [Google Scholar]
- Dawson JC, Legg JA, Machesky LM. BAR domain proteins: a role in tubulation, scission and actin assembly in clathrin-mediated endocytosis. Trends Cell Biol. 2006;16:493–498. doi: 10.1016/j.tcb.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Haeckel A, Ahuja R, Gundelfinger ED, Qualmann B, Kessels MM. The actin-binding protein Abp1 controls dendritic spine morphology and is important for spine head and synapse formation. J Neurosci. 2008;28:10031–10044. doi: 10.1523/JNEUROSCI.0336-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho HY, Rohatgi R, Lebensohn AM, Ma L, Li J, Gygi SP, Kirschner MW. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell. 2004;118:203–216. doi: 10.1016/j.cell.2004.06.027. [DOI] [PubMed] [Google Scholar]
- Itoh T, Erdmann KS, Roux A, Habermann B, Werner H, De Camilli P. Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev Cell. 2005;9:791–804. doi: 10.1016/j.devcel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Kakimoto T, Katoh H, Negishi M. Regulation of neuronal morphology by Toca-1, an F-BAR/EFC protein that induces plasma membrane invagination. J Biol Chem. 2006;281:29042–29053. doi: 10.1074/jbc.M604025200. [DOI] [PubMed] [Google Scholar]
- Kessels MM, Qualmann B. Syndapins integrate N-WASP in receptor-mediated endocytosis. EMBO J. 2002;21:6083–6094. doi: 10.1093/emboj/cdf604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels MM, Qualmann B. Syndapin oligomers interconnect the machineries for endocytic vesicle formation and actin polymerization. J Biol Chem. 2006;281:13285–13299. doi: 10.1074/jbc.M510226200. [DOI] [PubMed] [Google Scholar]
- Kessels MM, Engqvist-Goldstein AE, Drubin DG. Association of mouse actin-binding protein 1 (mAbp1/SH3P7), an Src kinase target, with dynamic regions of the cortical actin cytoskeleton in response to Rac1 activation. Mol Biol Cell. 2000;11:393–412. doi: 10.1091/mbc.11.1.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Tsuchiya D, Takata K, Shibagaki K, Taniguchi T, Smith MA, Perry G, Miki H, Takenawa T, Shimohama S. Possible involvement of Wiskott-Aldrich-Syndrome protein family in aberrant neuronal sprouting in Alzheimer's disease. Neurosci Lett. 2003;346:149–152. doi: 10.1016/s0304-3940(03)00506-8. [DOI] [PubMed] [Google Scholar]
- Kretzschmar E, Bui M, Rose JK. Membrane association of influenza virus matrix protein does not require specific hydrophobic domains or the viral glycoproteins. Virology. 1996;220:37–45. doi: 10.1006/viro.1996.0283. [DOI] [PubMed] [Google Scholar]
- Kumar V, Alla SR, Krishnan KS, Ramaswami M. Syndapin is dispensable for synaptic vesicle endocytosis at the Drosophila larval neuromuscular junction. Mol Cell Neurosci. 2009;40:234–241. doi: 10.1016/j.mcn.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung Y, Ally S, Goldberg MB. Bacterial actin assembly requires Toca-1 to relieve N-WASP autoinhibition. Cell Host Microbe. 2008;3:39–47. doi: 10.1016/j.chom.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SX, Grant B, Hirsh D, Maxfield FR. Rme-1 regulates the distribution and function of the endocytic recycling compartment in mammalian cells. Nat Cell Biol. 2001;3:567–572. doi: 10.1038/35078543. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Insall RH. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr Biol. 1998;8:1347–1356. doi: 10.1016/s0960-9822(98)00015-3. [DOI] [PubMed] [Google Scholar]
- Modregger J, Ritter B, Witter B, Paulsson M, Plomann M. All three PACSIN isoforms bind to endocytic proteins and inhibit endocytosis. J Cell Sci. 2000;113:4511–4521. doi: 10.1242/jcs.113.24.4511. [DOI] [PubMed] [Google Scholar]
- Parsons M, Monypenny J, Ameer-Beg SM, Millard TH, Machesky LM, Peter M, Keppler MD, Schiavo G, Watson R, Chernoff J, Zicha D, Vojnovic B, Ng T. Spatially distinct binding of Cdc42 to PAK1 and N-WASP in breast carcinoma cells. Mol Cell Biol. 2005;25:1680–1695. doi: 10.1128/MCB.25.5.1680-1695.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJG, Evans PR, McMahon HT. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- Pinyol R, Haeckel A, Ritter A, Qualmann B, Kessels MM. Regulation of N-WASP and the Arp2/3 complex by Abp1 controls neuronal morphology. PLoS One. 2007;2:e400. doi: 10.1371/journal.pone.0000400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu Rev Biophys Biomol Struct. 2007;36:451–477. doi: 10.1146/annurev.biophys.35.040405.101936. [DOI] [PubMed] [Google Scholar]
- Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules. Nat Rev Mol Cell Biol. 2004;5:133–147. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- Qualmann B, Kelly RB. Syndapin isoforms participate in receptor-mediated endocytosis and actin organization. J Cell Biol. 2000;148:1047–1062. doi: 10.1083/jcb.148.5.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qualmann B, Kessels MM. New players in actin polymerization—WH2 domain-containing actin nucleators. Trends Cell Biol. 2009;19:276–285. doi: 10.1016/j.tcb.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Qualmann B, Roos J, DiGregorio PJ, Kelly RB. Syndapin I, a synaptic dynamin-binding protein that associates with the neural Wiskott-Aldrich syndrome protein. Mol Biol Cell. 1999;10:501–513. doi: 10.1091/mbc.10.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca DL, Martin S, Jenkins EL, Hanley JG. Inhibition of Arp2/3-mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nat Cell Biol. 2008;10:259–271. doi: 10.1038/ncb1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SL, Wiedemann U, Stuurman N, Vale RD. Molecular requirements for actin-based lamella formation in Drosophila S2 cells. J Cell Biol. 2003;162:1079–1088. doi: 10.1083/jcb.200303023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Ho HY, Kirschner MW. Mechanism of N-WASP activation by Cdc42 and phosphatidylinositol 4, 5-bisphosphate. J Cell Biol. 2000;150:1299–1310. doi: 10.1083/jcb.150.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen A, Faix J, Resch GP, Linkner J, Wehland J, Small JV, Rottner K, Stradal TE. Filopodia formation in the absence of functional WAVE- and Arp2/3-complexes. Mol Biol Cell. 2006;17:2581–2591. doi: 10.1091/mbc.E05-11-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser GA, Rahim NA, VanderWaal KE, Gertler FB, Lanier LM. Arp2/3 is a negative regulator of growth cone translocation. Neuron. 2004;43:81–94. doi: 10.1016/j.neuron.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007;8:37–48. doi: 10.1038/nrm2069. [DOI] [PubMed] [Google Scholar]
- Tsujita K, Suetsugu S, Sasaki N, Furutani M, Oikawa T, Takenawa T. Coordination between the actin cytoskeleton and membrane deformation by a novel membrane tabulation domain of PCH proteins is involved in endocytosis. J Cell Biol. 2006;172:269–279. doi: 10.1083/jcb.200508091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Navarro MV, Peng G, Molinelli E, Lin Goh S, Judson BL, Rajashankar KR, Sondermann H. Molecular mechanism of membrane constriction and tubulation mediated by the F-BAR protein Pacsin/Syndapin. Proc Natl Acad Sci U S A. 2009;106:12700–12705. doi: 10.1073/pnas.0902974106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner AM, Nebhan CA, Hu L, Majumdar D, Meier KM, Weaver AM, Webb DJ. N-WASP and the Arp2/3 complex are critical regulators of actin in the development of dendritic spines and synapses. J Biol Chem. 2008;283:15912–15920. doi: 10.1074/jbc.M801555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigge P, Vallis Y, McMahon HT. Inhibition of receptor-mediated endocytosis by the amphiphysin SH3 domain. Curr Biol. 1997;7:554–560. doi: 10.1016/s0960-9822(06)00254-5. [DOI] [PubMed] [Google Scholar]
- Witte H, Bradke F. The role of the cytoskeleton during neuronal polarization. Curr Opin Neurobiol. 2008;18:479–487. doi: 10.1016/j.conb.2008.09.019. [DOI] [PubMed] [Google Scholar]
- Yamaji-Hasegawa A, Tsujimoto M. Asymmetric distribution of phospholipids in biomembranes. Biol Pharm Bull. 2006;29:1547–1553. doi: 10.1248/bpb.29.1547. [DOI] [PubMed] [Google Scholar]