

Abstract
In cases of neuronal injury when regeneration is restricted, functional recovery can occur through reorganization of the remaining neural circuitry. We found an example of such recovery in the central pattern generator (CPG) for the escape swim of the mollusc Tritonia diomedea. The CPG neurons are bilaterally represented and each neuron projects an axon through one of two pedal commissures. Cutting the posterior pedal commissure [pedal nerve 6 (PdN6)] in the animal or in the isolated brain caused a deficit in the swim behavior and in the fictive motor pattern, respectively, each of which recovered over the course of 20 h. Locally blocking spiking activity in PdN6 with sodium-free saline and/or tetrodotoxin disrupted the motor pattern in a reversible manner. Maintained blockade of PdN6 led to a functional recovery of the swim motor pattern similar to that observed in response to cutting the commissure. Among the CPG neurons, cerebral neuron 2 (C2) makes functional connection onto the ventral swim interneuron-B (VSI) in both pedal ganglia. Cutting or blocking PdN6 eliminated C2-evoked excitation of VSI in the pedal ganglion distal to the lesion. Associated with the recovery of the swim motor pattern, the synaptic action of C2 onto VSI in the proximal pedal ganglion changed from being predominantly inhibitory to being predominantly excitatory. These results show that the Tritonia swim CPG undergoes adaptive plasticity in response to the loss of critical synaptic connections; reversal of synaptic action in the CPG may be at least partially responsible for this functional recovery.
Introduction
Neuronal injury can result in partial or total loss of function because of the interruption of axonal conduction and consequent loss of synaptic communication. Such loss of function can be compensated by adaptive plasticity of the nervous system; however, little is known about the mechanisms of reorganization at a cellular level and their relationships to functional recovery. Here, we document an example of natural recovery of function that does not involve regeneration after a lesion.
Spinal or cortical injury can cause irreversible loss of movement control (Rossignol, 2000; Fouad and Pearson, 2004; Nudo, 2006). In some cases, recovery of function occurs because of regeneration of severed axons across the lesion or reinnervation by collateral axons (Cohen et al., 1986; Commissiong et al., 1991; Ide, 1996; Wieloch and Nikolich, 2006; Navarro et al., 2007). If such reconnection or reinnervation is physically impossible because of a large gap or other barriers, then the loss of function can be compensated by reorganization of the remaining neural circuitry (Barbeau et al., 1999; Antri et al., 2002; Navarro et al., 2007; Cramer, 2008). Several studies have shown that the plasticity of dendrites is enhanced near the damaged region and also in distant regions after cortical or spinal injury (Cai et al., 2006; Nudo, 2006; Rossignol, 2006). However, the cellular mechanisms underlying recovery of function in mammalian nervous systems are still speculative.
To understand the mechanisms underlying the functional recovery of a neuronal circuit, it is useful to examine a system in which the neurons in the circuit are easily identified and accessible before, during, and after a lesion. The nervous systems of gastropod molluscs fulfill these criteria; they have large identifiable neurons grouped into ganglia that are interconnected by commissural and connective nerves. In the present study, we examined the effect of a disruptive lesion of a well characterized central pattern generator (CPG) for the escape swim of a nudibranch, Tritonia diomedea.
The escape swim behavior of Tritonia consists of a series of whole-body flexions in response to a particular sensory stimulus (Getting, 1989; Katz, 2009). This fictive motor pattern can be elicited in isolated brain preparations by electrically stimulating a body wall nerve. The swim CPG consists of three uniquely identifiable neuronal types, each of which is bilaterally represented (Fig. 1 A,B): ventral swim interneuron-B (VSI-B) (http://neuronbank.org/Tri0002436), cerebral neuron 2 (C2) (http://neuronbank.org/Tri0002380), and three serotonergic dorsal swim interneurons (DSIs) (http://neuronbank.org/Tri0001043) (Getting et al., 1980; Getting, 1981, 1983, 1989). Their large somata (50–80 μm) permit stable intracellular microelectrode recordings to be maintained for several hours. This allows us to record the synaptic strength of identified synapses within a defined neural circuit and observe changes over time during the recovery from a lesion.
Figure 1.

The Tritonia swim network and swim motor program. A, A schematic diagram of the neural circuit underlying the swim motor pattern. All neurons have a contralateral counterpart that is not represented. The shaded circle indicates the neurons that comprise the swim CPG. The filled triangles represent excitatory synapses, and the filled circles represent inhibitory synapses. Combinations of triangle and circle are multicomponent synapses. The dotted lines indicate that the connection is either polysynaptic or not determined. C2, Cerebral cell 2; DFN-A,B, dorsal flexion neurons; DRI, dorsal ramp interneuron; DSI, dorsal swim interneuron; Tr1, trigger neuron 1; VFN, ventral flexion neuron; VSI, ventral swim interneuron [based on Getting et al. (1980), Getting (1981), Hume and Getting (1982), Frost and Katz (1996), and Frost et al. (2001)]. B, Schematic drawing of a dorsal view of the Tritonia brain, showing the location of the swim CPG neurons and their axonal projections. DSI and C2 are located on the dorsal surface of the cerebral ganglion. VSI is located on the ventral side of the pleural ganglion. C2 and VSI project their axons through PdN6, whereas DSI projects through PdN5. The dotted lines indicate PdN5 was removed on isolation of the brain. C, An example of the swim motor pattern. Simultaneous intracellular recordings from three CPG neurons, C2, DSI, and VSI, and extracellular recording from PdN6 are shown. The bursting pattern was elicited by stimulation of the left pedal nerve 3 by applying voltages pulses (8 V, 1 ms) at 5 Hz for 3 s (starting at arrow).
Here, we found that severing a set of connections between some CPG neurons impaired motor pattern production but that the system spontaneously recovered over the course of a few hours to a day. Furthermore, we observed corresponding changes in synaptic strength that can account for the functional recovery.
Materials and Methods
Animal surgery.
Specimens of the nudibranch, Tritonia diomedea, were obtained from Living Elements Ltd. and Friday Harbor Laboratories. For behavioral tests of lesions, animals were placed for 1 h in ice-chilled artificial sea water (Instant Ocean Sea Salt) containing 0.1% 1-phynoxy-2-propanol (Redondo and Murray, 2005; Wyeth et al., 2009). Then, a 2 cm incision was made in the skin between the rhinophores to expose the brain (see Fig. 4 A). In the experimental animals, both pedal–pedal commissures, which are also known as pedal nerve 5 and 6 (PdN5, PdN6) (Willows et al., 1973), were cut near the right pedal ganglion with fine scissors, whereas in the sham controls, the commissures were exposed but not cut. The incision was stitched with silk thread and sealed with cyanoacrylate glue (ethyl cyanoacrylate; WPI).
Figure 4.

Cutting PdN6 in the animal disrupted swimming. A, The animal was anesthetized and the body wall above the buccal mass was cut open. PdN6 was transected near the right pedal ganglion with scissors. Nerve-transected animals were paired with sham-operated animals. B, Fifty percent of nerve-transected animals often failed to swim after the surgery (black triangles). In contrast, all sham-operated animals swim (white circles). C, Cutting PdN6 reduced the number of body flexions during the swim behavior. Graph showing the average number of body flexions during the escape swim behavior for animals with PdN6 transected (black triangles) and sham-operated controls (white circles). Data for B and C were obtained from six animals. Error bars indicate SEM. Asterisks indicate significant difference (see Results).
Animals with lesions were paired with sham-operated animals for the behavioral assay. The observer was blind to the condition of each animal in a trial. The swim was induced by applying 5 m NaCl solution (0.5 ml) to the dorsal body surface, and the response was recorded with a video camera. The number of complete ventrally directed body flexions was counted as the number of swim cycles. A retraction of the body without a body flexion in response to the stimulus was considered as a swim failure. The swim tests were performed three times before the surgery (20, 2.5, and 1 h before the surgery). After the surgery, the swim tests were performed three times on the same day (2, 3.5, and 5 h after the surgery) and once the next day (20 h after the surgery).
Isolated brain preparations.
The brain, consisting of the fused cerebropleural and pedal ganglia, was removed from the animal and immediately pinned to the bottom of a Sylgard-lined chamber (1 ml) in which it was superfused with saline at 4°C. Physiological saline composition was as follows (in mm): 420 NaCl, 10 KCl, 10 CaCl2, 50 MgCl2, 10 d-glucose, and 10 HEPES, pH 7.4. The cell bodies of the neurons were exposed by removing the connective tissue sheath from the surface of the ganglia (Willows et al., 1973). Suction electrodes made from polyethylene tubing were placed on both left and right pedal nerves 3 (PdN3). A suction electrode made from a borosilicate glass tube (inner diameter, 0.75 mm; outer diameter, 1.50 mm) was placed in the en passant configuration on PdN6. In this study, the other pedal–pedal commissure, pedal nerve 5 (PdN5) (Willows et al., 1973), was cut in all isolated brain preparations during dissection as indicated by the dotted lines in Figure 1 B; cutting it after the dissection had no apparent effect on the swim motor pattern (N = 6). The preparation was left >3 h superfused in saline at 8–10°C to recover from possible damage during the dissection.
Neurons were identified by soma location, axonal projection (Fig. 1 B), coloration, synaptic connectivity, and activity pattern at rest and during the swim motor program as previously described (Getting, 1981, 1983). There are three types of CPG neurons: DSIs, C2, and VSI-B. For simplicity, we will refer to VSI-B as VSI for the remainder of this paper. C2 and DSI have cell bodies in the cerebral ganglion and project the axons toward the contralateral pedal ganglion, whereas VSI has its cell body in the pleural ganglion and projects its axon toward the ipsilateral pedal ganglion. The axons of C2, DSI, and VSI then exit the pedal ganglion through one of two commissural nerves (PdN5 and PdN6) and reach the other pedal ganglion (Newcomb et al., 2006; Hill and Katz, 2007). To identify neurons, the swim motor program was evoked by stimulating body wall nerve PdN3 with a train of voltage pulses (5–15 V, 1 ms) at 5 Hz for 3 s via a suction electrode. Unilateral electrical stimulation of PdN3 is sufficient to elicit the motor pattern (Fig. 1 C). Even after cutting two commissural nerves (PdN5 and PdN6), each bilateral pair of CPG neurons always burst synchronously presumably because of electrical coupling among C2s and DSIs through the cerebral commissure (Getting et al., 1980) and because of the bilateral projections of the gating interneuron, dorsal ramp interneuron (DRI) (Frost and Katz, 1996). Swim stimuli were given at intervals of >10 min.
In some experiments, the bathing medium was switched to saline containing a high concentration of divalent cations (Hi-Di saline), which raises the threshold for spiking and reduces spontaneous neural firing. The composition of the Hi-Di saline was as follows (in mm): 285 NaCl, 10 KCl, 25 CaCl2, 125 MgCl2, 10 d-glucose, and 10 HEPES, pH 7.4 (Sakurai and Katz, 2003). For all experiments conducted in the Hi-Di saline, the ganglia were superfused at 2 ml/min at 10°C.
Electrophysiological recordings and stimulations.
Neurons were impaled with glass microelectrodes filled with 3 m KCl (resistance, 8–15 MΩ). Axoclamp-2B amplifiers (Molecular Devices) were used for all electrophysiological experiments. Recordings were digitized at 3–20 kHz with a 1401plus A/D converter from Cambridge Electronic Design (CED). Data acquisition and analysis were performed with Spike2 software (CED) and SigmaPlot (SPSS Inc.). Statistical comparisons were made using Student's t test, paired t test, or a one-way or two-way repeated-measures ANOVA with post hoc pairwise multiple comparison by Fisher's least significant difference (LSD) method. In all cases, p < 0.05 was considered significant. Results are expressed as the mean ± SEM.
Tracer injection.
To determine its axonal projection pattern, VSI was filled with Neurobiotin tracer [N-(2-amino-ethyl)biotinamide hydrochloride; Vector Laboratories]. A microelectrode filled with 4% Neurobiotin solution (in 0.75 m KCl, 0.05 m HEPES, pH 7.4) was inserted into the cell body, and electrical current pulses (positive square pulses of 1–10 nA at 50% duty cycle) were applied at 1 Hz for >30 min. The preparation was then incubated in running physiological saline for 6 h at 8–10°C. After incubation, the brain was fixed in 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 m PBS, pH 7.4, for 5 h at 4°C. After rinsing with PBS several times, the brain treated with 4.0% Triton X-100 in PBS overnight, and then incubated with streptavidin, Alexa Fluor 594 conjugate (Invitrogen) diluted to 1:200 by PBS containing 0.5% Triton X-100 for 5 d at 4°C. The brain was rinsed in distilled water, dehydrated in a graded ethanol series, and mounted on a slide glass with Cytoseal 60 (Electron Microscopy Sciences) for confocal laser-scanning microscopy (LSM 510 mounted on an Axiovert 100 M; Carl Zeiss).
Results
Functional CPG connections are mediated by axons in the pedal–pedal connective
Anatomical and electrophysiological evidence suggests that the axons traveling through PdN6 mediate functional connections between CPG neurons. Dye fills of VSI (N = 6) revealed extensive arborizations in the pleural ganglion and pedal ganglion ipsilateral to its soma. The axon of VSI extended through PdN6 and some branches were observed in the contralateral pedal ganglion (Fig. 2 A).
Figure 2.
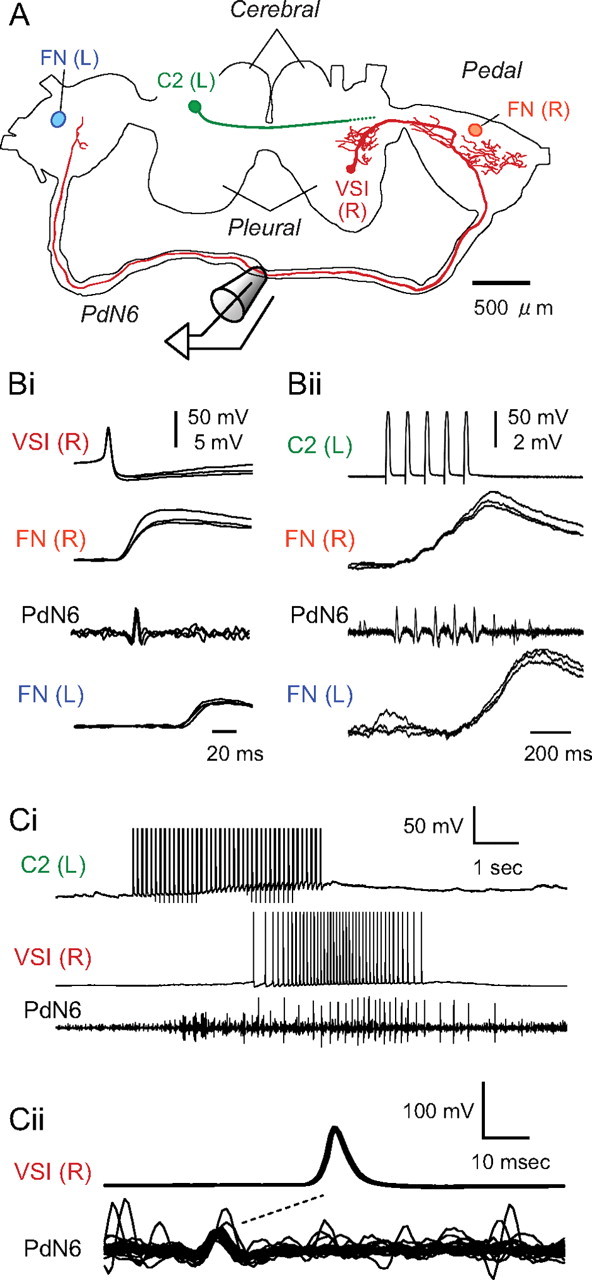
VSI and C2 made synaptic connections in both pedal ganglia. A, A drawing made from confocal images of VSI (red) labeled by Neurobiotin tracer (Vector Laboratories). Locations of C2 (green) and the follower neurons (FNs) (orange and blue) in the pedal ganglia are also shown. The VSI axon projects ipsilaterally through the posterior portion of the cerebral–pedal connective and then continues through the pedal–pedal connective (PdN6). The axon of C2 projects contralaterally toward the pedal ganglion as previously published (Getting et al., 1980). Bi, The VSI action potentials were recorded in PdN6 and evoked one-for-one EPSPs in FNs in the right and left pedal ganglia. Simultaneous intracellular recordings from right VSI, right VFN, and left VFN and extracellular recording from PdN6 are shown. Each sweep was triggered by a VSI action potential; three sweeps were superimposed. Bii, C2 spikes were recorded in PdN6 and C2 evoked summated EPSPs in FNs of both pedal ganglia. Overlaid traces were triggered by the first stimulus current pulse (7 nA, 20 ms) injected into C2 to evoke a train of action potentials (10 Hz). The EPSPs facilitated greatly and had a slow time course, making it difficult to distinguish individual EPSPs. Ci, C2 caused delayed excitation of VSI. Simultaneous intracellular recordings from left C2 and right VSI and extracellular en passant recording from PdN6 are shown. C2 was stimulated by injecting repetitive square current pulses (7 nA, 20 ms) at 10 Hz for 4 s, which induced a burst of action potentials in VSI. Cii, The C2-evoked action potentials in VSI were antidromic. Overlaid traces of VSI action potentials and corresponding PdN6 impulses evoked by C2 are shown. The traces were triggered by the peak of VSI action potentials. The nerve impulse preceded the somatic action potential in each trace, suggesting that these action potentials propagated antidromically from the distal pedal ganglion.
The left and right VSIs were electrically coupled. Injection of hyperpolarizing current pulses into one VSI produced a small hyperpolarization in its contralateral counterpart (data not shown). The ratio of the membrane potential changes (i.e., the coupling coefficient) was 0.038 ± 0.005 (N = 7). Cutting PdN6 eliminated the electrical coupling between VSIs in all preparations tested (N = 8), suggesting that the commissure is necessary for this coupling. The importance of this connection for motor pattern generation is not known because injecting depolarizing current into a VSI to evoke an intense burst of action potentials never evoked firing in the contralateral VSI (data not shown). However, electrical coupling between the two VSIs may still contribute to their synchrony during the swim motor pattern by conducting slower membrane potential changes.
VSI makes excitatory synapses onto the efferent flexion neurons (FNs) in both pedal ganglia (Fig. 2 Bi). In all preparations, the extracellular recording from PdN6 revealed impulses that were time-locked one-for-one to the action potentials recorded from VSI soma (N > 100). Spikes elicited in a VSI through intracellular current injection evoked one-for-one, constant-latency EPSPs in both ipsilateral and contralateral FNs. The axonal impulse recorded on PdN6 appeared after the EPSP in the ipsilateral FN but before the EPSP in the contralateral FN. Similar results were obtained in 14 preparations. Thus, PdN6 allows VSI to make synaptic connections in the pedal ganglion contralateral to its soma.
Interneuron C2 also makes synaptic connections in both pedal ganglia that are mediated via PdN6 (Fig. 2 Bii). Units on PdN6 that were time-locked to C2 soma spikes were recorded in >50 preparations. The EPSPs evoked by C2 action potentials were initially smaller than the VSI-evoked EPSPs but, as previously reported, increased in amplitude during a train because of facilitation and summation (cf. Katz and Frost, 1995). As with VSI, constant-latency EPSPs could be recorded in an ipsilateral FN and a contralateral FN that corresponded with time-locked spikes on PdN6.
C2 stimulation excited VSI in the pedal ganglion most distant from its soma. Figure 2 Ci shows that a train of C2 action potentials evoked by intracellular current pulses induced VSI to fire a burst of action potentials. The VSI firing response originated in the pedal ganglion contralateral to its soma and propagated in the antidromic direction; extracellular recordings from PdN6 showed that the axonal impulses preceded the soma action potentials (Fig. 2 Cii). This C2-evoked excitation of VSI was eliminated when PdN6 was cut (compare Fig. 7 Bi). These results suggest that VSI has spike initiation zones in both pedal ganglia and that the C2-evoked VSI action potentials are initiated in the more distant pedal ganglion.
Figure 7.

The change in the synaptic action of C2 onto VSI after cutting PdN6. In A–C, the membrane potential response of VSI to a C2 burst in normal saline (i) or in Hi-Di saline (ii). The locations and the strength of the C2 action onto VSI and the direction of action potential propagation in the VSI axon are shown schematically in iii. A, With PdN6 intact, C2 excited VSI in the distal pedal ganglion. C2 stimulation (10 Hz, 2 s) induced a delayed burst in VSI (i). Action potentials are truncated to show underlying potential. Inset, Overlaid traces of VSI spike and the nerve impulses recorded from PdN6, showing that the C2-evoked VSI action potentials were antidromic (iii, dotted arrow) (compare Fig. 2). When the action potential firing threshold was raised by Hi-Di saline (ii), C2 stimulation produced a biphasic membrane potential change in VSI with initial depolarization and subsequent hyperpolarization. B, Immediately after the cut, the effect of C2 was similar to A in Hi-Di saline (ii), but it did not evoke VSI firing in normal saline (i). The inhibitory action was dominant in the synaptic action of C2 onto VSI on the proximal side of the cut. C, The C2-evoked excitation recovered after 15 h (i). The action potentials were orthodromic (inset). In Hi-Di saline, the depolarizing component became larger in the C2-evoked response in VSI (ii). D, A bar graph showing the VSI spike frequency evoked by C2 stimulation (10 Hz for 2 s) before the nerve cut (N = 16), within 2 h after the cut, and the next day (15–20 h) after the cut (N = 5). VSI did not fire any action potentials in response to C2 stimulation within 1 h of cutting PdN6 in any of the preparations examined (N = 16). E, Increase of depolarizing component in the C2-evoked synaptic potential in VSI. A bar graph shows the amplitudes of the depolarizing and hyperpolarizing responses evoked by C2 stimulation (10 Hz for 2–4 s) in Hi-Di saline. The peak and trough values were measured before the nerve cut (N = 12), <2 h after the nerve cut (N = 8), and from 15 to 22 h after the nerve cut (N = 10). The mean value of the depolarizing responses recorded next day (15–22 h) was significantly larger than the previous two responses as indicated by asterisks (p < 0.05, by Student's t test). In contrast, there was no significant difference in the hyperpolarizing responses among the three (p > 0.1, by Student's t test). Error bars indicate SEM.
Both VSI spike initiation zones are functional during a swim motor pattern. This was shown by recording VSI action potentials at three loci: the soma and two points along PdN6 (Fig. 3 A). When VSI was made to fire action potentials by injecting depolarizing current into the soma (Fig. 3 Bi), time-locked axonal impulses were observed in PdN6 propagating away from the soma (Fig. 3 Ci). During a swim motor pattern, spikes could be observed propagating in both directions (Fig. 3 B,Cii,iii). In the first burst (Fig. 3 Bii), most of the VSI action potentials tended to propagate in the orthodromic direction, from the proximal pedal ganglion to the distal one (Fig. 3 Cii). However, in the fifth burst of the swim motor pattern (Fig. 3 Biii), the action potentials tended to propagate antidromically, from the distal pedal ganglion back to the soma (Fig. 3 Ciii). Having action potentials initiate at two sites during a swim motor pattern suggests that synaptic inputs to VSI in both pedal ganglia have functional significance.
Figure 3.

VSI action potentials initiated in either pedal ganglion during a swim motor pattern. A, A schematic diagram showing the recording and stimulus configuration. Intracellular microelectrodes were placed in the soma of the right VSI, and extracellular recordings were made by en passant suction electrodes placed onto PdN6 at two loci (proximal and distal). The nerve stimulation was given to the left PdN3 via a suction electrode. B, Intracellular activity recorded from VSI and extracellular impulses recorded from two loci of PdN6 when VSI was depolarized by current injection (left) and during the swim motor program (right). Upward and downward arrows under the left VSI soma trace indicate the duration of current injection controlled by hand (0∼2 nA). The upward arrow under the right traces indicates the start time of PdN3 stimulation (8 V, 1 ms pulses at 5 Hz for 3 s). i, ii, and iii indicate bursts used for overlaid spike-triggered traces shown in Ci–iii. C, Overlaid spike-triggered traces show that the location of the spike initiation zone switched during the swim motor pattern of the time periods in B. Schematic drawings under the traces show the presumptive spike initiation zones and the direction of action potential conduction in VSI. Note that there was a slight change in the latency from the somatic action potential with respect to the extracellular spikes. This indicates that the proximal spike initiation zone is closer to the PdN6 recording site during the swim motor pattern (Cii) than when spikes are initiated by current injection into the soma (Ci).
Cutting the pedal–pedal commissure temporarily impaired the swimming behavior
To test whether the impulse propagation through PdN6 has a functional significance for the swimming behavior, we compared the behavior of animals with the commissure surgically transected to those with sham surgery (Fig. 4 A). Before surgery, all animals responded to the salt stimulus with a swim (Fig. 4 B). The mean number of flexion cycles per swim before the surgery was not significantly different between the two groups (Fig. 4 C) (sham, 6.1 ± 0.46 cycles; transected, 6.0 ± 0.42 cycles; p = 0.84 by Student's t test; N = 6). After the surgery, the number of flexion cycles was significantly reduced in both groups (nerve cut, F (4,20) = 9.8, p < 0.001; sham, F (4,20) = 10.1, p < 0.001 by one-way repeated-measures ANOVA with Fisher's LSD method; N = 6). Two hours after the surgery, all animals in the sham group swam with two to five flexion cycles (3.0 ± 0.52 cycles; N = 6), whereas 50% (three of six animals) in the transected group failed to swim. The remainder of the transected group showed a swim with two to three flexion cycles (N = 3) (Fig. 4 B). At 3.5 h, 33% still failed to swim and the rest showed a swim with two to four flexion cycles (2.8 ± 0.58 cycles; N = 4), whereas all animals in the sham group showed a swim with four to five cycles (4.33 ± 0.21 cycles; N = 6). A two-way repeated-measures ANOVA with all pairwise multiple comparison (Fisher's LSD method) showed a significant difference between the nerve-cut group and the sham group at 2 and 3.5 h after the surgery in the number of cycles [at 2 h, 3.0 ± 0.52 cycles (sham) and 1.33 ± 0.61 cycles (transected), p < 0.05; at 3.5 h, 4.33 ± 0.21 (sham) and 2.0 ± 0.58 (transected), p < 0.01]. Even if the failed-swim animals were excluded, the nerve-cut animals still showed significantly fewer cycles at 3.5 h after the surgery [4.33 ± 0.21 (sham) and 2.8 ± 0.58 (transected), p < 0.05]. The swim response mostly recovered 5 h after surgery; there was no significant difference between the transected and the sham groups. By 15 h, all of the animals again swam to the stimulus and the mean number of cycles was nearly identical. Thus, cutting the pedal commissure initially impaired the swimming response, but it recovered spontaneously over the course of several hours. The recovery could not have involved regeneration across the cut because the two ends of the transected nerve were physically separated. Thus, functional recovery must involve a mechanism that does not require axonal regeneration.
The isolated brain recovered from nerve transection
The motor pattern for the Tritonia escape swim behavior can be reproduced by electrically stimulating a body wall nerve even when the brain is isolated in a dish. Therefore, we examined the effect of cutting PdN6 in an isolated brain preparation (Fig. 5). On cutting PdN6 while intracellularly recording from C2 and VSI, there was an immediate, but brief barrage of action potentials in VSI, presumably caused by the injury (Fig. 5 B). There was no long-term effect on the resting membrane potential or spontaneous spiking activity in either C2 or VSI. The responses varied among preparations; however, the injury firing never exceeded >90 s.
Figure 5.
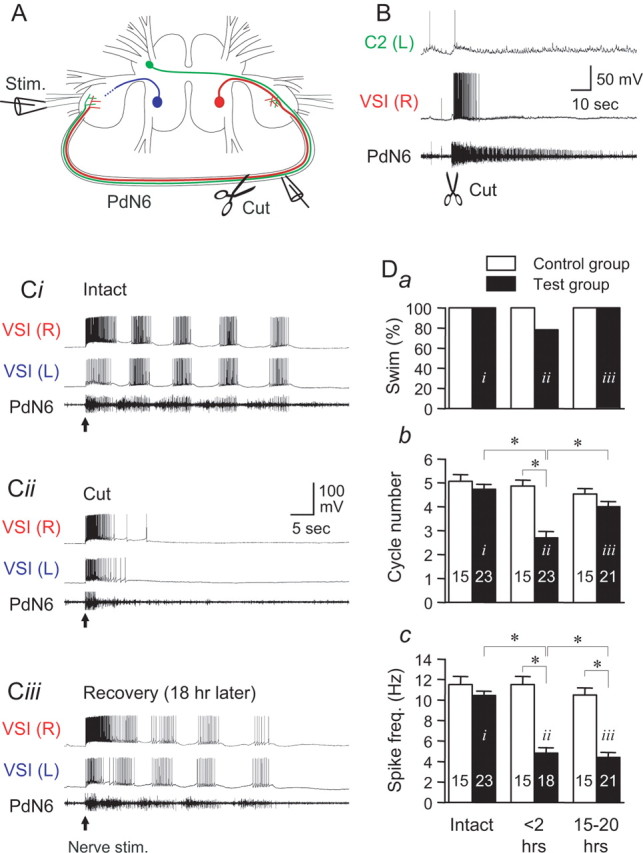
Cutting PdN6 in an isolated brain disrupted the swim motor pattern, which eventually recovered. A, A schematic drawing of the Tritonia brain, showing two VSIs, the location of the extracellular stimulation on the left PdN3, and en passant recording from PdN6. After recording the swim motor pattern, PdN6 was cut as indicated by scissors. B, Cutting PdN6 caused a brief firing of the CPG neurons. An example showing the responses of left C2 and right VSI to cutting PdN6. Immediately after the cut, C2 showed only a single action potential, whereas VSI showed tonic firing lasting for ∼10 s. Injury discharges from unknown axons in PdN6 were observed for ∼1 min after the cut. C, Simultaneous intracellular recording from both VSIs and extracellular recording from PdN6 were made 5 min before cutting PdN6 (Ci), 5 min after cutting PdN6 (Cii), and 18 h after the cut (Ciii). The arrows indicate the time of the nerve stimulation. D, The effect of cutting PdN6 on the following: the percentage of preparations showing more than one burst (a), the number of burst cycles (b), and the intraburst spike frequency (c). In b and c, the averaged number of the burst cycles (b) and the spike frequency in the second burst in the swim motor pattern (c) were plotted in bar graphs with error bars (mean ± SEM). The black bars are from experimental preparations, and the white bars show time-matched controls without the lesion. The data were obtained within 1 h before the cut (intact), within 2 h of the cut (<2 h), and the day after the cut (15–20 h). In each test, the averaged value of up to three swim responses evoked at the intervals of >10 min was measured. The label (i–iii) in each bar corresponds to the example traces shown in Ci–Ciii. The number in each bar shows the number of preparations tested. Each bracket with an asterisk indicates significant difference (Student's t test, p < 0.05; N = 18–23).
Cutting PdN6 impaired the fictive motor pattern in a manner similar to the effect on the swimming behavior. We ran two sets of isolated brain preparations, one set in which we cut PdN6 (N = 23) and the other in which the nerve was left intact (N = 15). Before cutting PdN6, nerve stimulation evoked a typical swim motor pattern in all 38 preparations tested (Fig. 5 Ci). When tested within 2 h of cutting PdN6, 21.7% of the test preparations produced a single burst in response to the nerve stimulation (Fig. 5 Cii,Da). This is presumably equivalent to a nonswimming, withdrawal response seen in vivo, although it is possible that a single VSI burst would also produce sufficient excitation of flexion neurons to produce a single body flexion (one cycle swim). However, the number of cycles was reduced in 87% of test preparations (20 of 23 preparations recorded). The mean number of cycles differed significantly from control preparations [2.7 ± 0.27 cycles (cut) and 4.9 ± 0.26 cycles (control); p < 0.05] (Fig. 5 Db). The intraburst spike frequency recorded in VSI also significantly decreased 2 h after cutting PdN6 [4.8 ± 0.55 Hz (cut) and 11.5 ± 0.81 Hz (control)] (Fig. 5 Dc). Thus, cutting PdN6 impaired the production of the fictive swim motor pattern in the isolated brain preparation by reducing both the cycle number and the VSI intraburst spike frequency.
We tested a variety of parameters to determine whether the impairment of the fictive swim motor response was caused by an increase in the threshold for eliciting the response. In all preparations tested, doubling the intensity of the electrical stimulus to PdN3 from 8 to 16 V failed to restore the reduced cycle number (N = 18). Also, the response did not depend on whether the left or right PdN3 was stimulated to elicit the swim; within 2 h of cutting PdN6, when the stimulation was applied to the nerve ipsilateral to the VSI there was a 45.5 ± 12.5% decrease in the number of cycles (from 4.8 ± 0.33 with PdN6 intact to 2.4 ± 0.53 with PdN6 cut; p < 0.01 by paired t test; N = 11) and a 42.2 ± 8.5% decrease with contralateral nerve stimulation (from 4.7 ± 0.26 with PdN6 intact to 2.7 ± 0.37 with PdN6 cut; p < 0.01 by paired t test; N = 15). Thus, there was no significant difference in the response produced by the stimulating either the left or right body wall nerves (p = 0.83 by Student's t test).
As we observed in vivo, the number of cycles in the swim motor pattern recovered the day after cutting PdN6 (Fig. 5 Ciii,D). The cycle number showed a significant recovery from the impairment (4.0 ± 0.22 cycles; p < 0.05 by Student's t test; N = 21), and there was no significant difference in the number of cycles compared with the control group at this time point (Fig. 5 Db) (p = 0.11). In contrast, the intraburst spike frequency of VSI remained depressed at 4.4 ± 0.49 Hz (N = 19), which is significantly lower than control (p < 0.001 by Student's t test) (Fig. 5 Dc). Thus, the CPG was compromised by the cut yet was able to make a functional recovery in the number of burst cycles. This indicates that the recovery of the swim behavior from injury is caused by a process that is within the CNS.
Reversibly blocking activity on PdN6 mimicked the effect of cutting the nerve
To test whether the impairment of the swim motor pattern was caused by injury to the neurons when their axons were severed, we used a reversible block of action potential propagation on PdN6 by locally applying low Na+ saline and/or TTX (10−4 m) to the extracellular recording pipette (Fig. 6, inset). Application of either solution successfully blocked action potential propagation and disconnected the membrane potential activity of the VSI soma from its distal spike initiation zone without causing any injury firing. Action potentials initiated by current injection in the soma failed to elicit EPSPs in follower neurons distal to block site (N = 6).
Figure 6.

Reversibly blocking PdN6 impaired the swim motor pattern and induced recovery. Action potential conduction on PdN6 was blocked by locally applying 0.1 mm TTX in reduced Na+ saline to a pipette into which the commissure had been drawn (inset illustration). A, Simultaneous intracellular recordings from both VSIs and extracellular recording from PdN6 are shown. Recordings show 5 min before blocking PdN6 (i), 5 min after initiating the block (ii), and a quick recovery by flushing the pipette with the normal saline (iii). The TTX saline was reapplied and left overnight. Recordings were made the next day with the TTX still in place (iv) and with the TTX flushed from the pipette (v). The arrows indicate the time of PdN3 stimulation. In each box are shown overlaid traces of the action potentials recorded from the right VSI and the axonal impulses in PdN6, both triggered by the somatic action potential. Calibration: 50 mV, 20 ms. B, Group data showing responses with saline in the pipette (white bars) and with the TTX block in place (black bars). a, The percentage of preparations showing more than one burst cycle. b, The mean number of burst cycles. c, The mean intraburst spike frequency from the second burst. The data were obtained within 1 h before the cut (intact) (i), within 2 h of blocking PdN6 with TTX (ii), and then after removal of the nerve block by flushing TTX (iii), and 15–20 h with TTX in place (iv), and then with the pipette flushed with normal saline (v). The labels (i–v) in each bar correspond to the example traces shown in Ai–Av. In each test, the averaged value of up to three swim responses evoked at the intervals of >10 min were measured. The plotted data were mean ± SEM over multiple preparations. The number in each bar shows the number of preparations tested. The bracket with an asterisk indicates significant difference (Student's t test, p < 0.01; N = 8–22).
In the experiment shown in Figure 6 A, blockade of axonal impulses was confirmed by examining the change in the impulse waveform. In normal saline, the impulse was triphasic with an apparent positive deflection. In contrast, when the action potential was blocked inside the pipette, the impulses became monophasic, spike-like, downward deflections (Fig. 6 A, insets), suggesting that the impulses came into the pipette and were blocked inside it. This was accompanied with the disappearance of the VSI-evoked EPSPs in the contralateral pedal ganglion (data not shown). Under intact conditions, antidromic action potentials were dominant (Fig. 6 Ai,iii,v, insets). Blocking action potential propagation on PdN6 caused VSI to produce only orthodromic action potentials (Fig. 6 Aii,iv). On washout, the antidromic impulses again became dominant in VSI.
Blocking action potential propagation on PdN6 consistently impaired the swim motor pattern (Fig. 6 A) (N = 22). In 14% of the preparations, VSI failed to fire repeated bursts shortly after the nerve cut (Fig. 6 Ba). The number of burst cycles decreased on average from 4.4 ± 0.20 to 2.5 ± 0.24 (p < 0.001 by paired t test; N = 22) (Fig. 6 Bb). The intraburst spike frequency decreased from 10.4 ± 0.61 to 4.1 ± 0.71 Hz (p < 0.001 by paired t test; N = 22) (Fig. 6 Bc).
When the low Na+/TTX block was removed by washing the inside of the pipette with saline, the motor pattern immediately returned to control levels (4.1 ± 0.29 cycles per swim and 10.6 ± 2.2 Hz intraburst spike frequency) (Fig. 6 Aiii,Bb). Thus, the decrease in the number of cycles and the intraburst spike frequency can be attributed to loss of action potential conduction and not to damage of the neurons.
When the nerve block was maintained overnight, the number of cycles returned to control levels in all preparations (Fig. 6 Aiv,Bb). VSI continued to produce orthodromic impulses in PdN6, indicating that the bursts were generated in the proximal pedal ganglion (Fig. 6 Aiv). Despite the functional recovery of the swim, as was observed when PdN6 was transected, the intraburst spike frequency did not fully recover (Fig. 6 Bc). These results indicate that the impairment and the recovery do not involve physical injury, but can be induced while simply blocking action potential propagation along the commissure.
Removing the conduction block after the recovery of the swim motor pattern did not further increase the number of cycles, but it did restore the intraburst spike frequency of VSI back to the original level (Fig. 6 Av,Bc). In addition, the VSI spikes during the swim bursts once again originated in the distal pedal ganglion. These results suggest that the compensation for the loss of the distal spike initiation zone is an increase in the excitation of VSI in its proximal spike initiation zone.
Synaptic changes can account for the recovery
We examined the synaptic connections among the CPG neurons to see whether there were changes that correspond to the recovery of bursting activity. The Tritonia swim CPG is a network oscillator; none of the CPG neurons exhibits endogenous bursting properties. Rather, rhythmic bursting is an emergent property of the network (Getting, 1989). A modeling study pinpointed the excitation of VSI by C2 as one of the key synapses that is necessary for the CPG to produce rhythmic bursting (Calin-Jageman et al., 2007). As noted before, in all preparations examined, C2 excited VSI in the distal pedal ganglion producing a burst of 2–26 action potentials with spike frequencies from 1 to 11 Hz (N = 16) (Figs. 2 Ci, 7 Ai). This excitation was lost in all preparations when PdN6 was blocked by TTX (N = 4) or cut (N = 12) (Fig. 7 Bi,D). Thus, during the time when the swim motor pattern is impaired, a key source of synaptic excitation to VSI is lost.
To better examine the C2-to-VSI synapse, we bathed the preparation in high divalent cation saline, which elevates the threshold for action potential firing. This had two relevant consequences: it decreased the contribution of polysynaptic components to the synaptic potential and it prevented the C2 and VSI spikes from traversing the intact PdN6. Under these conditions, with PdN6 intact, the C2-to-VSI synapse was found to be primarily inhibitory (Fig. 7 Aii). We often observed two synaptic components: an initial small depolarization followed by a larger, more prolonged hyperpolarization.
After cutting PdN6, the effect of C2 on VSI in high divalent cation saline was relatively unchanged (Fig. 7 Bii). The shape of the synaptic potential was very similar to what was observed in normal saline after cutting PdN6 (Fig. 7 Bi). Thus, the synaptic connection from C2 to VSI is primarily inhibitory before cutting PdN6 and this does not change immediately on cutting the commissure.
When the C2-to-VSI synapse was tested the day after the transection of PdN6, C2 once again depolarized VSI. In 5 of 15 preparations, C2 stimulation (10 Hz, 2 s) evoked VSI firing but its frequency is significantly lower than the average of control responses (p < 0.01 by Student's t test) (Fig. 7 Ci,D). Unlike the intact condition, the VSI spike arose in the proximal ganglion (Fig. 7 Ci, inset). In high divalent cation saline, the C2-evoked synaptic potential switched to a predominantly slow excitatory potential (Fig. 7 Cii). The peak amplitude of this depolarizing potential recorded the day after the transection was significantly larger than that evoked before or immediately after the cut (Fig. 7 E, brackets). Thus, the functional connection from C2 to VSI in the proximal pedal ganglion switched from being predominantly inhibitory to predominantly excitatory during the recovery period after disconnection of the distal pedal ganglion. This change in the sign of the synapse could be responsible for the recovery of the swim motor pattern.
The excitation of VSI by C2 in the proximal pedal ganglion was not as strong as the original excitation in the distal pedal ganglion. The spike frequency evoked by a C2 spike train (10 Hz, 2 s) was significantly lower after recovery from disconnection (p < 0.05 by Student's t test) (Fig. 7 D). This decrease in the frequency of the C2-evoked VSI firing appeared to reflect the decrease in VSI spike frequency during the recovered swim motor pattern (Figs. 5, 6).
Discussion
In this study, we severed connections within the CPG circuit for the escape swim behavior of Tritonia by cutting one of the pedal–pedal commissures and found that it impaired the ability of the animal to exhibit the swimming behavior. Cutting this commissure or blocking action potential propagation along it also prevented the production of the fictive motor pattern in isolated brain preparations. This functional impairment is at least partly attributable to the loss of excitation from C2 to VSI in the pedal ganglion distal to the lesion; modeling studies showed that this C2-evoked VSI excitation is critical for the motor pattern generation (Calin-Jageman et al., 2007). Both the behavior and the fictive motor pattern recovered within 20 h of creating the lesion. This recovery occurred without reconnection of the transected commissure but was associated with a change in the sign of the C2-to-VSI synaptic potential; going from inhibitory to excitatory in the ganglion proximal to the lesion. The present results suggest that synaptic plasticity within the Tritonia swim CPG may underlie the behavioral recovery after the lesion of the CPG.
There have been many studies describing recovery of behavioral function after injury in both peripheral and central nervous systems. In peripheral nerves, axonal regrowth through the injured area is critical for the recovery of peripheral function (Ide, 1996; Johnson et al., 1999). In the CNS, however, regeneration and regrowth generally do not occur. It has been suggested that, in mammalian cortex, the entire cortical network participates in the recovery process rather than depending on the axonal regrowth in the damaged area (Nudo, 2006; Cramer, 2008). In the transected spinal cord, the neural circuitry below the lesion can generally reproduce hindlimb locomotion after training (Barbeau and Rossignol, 1987; Rossignol et al., 2009). It has been suggested that biochemical changes at the cellular level underlie some degree of recovery (Edgerton et al., 2004). More recently, Barrière et al. (2008) showed that, after partial spinal cord lesion, the spinal CPG below the lesion also participates in the recovery of locomotion since, after an additional complete section, cats can reexpress locomotion within 24 h. Thus, the removal of certain pathways produces specific motor deficits, but the CPG and other intact pathways are still able to optimize remnant motor functions (Rossignol et al., 2009).
Our results indicate that functional recovery in the Tritonia swim CPG appears to involve network reorganization; there is a change in the synaptic action between the interneurons in the circuit and no regeneration across the lesion site. The time course of recovery (several hours) is much faster than reported in vertebrate systems, which recover over the course of days to weeks. Although significant axonal growth is unlikely in this system, there could be sprouting of fine processes during this time, which may underlie the synaptic changes we observed.
Role of multiple spike initiation zones in functional recovery
In this study, we found that VSI has at least two spike initiation zones, which can independently integrate synaptic input. During the course of a swim motor pattern, we observed action potentials originating in either zone. There have been many examples of neurons with multiple spike initiation zones in invertebrate nervous systems (Calabrese and Kennedy, 1974; Meyrand et al., 1992; Zecević, 1996; Baccus et al., 2001; Masino and Calabrese, 2002) and also in mammals (Kolta et al., 1995; Beloozerova and Rossignol, 1999; Lazarewicz et al., 2002).
We propose that recovery of function in Tritonia involves a change in functional connectivity such that the proximal synaptic integration site becomes more excitatory when the distal site is lost. The presence of multiple spike initiation zones makes the circuit more flexible by providing a locus of plasticity that can compensate the loss of synaptic connections. It is possible that neurons with multiple spike initiation zones in other systems may also be able to switch sites of excitation and that this might play a key role in the recovery of neural circuit functions after a loss of synaptic connections by injury.
Resolution of a discrepancy
In previous studies of the Tritonia swim CPG, little attention was paid to the multiple spike initiation zones or to the role of the pedal–pedal commissures, PdN5 or PdN6. In fact, in most experiments conducted before 1993, the commissures were not routinely preserved during the dissection (A. O. D. Willows and W. N. Frost, personal communication). However, all of the CPG neurons have axons that project through one of these two commissures (Newcomb et al., 2006; Hill and Katz, 2007). We have shown in this study that C2 and VSI not only make synaptic connections in both pedal ganglia through PdN6 but that VSI receives important excitatory input in the distal ganglion.
This may help resolve discrepancies in the literature regarding the nature of the synapse from C2 to VSI. When this synapse was first described, it was said to be purely excitatory, producing delayed excitation (Getting, 1983). However, when this synapse was remeasured in another study, it was found to be primarily inhibitory, with an initial excitatory component, similar to what we observed with the commissure intact in this study (Calin-Jageman et al., 2007). A parameter space analysis revealed that this synapse must be excitatory in order for the CPG to produce rhythmic activity. The resolution to the discrepancy is that the previous experiments by Getting (1983) were probably performed in preparations that had already recovered from cutting the commissure; the purely excitatory synaptic potentials [Getting (1983), their Fig. 6] look very similar to those that we observed 15 h after cutting PdN6 (Fig. 7 C). In contrast, the synaptic potentials reported in the later experiments by Calin-Jageman et al. (2007) were made with the commissure intact. Thus, the researchers may have been examining different states of the same circuit.
Potential mechanisms underlying the recovery
There is a growing realization that neural circuits maintain their activity patterns through homeostatic plasticity (Turrigiano, 1999; Davis, 2006; Marder and Goaillard, 2006). In the crustacean stomatogastric ganglion, it has been shown that decentralization caused by removing the extrinsic neuromodulatory inputs impairs the production of rhythmic activity, but a stable new activity pattern spontaneously recovers within hours to days (Thoby-Brisson and Simmers, 1998, 2002; Luther et al., 2003). Moreover, the long-term action of neuromodulatory inputs and the regulations of Ca+ and K+ conductances were suggested to underlie the spontaneous homeostatic recovery of oscillatory activity (Haedo and Golowasch, 2006; Zhang et al., 2009). In the case of Tritonia, it is not clear what homeostatic signal could convey the need to reorganize the CPG because the swim CPG is not persistently active. We do not know whether the disruption of ongoing nonrhythmic activity plays a role in triggering the recovery process or whether the absence of the swim motor pattern on nerve stimulation initiates the recovery. It is clear that injury itself is not a cue for the recovery because simply blocking action potential propagation through the commissure is sufficient to prevent the swim and initiate the recovery.
We showed in this study that the functional recovery of the swim CPG is much more rapid than the recovery from injury in other systems. Yet, several studies have reported similar quick switching of the synaptic input from inhibitory to excitatory or vice versa. For example, the synaptic terminals of sympathetic neurons in mammalian heart switch from excitatory to inhibitory by changing the dominant neurotransmitter from norepinephrine to acetylcholine in response to a neurotrophine action (Yang et al., 2002; Slonimsky et al., 2003). In immature mouse hippocampus, synaptic inputs onto pyramidal cells and interneurons exhibit a shift from phasic inhibitory synaptic input to a dominant phasic excitation before the epileptic seizure in a few seconds (Derchansky et al., 2008). Another potential mechanism for the synaptic reversal may be a change in the reversal potential for the ionic conductance. Inhibitory synapses can become excitatory if the reversal potential for Cl− is shifted to a more positive potential. This can occur by downregulation of Cl− transporters, as was observed in response to spinal cord injury (Lamsa and Taira, 2003; Payne et al., 2003; Lu et al., 2008; Vinay and Jean-Xavier, 2008). This could be a component of the recovery response because it recapitulates an earlier developmental state (Jean-Xavier et al., 2006, 2007). Additional studies are needed to test the cellular mechanisms of synaptic reorganization in the Tritonia swim CPG.
In conclusion, our results show that the Tritonia swim CPG undergoes adaptive plasticity in response to loss of important synaptic connections. Thus, the nature of the network properties of this CPG may be more plastic and less hardwired than previously realized. In this study, we examined only a specific synaptic interaction and suggested that the reversal in the polarity of its action may partly underlie this functional recovery by compensating the loss of synaptic excitation by the lesion. Similar changes may occur at other loci in the circuit in addition to the C2-to-VSI synapse. Understanding the mechanisms for recovery of behavioral function will provide insight into mechanisms underlying spontaneous recovery in more complicated nervous systems.
Footnotes
This work was supported by National Science Foundation Grant ISO-0814411 and Brains and Behavior Seed Grant Program from Georgia State University. We thank Dr. Russell Wyeth and Dr. James Murray for their valuable suggestions on the animal surgery.
References
- Antri M, Orsal D, Barthe JY. Locomotor recovery in the chronic spinal rat: effects of long-term treatment with a 5-HT2 agonist. Eur J Neurosci. 2002;16:467–476. doi: 10.1046/j.1460-9568.2002.02088.x. [DOI] [PubMed] [Google Scholar]
- Baccus SA, Sahley CL, Muller KJ. Multiple sites of action potential initiation increase neuronal firing rate. J Neurophysiol. 2001;86:1226–1236. doi: 10.1152/jn.2001.86.3.1226. [DOI] [PubMed] [Google Scholar]
- Barbeau H, Rossignol S. Recovery of locomotion after chronic spinalization in the adult cat. Brain Res. 1987;412:84–95. doi: 10.1016/0006-8993(87)91442-9. [DOI] [PubMed] [Google Scholar]
- Barbeau H, McCrea DA, O'Donovan MJ, Rossignol S, Grill WM, Lemay MA. Tapping into spinal circuits to restore motor function. Brain Res Brain Res Rev. 1999;30:27–51. doi: 10.1016/s0165-0173(99)00008-9. [DOI] [PubMed] [Google Scholar]
- Barrière G, Leblond H, Provencher J, Rossignol S. Prominent role of the spinal central pattern generator in the recovery of locomotion after partial spinal cord injuries. J Neurosci. 2008;28:3976–3987. doi: 10.1523/JNEUROSCI.5692-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloozerova I, Rossignol S. Antidromic discharges in dorsal roots of decerebrate cats. I. Studies at rest and during fictive locomotion. Brain Res. 1999;846:87–105. doi: 10.1016/s0006-8993(99)01967-8. [DOI] [PubMed] [Google Scholar]
- Cai LL, Courtine G, Fong AJ, Burdick JW, Roy RR, Edgerton VR. Plasticity of functional connectivity in the adult spinal cord. Philos Trans R Soc Lond B Biol Sci. 2006;361:1635–1646. doi: 10.1098/rstb.2006.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese RL, Kennedy D. Multiple sites of spike initiation in a single dendritic system. Brain Res. 1974;82:316–321. doi: 10.1016/0006-8993(74)90612-x. [DOI] [PubMed] [Google Scholar]
- Calin-Jageman RJ, Tunstall MJ, Mensh BD, Katz PS, Frost WN. Parameter space analysis suggests multi-site plasticity contributes to motor pattern initiation in Tritonia . J Neurophysiol. 2007;98:2382–2398. doi: 10.1152/jn.00572.2007. [DOI] [PubMed] [Google Scholar]
- Cohen AH, Mackler SA, Selzer ME. Functional regeneration following spinal transection demonstrated in the isolated spinal cord of the larval sea lamprey. Proc Natl Acad Sci U S A. 1986;83:2763–2766. doi: 10.1073/pnas.83.8.2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commissiong JW, Sauve Y, Csonka K, Karoum F, Toffano G. Recovery of function in spinalized, neonatal rats. Brain Res Bull. 1991;27:1–4. doi: 10.1016/0361-9230(91)90272-l. [DOI] [PubMed] [Google Scholar]
- Cramer SC. Repairing the human brain after stroke: I. Mechanisms of spontaneous recovery. Ann Neurol. 2008;63:272–287. doi: 10.1002/ana.21393. [DOI] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Derchansky M, Jahromi SS, Mamani M, Shin DS, Sik A, Carlen PL. Transition to seizures in the isolated immature mouse hippocampus: a switch from dominant phasic inhibition to dominant phasic excitation. J Physiol. 2008;586:477–494. doi: 10.1113/jphysiol.2007.143065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgerton VR, Tillakaratne NJ, Bigbee AJ, de Leon RD, Roy RR. Plasticity of the spinal neural circuitry after injury. Annu Rev Neurosci. 2004;27:145–167. doi: 10.1146/annurev.neuro.27.070203.144308. [DOI] [PubMed] [Google Scholar]
- Fouad K, Pearson K. Restoring walking after spinal cord injury. Prog Neurobiol. 2004;73:107–126. doi: 10.1016/j.pneurobio.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Frost WN, Katz PS. Single neuron control over a complex motor program. Proc Natl Acad Sci U S A. 1996;93:422–426. doi: 10.1073/pnas.93.1.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost WN, Hoppe TA, Wang J, Tian LM. Swim initiation neurons in Tritonia diomedea . Am Zool. 2001;41:952–961. [Google Scholar]
- Getting PA. Mechanisms of pattern generation underlying swimming in Tritonia. I. Neuronal network formed by monosynaptic connections. J Neurophysiol. 1981;46:65–79. doi: 10.1152/jn.1981.46.1.65. [DOI] [PubMed] [Google Scholar]
- Getting PA. Mechanisms of pattern generation underlying swimming in Tritonia. III. Intrinsic and synaptic mechanisms for delayed excitation. J Neurophysiol. 1983;49:1036–1050. doi: 10.1152/jn.1983.49.4.1036. [DOI] [PubMed] [Google Scholar]
- Getting PA. A network oscillator underlying swimming in Tritonia . In: Jacklet JW, editor. Neuronal and cellular oscillators. New York: Marcel Dekker; 1989. pp. 215–236. [Google Scholar]
- Getting PA, Lennard PR, Hume RI. Central pattern generator mediating swimming in Tritonia. I. Identification and synaptic interactions. J Neurophysiol. 1980;44:151–164. doi: 10.1152/jn.1980.44.1.151. [DOI] [PubMed] [Google Scholar]
- Haedo RJ, Golowasch J. Ionic mechanism underlying recovery of rhythmic activity in adult isolated neurons. J Neurophysiol. 2006;96:1860–1876. doi: 10.1152/jn.00385.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill ES, Katz PS. Role of membrane potential in calcium signaling during rhythmic bursting in Tritonia swim interneurons. J Neurophysiol. 2007;97:2204–2214. doi: 10.1152/jn.01244.2006. [DOI] [PubMed] [Google Scholar]
- Hume RI, Getting PA. Motor organization of Tritonia swimming. II. Synaptic drive to flexion neurons from premotor interneurons. J Neurophysiol. 1982;47:75–90. doi: 10.1152/jn.1982.47.1.75. [DOI] [PubMed] [Google Scholar]
- Ide C. Peripheral nerve regeneration. Neurosci Res. 1996;25:101–121. doi: 10.1016/0168-0102(96)01042-5. [DOI] [PubMed] [Google Scholar]
- Jean-Xavier C, Pflieger JF, Liabeuf S, Vinay L. Inhibitory postsynaptic potentials in lumbar motoneurons remain depolarizing after neonatal spinal cord transection in the rat. J Neurophysiol. 2006;96:2274–2281. doi: 10.1152/jn.00328.2006. [DOI] [PubMed] [Google Scholar]
- Jean-Xavier C, Mentis GZ, O'Donovan MJ, Cattaert D, Vinay L. Dual personality of GABA/glycine-mediated depolarizations in immature spinal cord. Proc Natl Acad Sci U S A. 2007;104:11477–11482. doi: 10.1073/pnas.0704832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Schroeder ML, Sánchez JA, Kirk MD. Axonal regeneration in the central nervous system of Aplysia californica determined by anterograde transport of biocytin. J Comp Neurol. 1999;406:476–486. doi: 10.1002/(sici)1096-9861(19990419)406:4<476::aid-cne5>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Katz PS. Tritonia swim network. Scholarpedia. 2009;4:3638. [Google Scholar]
- Katz PS, Frost WN. Intrinsic neuromodulation in the Tritonia swim CPG: the serotonergic dorsal swim interneurons act presynaptically to enhance transmitter release from interneuron C2. J Neurosci. 1995;15:6035–6045. doi: 10.1523/JNEUROSCI.15-09-06035.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolta A, Lund JP, Westberg KG, Clavelou P. Do muscle-spindle afferents act as interneurons during mastication? Trends Neurosci. 1995;18:441. doi: 10.1016/0166-2236(95)94493-o. [DOI] [PubMed] [Google Scholar]
- Lamsa K, Taira T. Use-dependent shift from inhibitory to excitatory GABAA receptor action in SP-O interneurons in the rat hippocampal CA3 area. J Neurophysiol. 2003;90:1983–1995. doi: 10.1152/jn.00060.2003. [DOI] [PubMed] [Google Scholar]
- Lazarewicz MT, Migliore M, Ascoli GA. A new bursting model of CA3 pyramidal cell physiology suggests multiple locations for spike initiation. Biosystems. 2002;67:129–137. doi: 10.1016/s0303-2647(02)00071-0. [DOI] [PubMed] [Google Scholar]
- Lu Y, Zheng J, Xiong L, Zimmermann M, Yang J. Spinal cord injury-induced attenuation of GABAergic inhibition in spinal dorsal horn circuits is associated with down-regulation of the chloride transporter KCC2 in rat. J Physiol. 2008;586:5701–5715. doi: 10.1113/jphysiol.2008.152348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther JA, Robie AA, Yarotsky J, Reina C, Marder E, Golowasch J. Episodic bouts of activity accompany recovery of rhythmic output by a neuromodulator- and activity-deprived adult neural network. J Neurophysiol. 2003;90:2720–2730. doi: 10.1152/jn.00370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Goaillard JM. Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci. 2006;7:563–574. doi: 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- Masino MA, Calabrese RL. Phase relationships between segmentally organized oscillators in the leech heartbeat pattern generating network. J Neurophysiol. 2002;87:1572–1585. doi: 10.1152/jn.00336.2001. [DOI] [PubMed] [Google Scholar]
- Meyrand P, Weimann JM, Marder E. Multiple axonal spike initiation zones in a motor neuron: serotonin activation. J Neurosci. 1992;12:2803–2812. doi: 10.1523/JNEUROSCI.12-07-02803.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro X, Vivó M, Valero-Cabré A. Neural plasticity after peripheral nerve injury and regeneration. Prog Neurobiol. 2007;82:163–201. doi: 10.1016/j.pneurobio.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Newcomb JM, Fickbohm DJ, Katz PS. Comparative mapping of serotonin-immunoreactive neurons in the central nervous systems of nudibranch molluscs. J Comp Neurol. 2006;499:485–505. doi: 10.1002/cne.21111. [DOI] [PubMed] [Google Scholar]
- Nudo RJ. Mechanisms for recovery of motor function following cortical damage. Curr Opin Neurobiol. 2006;16:638–644. doi: 10.1016/j.conb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Redondo RL, Murray JA. Pedal neuron 3 serves a significant role in effecting turning during crawling by the marine slug Tritonia diomedea (Bergh) J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 2005;191:435–444. doi: 10.1007/s00359-005-0604-1. [DOI] [PubMed] [Google Scholar]
- Rossignol S. Locomotion and its recovery after spinal injury. Curr Opin Neurobiol. 2000;10:708–716. doi: 10.1016/s0959-4388(00)00151-3. [DOI] [PubMed] [Google Scholar]
- Rossignol S. Plasticity of connections underlying locomotor recovery after central and/or peripheral lesions in the adult mammals. Philos Trans R Soc Lond B Biol Sci. 2006;361:1647–1671. doi: 10.1098/rstb.2006.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol S, Barriere G, Alluin O, Frigon A. Re-expression of locomotor function after partial spinal cord injury. Physiology (Bethesda) 2009;24:127–139. doi: 10.1152/physiol.00042.2008. [DOI] [PubMed] [Google Scholar]
- Sakurai A, Katz PS. Spike timing-dependent serotonergic neuromodulation of synaptic strength intrinsic to a central pattern generator circuit. J Neurosci. 2003;23:10745–10755. doi: 10.1523/JNEUROSCI.23-34-10745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slonimsky JD, Yang B, Hinterneder JM, Nokes EB, Birren SJ. BDNF and CNTF regulate cholinergic properties of sympathetic neurons through independent mechanisms. Mol Cell Neurosci. 2003;23:648–660. doi: 10.1016/s1044-7431(03)00102-7. [DOI] [PubMed] [Google Scholar]
- Thoby-Brisson M, Simmers J. Neuromodulatory inputs maintain expression of a lobster motor pattern-generating network in a modulation-dependent state: evidence from long-term decentralization in vitro . J Neurosci. 1998;18:2212–2225. doi: 10.1523/JNEUROSCI.18-06-02212.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoby-Brisson M, Simmers J. Long-term neuromodulatory regulation of a motor pattern-generating network: maintenance of synaptic efficacy and oscillatory properties. J Neurophysiol. 2002;88:2942–2953. doi: 10.1152/jn.00482.2001. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci. 1999;22:221–227. doi: 10.1016/s0166-2236(98)01341-1. [DOI] [PubMed] [Google Scholar]
- Vinay L, Jean-Xavier C. Plasticity of spinal cord locomotor networks and contribution of cation-chloride cotransporters. Brain Res Rev. 2008;57:103–110. doi: 10.1016/j.brainresrev.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Wieloch T, Nikolich K. Mechanisms of neural plasticity following brain injury. Curr Opin Neurobiol. 2006;16:258–264. doi: 10.1016/j.conb.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Willows AO, Dorsett DA, Hoyle G. The neuronal basis of behavior in Tritonia. I. Functional organization of the central nervous system. J Neurobiol. 1973;4:207–237. doi: 10.1002/neu.480040306. [DOI] [PubMed] [Google Scholar]
- Wyeth RC, Croll RP, Willows AO, Spencer AN. 1-Phenoxy-2-propanol is a useful anaesthetic for gastropods used in neurophysiology. J Neurosci Methods. 2009;176:121–128. doi: 10.1016/j.jneumeth.2008.08.028. [DOI] [PubMed] [Google Scholar]
- Yang B, Slonimsky JD, Birren SJ. A rapid switch in sympathetic neurotransmitter release properties mediated by the p75 receptor. Nat Neurosci. 2002;5:539–545. doi: 10.1038/nn0602-853. [DOI] [PubMed] [Google Scholar]
- Zecević D. Multiple spike-initiation zones in single neurons revealed by voltage-sensitive dyes. Nature. 1996;381:322–325. doi: 10.1038/381322a0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Khorkova O, Rodriguez R, Golowasch J, Golowaschi J. Activity and neuromodulatory input contribute to the recovery of rhythmic output after decentralization in a central pattern generator. J Neurophysiol. 2009;101:372–386. doi: 10.1152/jn.01290.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]