

Abstract
Recent studies suggest that bone marrow-derived macrophages can effectively reduce β-amyloid (Aβ) deposition in brain. To further elucidate the mechanisms by which macrophages degrade Aβ, we cultured murine macrophages on top of Aβ plaque-bearing brain sections from transgenic mice expressing PDAPP [human amyloid precursor protein (APP) with the APP717V>F mutation driven by the platelet-derived growth factor promoter]. Using this ex vivo assay, we found that macrophages from wild-type mice very efficiently degrade both soluble and insoluble Aβ in a time-dependent manner and markedly eliminate thioflavine-S positive amyloid deposits. Because macrophages express and secrete apolipoprotein E (apoE), we compared the efficiency of Aβ degradation by macrophages prepared from apoE-deficient mice or mice expressing human apoE2, apoE3, or apoE4. Macrophages expressing apoE2 were more efficient at degrading Aβ than apoE3-expressing, apoE4-expressing, or apoE-deficient macrophages. Moreover, macrophage-induced degradation of Aβ was effectively blocked by an anti-apoE antibody and receptor-associated protein, an antagonist of the low-density lipoprotein (LDL) receptor family, suggesting involvement of LDL receptors. Measurement of matrix metalloproteinase-9 (MMP-9) activity in the media from human apoE-expressing macrophages cocultured with Aβ-containing brain sections revealed greater levels of MMP-9 activity in apoE2-expressing than in either apoE3- or apoE4-expressing macrophages. Differences in MMP-9 activity appear to contribute to the isoform-specific differences in Aβ degradation by macrophages. These apoE isoform-dependent effects of macrophages on Aβ degradation suggest a novel “peripheral” mechanism for Aβ clearance from brain that may also, in part, explain the isoform-dependent effects of apoE in determining the genetic risk for Alzheimer's disease.
Introduction
β-Amyloid (Aβ) accumulation and senile plaque formation in brain are striking neuropathological hallmarks of Alzheimer's disease (AD). Excessive β-amyloid deposition may result from increased Aβ synthesis as occurs in familial early-onset AD and (or) decreased Aβ clearance in brain (Selkoe, 2001). There is, however, no compelling evidence that increased Aβ production occurs in the more common late-onset forms of AD, suggesting that insufficient or impaired Aβ clearance may drive Aβ deposition and amyloid plaque formation.
The ε4 allele of the apolipoprotein E (APOE) gene remains the most widely replicated genetic risk factor for late-onset AD, with ε4 carriers having a greater risk (3–15-fold), as well as an earlier age of disease onset (Saunders et al., 1993). In contrast, inheritance of the ε2 allele appears to be protective (Corder et al., 1994). By characterizing human amyloid precursor protein (APP) with the APP717V>F mutation driven by the platelet-derived growth factor promoter (PDAPP) transgenic mice expressing the three common human apoE isoforms, we have previously demonstrated an apoE isoform-specific effect on Aβ burden, independent of Aβ synthesis, indicating that the interactions between apoE and Aβ are critical for Aβ-associated neuropathology (Holtzman et al., 1999; Fagan et al., 2002).
In brain, apoE is mainly synthesized and secreted by astrocytes and microglia (Boyles et al., 1985; Xu et al., 2006), both of which are found to surround amyloid plaques. Recently, we reported that astrocytes promote Aβ clearance via an apoE-dependent mechanism (Koistinaho et al., 2004). In contrast, the precise role of microglia on AD pathology is unknown. Activation of microglia does trigger Aβ phagocytosis, but it is unclear if microglia can effectively degrade internalized Aβ (Paresce et al., 1996, 1997; Chung et al., 1999; Wegiel et al., 2001).
Macrophages are the peripheral counterpart of microglia and have attracted attention recently based on the findings that a subset of microglia surrounding and invading Aβ-containing plaques in transgenic AD mouse models are in fact bone marrow derived (Malm et al., 2005; Simard et al., 2006). Bone marrow-derived microglia (BMDM), but not resident microglia, have been reported to prevent the formation and even eliminate brain amyloid deposits (Simard et al., 2006). Circulating BMDM that are recruited to brain may, therefore, play an important role in enhancing Aβ clearance.
Although macrophages have been shown to readily internalize Aβ, less is known about their ability to effectively degrade various forms of Aβ. To further elucidate the mechanisms of Aβ clearance by BMDM and to explore the possible cellular mechanisms associated with apoE isoform-dependent Aβ deposition and amyloid formation, we characterized the ability of peritoneal macrophages prepared from wild-type and human apoE-targeted replacement mice to degrade Aβ. In addition to internalizing Aβ, we found that macrophages are extremely efficient at degrading soluble and insoluble Aβ, including amyloid itself. The ability of macrophages to degrade Aβ was facilitated by apoE expression, was also apoE isoform-dependent (E2 > E3 > E4), and blocked by the low-density lipoprotein (LDL) receptor antagonist receptor-associated protein (RAP). Importantly, we found that the apoE isoform-dependent macrophage-mediated Aβ degradation was, in part, mediated by secretion of matrix metalloproteinase-9 (MMP-9). Our data suggest that macrophage-mediated Aβ degradation in brain may constitute a novel peripheral clearance mechanism and delineates a previously unknown role for apoE in modulating Aβ-degrading proteases that may help explain the role of apoE as a genetic risk factor for AD.
Materials and Methods
Cell culture and reagents.
Peritoneal macrophages from the following mouse models, C57BL/6 mice [wild type (WT)], Apoe-targeted deletion (Apoe−/−), and APOE-targeted replacement mice (apoE2, apoE3, or apoE4), were prepared as described previously (Sullivan et al., 1997; Davies and Gordon, 2005). Briefly, WT or mice of the indicated APOE genotypes, at 8–12 weeks of age, were injected intraperitoneally with 1.5 ml of aged 4% Brewer thioglycolate solution (Difco Laboratories). Four days after injection, macrophages were harvested by lavage with 10 ml of growth medium [RPMI 1640 (containing 25 mm HEPES buffer and l-glutamine), 2% fetal bovine serum (FBS), 100 U/ml of penicillin, and 100 μg/ml of streptomycin (Invitrogen)]. Contaminating red blood cells were removed with blood lysis buffer (0.15 m NH4Cl, 0.01 m KHCO3, 0.1 mm EDTA, pH 7.3) if present. The cells were spun down and resuspended in fresh growth medium and seeded in cell culture plates or slide chambers. Two hours after seeding, the cultures were washed with PBS to remove unadherent cells, and fresh growth media was applied to establish primary cultures. When comparing macrophages harvested from Apoe−/−, apoE2, apoE3, and apoE4 mice, cultures were maintained in a medium supplemented with 2% lipoprotein-deficient FBS (Invitrogen). Experiments were performed 0–2 d after the cells were seeded and cultures were at least 90% pure as determined by staining with the macrophage specific markers F4/80, CD11b, or CD68. All animal experiments were approved by the Institutional Animal Care and Use Committee, Lilly Research Laboratory, Eli Lilly and Company.
Aβ degradation by macrophage condition media.
To prepare macrophage-conditioned media (MCM), macrophages were isolated as described above and incubated in media for 24 h that was then replaced with serum-free media [RPMI 1640, 0.3% BSA (Invitrogen), 100 U/ml of penicillin and 100 μg/ml of streptomycin] and cultured for an additional 96 h. The MCM was harvested and cleared by centrifugation at 1500 rpm. Brain sections from a very old PDAPP mouse or 1 ng/ml of freshly dissolved Aβ42 (AnaSpec) was then incubated with control media (prepared in parallel to MCM but without the addition of cells), MCM, or MCM heated to 90°C for 15 min for 96 h. Aβ42 levels in the media or remaining in the PDAPP brain section were assessed by ELISA as described below.
In vitro Aβ degradation assay.
Macrophages were plated in 48-well plates at a density of 3 × 105 cells/well. The cells were cultured overnight and then switched to serum-free media with freshly dissolved Aβ42 (3.0 μg/ml; AnaSpec) and the indicated treatment for the indicated times. After incubation, the culture media were collected, and Aβ42 levels were measured by ELISA (Johnson-Wood et al., 1997). The cells were washed with PBS and lysed in 70 μl of 5.5 m guanidine-HCI (GuHCl), and Aβ was measured by ELISA.
Aβ42 internalization assay.
Internalization of Aβ42 by macrophages was assayed as described previously (Chung et al., 1999) with the following modifications: fluorescently labeled Aβ42 (FAM-Aβ42; AnaSpec) was dissolved in DMSO, immediately aliquoted, and stored at −80°C. Macrophages were seeded in 4-well slide chambers at a density of 2 × 105 cells/well and incubated with 1 μg/ml FAM-Aβ42 and the indicated treatment in serum-free medium for the indicated times. Cells were rinsed with PBS to remove any Aβ42 that was not bound or taken up and incubated with a monoclonal antibody specific to F4/80, or LAMP-1 (BD Pharmingen; 1:100). Aβ uptake was determined by quantifying FAM-Aβ42 fluorescence that colocalized with the F4/80 or LAMP-1 markers by immunofluorescence or confocal microscopy in at least three randomized fields per treatment using Image-Pro Plus software.
Ex vivo Aβ degradation assay.
Homozygous transgenic PDAPP (Games et al., 1995), 22–26 months of age, were perfused with heparinized saline (2500 IU/L). The brains were removed and frozen in liquid nitrogen and stored at −80°C until sectioning. Sagittal sections (10 or 16 μm) were cut on a CM 3050-S cryostat (Leica), mounted on to poly-l-lysine coated coverslips, transferred to 2-well chamber slides, and used immediately or stored at −80°C until use. Freshly harvested peritoneal macrophages were seeded in the chamber on top of sections at a density of 1 × 106 cells in 1 ml of medium at 37°C for the indicated times (Koistinaho et al., 2004). This assay was further modified to physically separate macrophages from the PDAPP brain section by plating the cells on the bottom of culture dishes and placing the coverslip mounted sections in a Millicell-PCF culture plate insert (Millipore Bioscience Research Reagents). For inhibition of macrophage-mediated Aβ degradation by various reagents, cultures were coincubated for the indicated times with or without 75 μg/ml anti-apoE antibody (Biodesign), 50 μg/ml anti-MMP-9 antibody (G657; Cell Signaling Technology), 10 μm MMP-9 inhibitor (MMP-2/9 inhibitor I; Calbiochem), or 50 μg/ml RAP (a generous gift from Dr. G. Bu, Washington University, St. Louis, MO). After incubation, Aβ present in the media was measured by ELISA as described below. The sections were washed with 1 ml of PBS and analyzed by ELISA, immunohistochemistry (IHC), or thioflavine-S staining.
Human Aβ ELISA.
Aβ levels in media, or GuHCl lysates from cells or sections (200 μl of 5.5 m GuHCl/section) were assayed as described previously (Johnson-Wood et al., 1997; Bales et al., 1999). In brief, guanidine-solubilized samples were diluted with Casein blocking buffer (Pierce) to a final concentration of 0.5 m GuHCl or less. Samples were loaded onto plates coated with an antibody that specifically recognizes the C-terminal domain of Aβ42 (21F12) as a capture, and biotinylated 3D6 (Aβ42) or biotinylated 266 (Aβ x-42) was used for detection. The signal after incubation with HRP-conjugated streptavidin (Research Diagnostics), the signal was developed with TMB substrate (Pierce) and read on a SpectraMAX 190 plate reader (Molecular Devices).
Immunohistochemistry.
After the indicated treatment, the cells were fixed with 4% paraformaldehyde and blocked with 0.5% BSA in 0.2% Triton X-100/PBS for 30 min. Cells or sections were then incubated with anti-F4/80 (Serotec; 1:100), anti-CD68 (BD Pharmingen; 1:100), anti-MMP-9 (Santa Cruz Biotechnology; 1:50), or anti-Aβ antibody (3D6; 1:1000) overnight at 4°C, washed with PBS with 0.1% Tween-20 (PBST) three times, and incubated with secondary antibodies conjugated to AlexaFluor 488 or 594 (Invitrogen) for 1 h at room temperature. The slides were washed with PBST three times and then coverslipped with VECTASHIELD HardSet Mounting Medium with 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI; Vector Laboratories). Representative images were captured using a fluorescence imaging system (Leica), and images from three randomized fields per treatment group were quantified using an Image-Pro Plus software program (Media Cybernetics) written in Image-Pro scripting language and run under Image-Pro Plus 4.5.0.29 (Bales et al., 1999; Koistinaho et al., 2004).
To measure Aβ burden by IHC, sections were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) and probed with antibodies 3D6 (1:1000), pan β amyloid antibody (Biosource;1:200) or CD68 (1:100) and detected with fluorescent-conjugated secondary antibodies. Thioflavine-S staining was performed as described previously (Bales et al., 1997). Briefly, fixed sections were incubated with 1% freshly made thioflavine-S for 8 min and then cleared with 80% ethanol for 1 min, followed by washing with H2O two times for 1 min each. The percentage area of the hippocampus occupied by 3D6 label (Aβ burden) or thioflavine-S positive Aβ deposits (amyloid burden) was calculated from images captured from six to eight sections per treatment group using the Image-Pro Plus software program.
Mass spectrometry.
Aβ42 was measured in brain sections that were solubilized by GuHCl (20 μl) from the ex vivo assay, diluted in 1 ml of RPMI-1640, and spiked with an internal peptide standard [15NAβ40 (R-peptide)]. Immunoprecipitation and matrix-assisted laser desorption/ionization (IP-MALDI) mass spectrometry was performed as described previously (Gelfanova et al., 2007).
Gelatin-substrate zymography.
MMP-9 enzyme activity from culture media was assayed by zymography using Novex Zymogram Gelatin Gels (Invitrogen) according the manufacturer's instructions. For detection of MMP activity, the zymogram gels were stained with SimplyBlue SafeStain (Invitrogen) and then destained with water. Enzyme activity attributed to MMP-9 or MMP-2 was visualized (on the basis of molecular weight) in the gelatin-containing zymograms as clear bands against a blue background (Leber and Balkwill, 1997). The gels were imaged by MagicScan, and quantification was performed by scanning densitometry (KDSOD 2.0).
Intracerebral injection of macrophages.
Macrophages were prepared from apoE2 or apoE4 targeted replacement mice as described above. Immediately before intracerebral injection, macrophages were centrifuged at 1500 rpm for 5 min and resuspended in artificial CSF (Swanson et al., 2004) at a cell density of 0.5 × 106 cells/μl. Seven heterozygous mice (C57BL/6 × PDAPP, 28 months of age) were anesthetized with Avertin (0.025 ml/g body weight) and immobilized in a stereotaxic apparatus (Stoelting). Macrophages (2 μl) were injected at a rate of 0.2 μl/min into the cerebral cortex at the following coordinates from the bregma: anterior/posterior, 0.0 mm; mediolateral: ±1.5 mm; dorsoventral: 1.5 mm below skull surface. The apoE2 macrophages were injected into the right hemisphere and E4 macrophages into the contralateral side of the same mouse. Two minutes after injection, the needle was slowly withdrawn. Animals were monitored until they regained full consciousness. Three days after injection of cells, the mice were terminally anesthetized with Avertin and perfused with heparinized saline (2500 IU/L) for 2 min (Takata et al., 2007; Pihlaja et al., 2008). The brains were removed and frozen in liquid nitrogen and stored at −80°C until sectioning. Brain Aβ burden was determined as described above. The removal of Aβ from the cortex by injected macrophages was calculated as the difference between Aβ burden proximal to the injection site and the amount of Aβ remaining within the injection site.
Statistical analyses.
To compare differences between the experimental groups, a two-tailed t test or one-way ANOVA followed by the Tukey–Kramer test for multiple comparisons were performed using GraphPad Prism software.
Results
Macrophages efficiently phagocytose and degrade exogenous Aβ
To determine if macrophages can internalize and degrade Aβ, we coincubated peritoneal macrophages from WT mice with human Aβ 42 and measured the amount of cell-associated Aβ42 as well as the amount remaining in the media using a sensitive ELISA (Fig. 1). Cell-associated Aβ42 increased in a time-dependent manner with maximal levels measured at 4 h after incubation with human Aβ42 and decreased by >90% over the ensuing 96 h (Fig. 1A; supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). In contrast, Aβ42 that could be measured in the media decreased in a time-dependent manner, such that <15% remained after 96 h of incubation (Fig. 1B; supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Incubation of Aβ42 in media alone (without the addition of macrophages) over the same time course did not result in any significant decrease in the level of Aβ42 measured by our ELISA (over 90% of the Aβ42 that was measured in the media after incubation could be recovered) (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material). Aβ42 incubated with or without macrophages for up to 96 h primarily remains monomeric; however, a small amount of oligomeric Aβ (<17 kDa), but no fibrillar Aβ or amyloid, could be detected (supplemental Fig. 1E–G, available at www.jneurosci.org as supplemental material). Incubation of Aβ42 with peritoneal macrophages dramatically reduces the amount of both Aβ monomer and oligomers (supplemental Fig. 1E, available at www.jneurosci.org as supplemental material). These data demonstrate that macrophages are able to efficiently internalize and degrade human Aβ42.
Figure 1.
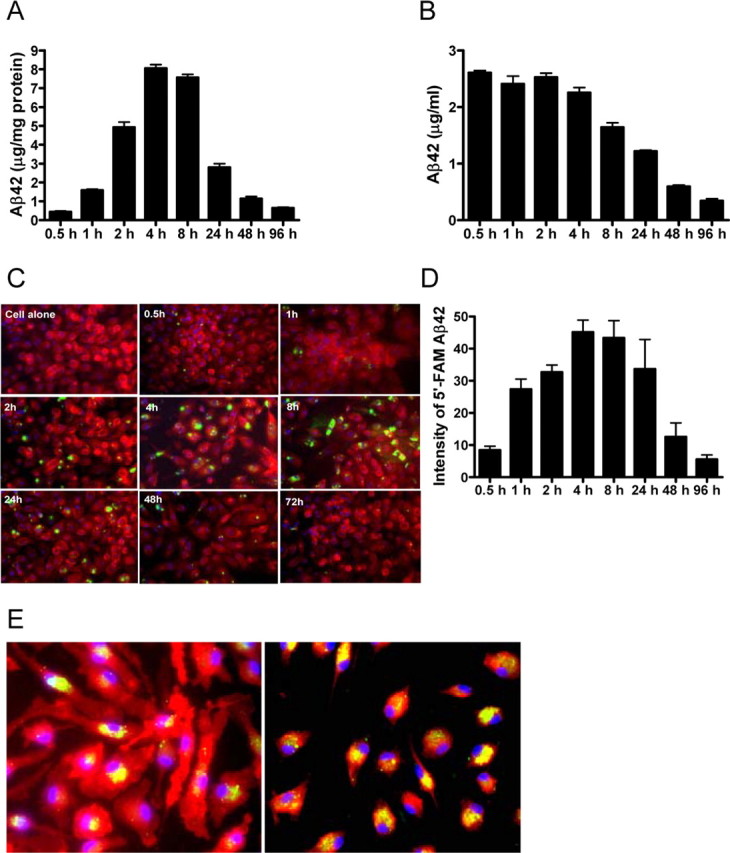
Macrophages degrade exogenous Aβ42. Human Aβ42 (3 μg/ml) was incubated with macrophages for the indicated times. A–C, Aβ42 levels in the cell pellet (A) or in the media (B) after incubation were measured by ELISA. FAM-Aβ42 (1 μg/ml; green) was incubated with macrophages (labeled red with F4/80) for the indicated times (C). Cell nuclei were visualized by DAPI (blue). Original magnification, 40×. D, The intensity of intracellular FAM-Aβ42 was quantified using Image-Pro Plus software. E, Colocalization (yellow) of FAM-Aβ42 (green) with macrophages (red; left panel) or the endosomal/lysosomal marker LAMP-1 (red; right panel). Macrophages were incubated with 1 μg/ml FAM-Aβ42 for 2 h and imaged with fluorescence microscopy. Cell nuclei were visualized with DAPI (blue). Original magnification, 40×. Representative data from one experiment that was repeated three times with similar results. Error bars represent the SE measurement.
We next investigated the intracellular localization of Aβ 42 after uptake by macrophages. Using FAM-Aβ42 coincubated with macrophages from WT mice, we observed a time-dependent uptake of Aβ42 and observed that the FAM-labeled Aβ42 was associated with the macrophage marker F4/80 (Fig. 1C–E). That the Aβ42 was internalized by the macrophages was confirmed by confocal microscopy (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material). Additionally, FAM-Aβ42 that was internalized by macrophages was clearly associated with the lysosomal marker LAMP-1 (Fig. 1E; supplemental Fig. 2C, available at www.jneurosci.org as supplemental material).
Macrophages efficiently degrade Aβ ex vivo
Because Aβ deposited in brain parenchyma represents a conformationally diverse set of peptides, we used an ex vivo assay in which we incubated peritoneal macrophages collected from WT mice on top of unfixed brain sections from old PDAPP transgenic mice to determine if macrophages could degrade various Aβ species that were deposited in brain (Fig. 2). Brain sections from very old PDAPP transgenic mice contain abundant Aβ deposits that are predominantly composed of nonfibriallar Aβ 42 and a small percentage (∼0.3%) of amyloid (thioflavine-S-positive Aβ deposits). A considerable amount of Aβ characterized as monomeric or oligomeric Aβ species present in sections can be solubilized by culture media or other aqueous solutions (data not shown). After incubation with macrophages, we observed a time-dependent decrease in the amount of Aβ42 in PDAPP brain sections [thoroughly solubilized by guanidine-HCl (supplemental Fig. 3, available at www.jneurosci.org as supplemental material)] such that only 58% of the Aβ42 remaining in the section could be measured 48 h after incubation (Fig. 2A). More striking, however, was the nearly complete loss of Aβ42 in brain sections when macrophages were incubated for 96 h (Fig. 2A). Importantly, there was a parallel time-dependent decrease in the amount of Aβ42 that could be measured in the media, indicating that macrophages can also efficiently degrade soluble forms of Aβ released from the brain section into the media (Fig. 2B). Additionally, the level of brain Aβ burden as measured by quantitative IHC decreased proportionately (Fig. 2C) (92.7 ± 9.5% decrease) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material) (n = 6 sections per treatment; p < 0.001). Similarly, when fibrillar Aβ deposits (thioflavine-S-positive amyloid) were quantified, the number of amyloid plaques remaining 96 h after PDAPP brain sections was <5% of that quantified in untreated sections (Fig. 2D) (98.1 ± 20.3% reduction; n = 4 sections per treatment; p < 0.01). Thus, macrophages very efficiently degrade virtually all deposited and soluble species of Aβ, including frank amyloid. We caution, however, that the various Aβ species present even in our ex vivo assay using PDAPP brain sections may still differ from those present in human AD brain.
Figure 2.
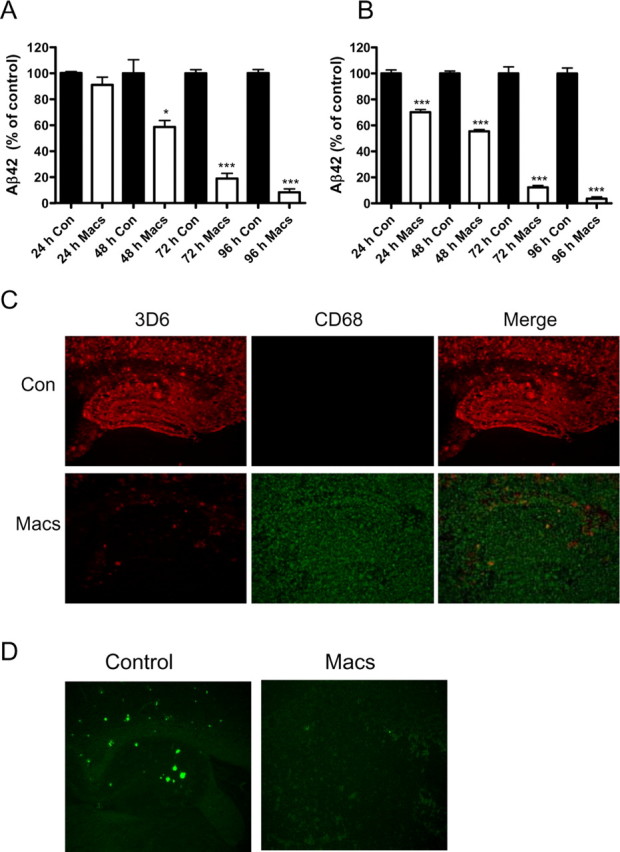
Macrophages efficiently degrade Aβ in situ. Brain sections from old PDAPP mice were incubated with (Macs) or without (Con) macrophages for the indicated times. A–C, Aβ42 remaining in brain sections (A) or released into the media (B) was measured by ELISA and plotted as a percentage of the control value. Representative PDAPP brain sections incubated with (Macs; bottom) or without (Con; top) macrophages for 96 h and immunostained for Aβ with the anti-Aβ antibody 3D6 (red) and the macrophage-selective marker CD68 (green; C). Original magnification, 5×. D, Representative photomicrograph demonstrating amyloid deposits stained with thioflavine-S (green) in the hippocampus of PDAPP mouse brain sections after incubation with (Macs) or without (Control) macrophages for 96 h. Original magnification, 5×. n = 6 sections/treatment; *p < 0.05, ***p < 0.001 versus control. Representative data from one experiment that was repeated three times with similar results. Error bars represent the SE measurement.
Macrophages degrade Aβ in situ in an apoE-dependent manner
We previously reported that astrocytes are able to degrade brain Aβ via an apoE-dependent mechanism (Koistinaho et al., 2004). Because macrophages also synthesize and secrete apoE, we next investigated whether macrophage-mediated Aβ degradation was dependent on apoE. When PDAPP brain sections were incubated for 96 h with macrophages prepared from apoE-deficient mice, we observed only a modest decrease in the levels of Aβ42 remaining in both the brain section and media (Fig. 3A,B). In contrast, and as previously observed, WT macrophages were able to efficiently degrade Aβ42 in both the PDAPP brain sections as well as media, and the differences between the apoE-deficient and WT mouse macrophages were statistically significant (Fig. 3A,B). Consistently, hippocampal Aβ immunoreactivity in old PDAPP brain sections was reduced by >80–90% after incubation for 96 h with WT mouse macrophages and only by ∼60% when adjacent brain sections were incubated with apoE-deficient macrophages (Fig. 3C,D).
Figure 3.
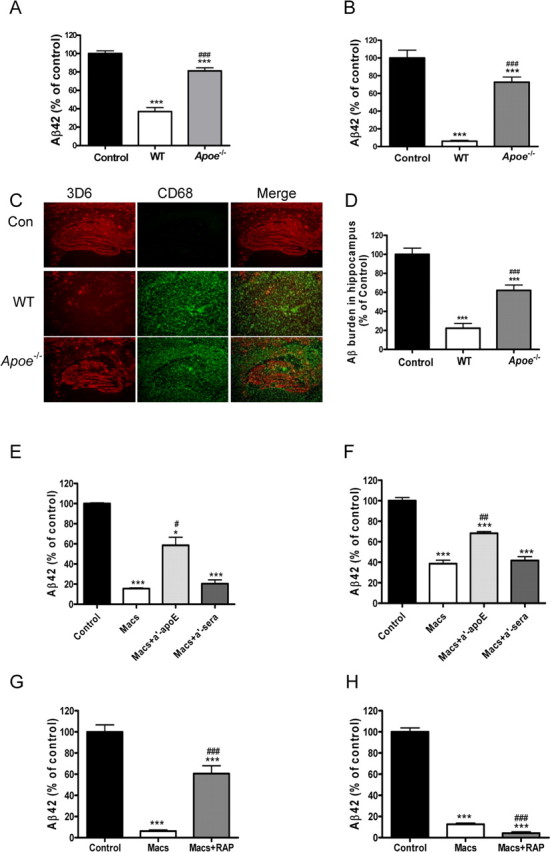
ApoE-dependent degradation of Aβ by macrophages. PDAPP brain sections were incubated with macrophages isolated from WT or Apoe-deficient (Apoe−/−) mice for 96 h. Adjacent PDAPP brain sections were incubated with media alone as control (Control). A–C, Aβ levels remaining in the sections (A) or released into the media (B) were measured by ELISA and are presented as a percentage of control value. Representative PDAPP brain sections incubated with WT macrophages (WT), ApoE-deficient macrophages (Apoe−/−), or media alone (Con) for 96 h and immunostained for Aβ with the anti-Aβ antibody 3D6 (red) and the macrophage-selective marker CD68 (green; C). Original magnification, 5×. D, Quantification of brain Aβ burden after incubation of PDAPP brain sections with macrophages. n = 6 sections per treatment; ***p < 0.001 versus control; ###p < 0.001 versus WT. E–H, PDAPP mouse brain sections were incubated with macrophages alone (Macs), macrophages plus a polyclonal rabbit anti-ApoE antibody (Macs + a′-ApoE; 75 μg/ml), an anti-rabbit sera (Macs + a′-sera; 75 μg/ml) (E, F), or RAP (Macs + RAP; 50 μg/ml) (G, H) for 96 h. Aβ remaining in the sections (E, G) or released into the media (F, H) were quantified by ELISA and plotted as a percentage of control value. n = 6 sections per treatment; *p < 0.05, ***p < 0.001 versus media only control; #p < 0.05, ##p < 0.01, ###p < 0.001 versus Macs. Representative data from one experiment that was repeated three times with similar results. Error bars represent the SE measurement.
To further confirm a role for apoE in macrophage-mediated Aβ degradation, we coincubated WT mouse macrophages with rabbit anti-mouse apoE antisera just before incubation with a PDAPP mouse brain section (Fig. 3E,F). In the presence of normal medium or normal rabbit sera, macrophages effectively reduced Aβ42 levels in the sections (Fig. 3E), as well as in the media (Fig. 3F). However, when macrophages were coincubated with sections and rabbit anti-mouse apoE antisera, the degradation of Aβ by macrophages was reduced by ≥50% (Fig. 3E,F).
Next, we investigated whether the LDL receptor antagonist, RAP, could block the ability of macrophages to degrade Aβ in PDAPP brain sections or Aβ released into the media. When macrophages were coincubated with brain sections from old PDAPP mice and RAP, there was a reduction in the ability of macrophages to degrade Aβ deposited in the brain section, but not in their ability to degrade soluble Aβ released into the media (Fig. 3G,H). When macrophages were incubated with FAM-labeled Aβ, RAP very modestly reduced the cell-associated/uptake of FAM-labeled Aβ after 2 h incubation and also significantly inhibited its degradation after 96 h (supplemental Fig. 5A,B, available at www.jneurosci.org as supplemental material). In contrast, incubation with RAP increased Aβ degradation in the media when macrophages were incubated with Aβ for shorter time points (4, 24, and 72 h) (supplemental Fig. 5C, available at www.jneurosci.org as supplemental material). Together, these data suggest that macrophages may employ independent mechanisms for degrading soluble and insoluble Aβ and that degradation of insoluble Aβ involves one or more LDL receptors.
Macrophages degrade Aβ42 via an apoE isoform-dependent mechanism
Since we established that macrophages can internalize and degrade soluble as well as deposited Aβ42 via an apoE-dependent mechanism, we next investigated whether there was an apoE isoform-dependent difference in the ability of macrophages to efficiently degrade Aβ (Fig. 4). Macrophages were prepared from apoE-targeted replacement mice expressing one of the three human apoE isoforms. When macrophages from apoE2-, apoE3-, or apoE4-targeted replacement mice were coincubated with PDAPP brain sections, there was a significant apoE isoform-dependent difference in the ability of macrophages to degrade Aβ42 in both brain sections and media such that apoE2-expressing macrophages degrade Aβ42 more efficiently than either apoE3- or apoE4-expressing macrophages (E2 ≫ E3 > E4) (Fig. 4). After 48 h of incubation with PDAPP brain sections and apoE2 macrophages, nearly 50% of the Aβ42 had been degraded, whereas only ∼20% of Aβ42 was degraded after coincubation with apoE3-expressing macrophages and virtually no Aβ42 was degraded when PDAPP brain sections were coincubated with macrophages collected from apoE4-expressing mice (Fig. 4A,B). By 96 h after incubation, all three apoE isoform-expressing macrophages were able to degrade both deposited and soluble Aβ42, but a similar isoform-dependent difference (E2 > E3 >E4) was observed (Fig. 4A,B). We further confirmed the apoE isoform-dependent degradation of Aβ42 by macrophages using a quantitative IP-MALDI analysis. Similar to the results observed when Aβ42 was measured by our sensitive ELISA, there was a significant apoE isoform-dependent decrease in the amount of Aβ42 that was recovered 96 h after macrophages derived from apoE-targeted replacement mice were incubated with PDAPP brain sections with the most robust and efficient degradation observed with macrophages expressing human apoE2 (Fig. 4C; supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Incubation of FAM-labeled Aβ with macrophages for 2 h did not reveal a significant difference in cell-associated Aβ42 in apoE2- versus apoE4-expressing macrophages, suggesting that the initial uptake of Aβ by macrophages is not affected by apoE isoforms. However, there was much more degradation of Aβ in apoE2-expressing versus apoE4-expressing macrophages after 96 h of incubation (supplemental Fig. 7, available at www.jneurosci.org as supplemental material), consistent with our ex vivo assay results, indicating the contribution of a mechanism independent of Aβ internalization involved in apoE isoform-specific effects in Aβ degradation by macrophages.
Figure 4.

Macrophages degrade Aβ42 via an apoE isoform-dependent manner. Macrophages isolated from targeted replacement mice expressing human apoE2 (E2), apoE3 (E3), or apoE4 (E4) were incubated with PDAPP mouse brain sections for 48 or 96 h. Adjacent sections were incubated with media alone as control (Con). A, B, Aβ42 remaining in the sections (A) or released into the media (B) after incubation was measured by ELISA and plotted as a percentage of control value. n = 6 sections per treatment. Representative data from one experiment that was repeated three times with similar results. *p < 0.05, ***p < 0.001 versus control; #p < 0.05, ##p < 0.01, ###p < 0.001 versus E2 macrophages. C, The degradation of Aβ42 in PDAPP brain sections after incubation with E2, E3, or E4 macrophages for 96 h was verified by quantitative IP-MALDI analysis. n = 6 sections per treatment; ***p < 0.001 versus control; #p < 0.05, ##p < 0.01 versus E2 macrophages. E2 or E4 macrophages were administered bilaterally (0.5 × 106 cells) via intracerebral injection into the right or left cerebral cortices, respectively, of old heterozygous PDAPP mice (28 months). D, Three days after injection, brain Aβ was calculated as the difference between Aβ burden proximal to the injection site and the amount of Aβ remaining within the injection site. n = 7 animals. **p < 0.01 versus E2 injection sites. Representative data from one experiment that was repeated two times with similar results. Error bars represent the SE measurement.
Macrophages can degrade Aβ in vivo
We next examined whether apoE2-expressing macrophages differ from apoE4-expressing macrophages in their ability to degrade human Aβ after direct intracerebral injection into mouse brain. Macrophages expressing human apoE2 or apoE4 were administered bilaterally (0.5 × 106 cells) via intracerebral injection into the right or left cortices of old PDAPP mice. Three days after intracerebral injection, Aβ immunoreactivity in the area injected with apoE2-expressing macrophages was significantly lower when compared with the contralateral side injected with apoE4-expressing macrophages (Fig. 4D; supplemental Fig. 8, available at www.jneurosci.org as supplemental material).
Macrophages secrete Aβ degrading enzymes
To first address whether Aβ42 degradation induced by macrophages requires a physical association with the cells, we cultured macrophages on the bottom of a culture dish that was physically separated from the PDAPP brain section by a filter. We then measured Aβ42 remaining in the brain section and released into the media (Fig. 5A,B). A significant decrease in the level of Aβ42 remaining in the brain section or released into the media was observed. Less than 50% of the Aβ42 could be detected in the brain section, and nearly 90% of the Aβ42 that was released into the media was degraded even when macrophages were prevented from physically associating with the brain section itself, suggesting that Aβ42 degradation induced by macrophages does not require a physical association with the cells (Fig. 5A,B). We next collected conditioned media from macrophages (MCM) that had been in culture for 96 h and incubated the MCM with either human Aβ42 or PDAPP brain sections (Fig. 5C–E). When MCM was incubated with human Aβ42 or PDAPP brain sections, there was a significant decrease in the amount of Aβ42 that could be measured. Heat treatment of MCM (90°C, 15 min) completely eliminated the observed Aβ degrading activity (Fig. 5E).
Figure 5.

Macrophages secrete Aβ degrading enzymes. Macrophages were physically blocked from associating with PDAPP brain sections by using the modified ex vivo assay (as described in Materials and Methods). A, B, Aβ42 remaining in the sections (A) or released into the media (B) after 96 h of incubation with (Macs) or without (Control) macrophages was measured by ELISA and are presented as a percentage of control value. n = 6 sections per treatment; ***p < 0.001 versus control. Representative data from one experiment that was repeated three times with similar results. PDAPP mouse brain sections (10 μm) were incubated with conditioned media from macrophages (MCM) or serum-free media (Control) for 96 h. C, D, Aβ remaining in the brain sections (C) or released into the media (D) was measured by ELISA and plotted as a percentage of control value. E, MCM spiked with human Aβ42 (Anaspec; 1 ng/ml) efficiently degrades Aβ42. The Aβ42 degradation activity of MCM is lost after boiling (B-MCM). n = 6 sections per treatment; ***p < 0.001 versus medium only control; ###p < 0.001 versus MCM plus Aβ. Representative data from one experiment that was repeated three times with similar results. Error bars represent the SE measurement.
Secretion of matrix metalloproteinase-9 is associated with macrophage-dependent Aβ degradation
Multiple proteases have been reported to degrade Aβ, and each protease appears to have cleavage sites that generate unique Aβ fragments (Roher et al., 1994; Howell et al., 1995; Backstrom et al., 1996; Eckman et al., 2001; Kurochkin, 2001; Carson and Turner, 2002). We incubated freshly dissolved synthetic human Aβ42 with macrophages for 2 and 72 h and then analyzed the culture medium and cell pellets by MALDI time-of-flight mass spectrometry. Incubation of Aβ42 with macrophages resulted in a significant decrease in the levels of full-length Aβ42 and with the concomitant appearance of several Aβ fragments in the media (Aβ1–19, 1–20, 1–22, 1–23, 1–28, 1–34, 1–38) and cell pellet (Aβ1–38, 4–42). These Aβ fragments represent characteristic cleavage patterns resulting from the activity of MMP-9, neprilysin, insulin-degrading enzyme, endothelin-converting enzyme, and other enzymes (supplemental Fig. 9, available at www.jneurosci.org as supplemental material). Given the pattern and sequence of Aβ peptides generated by macrophages after incubation with full-length Aβ42 and a recent report demonstrating that MMP-9 can degrade fibrillar Aβ (Yan et al., 2006), we investigated whether MMP-9 could play a role in the macrophage-dependent degradation of Aβ42 that we observed. When macrophages were coincubated with an anti-MMP-9 antibody and then plated on top of PDAPP brain sections, there was a significant reduction in the ability of macrophages to degrade Aβ, both in the section and in the media (Fig. 6A,B). Coincubation with an anti-MMP-9 antibody reduced the ability of macrophages to degrade Aβ by ∼50% (Fig. 6). Additionally, when an MMP-9 inhibitor (MMP-2/9 inhibitor I, 10 μm, 96 h) was added to the cultures, there was a modest but significant inhibition in the ability of macrophages to degrade Aβ (Fig. 6C,D.)
Figure 6.
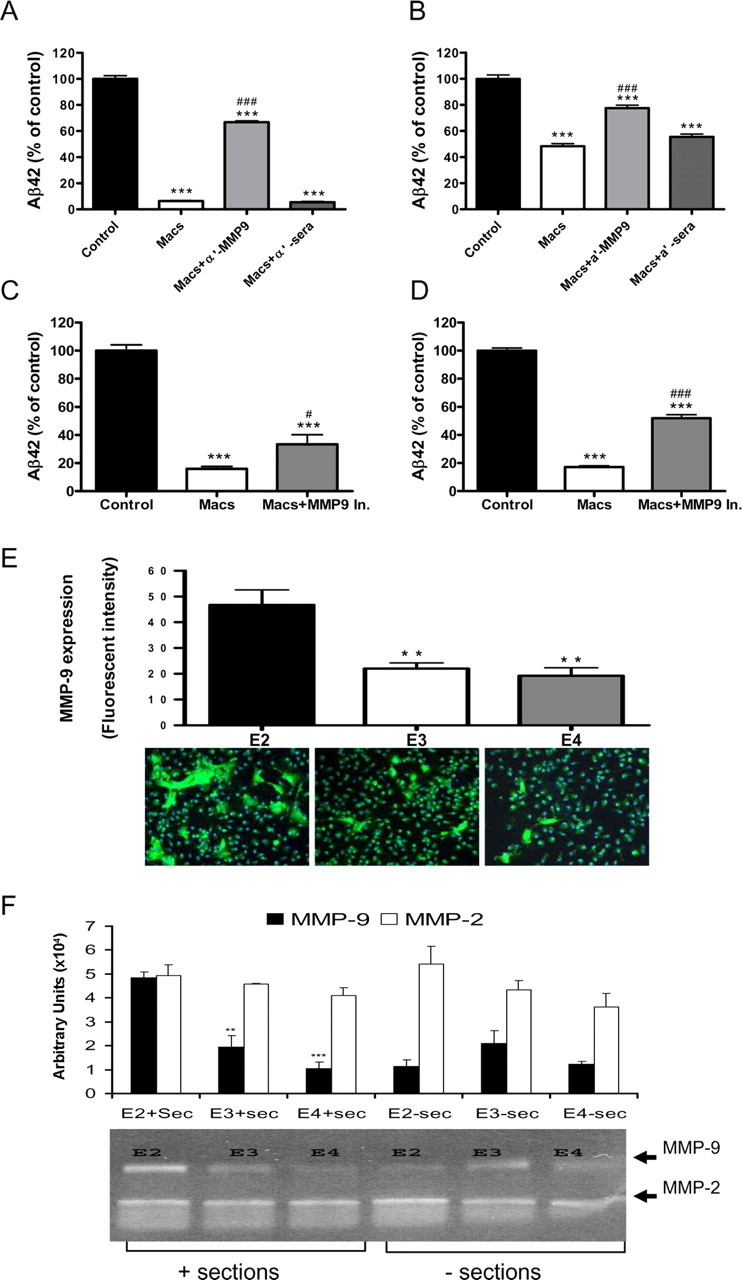
Secretion of MMP-9 is associated with macrophage-dependent Aβ degradation. A, B, PDAPP brain sections were incubated with media alone (Control), macrophages (Macs), macrophages plus a polyclonal rabbit anti-mouse MMP-9 antibody (Macs + a′-MMP-9; 50 μg/ml), or macrophages plus an anti-rabbit sera (Macs + a′-sera; 50 μg/ml) for 96 h. C, D, PDAPP brain sections were incubated with media alone (Control), macrophages (Macs), or macrophages plus an MMP-9 inhibitor (Macs + MMP-9 In.; 10 μm) for 96 h. Aβ42 levels in the sections (A, C) or media (B, D) were measured by ELISA and are presented as a percentage of control value. n = 6 sections per treatment; ***p < 0.001 versus Control; #p < 0.05, ###p < 0.001 versus Macs. Expression of MMP-9 (green) in macrophages isolated from mice expressing human apoE2 (E2), apoE3 (E3), or apoE4 (E4). E, MMP-9 immunoreactive cells were quantified using Image-Pro Plus software. Cell nuclei visualized by DAPI (blue). Original magnification, 20×. **p < 0.01 versus E2. F, Gelatin-substrate zymography and quantitative analysis of MMP-9 and MMP-2 activity in media from apoE2 (E2)-, apoE3 (E3)-, or apoE4 (E4)-expressing macrophages incubated with (+ sections) or without (− sections) PDAPP brain sections. n = 6 sections per treatment; **p < 0.01, ***p < 0.001 versus apoE2 macrophages. Representative data from one experiment that was repeated three times with similar results. Error bars represent the SE measurement.
We next determined if there was a difference in the level of MMP-9 protein expressed by macrophages expressing different human apoE isoforms using IHC. We observed significantly more MMP-9 protein in apoE2-expressing macrophages when compared with apoE3- or apoE4-expressing macrophages (Fig. 6E). We next measured the activity of MMP-9 in the media after macrophages from each of the three human apoE isoforms were incubated with or without a brain section from a very old PDAPP mouse using gelatin substrate zymography (Fig. 6F). Although the activity of MMP-9 was similar when measured in the media of cultures prepared from human apoE 2, 3, or 4 mice, there was a significant increase in the activity of MMP-9 in apoE2-expressing macrophages that had been coincubated with brain sections from very old PDAPP transgenic mice (Fig. 6F). In contrast, there was no significant difference in MMP-2 activity under the same incubation conditions (Fig. 6F).
Discussion
We have demonstrated that peritoneal macrophages very efficiently degrade soluble, insoluble/aggregated as well as more fibrillar forms of Aβ (i.e., thioflavine-S positive amyloid). Our findings confirm and extend a recent report by Majumdar et al. (2008), contrasting the ability of microglia which incompletely degrade fibrillar Aβ with that of macrophages. Like microglia, macrophages actively internalize Aβ42 via a phagocytic mechanism and deliver it to lysosomes, where in contrast to microglia they nearly completely degrade all of the internalized Aβ (Fig. 1A). However, using our ex vivo assay it is also clear that macrophage-induced Aβ degradation is due in good measure to secretion of one or more proteases (see below), because physical contact of macrophages with the Aβ-containing tissue sections is not required for the robust Aβ/amyloid degradation. Moreover, MCM (devoid of intact macrophages) is quite efficient at degrading Aβ, and this Aβ-degrading activity is completely heat sensitive (Fig. 5).
We also delineated an important role for macrophage apoE expression in the ability of macrophages to degrade soluble and insoluble Aβ. Compared with macrophages prepared from WT mice, those prepared from apoE-deficient mice displayed a markedly reduced ability to degrade Aβ over an identical time course. Moreover, the addition of anti-apoE antisera also reduced Aβ degradation in our ex vivo assay (Fig. 3E,F). It is important to emphasize that apoE-deficient macrophages can still degrade Aβ but at a markedly reduced rate. Finally, the ability of wild-type mouse macrophages to degrade Aβ is blocked by coincubation with RAP, suggesting that one or more LDL receptors, which are well known receptors for apoE, are involved. Moreover, RAP was more effective at inhibiting the degradation of insoluble Aβ present in the PDAPP brain sections when compared with Aβ released into the media (Fig. 3G,H), indicating a possible difference in the mechanism(s) by which macrophages degrade Aβ released into the media and Aβ remaining in sections (e.g., cellular uptake vs secreted protease).
Because the ε4 allele of APOE represents a major risk factor for late-onset AD, we investigated the ability of macrophages prepared from human apoE-targeted replacement transgenic mice to degrade Aβ. Our data demonstrate that apoE2-expressing macrophages were substantially more efficient at degrading Aβ than either apoE3- or apoE4-expressing macrophages (E2 ≫ E3 > E4) (Fig. 4A–C). ApoE2-expressing macrophages also more efficiently degrade Aβ when directly injected into the cerebral cortex of old PDAPP mice, confirming the apoE isoform-dependent nature of the underlying mechanism in vivo. Finally, given the ability of macrophages to degrade both nonfibrillar as well as fibrillar Aβ plaques/deposits, along with the specific Aβ peptides produced after incubation of Aβ42 with macrophages or MCM, we examined the possible role of known Aβ-degrading enzymes, but especially MMP-9, which has in contrast to neprilysin and insulin-degrading enzyme, recently been shown to degrade fibrillar Aβ42 (Yan et al., 2006) and is known to be secreted from various cells including macrophages, astrocytes, and microglia (Gottschall et al., 1995; Muir et al., 2002). Additionally, the expression of MMP-9 has also been observed to be increased in the brain of AD patients (Backstrom et al., 1992; Lorenzl et al., 2003), indicating that MMP-9 may play an important role in AD pathology. We observed that cellular MMP-9 expression measured by IHC was indeed apoE isoform-dependent (E2 > E3 > E4) (Fig. 6F). In contrast, MMP-9 activity measured in the media of cultured macrophages was similar regardless of the apoE isoform expressed (Fig. 6F). However, when the three human apoE isoform-expressing macrophages were incubated with Aβ-containing PDAPP brain sections, we observed a marked apoE isoform-dependent difference in MMP-9 activity in the media measured by gelatin zymography (Fig. 6F). There was no difference in MMP-2 activity under these same incubation conditions (Fig. 6F). Both an anti-MMP-9 antibody and MMP-9 inhibitor partially, but substantially, inhibited Aβ degradation induced by macrophages in both PDAPP sections and media. Thus, the apoE isoform-dependent degradation of Aβ appears to be due, at least in part, to MMP-9; and MMP-9 secretion from macrophages appears to be enhanced in an apoE isoform-dependent manner (Fig. 6F) after exposure to Aβ. Our finding that Aβ can induce MMP-9 secretion from macrophages is reminiscent of earlier work by Deb et al. (2003), demonstrating that Aβ stimulates MMP-9 production in astrocytes. Although the exact mechanism(s) underlying the apoE-dependent induction of MMP-9 expression and secretion observed in our experiments is unknown, we have recently found that direct exposure of BV2 cells (a microglia-like cell line) to exogenous apoE increases the expression and secretion of MMP-9 (F. Liu and S. Paul, unpublished observations).
A recent report by Guo et al. (2006) confirms that Aβ exposure stimulates MMP-9 secretion from neonatal astrocytes and further confirms that the addition of exogenous apoE4 attenuates Aβ-induced secretion of MMP-9. There is increasing evidence that MMPs, such as MMP-9, play important roles in regulating extracellular Aβ levels in brain (Yin et al., 2006), and our data demonstrating that apoE isoforms differentially determine the level of MMP-9 secretion from macrophages suggests that the reported effects of various apoE isoforms on amyloid deposition in vivo (Holtzman et al., 1999, 2000; Fagan et al., 2002; DeMattos et al., 2004; Dodart et al., 2005) may be due in part to differences in MMP-9 activity or secretion from apoE-expressing cells, including astrocytes, microglia, or macrophages. However, additional yet to be determined mechanisms may underlie the ability of macrophages to robustly degrade Aβ either in vitro or in vivo. Further work will be required to elucidate exactly how the three apoE isoforms differentially impact the expression and secretion of MMP-9 and potentially other Aβ-degrading enzymes.
Finally, our data further confirm that circulating macrophages are incredibly efficient Aβ-degrading cells and establish an important role for apoE and MMP-9 in mediating Aβ degradation and clearance by macrophages. Given recent data suggesting that bone marrow-derived macrophages and microglia play an important role in β-amyloid deposition and senile plaque formation in APP transgenic mouse models of AD (Malm et al., 2005; Simard et al., 2006), it is tempting to speculate that macrophage-mediated Aβ clearance in brain may explain, at least in part, the important role of apoE in determining the genetic risk and pathogenesis of AD.
Footnotes
We thank Prof. Guojun Bu for the generous gift of RAP, Dr. Shaoyou Chu and Michail Esterman for assistance with confocal microscopy and image analysis, Dr. Ronald DeMattos and Dr. Xiyun Chai for manuscript discussion, and Jeff Hanson for assistance with Image-Pro Plus software.
References
- Backstrom JR, Miller CA, Tökés ZA. Characterization of neutral proteinases from Alzheimer-affected and control brain specimens: identification of calcium-dependent metalloproteinases from the hippocampus. J Neurochem. 1992;58:983–992. doi: 10.1111/j.1471-4159.1992.tb09352.x. [DOI] [PubMed] [Google Scholar]
- Backstrom JR, Lim GP, Cullen MJ, Tökés ZA. Matrix metalloproteinase-9 (MMP-9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid-β peptide (1–40) J Neurosci. 1996;16:7910–7919. doi: 10.1523/JNEUROSCI.16-24-07910.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM. Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition. Nat Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B, Paul SM. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 1999;96:15233–15238. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest. 1985;76:1501–1513. doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson JA, Turner AJ. β-amyloid catabolism: roles for neprilysin (NEP) and other metallopeptidases? J Neurochem. 2002;81:1–8. doi: 10.1046/j.1471-4159.2002.00855.x. [DOI] [PubMed] [Google Scholar]
- Chung H, Brazil MI, Soe TT, Maxfield FR. Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer's amyloid β-peptide by microglial cells. J Biol Chem. 1999;274:32301–32308. doi: 10.1074/jbc.274.45.32301. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Jr, Rimmler JB, Locke PA, Conneally PM, Schmader KE. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- Davies JQ, Gordon S. Isolation and culture of murine macrophages. Methods Mol Biol. 2005;290:91–103. doi: 10.1385/1-59259-838-2:091. [DOI] [PubMed] [Google Scholar]
- Deb S, Wenjun Zhang J, Gottschall PE. β-Amyloid induces the production of active, matrix-degrading proteases in cultured rat astrocytes. Brain Res. 2003;970:205–213. doi: 10.1016/s0006-8993(03)02344-8. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW, Harmony JA, Aronow BJ, Bales KR, Paul SM, Holtzman DM. ApoE and clusterin cooperatively suppress Aβ levels and deposition: evidence that ApoE regulates extracellular Aβ metabolism in vivo. Neuron. 2004;41:193–202. doi: 10.1016/s0896-6273(03)00850-x. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Marr RA, Koistinaho M, Gregersen BM, Malkani S, Verma IM, Paul SM. Gene delivery of human apolipoprotein E alters brain Aβ burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2005;102:1211–1216. doi: 10.1073/pnas.0409072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer's amyloid β peptide by endothelin-converting enzyme. J Biol Chem. 2001;276:24540–24548. doi: 10.1074/jbc.M007579200. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine ApoE markedly alters A β metabolism before and after plaque formation in a mouse model of Alzheimer's disease. Neurobiol Dis. 2002;9:305–318. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogueet L, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Gelfanova V, Higgs RE, Dean RA, Holtzman DM, Farlow MR, Siemers ER, Boodhoo A, Qian YW, He X, Jin Z, Fisher DL, Cox KL, Hale JE. Quantitative analysis of amyloid-β peptides in cerebrospinal fluid using immunoprecipitation and MALDI-Tof mass spectrometry. Brief Funct Genomic Proteomic. 2007;6:149–158. doi: 10.1093/bfgp/elm010. [DOI] [PubMed] [Google Scholar]
- Gottschall PE, Yu X, Bing B. Increased production of gelatinase B (matrix metalloproteinase-9) and interleukin-6 by activated rat microglia in culture. J Neurosci Res. 1995;42:335–342. doi: 10.1002/jnr.490420307. [DOI] [PubMed] [Google Scholar]
- Guo S, Wang S, Kim WJ, Lee SR, Frosch MP, Bacskai BJ, Greenberg SM, Lo EH. Effects of apoE isoforms on β-amyloid-induced matrix metalloproteinase-9 in rat astrocytes. Brain Res. 2006;1111:222–226. doi: 10.1016/j.brainres.2006.06.041. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, Chang LK, Sun Y, Paul SM. Expression of human apolipoprotein E reduces amyloid-β deposition in a mouse model of Alzheimer's disease. J Clin Invest. 1999;103:R15–R21. doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell S, Nalbantoglu J, Crine P. Neutral endopeptidase can hydrolyze β-amyloid(1–40) but shows no effect on β-amyloid precursor protein metabolism. Peptides. 1995;16:647–652. doi: 10.1016/0196-9781(95)00021-b. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and A β42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat Med. 2004;10:719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- Kurochkin IV. Insulin-degrading enzyme: embarking on amyloid destruction. Trends Biochem Sci. 2001;26:421–425. doi: 10.1016/s0968-0004(01)01876-x. [DOI] [PubMed] [Google Scholar]
- Leber TM, Balkwill FR. Zymography: a single-step staining method for quantitation of proteolytic activity on substrate gels. Anal Biochem. 1997;249:24–28. doi: 10.1006/abio.1997.2170. [DOI] [PubMed] [Google Scholar]
- Lorenzl S, Albers DS, Relkin N, Ngyuen T, Hilgenberg SL, Chirichigno J, Cudkowicz ME, Beal MF. Increased plasma levels of matrix metalloproteinase-9 in patients with Alzheimer's disease. Neurochem Int. 2003;43:191–196. doi: 10.1016/s0197-0186(03)00004-4. [DOI] [PubMed] [Google Scholar]
- Majumdar A, Chung H, Dolios G, Wang R, Asamoah N, Lobel P, Maxfield FR. Degradation of fibrillar forms of Alzheimer's amyloid β-peptide by macrophages. Neurobiol Aging. 2008;29:707–715. doi: 10.1016/j.neurobiolaging.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm TM, Koistinaho M, Pärepalo M, Vatanen T, Ooka A, Karlsson S, Koistinaho J. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to β-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–142. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Muir EM, Adcock KH, Morgenstern DA, Clayton R, von Stillfried N, Rhodes K, Ellis C, Fawcett JW, Rogers JH. Matrix metalloproteases and their inhibitors are produced by overlapping populations of activated astrocytes. Brain Res Mol Brain Res. 2002;100:103–117. doi: 10.1016/s0169-328x(02)00132-8. [DOI] [PubMed] [Google Scholar]
- Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid β-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Paresce DM, Chung H, Maxfield FR. Slow degradation of aggregates of the Alzheimer's disease amyloid β-protein by microglial cells. J Biol Chem. 1997;272:29390–29397. doi: 10.1074/jbc.272.46.29390. [DOI] [PubMed] [Google Scholar]
- Pihlaja R, Koistinaho J, Malm T, Sikkilä H, Vainio S, Koistinaho M. Transplanted astrocytes internalize deposited β-amyloid peptides in a transgenic mouse model of Alzheimer's disease. Glia. 2008;56:154–163. doi: 10.1002/glia.20599. [DOI] [PubMed] [Google Scholar]
- Roher AE, Kasunic TC, Woods AS, Cotter RJ, Ball MJ, Fridman R. Proteolysis of A β peptide from Alzheimer disease brain by gelatinase A. Biochem Biophys Res Commun. 1994;205:1755–1761. doi: 10.1006/bbrc.1994.2872. [DOI] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roseset AD. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Clearing the brain's amyloid cobwebs. Neuron. 2001;32:177–180. doi: 10.1016/s0896-6273(01)00475-5. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, Maeda N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem. 1997;272:17972–17980. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- Swanson CJ, Perry KW, Schoepp DD. The mGlu2/3 receptor agonist, LY354740, blocks immobilization-induced increases in noradrenaline and dopamine release in the rat medial prefrontal cortex. J Neurochem. 2004;88:194–202. doi: 10.1046/j.1471-4159.2003.02125.x. [DOI] [PubMed] [Google Scholar]
- Takata K, Kitamura Y, Yanagisawa D, Morikawa S, Morita M, Inubushi T, Tsuchiya D, Chishiro S, Saeki M, Taniguchi T, Shimohama S, Tooyama I. Microglial transplantation increases amyloid-β clearance in Alzheimer model rats. FEBS Lett. 2007;581:475–478. doi: 10.1016/j.febslet.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Wegiel J, Wang KC, Imaki H, Rubenstein R, Wronska A, Osuchowski M, Lipinski WJ, Walker LC, LeVine H. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APP(SW) mice. Neurobiol Aging. 2001;22:49–61. doi: 10.1016/s0197-4580(00)00181-0. [DOI] [PubMed] [Google Scholar]
- Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26:4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan P, Hu X, Song H, Yin K, Bateman RJ, Cirrito JR, Xiao Q, Hsu FF, Turk JW, Xu J, Hsu CY, Holtzman DM, Lee JM. Matrix metalloproteinase-9 degrades amyloid-β fibrils in vitro and compact plaques in situ. J Biol Chem. 2006;281:24566–24574. doi: 10.1074/jbc.M602440200. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, Bateman R, Song H, Hsu FF, Turk J, Xu J, Hsu CY, Mills JC, Holtzman DM, Lee JM. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-β peptide catabolism. J Neurosci. 2006;26:10939–10948. doi: 10.1523/JNEUROSCI.2085-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]