

Abstract
Neurodegeneration is the irremediable pathological event occurring during chronic inflammatory diseases of the CNS. Here we show that, in experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis, inflammation is capable in enhancing glutamate transmission in the striatum and in promoting synaptic degeneration and dendritic spine loss. These alterations occur early in the disease course, are independent of demyelination, and are strongly associated with massive release of tumor necrosis factor-α from activated microglia. CNS invasion by myelin-specific blood-borne immune cells is the triggering event, and the downregulation of the early gene Arc/Arg3.1, leading to the abnormal expression and phosphorylation of AMPA receptors, represents a culminating step in this cascade of neurodegenerative events. Accordingly, EAE-induced synaptopathy subsided during pharmacological blockade of AMPA receptors. Our data establish a link between neuroinflammation and synaptic degeneration and calls for early neuroprotective therapies in chronic inflammatory diseases of the CNS.
Introduction
Glutamate-mediated excitotoxicity is emerging as a crucial determinant of neuronal injury in multiple sclerosis (MS). Accordingly, glutamate levels have been found to be significantly higher in the CSF (Stover et al., 1997; Sarchielli et al., 2003) and in the brains of MS patients (Srinivasan et al., 2005). Furthermore, glutamate clearance and receptor expression are impaired in MS brains (Pitt et al., 2000; Geurts et al., 2003, 2005; Vallejo-Illarramendi et al., 2006) and in animal models of the disease (Hardin-Pouzet et al., 1997; Pitt et al., 2000; Smith et al., 2000; Ohgoh et al., 2002), whereas glutamate receptor antagonists exert beneficial effects in experimental autoimmune encephalomyelitis (EAE) (Wallstrom et al., 1996; Bolton and Paul, 1997; Pitt et al., 2000; Smith et al., 2000) and in MS (Plaut, 1987) by limiting not only oligodendrocyte but also neuronal damage (Pitt et al., 2000; Smith et al., 2000). These findings, therefore, suggest that glutamate-mediated excitotoxicity may play a role in the pathogenesis of MS, as proposed in primarily neurodegenerative disorders (Choi, 1988).
Despite the critical importance attributed to synaptic changes in the pathophysiology of MS, to date no study investigated in a straightforward manner the effects of the immuno-mediated attack of myelin antigens on synaptic properties of central neurons. In the present work, therefore, we wanted to study synaptic functioning in the brains of mice with myelin oligodendrocyte glycoprotein (MOG)-induced EAE, a well established animal model of MS. We selected the striatum for our investigation because this subcortical brain area is particularly prone to develop neurodegenerative damage in the course of MS (Bakshi et al., 2002; Bermel et al., 2003).
Materials and Methods
EAE induction and clinical score.
As described previously (Pluchino et al., 2003), chronic-progressive EAE was induced in 6- to 8-week-old C57BL/6 female mice by subcutaneous immunization with MOG 35–55 peptide. Body weight and clinical score (0, healthy; 1, limp tail; 2, ataxia and/or paresis of hindlimbs; 3, paralysis of hindlimbs and/or paresis of forelimbs; 4, tetraparalysis; 5, moribund or death) were recorded daily. In some experiments, EAE mice received twice daily intraperitoneal injections of NBQX (30 mg/kg, dissolved in saline, 0.9% NaCl) or of vehicle (n = 8 mice per group) for 10 consecutive days starting from 10 d post-immunization (dpi). Additional information is provided in supplemental Methods (available at www.jneurosci.org as supplemental material).
Electrophysiology.
Mice were killed by cervical dislocation under halothane anesthesia, and corticostriatal coronal slices (200 μm) were prepared from fresh tissue blocks of the brain using a vibratome (Centonze et al., 2005, 2007). A single slice was then transferred to a recording chamber and submerged in a continuously flowing artificial CSF (34°C, 2–3 ml/min) gassed with 95% O2–5% CO2. The composition of the control solution contained the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose, and 25 NaHCO3.
Whole-cell patch-clamp recordings were made with borosilicate glass pipettes (1.8 mm outer diameter; 2–4 MΩ), in voltage-clamp mode, at the holding potential of −80 mV. The recording pipettes were filled with internal solution of the following composition (in mm): 125 K+-gluconate, 10 NaCl, 1.0 CaCl2, 2.0 MgCl2, 0.5 BAPTA, 19 HEPES, 0.3 GTP, and 1.0 Mg-ATP, adjusted to pH 7.3 with KOH. Bicuculline (10 μm) was added to the perfusing solution to block GABAA-mediated transmission.
To study the effects of microglia on striatal excitatory and inhibitory transmission, microglia was gently placed onto the surface of a striatal slice, submerged in a continuously oxygenated recording chamber for 30–60 min before the electrophysiological recordings. In the case of BV2 microglia cell line, the presence of microglial cells over the slices was monitored with a 40× water-immersion objective combined with an infrared filter and, in some instances at the end of the experiment, by using a fluorescent microscope. Additional information is provided in supplemental Methods (available at www.jneurosci.org as supplemental material).
GluR1 and GluR1 pSer845 detection in postsynaptic membranes.
For postsynaptic density (PSD) isolation, a previously described method was slightly modified (Gardoni et al., 2006). Brains were removed within 30 s after decapitation, and hippocampus and striatum were dissected and separately homogenized in homogenization buffer (in mm: 320 sucrose, 1 HEPES, pH 7.4, 1 MgCl2, 1 EDTA, 1 NaHCO3, 1 PMSF, 1 Na3VO4, 20 β-glycerophosphate, 5 NaF, and protease inhibitor cocktail; Sigma) with 10 strokes of a tight-fitting glass Dounce tissue grinder (7 ml; Wheaton). Cell debris and nuclei were removed by centrifugation at 1000 × g. The resulting supernatant was centrifuged at 3000 × g for 15 min to obtain a crude membrane fraction. The pellet was resuspended in 1 mm HEPES containing proteases inhibitors in a tight-fitting glass Dounce tissue grinder (7 ml; Wheaton) and centrifuged at 100,000 × g for 1 h. The resultant pellet was resuspended in buffer containing 75 mm KCl, 1% Triton X-100, and proteases inhibitors and centrifuged at 100,000 × g for 1 h. The supernatant was stored and referred to as Triton X-100-soluble fraction (TSF). The final pellet was resuspended in a glass–glass potter in 20 mm HEPES and proteases inhibitors. This last fraction was referred as Triton X-100-insoluble fraction. Additional information is provided in supplemental Methods (available at www.jneurosci.org as supplemental material).
Crude synaptosomal pellet (P-2) preparation.
Mice were killed by cervical dislocation, and the brains were rapidly removed to prepare the P-2 fraction. The striata were pooled (two mice for each experimental condition) and homogenized in 3.6 ml of homogenization buffer (0.32 m sucrose, 4 mm HEPES, pH 7.4, 1 mm EGTA, and 1 mm PMSF with protease inhibitors). The homogenate was spun at 1000 × g at 4°C for 10 min to remove nuclei and cell debris. The resulting supernatant was centrifuged at 12,000 × g at 4°C for 15 min. The pellet was resuspended in 2.5 ml of homogenization buffer and centrifuged (15 min, 13,000 × g, 4°C) to obtain the final P-2 fraction.
Flow cytometry in crude synaptosomal preparation (P-2 fraction).
The distribution of phospholipids in the plasma membrane changes during early steps of apoptosis, and this involves the translocation of the membrane phospholipid phosphatidylserine (PS) from the internal layer to the external layer of cell membrane. In presence of Ca2+, Annexin V has a high specificity and affinity for PS. Thus, the binding of Annexin V to membrane with exposed PS provides a highly sensitive method for detecting early cellular apoptosis. Emerging data from neurological studies indicate that, in neuron, sublethal apoptotic activity can lead to a limited form of apoptosis in synapses, thus causing a synaptic failure without neuronal cell death.
With this preamble, we have applied a flow cytometry method on P-2 fraction preparation, and we have used the Annexin-V to detect synaptosomal particles with exposed PS and viability marker calcein AM to analyze only “viable synaptosomes.”
The P-2 fraction was resuspended in binding buffer (in mm: 10 HEPES, pH 7.4, 150 NaCl, 5 KCl, 5 MgCl2, and 1.8 CaCl2). For dye labeling, dye diluted in binding buffer was added to 0.1 ml aliquots of P-2, incubated for 10 min at room temperature, and then diluted in PBS (final volume, 0.5 ml) for immediate flow cytometry analysis. For calcein AM, final dye concentration was 100 nm, and, for Annexin V-phycoerythrin (PE), final concentration was 1 mg/ml.
P-2 particle fluorescence was acquired on a fluorescence-activated cell sorter, and analyzed with logarithmic amplification. Polystyrene beads (1.4 and 4.5 μm) were run on the instrument to standardize forward scatter measurements. Particles whose sizes were 1.4 and 4.5 μm were analyzed according to previous studies (Gylys et al., 2004a,b). Analysis was performed using Flowjo software, version 8.4.
Annexin-V-PE was purchased from BD Pharmingen(catalog #TM 556421), and calcein AM was obtained from Invitrogen (catalog #C3100MP).
Preparation of CD3+ cells from EAE mice.
After aseptically removing spleens from donor EAE mice (25 dpi), splenocyte suspensions were prepared by mechanically teasing the spleen with the blunt end of a 10 ml plastic syringe plunger in a Petri dish, then passing suspensions through a BD Falcon 70 μm cell strainer (BD Biosciences-Discovery Labware) to remove debris, and finally suspending the cells in 1% fetal bovine serum, Roswell Park Memorial Institute 1640. From unfractionated splenocytes, CD3+ T-cell lymphocytes were obtained by magnetic-activated cell sorting (Miltenyi Biotec; mCD3 isolation kit). Pure T cells, 5 × 103, were then placed onto a single slice and incubated for 30–60 min before the electrophysiological experiments.
Preparation and activation of BV2 microglia cell line.
The BV2 immortalized murine microglial cell line was provided by Dr. F. Aloisi (Department of Cell Biology and Neuroscience, Istituto Superiore di Sanità, Rome, Italy). BV2 cells were infected with a retrovirus codifying the green fluorescent protein gene (supplemental Methods, available at www.jneurosci.org as supplemental material) and treated for 24 h with Th1-specific proinflammatory cytokines [100 U/ml IL1-β (Euroclone), 200 U/ml tumor necrosis factor α (TNFα) (Peprotech), and 500 U/ml interferon γ (IFNγ) (Becton Dickinson) (Th1 mix)] known to peak during the acute phase of EAE (Furlan et al., 1999). Then, cells were placed onto a single slice (30–60 min), and whole-cell patch-clamp recordings were made as above. Additional information is provided in supplemental Methods (available at www.jneurosci.org as supplemental material).
Preparation and activation of primary microglia cell line.
Primary microglia cultures were obtained from C57BL/6 mice (postnatal days 0–2) (supplemental Methods, available at www.jneurosci.org as supplemental material). Cells were tested for purity by flow cytometry in each experiment. Briefly, primary microglial cells were stained for CD11b (BD Biosciences) detection. Then, cells were analyzed on a FacsCanto (BD Biosciences), and 50,000 events were acquired. For quantitative real-time experiments (n = 3 independent preparations), cells were plated on 35 mm dishes and treated with cytokines for 24 h. Cells were activated with the Th1 mix (see above). Cells treated with anti-inflammatory cytokines (in ng/ml: 10 IL4, 10 IL5, and 10 IL13; all R & D Systems) (Th2 mix) and saline were used as additional controls. Then, total mRNA was extracted as described, and the TNFα and Iba1 mRNA levels were evaluated by using the LightCycler 480 System (Roche) as described. Histogram showed the mean ± SEM of three independent preparations.
In situ hybridization.
In situ hybridization was performed according to standard method as described previously (Muzio et al., 2002). Briefly, 10-μm-thick brain sections were postfixed 15 min in 4% paraformaldehyde and then washed three times in PBS. Slides were incubated in 0.5 mg/ml Proteinase K in 100 mm Tris-HCl, pH 8, 50 mm EDTA for 10 min at 30°C. This was followed by 15 min in 4% paraformaldehyde. Slices were then washed three times in PBS and then washed in H2O. Sections were incubated in 0.1 m triethanolamine, pH 8, for 5 min. Finally, 400 μl of acetic anhydride was added two times for 5 min each. Then, sections were rinsed in H2O for 2 min and air dried. Hybridization was performed overnight at 60°C with α-UTP33-labeled riboprobe at a concentration of 106 cpm/slide. The following day, sections were rinsed in 5× SSC for 5 min and then washed in 50% formamide, 2× SSC for 30 min at 60°C. Then slides were incubated in 20 mg/ml ribonuclease-A (Roche) in 0.5 m NaCl, 10 mm Tris-HCl, pH 8, 5 mm EDTA for 30 min at 37°C. Sections were washed in 50% formamide, 2× SSC for 30 min at 60°C, and then slides were rinsed two times in 2× SSC. Finally, slides were dried by using ethanol series. LM1 (GE Healthcare) emulsion was applied in a dark room, according to the instructions of the manufacturer. After 3 weeks, sections were developed in a dark room, counterstained with 4′,6′-diamidino-2-phenylindole (DAPI), and mounted with DPX (BDH). Arc/Arg3.1 riboprobe was kindly provided by Dr. Mucke (Gladstone Institute of Neurological Disease, San Fransisco, CA) (Palop et al., 2005). EAE and healthy control (HC) brains (n = 3 per each group) were used for the in situ hybridization study. Microphotographs of one section every 280 μm, ranging from bregma +1.2 to bregma −0.4, were digitalized in dark-field light microscopy (Olympus BX51 and 4× objective) by using a CCD camera (Leica). To confirm the specificity of the different RNA probes, sense strand RNA probes (showing no signal) were used as negative controls.
Laser microdissection and real-time PCR.
Laser microdissection of the striatum was performed on three EAE 20 dpi and three control brains by using the laser-capture microdissector (AS LMD; Leica). Briefly, 25 μm coronal cryosections were generated starting from bregma +1.4 to bregma −0.4 of the brain. Sections were dehydrated after the ethanol series. Then, four rectangular sections (200 × 300 μm) of the caudate–putamen were microdissected from each slice. Finally, we collected a mean of 107 ± 10 dissections from each EAE 20 dpi brain and 117 ± 12 dissections from each control brain. Total RNA was extracted by using RNeasy Mini kit (Qiagen) according to the recommendations of the manufacturer, including DNase digestion. cDNA synthesis were performed using ThermoScript RT-PCR System (Invitrogen) and Random Hexamer (Invitrogen) according the instructions of the manufacturer in a volume of 20 μl. The LightCycler 480 System (Roche) and SYBR Green JumpStart Taq ReadyMix for High Throughput QPCR (Sigma) were used for the quantification of Arc/Arg3.1 gene by using the following primers: Arc/Arg3.1 forward, 5′GGCAGCGGCTGGAGCCTACAGAG3′; Arc/Arg3.1 reverse, 5′GCTCTTGGGCTGAGCTGGGGTG CT3′; CD45 forward, 5′CTCTCCTTGATTTCCAATGTGCTGTGTC3′ CD45 reverse, 5′GCTAAGGCGACAGA GGTGTCTGATGG3′; Iba1 forward, 5′GCAGGAAGAGAGGCTGGAGGGGATC3′ Iba1 reverse, 5′CTCT TAGCTCTAGGTGGGTCTTGGG3′; Tnfα forward, 5′GCCTCTTCTCATTCCTGCTTGTGGCAG3′; Tnfα reverse, 5′GACGTGGGCTACA GGCTTGTCACTCG3′. The following were used as housekeeping gene: Histone H3 gene H3 forward, 5′GGTGA AGAAACCTCATCGTTACAGGCCTGGTAC3′; H3 reverse, 5′CTGCAAAGCACCAATAGC TGCACTCTGGAAGC3′. Histogram showed the mean ± SEM of two independent experiments. The ABI prism 7700 Sequence detection system and TaqMan gene expression assay (Applied Biosystems) were used for the quantification of the following genes: IL13 (Applied Biosystems: Mm00434204_m1), IL4 (Applied Biosystems: Mm004452259_m1), and IL10 (Applied Biosystems: Mm00439616_m1) on laser-captured microdissections and TNFα (Applied Biosystems: Mm00443258_m1) on BV2 cells treated with cytokines. In these experiments, we used glyceraldehyde-3-phosphate dehydrogenase (Applied Biosystems: 4352339E) as housekeeping gene. Statistical analysis was performed using unpaired Student's t test.
Preparation of primary neuronal cell line.
Primary neuronal cultures were obtained from embryonic day 16.5 (E16.5) CD1 forebrains. Experimental details about the Western blot analysis, the PSD95 staining, and the real-time PCR experiments are provided in supplemental Methods (available at www.jneurosci.org as supplemental material).
5′-Lodo-2′-deoxyuridine administration and detection.
During DNA replication, in the S-phase of the cell cycle, proliferating cells are easily traceable by injecting mice with the thymidine analog 5′-lodo-2′-deoxyuridine (IddU) (Muzio et al., 2005). By varying the pulsing paradigm and the examination time points after pulsing, it is possible to quantify the percentage of proliferating cells located within the tissue (Takahashi et al., 1992, 1993). Indeed, for an asynchronously proliferating population of cells, the number of cells distributed along equivalent intervals of the cell cycle will be the same. Thus, a short pulse of IddU labels cells that are in the S-phase, but, with prolonged IddU administration, it is possible to label all the proliferating cells that enter the S-phase. The first intraperitoneal injection was done at the concentration of 100 mg/kg (IddU stock solution was done by diluting IddU in 0.007N NaOH at the concentration of 50 mg/ml) in 0.9% saline, and the following injections were done at the concentration of 70 mg/kg in 0.9% saline and then every 2 h. Animals were divided in three groups: the first one received IddU for 1 h, the second group for 10 h, and the last one for 12 h. A total of at least three EAE and three HC females per group were killed at each time point by anesthetic overdose and transcardially perfused with 4% paraformaldehyde in PBS, pH 7.2. Dissected brains were postfixed in the same solution for 12 h at +4°C and then cryoprotected for at least 24 h in 30% sucrose in PBS at +4°C. Serial 10 μm coronal cryosections were mounted directly onto Superfrost plus (Menzel-Glaser) slides. Then, sections were postfixed in 4% paraformaldehyde in PBS, pH 7.2, and washed three times with PBS. Samples were boiled in 10 mm sodium citrate, pH 6, for 5 min and allowed to cool down slowly. The sections were incubated in 2 m HCl for 20 min at room temperature to denature the DNA. This was followed by 10 min washing in buffer borate, pH 8.5, to equilibrate the pH. Then, sections were washed three times in PBS and blocked in PBS, 1 mg/ml BSA, 10% FBS, and 0.1% Triton X-100. Single and double immunofluorescence experiments were performed according the standard protocol. Briefly, we used the anti-pan halogen antibody (mouse clone B44; BD Biosciences) at 1:75 for IddU detection and the following antibodies for the double immunofluorescence experiments: anti-Iba1 (rabbit; Wako Bioproducts) at 1:400, anti-GFAP (rabbit; Dako) at 1:1000, and anti-neuronal-specific nuclear protein (NeuN) (mouse; Millipore Bioscience Research Reagents) at 1:1000. Antibodies were applied overnight at +4°C. Sections were rinsed three times in PBS, and anti-mouse Alexa fluor 488 or anti-rabbit Alexa fluor 546 were incubated for 2 h at room temperature. Finally, sections were washed three times with PBS, counterstained with DAPI at 1:15000, and coverslipped with Fluorsave (Calbiochem) before imaging. For the NeuN/IddU double immunofluorescence experiments, IddU was coupled with Alexa flour 488 fluorochrome by using the Invitrogen zenon labeling kit according to the recommendations of the manufacturer. One section every 280 μm of a brain region ranging from the anterior bregma +1.3 to the posterior bregma −0.3 was used to count single IddU and double-positive cells located within the caudate–putamen. Confocal (SP2; Leica) and light (BX51; Olympus) microscopy were used to analyze tissue staining. IddU-positive (IddU+) cells were counted within the caudate–putamen, and histograms showed the mean ± SEM value. IddU/Iba1, IddU/GFAP-double positive cells were counted on parallel sections, and histograms showed their percentage. Statistical analysis was performed using unpaired Student's t test.
Immunohistochemistry and immunofluorescence.
For immunohistochemistry experiments, sections were postfixed 5 min in 4% paraformaldehyde, followed by three washes in PBS. Antigen retrieval, when appropriate, was performed as indicated previously (Muzio et al., 2002). Endogenous peroxidase blocking reaction was obtained by incubating sections 20 min in the 3% methanol/H2O2 solution. Then, sections were washed for 5 min for three times in PBS and incubated in the blocking mix (1× PBS, 10% FBS, 1 mg/ml BSA, and 0.1% Triton X-100), for 1 h at room temperature. Anti-phospho-hystone3 (pH3) (rabbit; Millipore) at 1:200 was diluted in blocking mix and incubated at +4°C overnight. The following day, sections were washed in PBS for 5 min for three times. Anti-rabbit biotinylated (Vector Laboratories) at 1:200 was diluted in blocking mix and applied. Slides were washed three times in 1× PBS for 5 min and incubated in ABC (Vector Laboratories) for 2 h. Then the slides washed three times in 1× PBS and developed in 0.1 m Na-acetate buffer containing 0.25% of 3-amino-9ethyl-carbazole (Sigma). Sections were mounted, and light microscopy (BX51; Olympus) were used to analyze tissue staining. pH3+ cells were analyzed on three independent EAE 20 dpi brains. Double and single immunofluorescence experiments were done according to standard laboratory protocols, and the following antibodies were used on three independent EAE and three independent HC: α-CD3 (rat; Serotec) at 1:100, α-Iba1 (rabbit; Wako Bioproducts) at 1:400, and α-NeuN (mouse; Millipore Bioscience Research Reagents) at 1:1000. Primary antibodies were incubated overnight at +4°C. Then, sections were washed three times in PBS and incubated with appropriate secondary antibodies conjugated with Alexa fluor fluorochrome for 2 h at room temperature. Sections were rinsed three times in PBS, counterstained with DAPI, and coverslipped. One section every 280 μm of a brain region ranging from the anterior bregma +1.3 to the posterior bregma −0.3 was digitalized by using the confocal microscopy (SP2; Leica) and used for our analysis.
Golgi staining.
The single-section Golgi technique (Izzo et al., 1987) was performed on coronal sections (100 μm) containing striata from HC (n = 5), EAE (30 dpi; n = 5), and EAE mice chronically treated with NBQX (n = 5). Additional information is provided in supplemental Methods (available at www.jneurosci.org as supplemental material).
Results
Glutamate transmission in EAE mice
To address excitatory transmission in experimental MS, we recorded synaptic activity from EAE mice. The duration of glutamate-mediated spontaneous (sEPSCs) and miniature (mEPSCs) EPSCs was increased in striatal neurons of EAE mice. This alteration was evident both in the presymptomatic (7–10 dpi; n = 6 mice) and clinical phase of the disease (20–30 dpi; mean clinical score, 2.8; n = l4 mice) (n = at least 20 neurons for each electrophysiological parameter and experimental group). A slower decay phase accounted for increased sEPSC duration (Fig. 1 a–d), whereas sEPSC rise time and amplitude were unchanged in EAE (n = at least 20 neurons for each time point and experimental group; p > 0.05) (Fig. 1 e,f). Statistical analysis showed a direct correlation between sEPSC duration and sEPSC decay time (10 dpi, n = 20, r = 0.53, p = 0.01; 25 dpi, n = 21, r = 0.69, p = 0.0004). Also, the frequency of sEPSC increased at 10 and 25 dpi in EAE mice (n = at least 20 neurons for each time point and experimental group; p < 0.01) (Fig. 1 g,h).
Figure 1.
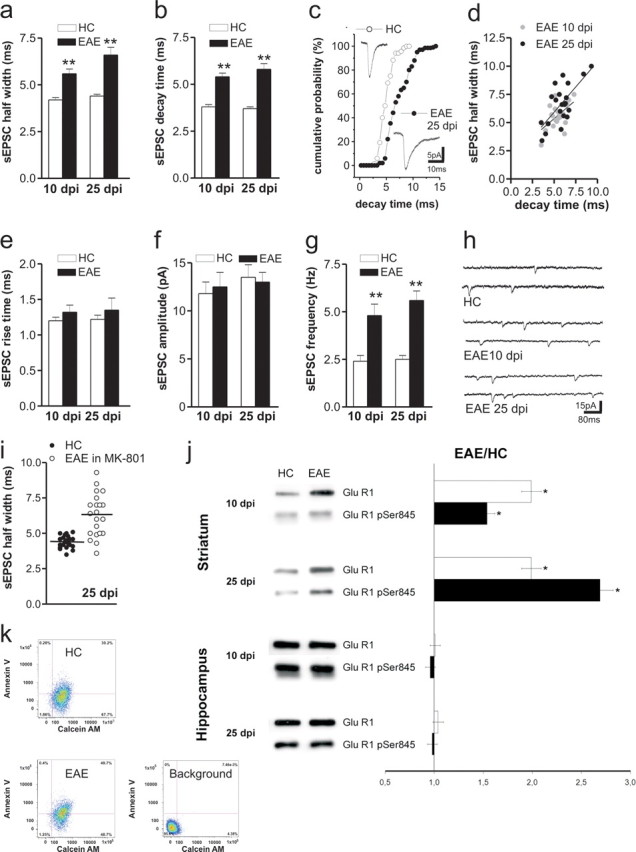
EAE alters glutamatergic transmission in the striatum. a, b, Half-width and decay time of glutamatergic sEPSCs recorded from striatal neurons increased in the presymptomatic (10 dpi) and in the acute (25 dpi) phase of EAE. **p < 0.01. c, Cumulative distributions of sEPSC decay time recorded in HC and in the acute stage of EAE. The electrophysiological traces are examples of sEPSC mean peak obtained by group analysis. d, Scatter plot of sEPSC half-width and sEPSC decay time recorded from neurons of EAE mice at 25 dpi. Pearson's test revealed a significant positive correlation. e, f, The graphs show that the sEPSC rise time and amplitude were unchanged in EAE at 10 and 25 dpi. g, The frequency of sEPSCs was increased at both preclinical and clinical stages of EAE. **p < 0.01. h, The electrophysiological traces are examples of sEPSCs (downward deflections) recorded from striatal neurons in control conditions and 10 and 25 dpi with MOG. i, The graph shows that pharmacological inhibition of NMDA receptors with MK-801 failed to normalize sEPSC half-width in EAE mice. j, EAE pathological process causes changes in GluR1 protein composition of PSD preparations. Left, Immunoblot of PSD proteins obtained from hippocampus and striatum of EAE mice at 10 and 25 dpi and control group animals (n = 3 for each experiment). Right, Densitometric quantification changes in gray values and SEM (EAE group/control group values). The histogram shows that the expression of GluR1 subunit of AMPA receptors and its phosphorylation at the Ser845 in isolated striatal postsynaptic densities of presymptomatic (10 dpi) and symptomatic (25 dpi) EAE mice was increased. *p < 0.01. k, Representative density plot showing calcein AM fluorescence versus Annexin V fluorescence for large synaptosomal particles (data were collected for 20,000 particles from each sample; 10,000 events are plotted). Percentage of total particles is shown for each quadrant. Particles positive for both markers are in the top right quadrant. Increase in Annexin V labeling was observed in striatum synaptosomes taken from EAE mice (25 dpi). Background, Untreated P-2 double labeled for Annexin V and calcein AM.
To see whether NMDA receptors mediated the observed changes of sEPSCs, we incubated striatal slices with MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate]. This NMDA receptor antagonist failed to affect sEPSCs in neurons from control mice (n = 8, p > 0.05 compared with pre-drug values for each sEPSC parameters), and to normalize sEPSC duration (n = 11, 96.4 ± 3%, p > 0.05 compared with pre-drug values and p < 0.01 compared with the healthy control (HC) group) and frequency (n = 11, 73.3 ± 3.1%, p < 0.05 compared with pre-drug values and to HC) in EAE mice at 25 dpi (Fig. 1 i). In contrast, application of the AMPA receptor antagonists NBQX (n = 5) or CNQX (n = 7) fully suppressed sEPSCs in both EAE and control mice (data not shown).
The changes in the shape of sEPSCs indicate a change in the number or in the kinetic properties of AMPA receptor channel complex. Of note, the activity of AMPA receptors is regulated by both expression and phosphorylation state of specific receptor subunits (Banke et al., 2000). Thus, we measured expression of GluR1 subunit of AMPA receptors and its phosphorylation at the Ser845 residue in isolated striatal postsynaptic densities of pre-symptomatic (10 dpi) and symptomatic (25 dpi) EAE mice (n = 3 mice for each time point). Western blot analysis showed that both total GluR1 and GluR1-p-Ser845 expression were up-regulated at 10 dpi and at 25 dpi in the striatum, whereas no difference was detectable for both GluR1 and GluR1-p-Ser845 expression in the hippocampus at both time points (Fig. 1 j). Similar findings were obtained when analyzing whole dissected striata at the following time points: 10 dpi, 25 dpi and 50 dpi (n = 3 mice for each time point) (supplemental Fig. 1 and Methods, available at www.jneurosci.org as supplemental material). We next asked whether the presynaptic compartment may be affected during EAE. To investigate presynaptic vesicles, we measured the synaptophysin expression levels on striatal micro-dissected tissue by Western blot. Synaptophysin expression levels were unchanged in 10 dpi and 25 dpi striatal extracts (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
To better characterize the synaptic degeneration in HC and EAE mice (25 dpi) we applied a cytofluorimetry-based method for analyzing in vitro nerve terminals to synaptosome preparation. By labeling crude striatal synaptosome preparations with Annexin V and with calcein AM before flow cytometry, we found a significant (p < 0.05) increase of synaptosomes positive for both markers in EAE (58 ± 9 vs 38 ± 8%; n = 4 mice), consistent with the occurrence of synaptic degeneration in these mice (Fig. 1 k). To rule out the possibility that PS exposure was a phenomenon related to neuronal death, we quantified the total number of striatal neurons. No significant difference was observed in EAE mice compared with HC (25 and 60 dpi; n = 3 for each time point) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Role of infiltrating lymphocytes in striatal synaptic alterations
The evidence that striatal glutamate transmission was altered since the presymptomatic stage of EAE was surprising, because peripheral immunization with MOG is reported to cause limited inflammatory involvement of the striatum (Politi et al., 2007). However, in the presymptomatic (10 dpi) and symptomatic phase of EAE, we found sparse inflammatory infiltrates in the striatum, mainly composed by CD3+ cells (Fig. 2 a). Of note, at 20–30 dpi, their number was higher within the striatum than within the cortex (20 dpi striatal infiltrates 11.4 ± 2.3 vs 5.1 ± 1.2 within the cortex; p < 0.05; n = 4 mice). In contrast, few or any infiltrating CD3+ cells were present during the chronic phase of EAE (60 and 90 dpi) (data not shown). Thus, we first investigated the potential involvement of CD3+ cells in the synaptic alterations of EAE. To this end, we preincubated striatal slices from control animals in the presence of CD3+ cells extracted from the lymph nodes of EAE (20–30 dpi) and HC mice. In slices incubated in the presence of CD3+ cells from EAE mice, we observed a dramatic alteration of sEPSCs (n = 13 for both groups), reminiscent of the defects seen in EAE. However, in EAE CD3+-treated slices, not only increased decay time but also increased rise time contributed to alter the kinetic of sEPSCs (p < 0.01 for both parameters). These data, therefore, suggest that these inflammatory cells induce changes of glutamate receptor function that are different from those observed in EAE brains, possibly involving both AMPA and NMDA receptors (Fig. 2 b).
Figure 2.

T lymphocytes alter synaptic transmission and cause microglia/macrophage activation in EAE. a, Inflammatory lesions in the striatum of EAE mice (20 dpi) were evaluated by double staining for CD3 (green in the box) or Iba1 (red in the box). b, The histogram shows that both rise and decay time of sEPSCs recorded from neurons in the presence of CD3+ cells were altered. The electrophysiological traces on the right are examples of sEPSC mean peak obtained by group analysis in control conditions (CD3+ cells from healthy controls) and in the presence of CD3+ cells extracted from EAE mice. **p < 0.01. c, Primary microglial cell cultures were generated starting from P2 C57BL/6 newborn. Primary microglia cells were shaken off and treated with either the Th1 or the Th2 mix for 24 h (n = 3 independent cell cultures). Then, total mRNA was extracted and used for the real-time PCR analysis. The histogram shows that Th1 mix treatment resulted in a dramatic upregulation of either Iba1 (4.3 ± 0.6; ***p < 0.001) or TNFα (3.15 ± 1.7; *p < 0.05) mRNA levels, markers of microglia/macrophage cell activation. Data represent the log2 of the fold changes (±SEM) with respect to the vehicle-treated cells. d, Striatal sections from HC and EAE mice (20, 30, and 60 dpi) were probed for IddU detection during 10 h of IddU administration. IddU+ cells (green) were distributed within either the dorsolateral or ventrolateral SVZ in both HC and EAE 60 dpi (arrows in first and fourth panels). However, EAE 20 dpi showed many IddU+ cells located within the striatal parenchyma (second panel). This phenotype was reduced at later time points as shown at either EAE 30 or 60 dpi. Scale bar, 150 μm. e, The mean ± SEM number of IddU+ cells were counted at each time point and plotted on histogram. ***p < 0.001. f, g, Cell counts revealed a mean number per section of 20 ± 2 (p < 0.0001) IddU+ cells at 8 dpi, 47 ± 2 (p < 0.0001) IddU+ cells at 14 dpi, 73 ± 16 (p < 0.01) IddU+ cells at 18 dpi, 280 ± 45 (p < 0.001) IddU+ cells at 20 dpi, 30 ± 14 (NS) IddU+ cells at 30 dpi, and 7 ± 2 (NS) IddU+ cells at 60 dpi. f, Confocal optical sections of parallel sections probed for Iba1 (red) and IddU (green) detection revealed that 45 ± 8% of the proliferating cells were Iba1+ at 8 dpi, 65 ± 8% of the proliferating cells were Iba1+ at either 12 or 14 dpi, 91 ± 3% of the proliferating cells were Iba1+ at either at 18 or 20 dpi, and 68 ± 11% of the proliferating cells were Iba1+ at either 25 or 30 dpi. Scale bar, 10 μm. h, Histogram showed mRNA fold changes ± SEM for Iba1 (4.3 ± 0.7; p < 0.01), CD45 (8.3 ± 2.7; p < 0.05), and TNFα (10.22 ± 0.9; p < 0.01). Total RNA was extracted from laser-captured microdissection of striatum, respectively, from HC and EAE 20 dpi brains and then used for quantitative real-time PCR. *p < 0.05; **p < 0.01.
Activated microglia reproduces the synaptic defects of EAE mice
Because sEPSCs alterations by activated CD3+ only partially resembled those induced by EAE mice, we investigated the effect of activated microglia on glutamate transmission. Indeed, activated microglia (BV2 microglia or primary microglia; 30–60 min before the electrophysiological recordings) deposited on striatal slices from control mice mimicked the alterations of glutamate transmission seen in EAE. Accordingly, activated microglia significantly (p < 0.01) increased the duration of sEPSCs (BV2 microglia, n = 16; primary microglia, n = 10) and of mEPSCs (BV2 microglia, n = 12, 5.8 ± 0.3 ms; primary microglia, n = 8, 6.2 ± 0.4 ms) by slowing their decay phases (Fig. 3 a–d). As also seen in EAE mice, MK-801 failed to normalize the duration of sEPSCs and mEPSCs recorded in the presence of activated microglia, implying changes of AMPA receptor function at the basis of this synaptic alteration (BV2 microglia, n = 8; primary microglia, n = 4; p > 0.05 compared with predrug values) (data not shown). Notably, non-activated BV2 or primary microglia produced no effect on the physiological properties of striatal sEPSCs recorded from control slices (n = at least 10 and p > 0.05 for each measure) (Fig. 3 a–d), whereas activated BV2 microglia applied on slices from EAE mice (25 dpi) failed to further increase sEPSC decay phase, indicating that EAE occluded the effects of activated microglia on this parameter (n = 8; p > 0.05) (Fig. 3 e).
Figure 3.

Activated microglia reproduces the synaptic defects of EAE mice. a, b, The histograms show that both sEPSC half-width and decay time increased in the presence of activated microglia (BV2 microglia and primary microglia) but not in the presence of nonactivated microglia. **p < 0.01. c, The electrophysiological traces are examples of sEPSC mean peak obtained by group analysis in the presence of activated and nonactivated BV2 microglia. d, Microglia failed to alter sEPSC rise time. e, Activated BV2 microglia failed to further increase sEPSC half-width and decay time in EAE mice.
Early and selective microglia/macrophage proliferation and activation in the striatum of EAE mice
Activated microglial/macrophage cells have been implicated recently in the pathophysiology of classical neurodegenerative diseases (Schwartz et al., 2006; Muzio et al., 2007). We hypothesized that infiltrating CD3+ T cells or macrophages may activate, through the release of cytokines, endogenous microglia cells, in turn contributing to synaptic alterations of EAE mice. Indeed, stimulation of microglia primary cultures with a proinflammatory Th1 cytokine mix (IL-1β, IFNγ, and TNFα), typical of EAE, but not with a Th2 cytokine mix (IL-4, IL-10, and IL-13), virtually absent in the striatum of EAE mice at the peak of inflammation (data not shown), resulted in a dramatic upregulation of both Iba1 (ionized calcium-binding adaptor molecule-1) and TNFα mRNA levels, markers of microglia/macrophage activation (Fig. 2 c). We then studied microglia/macrophage cell activation during ongoing EAE (starting from 8 to 60 dpi), by assaying cell proliferation within the striatum after administration of the S-phase tracer IddU. Because we cannot distinguish endogenous microglia from infiltrating macrophages, our cell counts are referred to the microglia/macrophage cell population. IddU was intraperitoneally administered (n = 3 mice for each group at each time point), and mice were killed 1, 10, or 12 h later (Fig. 2 d) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Cell proliferation was assayed on coronal sections in a region spanning from the anterior bregma +1.3 to the bregma −0.3. As described previously, control brains showed IddU+ proliferating cells mainly confined to the striatal ventricular lining in which adult neural precursors cells normally reside (Doetsch et al., 1999, 2002a,b). However, EAE brains revealed many IddU+ cells located throughout the entire striatal parenchyma (Fig. 2 d) and septum (data not shown). The mean number of IddU+ cells per sections showed that striatal cell proliferation greatly enhanced at 20 dpi (p < 0.001) (Fig. 2 d,e). Nevertheless, a significant number of proliferating striatal cells was detected also during the presymptomatic phase of the disease (8 dpi; p < 0.001) (Fig. 2 e). IddU incorporation reflects DNA duplication, but it is not a marker for cell division (Rakic, 2002). Thus, control and EAE mice at 20 dpi were injected for either 1 or 12 h, and the numbers of total labeled cells per section were compared with those obtained from 10 h experiments. Increased IddU+ cells per sections in the 10 and 12 h experiments indeed suggest that striatal cells underwent effective proliferation. Moreover, striatal cell proliferation was confirmed by the presence of the mitotic marker pH3 within the striatal parenchyma (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
The phenotype of proliferating cells was detected at each time point by double probing parallel sections with, respectively, Iba1 (a microglia/macrophage marker)/IddU, GFAP (an astrocyte marker)/IddU, and NeuN (a neuronal marker)/IddU. Double immunofluorescence revealed that the vast majority of striatal proliferating cells belonged to the microglia/macrophage lineage (Fig. 2 f,g). EAE 20 dpi brains showed few striatal GFAP/IddU double-positive cells (supplemental Fig. 5, available at www.jneurosci.org as supplemental material), whereas NeuN/IddU double-positive cells were never detected (data not shown).
Activation of microglial/macrophage cells is associated with the upregulation of surface receptors such as CD45, the major histocompatibility complex II, CD68, Iba1, and CD11c (Ponomarev et al., 2005). Activated microglia/macrophage cells are also able to release potentially cytotoxic molecules such as oxygen radicals and proinflammatory cytokines (Colton and Gilbert, 1987). Laser-captured microdissections of the striatum from EAE mice (20 dpi; n = 3 for each group) showed a statistically significant increase of Iba1, CD45, and TNFα mRNA levels (p < 0.01, p < 0.05, and p < 0.01 respectively) (Fig. 2 h) compared with control mice.
Together, these results suggest an early and transient microglia/macrophage cell activation/proliferation within striatal parenchyma after MOG immunization, whose onset precedes the appearance of clinical deficits and peaking during the clinical phase of the disease.
Role of TNFα in the synaptic defects of EAE mice
The effects of proliferating microglia/macrophage cells on striatal glutamate transmission might be mediated by a number of soluble factors released during activation, including inflammatory cytokines. Among these factors, TNFα has been shown recently to induce significant changes of glutamate transmission in the hippocampus, by altering the composition of glutamate AMPA receptor subunit (Stellwagen et al., 2005; Stellwagen and Malenka, 2006; Leonoudakis et al., 2008). TNFα mRNA content increased in coincidence with immunization-dependent microglia/macrophage cell activation (Fig. 2 h), suggesting that this cytokine may play a role in the synaptic defects induced by EAE and activated microglia/macrophage. Thus, we investigated whether TNFα could replicate in control slices these alterations. TNFα (3 h incubation) mimicked the effects of EAE and of activated microglia on sEPSC kinetic properties, by increasing decay time and duration of these synaptic events (n = 16; p < 0.01) (Fig. 4 a,b). As with activated primary microglia, TNFα failed to alter other parameters of glutamate transmission (data not shown) and did not increase further sEPSC duration in slices from acute EAE mice (n = 6; p > 0.05) (Fig. 4 c). Finally, to confirm the role of TNFα in the synaptic effects of activated microglia, we performed recordings in slices incubated with both activated primary microglia and TNF receptor (TNFR)–Ig (courtesy of Dr. Angelo Corti, San Raffaele Scientific Institute, Milan, Italy), to block the activity of endogenous TNFα. In this set of experiments, sEPSC frequency (HC, 2.4 ± 0.3 Hz; TNFα, 2.6 ± 0.4 Hz), amplitude (HC, 12.3 ± 1.1 pA; TNFα, 11.4 ± 1.4 pA), and kinetic properties (Fig. 4 d) were indistinguishable from those recorded in control conditions (n = 8; p > 0.05 for each parameter).
Figure 4.

TNFα reproduces the synaptic defects of EAE mice. a, The electrophysiological traces are examples of sEPSC mean peak obtained by group analysis in control conditions and in the presence of TNFα. b, The histogram shows that both sEPSC half-width and decay time were increased in the presence of TNFα. **p < 0.01. c, TNFα failed to further increase sEPSC half-width and decay time in EAE mice. d, Activated microglia failed to alter sEPSC kinetic properties after the blockade of endogenous TNFα activity with TNFR–Ig.
Role of Arc/Arg3.1 gene in striatal synaptic defects of EAE mice
We next tried to understand how inflammation could induce such a rapid change in synaptic transmission. We focused our attention on the group of the immediate early genes, because they are rapidly regulated during stimulation (Lyford et al., 1995). Among them, Arc/Arg3.1 gene has been shown to be regulated in the brain by neuroinflammation (Bonow et al., 2008) and to be implicated in the regulation of the AMPA receptor subunit GluR1 and in AMPA receptor-mediated synaptic transmission. Accordingly, downregulation of Arc/Arg3.1 gene expression has been convincingly associated with increased AMPA receptor-mediated synaptic transmission (Chowdhury et al., 2006; Shepherd et al., 2006).
To study whether inflammation could affect the Arc/Arg3.1 gene expression, primary neuronal cell cultures were generated from embryonic E16.5 cortical domains. Arc/Arg3.1 expression is relatively low in young neurons but increases markedly in older neurons (Shepherd et al., 2006). Twelve days after plating, neurons were treated with either a Th1 cytokine mix or saline for 3–24 h. After the removal of the stimulus, treated neurons did not show any morphological alteration when compared with sham-treated ones (data not shown) and maintained IFNγ and TNFα receptor expression (Fig. 5 a). Moreover, total protein extracts from Th1-treated neurons significantly showed the upregulation of GluR1, GluR1 Ser 845, and the postsynaptic scaffolding protein PSD95 (Fig. 5 b). Consistent with this result, acute Th1 treatments (n = 2 independent cultures) substantially increased PSD-95 surface expression (Fig. 5 c,e). Noticeably, Arc/Arg3.1 mRNA levels were significantly downregulated by the Th1 cytokine mix (Fig. 5 d). To confirm this result, Arc/Arg3.1 mRNA levels were also analyzed in the course of EAE. Coronal sections obtained from EAE (20–30 dpi) and matched HC groups were probed for Arc/Arg3.1 detection by in situ hybridization (n = 3 for each group). Sections from control animals showed Arc/Arg3.1 mRNA distributed within the cortical plate, hippocampus, and striatum (Fig. 5 f, left, and data not shown) as described previously (Chowdhury et al., 2006). However, Arc/Arg3.1 mRNA striatal expression levels were dramatically downregulated within EAE 20–30 dpi striata (Fig. 5 f, middle, right), whereas Arc/Arg3.1 cortical expression remained unchanged. Consistent with the previous results, coronal sections obtained from both HC and EAE 20 dpi brains were microdissected to obtain striatal RNA extracts (n = 3 for each group). Using real-time reverse transcription (RT)-PCR, we found significant downregulation of Arc/Arg3.1 mRNA levels, accounting for only 48.3 ± 14.4% of the levels found in healthy control striata (n = 3 per group; p < 0.001).
Figure 5.

Arc/Arg3x.1 mRNA levels were downregulated in Th1-treated primary neuronal cell cultures and in EAE. a, Primary neurons obtained from CD1 E16.5 embryos were treated with a Th1 mix or saline [control (C)] for, respectively, 3, 6, and 24 h (n = 5 independent cell cultures). After the removal of the stimulus, neurons did not show any morphological changes with respect to untreated ones (data not shown). RT-PCR analysis showed that CD119 and p55 expression was maintained in Th1-treated neurons. b, Then, total extracts from Th1 at 3, 6, and 24 h and vehicle were applied on 10% SDS-PAGE, followed by Western analysis with, respectively, α-GluR1, α-pSer845 GluR1, α-PSD95, and α-TuJ1 as housekeeping gene. Histograms to the right of blots show fold induction (FI) over vehicle (±SD). GluR1 was significantly upregulated during either 6 or 24 h of Th1 treatment: 3 h, 1.6 ± 1.7, NS; 6 h, 1.7 ± 0.4, *p < 0.05; 24 h, 2.6 ± 0.6, *p < 0.05. GluR1 Ser845 was significantly upregulated after either 6 or 24 h of Th1 treatment: 3 h, 0.1 ± 0.01, NS; 6 h, 3.3 ± 0.3, *p < 0.05; 24 h, 3.5 ± 0.2, *p < 0.05. PSD95 was significantly upregulated during either 6 or 24 h of Th1 treatment: 3 h,1.0 ± 0.07, NS; 6 h, 3.1 ± 0.2, *p < 0.05; 24 h, 2.7 ± 0.5, *p < 0.05. c, Representative micrograph of vehicle-treated neurons stained for PSD95 (green). Application of Th1 treatment increases the number of PSD95-expressing synaptic boutons in cultured neurons. d, Arc/Arg3.1 mRNA levels were measured in neuronal cultures at 3, 6, 12, and 24 h after Th1 treatment by using a real-time RT-PCR assay and the mRNA levels, expressed as percentages ± SEM. *p < 0.05. Histogram in e shows bouton fold change over controls (3 h, 2.0 ± 1.1, *p < 0.05; 6 h, 2.5 ± 1.6, *p < 0.05; 24 h, 2.45 ± 1.1, *p < 0.05). f, HC and EAE brain coronal sections probed by in situ hybridization for Arc/Arg3.1 mRNA detection display Arc/Arg3.1 mRNA expression within the cortex and within the striatum. Right panels show Arc/Arg3.1 mRNA expression, respectively, in EAE 20 and 30 dpi brains. EAE basal neocortical Arc/Arg3.1 mRNA levels are conserved but were strongly downregulated within EAE striatum. Scale bars: c, 10 μm; f, 150 μm.
Role of AMPA receptors in synaptic pathology of EAE mice
TNFα-induced AMPA receptor upregulation has been convincingly associated with excitotoxic neuronal damage (Ferguson et al., 2008; Leonoudakis et al., 2008). Thus, we next investigated the role of increased AMPA receptor-mediated transmission in EAE-induced clinical deficits and synaptic pathology. In vivo blockade of AMPA receptors with NBQX (30 mg/kg; n = 8 mice) greatly ameliorated the clinical score of EAE mice (p < 0.05) (Fig. 6 a). Furthermore, single-section Golgi preparations showed that second dendritic branches of striatal neurons had a marked reduction of spine density along the whole extent of the dendrites in EAE (25 dpi, n = 5) compared with HC mice, although cell somata and primary dendrites were similar in the two groups. EAE mice receiving chronic intraperitoneal administration of NBQX (25 dpi, n = 5) exhibited the same unaltered morphological features of the somata of striatal neurons but presented increased spine density compared with EAE receiving vehicle (F = 79.06; p < 0.0001) (Fig. 6 b,c). These results strongly support the involvement of AMPA receptors in EAE-induced dendritic spine degeneration, although it should be remembered that kainate receptors are also blocked by NBQX.
Figure 6.

Blockade of AMPA receptors ameliorates the clinical and synaptic deficits in EAE. a, Time course of the clinical score of EAE mice treated or not with NBQX, blocker of AMPA/kainite receptors. b, The histogram shows the quantification of spine density in the three experimental conditions. **p < 0.01 versus EAE; # p < 0.05 versus HC. c, Examples of single-section Golgi preparations showing dendrites from HC and EAE mice receiving intraperitoneal injections of NBQX or vehicle. Scale bar: all three panels, 8 μm.
Discussion
The present study is the first thorough investigation of synaptic transmission in an experimental model of MS. Our data demonstrated an early alteration of excitatory transmission in the striatum of EAE mice. This alteration was responsible for synaptic degeneration in this model and support the emerging notion that the neuronal compartment of the CNS suffers in parallel and even independently of white matter damage in MS. Abnormal glutamate transmission involved AMPA receptors and were likely secondary to the downregulation of Arc/Arg3.1 gene in striatal neurons. Evidence has already been provided that Arc/Arg3.1 gene is downregulated also in a lipopolysaccharide model of neuroinflammation (Bonow et al., 2008).
Arc/Arg3.1 early gene has been heavily implicated in the regulation of glutamate transmission mediated by AMPA receptors, because it controls AMPA receptor trafficking via its interaction with components of the endocytic machinery (Chowdhury et al., 2006). Furthermore, Arc/Arg3.1 overexpression results in a cell-wide decrease in the surface expression of GluR1-containing AMPA receptors (Shepherd et al., 2006), whereas Arc/Arg3.1 null primary neurons show an increased surface expression of AMPA receptors (Shepherd et al., 2006). In line with these findings, we have observed that Arc/Arg3.1 was downregulated during inflammatory cues and in the striatum of EAE mice, in coincidence with increased expression, phosphorylation, and activity of AMPA receptors. Increase of surface expression of GluR1-containing AMPA receptors by Arc/Arg3.1 downregulation does not explain the increase of total GluR1 observed by Western blot on whole striatal extracts, pointing to additional regulatory mechanisms, including altered trafficking (Ferguson et al., 2008; Leonoudakis et al., 2008), not explored in the present study.
Microglia/macrophages can be activated by different means to display protective or detrimental effects (Butovsky et al., 2006; Schwartz et al., 2006). In the striatum, at the peak of EAE, we found the presence of high levels of proinflammatory Th1 but not Th2 cytokines. We also found that microglial activation by Th1 cytokines and the resulting release of TNFα are early events in EAE, occurring even before the appearance of the clinical deficits. TNFα released from activated microglia is a likely candidate for the induction of the synaptic deficits in EAE, because both activated microglia and TNFα mimicked the effects of EAE induction on the sensitivity of AMPA receptors to synaptically released glutamate. The role of activated microglia and TNFα in EAE-triggered glutamate transmission changes was further confirmed by the occlusion experiments showing that incubation with either microglia or TNFα did not potentiate the effects of EAE induction on sEPSC kinetic properties. Notably, microglia activation and changes in the expression and physiological properties of glutamate AMPA receptors preceded motor disturbances in EAE mice, strengthening the conclusion that these phenomena do not reflect secondary adaptive processes to the disease.
We proved an increase of apoptotic-like events at the synapse level, as revealed by an increase of Annexin V labeling in synaptosomes obtained from EAE mice compared with HC. This last finding suggests that synaptic PS exposure might be involved in the synapse–glia interaction in which the glial cells might play a critical role in controlling of synaptic loss (Barres, 2008).
Finally, we also observed that the effects of activated microglia were prevented by blocking TNFα signaling, and evidence exists that this cytokine is involved in specific forms of AMPA receptor-mediated synaptic plasticity and neuronal damage in other brain areas. Previous experiments, in fact, showed that TNFα was able to modulate AMPA receptor-mediated synaptic transmission (Beattie et al., 2002; Stellwagen et al., 2005; Stellwagen and Malenka, 2006) and excitotoxicity (Bernardino et al., 2005) by inducing the upregulation of the GluR1 subunit in vitro.
The involvement of AMPA receptors in the neurodegenerative damage of EAE has been suggested in previous works (Kanwar et al., 2004) and further demonstrated in the present study, showing that clinical deficits and dendritic spine loss triggered by neuroinflammation are prevented by NBQX, a selective inhibitor of AMPA receptors.
Activated microglia, TNFα, and glutamate AMPA receptors might represent valuable targets to contrast synaptic alterations and neurodegeneration in MS.
Footnotes
This investigation was supported by Italian National Ministero dell'Università e della Ricerca (D.C.), Italian National Ministero della Salute (D.C., F.C., L.B.), Fondazione Italiana Sclerosi Multipla (D.C., L.B., R.F., F.C., G.M.), and BMW (G.M.).
References
- Bakshi R, Benedict RH, Bermel RA, Caruthers SD, Puli SR, Tjoa CW, Fabiano AJ, Jacobs L. T2 hypointensity in the deep gray matter of patients with multiple sclerosis: a quantitative magnetic resonance imaging study. Arch Neurol. 2002;59:62–68. doi: 10.1001/archneur.59.1.62. [DOI] [PubMed] [Google Scholar]
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bermel RA, Innus MD, Tjoa CW, Bakshi R. Selective caudate atrophy in multiple sclerosis: a 3D MRI parcellation study. Neuroreport. 2003;14:335–339. doi: 10.1097/00001756-200303030-00008. [DOI] [PubMed] [Google Scholar]
- Bernardino L, Xapelli S, Silva AP, Jakobsen B, Poulsen FR, Oliveira CR, Vezzani A, Malva JO, Zimmer J. Modulator effects of interleukin-1β and tumor necrosis factor-α on AMPA-induced excitotoxicity in mouse organotypic hippocampal slice cultures. J Neurosci. 2005;25:6734–6744. doi: 10.1523/JNEUROSCI.1510-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton C, Paul C. MK-801 limits neurovascular dysfunction during experimental allergic encephalomyelitis. J Pharmacol Exp Ther. 1997;282:397–402. [PubMed] [Google Scholar]
- Bonow RH, Aïd S, Zhang Y, Becker KG, Bosetti F. The brain expression of genes involved in inflammatory response, the ribosome, and learning and memory is altered by centrally injected lipopolysaccharide in mice. Pharmacogenomics J. 2008 doi: 10.1038/tpj.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Landa G, Kunis G, Ziv Y, Avidan H, Greenberg N, Schwartz A, Smirnov I, Pollack A, Jung S, Schwartz M. Induction and blockage of oligodendrogenesis by differently activated microglia in an animal model of multiple sclerosis. J Clin Invest. 2006;116:905–915. doi: 10.1172/JCI26836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Rossi S, Prosperetti C, Tscherter A, Bernardi G, Maccarrone M, Calabresi P. Abnormal sensitivity to cannabinoid receptor stimulation might contribute to altered gamma-aminobutyric acid transmission in the striatum of R6/2 Huntington's disease mice. Biol Psychiatry. 2005;57:1583–1589. doi: 10.1016/j.biopsych.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Centonze D, Bari M, Rossi S, Prosperetti C, Furlan R, Fezza F, De Chiara V, Battistini L, Bernardi G, Bernardini S, Martino G, Maccarrone M. The endocannabinoid system is dysregulated in multiple sclerosis and in experimental autoimmune encephalomyelitis. Brain. 2007;130:2543–2553. doi: 10.1093/brain/awm160. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52:445–459. doi: 10.1016/j.neuron.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA, Gilbert DL. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987;223:284–288. doi: 10.1016/0014-5793(87)80305-8. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Caillé I, Lim DA, García-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Petreanu L, Caille I, Garcia-Verdugo JM, Alvarez-Buylla A. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron. 2002a;36:1021–1034. doi: 10.1016/s0896-6273(02)01133-9. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Verdugo JM, Caille I, Alvarez-Buylla A, Chao MV, Casaccia-Bonnefil P. Lack of the cell-cycle inhibitor p27Kip1 results in selective increase of transit-amplifying cells for adult neurogenesis. J Neurosci. 2002b;22:2255–2264. doi: 10.1523/JNEUROSCI.22-06-02255.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, Bresnahan JC, Beattie MS. Cell death after spinal cord injury is exacerbated by rapid TNF α-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J Neurosci. 2008;28:11391–11400. doi: 10.1523/JNEUROSCI.3708-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, Bergami A, Desina G, Comi G, Flavell R, Su MS, Adorini L. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol. 1999;163:2403–2409. [PubMed] [Google Scholar]
- Gardoni F, Picconi B, Ghiglieri V, Polli F, Bagetta V, Bernardi G, Cattabeni F, Di Luca M, Calabresi P. A critical interaction between NR2B and MAGUK in l-DOPA induced dyskinesia. J Neurosci. 2006;26:2914–2922. doi: 10.1523/JNEUROSCI.5326-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts JJ, Wolswijk G, Bö L, van der Valk P, Polman CH, Troost D, Aronica E. Altered expression patterns of group I and II metabotropic glutamate receptors in multiple sclerosis. Brain. 2003;126:1755–1766. doi: 10.1093/brain/awg179. [DOI] [PubMed] [Google Scholar]
- Geurts JJ, Wolswijk G, Bö L, Redeker S, Ramkema M, Troost D, Aronica E. Expression patterns of group III metabotropic glutamate receptors mGluR4 and mGluR8 in multiple sclerosis lesions. J Neuroimmunol. 2005;158:182–190. doi: 10.1016/j.jneuroim.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Wiley DJ, Cole GM. Rapid annexin-V labeling in synaptosomes. Neurochem Int. 2004a;44:125–131. doi: 10.1016/s0197-0186(03)00146-3. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Cole GM. Enrichment of presynaptic and postsynaptic markers by size-based gating analysis of synaptosome preparations from rat and human cortex. Cytometry A. 2004b;60:90–96. doi: 10.1002/cyto.a.20031. [DOI] [PubMed] [Google Scholar]
- Hardin-Pouzet H, Krakowski M, Bourbonnière L, Didier-Bazes M, Tran E, Owens T. Glutamate metabolism is down-regulated in astrocytes during experimental allergic encephalomyelitis. Glia. 1997;20:79–85. doi: 10.1002/(sici)1098-1136(199705)20:1<79::aid-glia8>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Izzo PN, Graybiel AM, Bolam JP. Characterization of substance P- and [Met]enkephalin-immunoreactive neurons in the caudate nucleus of cat and ferret by a single section Golgi procedure. Neuroscience. 1987;20:577–587. doi: 10.1016/0306-4522(87)90111-4. [DOI] [PubMed] [Google Scholar]
- Kanwar JR, Kanwar RK, Krissansen GW. Simultaneous neuroprotection and blockade of inflammation reverses autoimmune encephalomyelitis. Brain. 2004;127:1313–1331. doi: 10.1093/brain/awh156. [DOI] [PubMed] [Google Scholar]
- Leonoudakis D, Zhao P, Beattie EC. Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J Neurosci. 2008;28:2119–2130. doi: 10.1523/JNEUROSCI.5159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyford GL, Yamagata K, Kaufmann WE, Barnes CA, Sanders LK, Copeland NG, Gilbert DJ, Jenkins NA, Lanahan AA, Worley PF. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron. 1995;14:433–445. doi: 10.1016/0896-6273(95)90299-6. [DOI] [PubMed] [Google Scholar]
- Muzio L, DiBenedetto B, Stoykova A, Boncinelli E, Gruss P, Mallamaci A. Conversion of cerebral cortex into basal ganglia in Emx2(−/−) Pax6(Sey/Sey) double-mutant mice. Nat Neurosci. 2002;5:737–745. doi: 10.1038/nn892. [DOI] [PubMed] [Google Scholar]
- Muzio L, Soria JM, Pannese M, Piccolo S, Mallamaci A. A mutually stimulating loop involving emx2 and canonical wnt signalling specifically promotes expansion of occipital cortex and hippocampus. Cereb Cortex. 2005;15:2021–2028. doi: 10.1093/cercor/bhi077. [DOI] [PubMed] [Google Scholar]
- Muzio L, Martino G, Furlan R. Multifaceted aspects of inflammation in multiple sclerosis: the role of microglia. J Neuroimmunol. 2007;191:39–44. doi: 10.1016/j.jneuroim.2007.09.016. [DOI] [PubMed] [Google Scholar]
- Ohgoh M, Hanada T, Smith T, Hashimoto T, Ueno M, Yamanishi Y, Watanabe M, Nishizawa Y. Altered expression of glutamate transporters in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002;125:170–178. doi: 10.1016/s0165-5728(02)00029-2. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Bien-Ly N, Massaro C, Yeung BZ, Yu GQ, Mucke L. Vulnerability of dentate granule cells to disruption of arc expression in human amyloid precursor protein transgenic mice. J Neurosci. 2005;25:9686–9693. doi: 10.1523/JNEUROSCI.2829-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- Plaut GS. Effectiveness of amantadine in reducing relapses in multiple sclerosis. J R Soc Med. 1987;80:91–93. doi: 10.1177/014107688708000210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluchino S, Quattrini A, Brambilla E, Gritti A, Salani G, Dina G, Galli R, Del Carro U, Amadio S, Bergami A, Furlan R, Comi G, Vescovi AL, Martino G. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422:688–694. doi: 10.1038/nature01552. [DOI] [PubMed] [Google Scholar]
- Politi LS, Bacigaluppi M, Brambilla E, Cadioli M, Falini A, Comi G, Scotti G, Martino G, Pluchino S. Magnetic-resonance-based tracking and quantification of intravenously injected neural stem cell accumulation in the brains of mice with experimental multiple sclerosis. Stem Cells. 2007;25:2583–2592. doi: 10.1634/stemcells.2007-0037. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005;81:374–389. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- Rakic P. Adult neurogenesis in mammals: an identity crisis. J Neurosci. 2002;22:614–618. doi: 10.1523/JNEUROSCI.22-03-00614.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarchielli P, Greco L, Floridi A, Floridi A, Gallai V. Excitatory amino acids and multiple sclerosis: evidence from cerebrospinal fluid. Arch Neurol. 2003;60:1082–1088. doi: 10.1001/archneur.60.8.1082. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Butovsky O, Brück W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci. 2006;29:68–74. doi: 10.1016/j.tins.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med. 2000;6:62–66. doi: 10.1038/71548. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain. 2005;128:1016–1025. doi: 10.1093/brain/awh467. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover JF, Lowitzsch K, Kempski OS. Cerebrospinal fluid hypoxanthine, xanthine and uric acid levels may reflect glutamate-mediated excitotoxicity in different neurological diseases. Neurosci Lett. 1997;238:25–28. doi: 10.1016/s0304-3940(97)00840-9. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr BUdR as an S-phase marker for quantitative studies of cytokinetic behaviour in the murine cerebral ventricular zone. J Neurocytol. 1992;21:185–197. doi: 10.1007/BF01194977. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr Cell cycle parameters and patterns of nuclear movement in the neocortical proliferative zone of the fetal mouse. J Neurosci. 1993;13:820–833. doi: 10.1523/JNEUROSCI.13-02-00820.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo-Illarramendi A, Domercq M, Pérez-Cerdá F, Ravid R, Matute C. Increased expression and function of glutamate transporters in multiple sclerosis. Neurobiol Dis. 2006;21:154–164. doi: 10.1016/j.nbd.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Wallström E, Diener P, Ljungdahl A, Khademi M, Nilsson CG, Olsson T. Memantine abrogates neurological deficits, but not CNS inflammation, in Lewis rat experimental autoimmune encephalomyelitis. J Neurol Sci. 1996;137:89–96. doi: 10.1016/0022-510x(95)00339-4. [DOI] [PubMed] [Google Scholar]