

Abstract
Synchronous calcium oscillations are observed in primary cultures of rat cortical neurons when mature networks are formed. This spontaneous neuronal activity needs an accurate control of calcium homeostasis. Alteration of intraneuronal calcium concentration is described in many neurodegenerative disorders, including Alzheimer disease (AD). Although processing of amyloid precursor protein (APP) that generates Aβ peptide has critical implications for AD pathogenesis, the neuronal function of APP remains unclear. Here, we report that expression of human APP (hAPP) in rat cortical neurons increases L-type calcium currents, which stimulate SK channels, calcium-dependent K+ channels responsible for medium afterhyperpolarization (mAHP). In a neuronal network, increased mAHP in some neurons expressing hAPP leads to inhibition of calcium oscillations in all the cells of the network. This inhibition is independent of production and secretion of Aβ and other APP metabolites. In a neuronal network, reduction of endogenous APP expression using shRNA increases the frequency and reduces the amplitude of calcium oscillations. Altogether, these data support a key role for APP in the control of neuronal excitability.
Introduction
Alzheimer disease (AD) is a neurodegenerative disorder characterized by accumulation of neurofibrillary tangles and senile plaques in the brain. Senile plaques contain an amyloid core of Aβ peptide, derived from the amyloid precursor protein (APP). APP, a type I transmembrane protein, is encoded by the APP gene, a member of a family that also includes APLP1 and APLP2 genes (Wasco et al., 1993; Slunt et al., 1994). APP knock-out mice have a normal phenotype presumably reflecting complementation by APLP1 and 2 proteins (Heber et al., 2000). In the absence of abnormal phenotype in APP knock-out mice, the function of the protein remains elusive. To investigate the neuronal function of APP, we studied the effect of human APP (hAPP) expression on spontaneous neuronal activity of rat cortical neurons in culture, by focusing on synchronous calcium oscillations, an established feature of mature networks of cortical neurons in vitro (Murphy et al., 1992; Opitz et al., 2002; Dravid and Murray, 2004). Synchronous calcium oscillations are also observed in neocortical slices (Silva et al., 1991; Flint et al., 1999) as well as in cat neocortex in vivo (Steriade et al., 1993). In the nervous system, the physiological role of calcium oscillations includes regulation of neuronal plasticity in developing neurons (Spitzer et al., 1995), promotion of neurotransmitter receptor expression in Xenopus embryonic neurons (Gu and Spitzer, 1995) and regulation of migration in developing cerebellar granule neurons (Komuro and Rakic, 1996).
Using adenovirus encoding hAPP, we showed that focal expression of hAPP in some neurons of a network increased the activity of L-type voltage-dependent calcium channels. This in turn activated calcium-activated K+ (SK) channels, responsible for an increased medium afterhyperpolarization (mAHP) phase after action potentials. This hAPP-mediated increase of mAHP interrupted spontaneous synchronous calcium oscillations in the entire network. In addition, silencing endogenous APP expression increased the frequency and decreased the amplitude of calcium oscillations. Altogether, these results demonstrate a novel function of APP in the control of neuronal activity.
Materials and Methods
Neuronal cultures and reagents
Primary cultures of cortical neurons were prepared from 17- to 18-d-old Wistar rat embryos as described previously (Macq et al., 1998). All cell culture reagents were purchased from Invitrogen. Cells were plated in culture dishes pretreated with 10 μg/ml poly-l-lysine in PBS and cultured for 7 d in vitro in NEUROBASAL medium supplemented with 2% (v/v) B-27 medium and 0.5 mm l-glutamine before infection with recombinant adenoviruses. Under these conditions, neuronal cultures display high differentiation and survival rates (Brewer, 1995).
Recombinant viruses and neuronal infection
The construction and purification of a recombinant adenovirus encoding β-galactosidase (Adβgal) or human APP carrying the Swedish mutation (AdAPPswe) was described previously (Lemarchand et al., 1992). AdAPPGFP and AdGFP were constructed and purified using the AdEasy XL Adenoviral Vector System (Stratagene). Briefly, either the pShuttle-internal ribosomal entry site (IRES)-hrGFP-2 vector (for AdGFP) or the same vector containing the cDNA encoding human APP695 (for AdAPPGFP) was inserted in the pAdEasy-1 vector. Adenoviruses were purified according to the manufacturer's instructions.
After 7 d in vitro, neurons were infected at a multiplicity of infection of 10 (for calcium measurements and protein analyses) or 70 (for electrophysiological, calcium imaging and immunocytochemical experiments) for 4 h.
Five different plasmids encoding shRNA raised against APP mRNA were obtained from Sigma-Aldrich and used for construction of recombinant lentiviruses, as previously described (Salmon and Trono, 2007). The most efficient was used to downregulate APP expression in 7 days in vitro (DIV) neurons incubated in the presence of the lentivirus for 6 d before analysis. A recombinant lentivirus encoding the neomycin resistance gene was used as a negative control.
Pharmacological treatment of cultured neurons
Tetrodotoxin (TTX) was purchased from Alomone Labs. CNQX and diltiazem were from Tocris, and nimodipine, nifedipine, and apamin were from Sigma-Aldrich. BayK6844 was a gift from Bayer. N-[N-(3,5-Difluorophenacetyl)-l-alanyl]-(S)-phenylglycine t-butyl ester (DAPT) was a gift from Luc Mercken (Aventis, Paris, France).
Acute treatment of neurons by TTX (1 μm), CNQX (50 μm), nimodipine (5 μm), apamin (200 nm), and diltiazem (50 μm) was performed using perfusion of the incubation chamber. Neurons were also incubated in the presence of 1 μm BayK6844 for 60 min, washed, and reincubated without BayK6844 for analysis.
DAPT (250 nm) was added to the culture medium 48 h after adenoviral infection. Medium containing DAPT was replaced every day, for 3–4 d, before calcium measurements were carried out.
Protein analysis by Western blotting
Cell culture media (10 μl) and cell lysates (10 μg of protein) were analyzed by Western blotting as described previously (Macq et al., 1998). Membranes were incubated overnight at 4°C with the human APP-specific WO-2 monoclonal antibody (1:1000) (The Genetics Company), the polyclonal anti-APPCterminal antibody (1:1000) (Sigma-Aldrich) or the rabbit polyclonal anti-actin antibody (1:2000) (Sigma-Aldrich).
Quantification of Aβ production
The quantification of extracellular Aβ40 was performed by ELISA as described previously (Kienlen-Campard et al., 2002). Fluorescence emission was measured on a HTS 7000 Plus plate reader (PerkinElmer) at excitation/emission wavelengths of 485 nm/535 nm, respectively. Aβ40 concentrations were expressed as picograms per milliliter.
Immunocytochemical analysis
Neurons were prepared for immunocytochemical analysis as described previously (Tilleux and Hermans, 2008). Fixed cells were incubated for 1 h at room temperature in the presence of the mouse WO-2 antibody (0,5 μg/ml), the anti-APPCterminal antibody (1:500) or the rabbit MAP-2 antibody (B9; kind gift from J.-P. Brion, Université Libre de Bruxelles, Brussels, Belgium) After washing, cells were incubated for 1 h with a secondary anti-mouse Cy3-conjugated IgG (Jackson ImmunoResearch Laboratory) (1:500) or a secondary anti-rabbit Cy3-conjugated IgG (1:500). Nuclei were stained with DAPI (Invitrogen) (1:5000). Preparations were mounted in Fluoprep medium (BioMerieux) and examined with an Olympus IX70 inverted microscope coupled to a charged-coupled device sensor (CCD) camera. Digital images were acquired using the TILLvisION software (T.I.L.L. Photonics).
Cytosolic free Ca2+ measurement
Calcium measurements on neuronal populations.
Neurons were plated at a density of 2.105 cells/cm2 on 22 mm glass coverslips. Four days after adenoviral infection, neurons were incubated in the presence of the Ca2+ indicator fura-2 acetoxymethylester (Fura-2 AM; Calbiochem) at a final concentration of 2 μm in Krebs-HEPES buffer (10 mm HEPES, 135 mm NaCl, 6 mm KCl, 2 mm CaCl2, 1.2 mm MgCl2, 10 mm d-glucose, pH 7.4) for 30 min at room temperature.
The glass coverslips were mounted in a perfusion chamber placed on the thermostated platform of a fluorimeter (CAF; Jasco) allowing measurement of fluorescence signal at 550 nm upon excitation at 340 nm (F340) and at 380 nm (F380). Cells were superfused with Krebs-HEPES buffer at 37°C until stabilization of the baseline. BayK6844 response was obtained by superfusing cells with Krebs-HEPES (10 mm HEPES, 126 mm NaCl, 2 mm CaCl2, 1.2 mm MgCl2, 10 mm d-glucose, pH 7.4) containing 15 mm KCl for 2 min before 1 μm BayK6844 application. Afterward, neurons were depolarized by superfusion with Krebs-HEPES buffer (10 mm HEPES, 91 mm NaCl, 2 mm CaCl2, 1.2 mm MgCl2, 10 mm d-glucose, pH 7.4) containing 50 mm KCl.
The cytosolic calcium concentration ([Ca2+]cyt) was estimated using the following calibration. The minimal F340/F380 ratio was determined by cell permeabilization with 1 μm ionomycin and incubation in a Krebs-HEPES buffer without Ca2+, containing 10 mm EGTA. The maximal F340/F380 ratio was determined by superfusion of Krebs-HEPES buffer containing 20 mm Ca2+. Autofluorescence, measured at the end of experiment by the addition of 10 mm MnCl2, was subtracted from all fluorescence values. [Ca2+]cyt was estimated as described previously (Grynkiewicz et al., 1985). Data were analyzed with the GraphPad Prism software. Statistics are presented as means ± SEM. Statistical significance was assessed by unpaired Student's t test, at a p value of < 0.05.
Calcium measurements in single neurons using calcium imaging.
Changes in [Ca2+]i were measured in isolated cells using the [Ca2+]i indicator Fura-2 AM. Cells, cultured on coated 15 mm round glass coverslips at a density of 7.5.105 cells/coverslip, were incubated in the dark with 2 μm Fura-2 AM for 30 min at room temperature in Krebs-HEPES buffer. Coverslips were then washed and mounted in a heated (37°C) and perfused (100 μl) microscope chamber (Warner Instrument Corporation). Cells were excited successively (1 or 2 Hz) at 340 and 380 nm for 200 ms and fluorescence was monitored at 510 nm using a CCD camera coupled to an inverted Olympus IX70 microscope (TILL Photonics). Fluorescence intensities from single cells excited at the two wavelengths (F340 and F380) were recorded separately and combined (fluorescence ratio: r = F340/F380) after background subtraction (fluorescence of a cell-free area), using the software TILLvisION version 3.3. Baseline mean ratio value (Rmean) was the mean ratio value after a 2 min recording, at the beginning of the experiment.
Calcium oscillations expressed as R/Rmean were defined as variations of >10% from Rmean, occurring synchronously in several cells of the field. All uninfected or Adβgal infected networks responded similarly to experimental procedures.
Electrophysiology
Calcium currents recording.
Standard whole-cell patch-clamp recording was used to measure whole-cell Ca2+ current from visually identified cortical neurons. The bath solution contained (in mm) 4.8 KCl, 10 BaCl2, 125 NaCl, 5 TEA, 5 HEPES, 15 glucose, 1.2 MgCl2, 0.001 TTX (all from Sigma), at a pH adjusted to 7.4 with TEA-OH and osmolarity of 310 mOsm/L. Barium was used as charge carrier, instead of calcium, to prevent inhibition of calcium currents by calcium itself. The internal pipette solution contained (in mm) 125 CsCl, 10 EGTA, 1 MgCl2, 5 HEPES, 30 KOH, 3 Na2ATP, 0.2 Na3GTP (pH adjusted to 7.2 with HCl; osmolarity: 260–280 mOsm/L; all components from Sigma). This solution was allowed to dialyze the cell for at least 5 min before recording. The inclusion of TTX, to block Na+ channels, Cs+ and TEA, to block K+ channels, insured that the voltage-gated calcium current was isolated for recording. The current recorded under these conditions was completely blocked by the application of 200 μm Cd2+, indicating that it was mediated by voltage-dependent Ca2+ channels. Nimodipine (5 μm) was applied after recording of basal current. L-type current was obtained by subtracting the remaining current (nimodipine-resistant) from the total current. Currents were leak-subtracted after off-line analysis of leak-current. Currents were normalized for cell capacitance (Cm, in pF), and expressed as current density (pA/pF).
Data were collected using standard voltage step protocols using a Axopatch 200B amplifier. Whole-cell currents were elicited by series of 500 ms depolarizing test pulses in the range of −60 to 50 mV from a holding potential of −100 mV. They were recorded at a frequency of 5 kHz and filtered at 1 kHz through a low-pass Bessel filter. Conductance (G) was calculated from peak current according to the equation G(V) = I(V − Vrev), where I was the current, V the test pulse voltage, and Vrev was the measured reversal potential. Conductance-voltage curves were fitted to a Boltzmann function of the form: G(V) = Gmax/{1+exp[(V − V0.5)/k]}, where V0.5 was the half-activation voltage, k was the slope factor and Gmax was the maximum conductance.
Resting membrane potential and mAHP.
Whole-cell current-clamp recordings of resting membrane potential (RMP) and mAHP were obtained using the amphotericin-perforated patch-clamp technique. The bath solution contained 148 mm NaCl, 2.5 mm KCl, 2.4 mm CaCl2, 1.3 mm MgCl2, 5 mm HEPES and 11 mm glucose (pH adjusted to 7.4 with NaOH). The internal pipette solution contained 120 mm K-aspartate, 10 mm NaCl, 3 mm MgCl2, 0.5 mm EGTA, 0.25 mm CaCl2, 2 mm Na2ATP and 5 mm HEPES (pH adjusted to 7.25 with KOH, osmolarity: 300mOsm/L). Amphotericin (250 μm) was added to the internal solution before the experiment.
Data were collected at a frequency of 10 kHz and filtered at 5 kHz through a low-pass Bessel filter. The RMP was recorded in current clamp configuration without current injection immediately after establishing the whole-cell configuration. Peak amplitude of afterhyperpolarization was measured as the difference between baseline and the membrane potential 15 ms after an action potential (timing corresponding to peak amplitude of mAHP) (Pineda et al., 1998) elicited by the injection of a short depolarizing current (500pA, 2 ms). Mean amplitude was calculated from three consecutive sweeps. The kinetics of mAHP decay was analyzed and fitted with pClamp software (Axon Instruments). Traces were considered valid if RMP, Cm, or leak-current was stable throughout the whole experiment.
Electrodes were pulled from capillary glass (Harvard Apparatus) on a DMZ puller (Zeitz Instrumente Vertriebs) and had a resistance of 3–6MΩ when filled with the intracellular solution. All data were recorded at room temperature using the Axopatch 200B amplifier and pClamp software (Axon Instruments) as previously described (Buryi et al., 1995).
Least-squares curve fitting and statistical analysis were performed using Graph Pad Prism software. Statistics are presented as means ± SEM. Statistical significance was established by Student's t test, at a p value of <0.05.
[3H]PN200–110 binding experiments
[3H]PN200–110 binding was measured in intact neurons plated in 12-well plates at 3.75.105 cells/cm2 as previously described (Morel et al., 1998). Briefly, cells were incubated for 90 min at 37°C in the presence of [3H]PN200–110 (82 Ci mmol−1). Nonspecific binding was determined in the presence of Nifedipine 1 μm, and subtracted from the total binding to obtain the specific binding. Binding data were analyzed by nonlinear regression. Statistical significance was estimated with an unpaired Student's t test.
Results
Spontaneous synchronous calcium oscillations are abolished by human APP expression
Cortical neurons cultured from rat embryos are able to develop mature networks within a few DIV. Neuronal processes extend and form functional synapses with other neurons, and excitatory synaptic inputs generate spontaneous synchronous calcium oscillations (Murphy et al., 1992; Opitz et al., 2002; Dravid and Murray, 2004). In agreement with this, single-cell calcium imaging showed calcium oscillations in our cortical neurons cultured for >10 DIV but not in neurons cultured for 7 DIV (Fig. 1A,B). At 13 DIV, 65 ± 6% of cells (analysis of 252 cells in 8 different experiments) showed spontaneous synchronous calcium oscillations at a frequency of 13 ± 2 oscillations/min. Therefore, all results were obtained on 13 DIV neurons. We observed that inhibition of AMPA receptors by CNQX abolished calcium oscillations (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Because glutamate transmission generates action potentials leading to calcium influx through voltage-dependent calcium channels, we tested the effect of TTX and diltiazem on spontaneous calcium oscillations. Results presented in Figure S1, available at www.jneurosci.org as supplemental material, show that both inhibition of Na+ channels and L-type calcium channels completely inhibited calcium oscillations, confirming that L-type calcium channels play a key role in the generation of synchronous calcium oscillations observed in cultured cortical neurons (Wang and Gruenstein, 1997).
Figure 1.
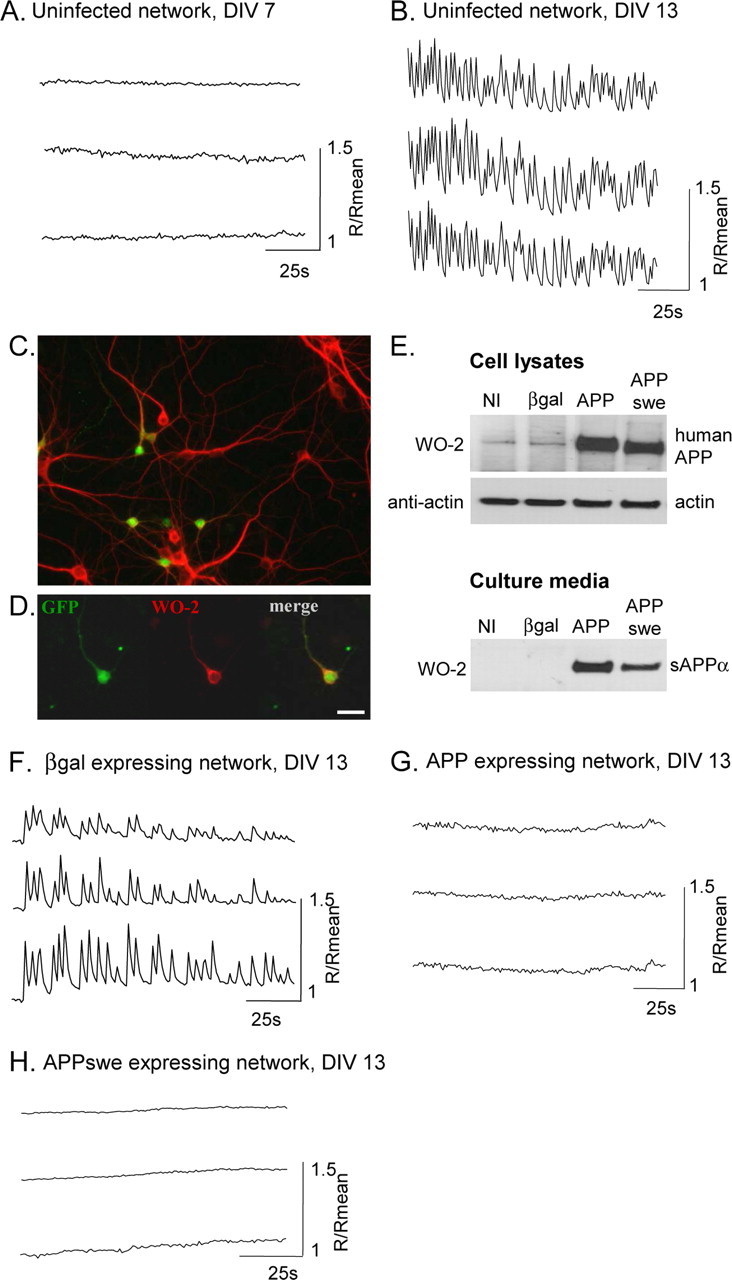
Spontaneous synchronous calcium oscillations are abolished by hAPP expression. A, B, F, G, Simultaneous traces of [Ca2+]i in three cells of a coverslip, measured with Fura-2 AM expressed as R/Rmean, were R is the ratio of F340/F380 and Rmean is the mean ratio value after 2 min recording. A, At 7 DIV, rat cortical neurons do not present calcium oscillations. B, After 13 DIV, neurons show spontaneous, synchronized calcium oscillations. C, In a neuronal network labeled by an anti-MAP-2 antibody (red), infection by AdAPPGFP allows APP and GFP (green) expression in a subset of neurons. D, Primary cortical neuron infected by AdAPPGFP, coexpressing GFP (green) and hAPP, labeled with hAPP specific monoclonal antibody, WO-2 (red). Scale bar, 20 μm. E, Western blot analysis of cell lysates (top panel) or culture media (bottom panel), from rat cortical neurons infected or not (NI) by Adβgal (βgal), AdAPPGFP (APP), or AdAPPswe (APPswe). Protein loading was controlled using rabbit actin antiserum. F, Neuronal network infected by Adβgal show spontaneous calcium oscillations, whereas in a network infected by AdAPPGFP or AdAPPswe, calcium oscillations are abolished (G, H).
hAPP expression in rat cortical neurons was achieved using a recombinant adenovirus encoding the hAPP sequence followed by an IRES and the humanized recombinant green fluorescent protein (hrGFP) gene (Fig. 1C). Adenoviral expression of hAPP and hrGFP remained stable for several days, and hAPP was processed in both the non-amyloidogenic and the amyloidogenic pathways (see Figs. 1E, 5B). Expression of hrGFP allowed the detection of infected cells (Fig. 1D), for analysis in patch-clamp studies.
Figure 5.

Inhibition of calcium oscillations by human APP is independent of γ-cleavage and extracellular APP metabolites. A, BayK6844 stimulated calcium entry was significantly higher in APP-expressing neurons, despite inhibition of γ-cleavage by DAPT treatment (as described in C). Error bars represent mean values ± SEM of normalized Δ[Ca2+]i where 100% is Δ[Ca2+]i in βgal-expressing neurons (βgal: n = 7; APP: n = 7; βgal+DAPT: n = 3; APP+DAPT: n = 3; **p < 0.01). B, Aβ40 production in 24 h (mean ± SEM; n = 4). C, Simultaneous traces of [Ca2+]i in three cells in a hAPP-expressing network show that cells do not recover synchronous calcium oscillations after DAPT treatment for 3–4 d. D, Simultaneous traces of [Ca2+]i in three cells in a βgal-expressing network show that conditioning cells in medium collected from hAPP-expressing network (for 24 h) does not abolish calcium oscillations.
Neuronal cultures were infected by Adβgal (control), AdAPPGFP, or AdAPPswe recombinant adenoviruses. Calcium imaging (Fig. 1F) showed that, as in uninfected neurons, networks expressing β-galactosidase displayed spontaneous synchronous calcium oscillations. Indeed, 64 ± 5% of cells (analysis of 332 cells in 11 different experiments) showed calcium oscillations at a frequency of 12 ± 2 oscillations/min. In contrast, expression of hAPP in a subset of neurons (23 ± 3%, n = 13) completely abolished calcium oscillations in all neurons of the network (analysis of 198 cells in 8 different experiments) (Fig. 1G). Neurons were also infected with AdAPPswe encoding a human APP mutant which induces hereditary AD and produces more extracellular Aβ (Johnston et al., 1994). In APPswe-expressing networks, no calcium oscillations were observed in all the cells of the network (analysis of 90 cells in 5 different experiments) (Fig. 1H).
As expression of hAPP in a small proportion of neurons was sufficient to inhibit calcium oscillations in the whole network, we wondered whether hAPP-expressing neurons could be hyperpolarized and thereby refractory to synaptic mediation of calcium oscillations.
Human APP expression increases the amplitude of medium AHP
We first compared the RMP in hAPP-expressing neurons and in GFP-expressing neurons. Results clearly demonstrate that hAPP expression did not modify the neuronal RMP (Fig. 2B).
Figure 2.

hAPP expression increases the peak amplitude of mAHP. A, Representative trace for mAHP in a cultured cortical neuron. An action potential (AP) was elicited by a 2 ms depolarizing current injection, followed by an hyperpolarization phase corresponding to mAHP. The mAHP peak amplitude was measured 15 ms after the AP (arrow) (τ: time constant of decay). B, Resting membrane potential was similar in hAPP- or GFP-expressing neurons, whereas mAHP peak amplitude was higher in hAPP-expressing neurons when compared with GFP-expressing neurons (mean values ± SEM of n cells; *p < 0.05 was considered significant).
To test whether excitability could be reduced due to an increased AHP, we measured the AHP by patch-clamping neurons in the current-clamp mode. Perforated patch (with amphotericin) was used to preserve intracellular calcium dynamics. After injecting a suprathreshold depolarizing current, we consistently recorded an action potential followed by an hyperpolarization. The magnitude and kinetics of hyperpolarization decay indicated that it corresponded to mAHP (Schwindt et al., 1988) (Fig. 2A). Its peak amplitude was calculated by measuring the difference between baseline and the voltage recorded 15 ms after the action potential. There was a significant increase of mAHP peak amplitude in hAPP-expressing versus GFP-expressing neurons (Fig. 2B). The kinetics of decay were similar in both groups.
L-type calcium channels stimulation inhibits calcium oscillations through activation of SK channels
Since mAHP is mediated by SK channels, members of the calcium-activated K+ channels family (Villalobos et al., 2004), we wondered whether these channels could be involved in the APP-mediated inhibition of synchronous calcium oscillations. To test that hypothesis, SK channels were inhibited by the specific antagonist, apamin. Single-cell calcium imaging showed that treatment of hAPP-expressing neurons by 200 nm apamin restored spontaneous calcium oscillations in 74 ± 8% of cells (analysis of 214 cells in 6 different experiments) (Fig. 3A), at a frequency of 6 ± 1 oscillations/min, suggesting strongly that SK channels are stimulated in hAPP-expressing neurons. In APPswe-expressing neurons, apamin also restored calcium oscillations in 54 ± 8% of cells (analysis of 79 cells in 3 different experiments; data not shown).
Figure 3.

Long term L-type calcium channels stimulation inhibits calcium oscillations through activation of SK channels. A, Simultaneous traces of [Ca2+]i in three cells of a hAPP- expressing network show that cells recover synchronous calcium oscillations after bath application of apamin (200 nm), an SK channel inhibitor. B–C, A 60 min pretreatment of a βgal- expressing neuronal network by BayK6844 inhibits spontaneous calcium oscillations (B), which can be recovered by bath application of apamin (C).
L-type calcium channels colocalize with SK channels, and this spatial proximity is thought to influence the regulation of SK channels during action potentials (Marrion and Tavalin, 1998). The effect of L-type calcium channels activation on calcium oscillations was assessed by single-cell calcium imaging, using the L-type calcium channel agonist BayK6844. A 60 min treatment of neurons by 1 μm BayK6844 completely abolished synchronous calcium oscillations (analysis of 73 cells in 3 different experiments) (Fig. 3B). Interestingly, apamin was able to restore calcium oscillations in BayK6844 treated neurons since 60 ± 7% of cells oscillated with a frequency of 7 ± 2 oscillations/min (Fig. 3C). Altogether, these results indicate that a chronic activation of L-type calcium channels leads to hyperactivation of SK channels, resulting in abolition of calcium oscillations. Therefore, we investigated whether hAPP expression was able to increase L-type calcium currents.
Increase of L-type calcium currents by hAPP
First, we measured cytosolic calcium concentration ([Ca2+]i) in response to BayK6844 application in a population of neurons. To increase affinity of BayK6844 for L-type calcium channels, βgal- or hAPP-expressing neurons were perfused with a slightly depolarizing solution containing 15 mm KCl. This slight depolarization also inhibited calcium oscillations in control or βgal-expressing neurons (Fig. 4A). The effect of KCl-mediated depolarization on calcium oscillations has been previously described (Wang and Gruenstein, 1997), and is illustrated in figure S1D. In basal conditions, in the presence of 15 mm KCl, or after depolarization by 50 mm KCl, [Ca2+]i was similar in βgal- or hAPP-expressing neurons (Fig. 4B). Opening L-type calcium channels with BayK6844 (1 μm) induced an increase in [Ca2+]i (Fig. 4A,B). Importantly, the BayK6844-mediated increase in [Ca2+]i was greater in hAPP-expressing neurons than in βgal-expressing neurons (Fig. 4B). (Δ[Ca2+]i: 65 ± 5.7 nm in APP neurons versus 45 ± 2.6 nm in βgal neurons; p < 0.05). These results suggest that expression of hAPP in rat cortical neurons modulates L-type calcium channels function.
Figure 4.
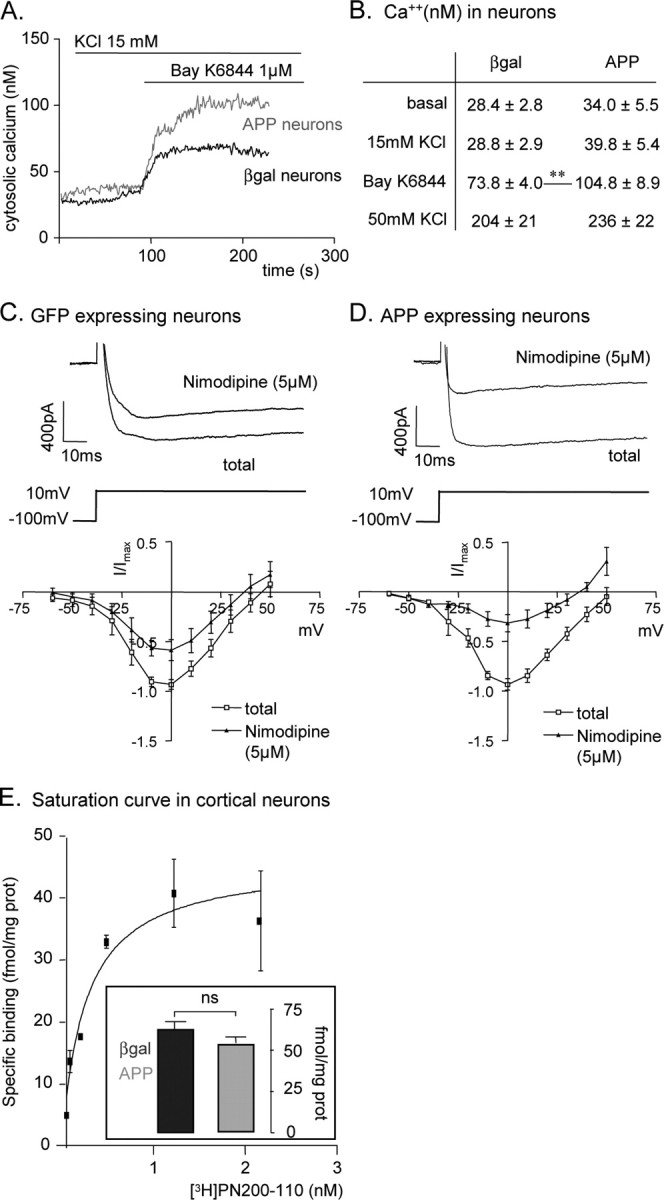
hAPP expression increases L-type calcium channel activity in neurons. A, [Ca2+]i in a population of neurons. Response to Bayk6844 is higher in hAPP-expressing neurons, when compared with βgal-expressing neurons. B, Mean values ± SEM of [Ca2+]i in neurons expressing hAPP (n = 7) or βgal (n = 7); (**p < 0.01). C, D, Top, Representative traces of whole-cell calcium current elicited by a voltage step from −100 mV to + 10 mV, before and after nimodipine (5 μm) application. Bottom, I-V plot of whole-cell calcium currents (average of n cells; APP: n = 6; GFP: n = 5) before and after nimodipine (5 μm) application, where I/Imax is the current normalized to the maximal current of each cell. E, Saturation curve for specific binding of [3H]PN200–110 radioligand to intact cultured cortical neurons. Insert, Specific binding of [3H]PN200–110 is similar in APP (n = 15) or βgal- expressing neurons (n = 6; ns, not significant).
To further investigate the possible influence of hAPP expression on L-type calcium channels activity, whole-cell calcium currents were analyzed in patch-clamp experiments. The maximal amplitude of total calcium currents was similar in hAPP-expressing and in GFP-expressing neurons (Maximal current, Imax: 17.3 ± 1.9 pA/pF for APP neurons; 13.8 ± 2.3 pA/pF for GFP neurons; not significant) (Fig. 4C,D). L-type current was measured after treatment with 5 μm nimodipine, a selective L-type channel antagonist. Nimodipine-resistant calcium currents corresponded to 58% of Imax in GFP-expressing neurons, versus only 31% in hAPP-expressing neurons (Fig. 4C,D). Therefore, maximal L-type currents were significantly increased in hAPP-expressing neurons (L-type Imax: 12.4 ± 1.8 pA/pF for hAPP-expressing neurons versus 4.9 ± 0.68 pA/pF for GFP-expressing neurons; p < 0.05). Further analysis showed that the voltage dependence of both total and L-type calcium currents was shifted to more positive potentials in hAPP-expressing neurons (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Half-activation voltage (V0.5) of L-type calcium current was higher for hAPP-expressing than for GFP-expressing neurons [V0.5 L-type calcium current: −10 ± 3 mV (n = 5) vs −19 ± 4 mV (n = 5)].
Altogether, these results show that hAPP expression in rat cortical neurons leads to an increase of L-type calcium currents in response to maximal stimulation. This could be mediated by an increase in the number of L-type calcium channels present at the plasma membrane, or by an increased activity of existing channels. To differentiate between these possibilities, we measured the binding of the L-type calcium channel antagonist [3H]PN200–110 to intact neurons. A saturation analysis showed that a concentration of [3H]PN200–110 >1 nm was sufficient to saturate all binding sites (Fig. 4E). The specific binding of [3H]PN200–110 at a saturating concentration (1.4 nm) was similar in hAPP- versus GFP-expressing neurons (respectively, 54.7 ± 3 fmol/mg prot and 62.5 ± 5 fmol/mg prot). These results strongly suggest that hAPP increases L-type calcium currents through their activation rather than via increased number of channels at the cell surface.
Inhibition of calcium oscillations by human APP is independent of Aβ production
To test whether hAPP-mediated inhibition of calcium oscillations could result from production of Aβ, neurons were treated by DAPT, a functional inhibitor of γ-secretase. hAPP- or βgal-expressing neurons were incubated in the presence of 250 nm DAPT, a concentration that completely inhibits the production of extracellular Aβ40 (Fig. 5B). [Ca2+]i was then measured after opening of L-type calcium channels by BayK6844. In the presence of DAPT, hAPP-expressing neurons maintained a larger increase in calcium entry in response to BayK6844 than βgal-expressing neurons, indicating that the effect of hAPP was independent of Aβ production or γ-cleavage (Fig. 5A). In addition, single-cell calcium imaging showed that inhibition of Aβ production by DAPT did not restore calcium oscillations in hAPP-expressing neurons (analysis of 74 cells in 3 different experiments) (Fig. 5C). Moreover, incubation of uninfected or βgal-expressing neurons in the presence of culture medium collected from hAPP-expressing neurons did not inhibit calcium oscillations since 77 ± 5% of cells (analysis of 194 cells in 6 different experiments) (Fig. 5D) showed calcium oscillations with a frequency of 12 ± 2 oscillations/min. This indicates that secreted metabolites of hAPP, including Aβ and sαAPP, are not responsible for the abolition of calcium oscillations.
Silencing endogenous APP expression affects calcium oscillations
Neuronal networks were infected by recombinant lentiviruses encoding APPshRNA (APPshRNA) or the neomycin resistance gene (Neo). Endogenous APP expression was drastically reduced by APPshRNA (Fig. 6A,B). The Neo lentivirus did not modify APP expression whereas a 95 ± 5% (n = 3) reduction was measured in Western blotting, after APPshRNA lentiviral expression (Fig. 6A). In neurons infected by Neo lentivirus, 89 ± 9% of cells (analysis of 148 cells in 4 different experiments) showed calcium oscillations at a frequency of 15 ± 3 oscillations/min, similar to uninfected neurons. In neurons infected by APPshRNA lentivirus, amplitudes of calcium oscillations were <10% variation from Rmean. However, 79 ± 10% of cells (analysis of 113 cells in 3 different experiments) showed these oscillations with a frequency of 34 ± 2 oscillations/min, which is twice as high as the frequency of uninfected or Neomycin-expressing networks (p < 0.001). Basal [Ca2+]i or calcium entry after BayK6844 stimulation were similar for uninfected, Neo-, or APPshRNA-expressing neurons (data not shown).
Figure 6.

Silencing endogenous APP affects spontaneous synchronous calcium oscillations. A, Western blot analysis of cell lysates from rat cortical neurons infected or not (NI) by APPshRNA (APPshRNA) or Neomycin (Neo) lentiviruses. Protein loading was controlled using rabbit actin antiserum. B, In a neuronal network labeled by an anti-MAP-2 antibody (red) and an anti-APPCterminal antibody (APPCter, green), infection by APPshRNA lentivirus (bottom) allows an efficient reduction of endogenous APP expression when compared with uninfected neurons. Scale bar, 20 μm. C, Simultaneous traces of [Ca2+]i in three cells in a Neo-expressing network show that calcium oscillations are not affected by lentiviral infection. D, Simultaneous traces of [Ca2+]i in three cells in a APPshRNA-expressing network show that reduction of endogenous APP expression increases frequency and decreases amplitude of calcium oscillations.
Discussion
hAPP expression in rat cortical neurons increases L-type calcium currents
We provide evidence that adenovirus-mediated expression of hAPP in rat cultured cortical neurons increases L-type calcium currents, and that this is independent of Aβ production. An increase in neuronal L-type calcium currents was previously reported to result from increased Aβ production (Ueda et al., 1997; Ekinci et al., 1999; Scragg et al., 2005; Webster et al., 2006). In contrast, in our study, DAPT completely inhibited Aβ production but did not affect the increase in L-type calcium currents, indicating that this increase is independent of γ-cleavage of APP generating Aβ or the amyloid intracellular domain (AICD). In addition, the expression of hAPP did not affect basal cytosolic calcium concentration and total calcium currents, which were previously reported to be increased by extracellular Aβ (Mattson et al., 1993; Ueda et al., 1997; Demuro et al., 2005; Webster et al., 2006; Kuchibhotla et al., 2008). Moreover, many studies looking at the effects of exogenous Aβ were performed using very high concentrations of peptide that are less physiological than the concentrations found in the culture medium of hAPP-expressing neurons.
Aβ was reported to promote trafficking and insertion of L-type calcium channels at the plasma membrane (Scragg et al., 2005). An increase in L-type calcium channels density was also observed in aged hippocampal neurons (Thibault and Landfield, 1996). In binding experiments, we did not measure any increased density of L-type calcium channels at the cell surface of hAPP-expressing neurons. Therefore, we suggest that hAPP expression increases L-type calcium channel activity, rather than the number of channels at the cell surface.
Activation of L-type calcium channels can result from increased mean open time or from increased probability of channel opening (Catterall, 2000). Activation of L-type calcium channels can result from PKA mediated phosphorylation of the α-1 subunit, Cav1.2 (Gross et al., 1990; Fisher and Johnston, 1990; Surmeier et al., 1995), and this phosphorylation is increased in aging brain (Davare and Hell, 2003). It will be interesting to investigate whether hAPP expression can increase Cav1.2 phosphorylation in our model. Interestingly, in neuronal cells, PKA activation of L-type current leads to a shift of the voltage activation curve toward more positive potentials (Gross et al., 1990), a feature that we also observed in hAPP-expressing neurons (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
hAPP expression in rat cortical neurons increases mAHP
Although the resting membrane potential was not affected by hAPP expression, a significant increase in mAHP was observed. AHP is mediated by calcium-activated K+ channels, responsible for repolarization after an action potential in excitable cells. According to their pharmacological and biophysical characteristics, different calcium-activated K+ channels mediate fast, medium or slow AHP. BK channels are large-conductance K+ channels, activated by large increases in cytosolic calcium and by voltage changes. They mediate the fast AHP. SK channels have small or intermediate K+ conductance, are highly sensitive and fast Ca2+ sensors but insensitive to voltage changes. They are specifically inhibited by apamin and mediate a K+ current named IAHP that contributes to the generation of medium AHP that activates within milliseconds and decays with a time-constant of hundreds of milliseconds (Schwindt et al., 1988). Whether SK channels also mediate the slow AHP, which decays within seconds after a burst of action potentials but is typically insensitive to apamin (Lancaster and Adams, 1986; Storm, 1989) is still a matter of debate. The contribution of apamin-sensitive SK channels to mAHP differs according to neuronal subtypes. In cortical neurons, the apamin-sensitive K+ current accounts for most of the mAHP (Villalobos et al., 2004), whereas the apamin-sensitive portion of mAHP is less important in hippocampal neurons (Storm, 1989; Stocker et al., 1999).
The molecular mechanisms by which SK channels are sensitive to calcium are well established. Calcium binding to calmodulin (CaM), constitutively attached to the CaM binding domain of SK channels, leads to a conformational change and channel opening (Schumacher et al., 2001). In addition, a functional coupling of L-type calcium channels to SK channels, which colocalize, was demonstrated in excised membrane patches of hippocampal neurons (Marrion and Tavalin, 1998). In this model, L-type calcium channels specifically regulate the SK-mediated AHP, leaving fast AHP unaltered. These mechanisms may explain why we observe an effect of L-type calcium currents on SK channels activity without any change in basal cytosolic calcium concentration.
hAPP expression inhibits spontaneous synchronous calcium oscillations in the network
hAPP expression by some neurons is sufficient to inhibit spontaneous synchronous calcium oscillations of the entire network that are rescued in the presence of apamin. The proposed molecular mechanisms are that hAPP expression increases the activity of L-type calcium channels, thereby stimulating apamin-sensitive SK channels responsible for mAHP. Increased mAHP lowers excitability of hAPP-expressing neurons (Sourdet et al., 2003; Stocker, 2004), which results in inhibition of synaptically propagated synchronous calcium oscillations in the entire network.
High concentrations of extracellular Aβ42, but not Aβ40, abolish neuronal calcium oscillations (Rui et al., 2006). In our culture system, a low concentration of extracellular Aβ was measured, but inhibition of Aβ production by DAPT did not restore calcium oscillations, indicating that inhibition of oscillations was not related to Aβ production.
Previous studies showed that recombinant sαAPP activates high-conductance, charybdotoxin-sensitive K+ channels resulting in hyperpolarization and reduced excitability in hippocampal neurons (Furukawa et al., 1996a; Furukawa et al., 1996b; Furukawa and Mattson, 1998). Since the conditioned medium of hAPP-expressing neurons does not affect calcium oscillations of control neurons, we conclude that sαAPP is not involved in our observations. In addition, we demonstrate that the APP effect is mediated by apamin-sensitive K+ channels which are not affected by sαAPP, as shown in (Furukawa et al., 1996a).
APP mutants responsible for inherited AD are known to increase the production of Aβ or the Aβ42/Aβ40 ratio. In our model, neuronal expression of APPswe, which increases the production of both Aβ40 and Aβ42 inhibited calcium oscillations, as did the wild-type form of hAPP. Contrary to our observations, an increased frequency of calcium oscillations was observed in hippocampal neurons from transgenic rats expressing human APP with the Swedish mutation (hAPPswe), compared with normal neurons, and this was independent of Aβ production (Kloskowska et al., 2008). This discrepancy may be due to chronic adaptations in transgenic animals, which cannot occur during acute expression of hAPP. In addition, as mentioned above, the pattern of SK channels expression is different in hippocampal and cortical neurons.
Disruption of neuronal excitability and abnormal AHP might contribute to neurodegeneration, in aging and AD brain (Ohno et al., 2004; Disterhoft and Oh, 2007; Driver et al., 2007; Ye et al., 2008). Transgenic mice overexpressing hAPPswe have an impairment of hippocampal gamma-frequency oscillations (Driver et al., 2007). Slow AHP is increased in these mice, and activation of L-type calcium channels, leading to hyperactivation of calcium-activated K+ channels, was proposed as a possible mechanism (Driver et al., 2007). Interestingly, treatment with dihydropyridines, L-type calcium channels antagonists, may improve memory in aging animals (Deyo et al., 1989; Straube et al., 1990; Solomon et al., 1995; Veng et al., 2003; Rose et al., 2007) and cognitive function in AD patients. (Lopez-Arrieta and Birks, 2002).
hAPP expression and calcium signaling
Calcium oscillations play an important role in a variety of cells. The frequency of oscillations controls gene expression by selectively activating transcription factors such as NF-AT, NFκB or Oct/OAP (Dolmetsch et al., 1998; Tomida et al., 2003). In neurons, the frequency of oscillations determines induction of long term potentiation or long term depression. Our results show that acute expression of hAPP disturbs calcium homeostasis by increasing L-type calcium currents, leaving cytosolic concentration of calcium and intracellular calcium stores unaltered (Pierrot et al., 2004). Moreover, we show that acute inhibition of endogenous APP expression alters the pattern of calcium oscillations, by reducing their amplitude and increasing their frequency. This demonstrates a key role of APP in the control of neuronal calcium signaling.
Neuronal injuries such as ischemia (Tomimoto et al., 1995; Popa-Wagner et al., 1998; Shi et al., 2000; Hellweg et al., 2003), trauma (Dewji et al., 1995; Van Den Heuvel et al., 2000) or neuroinflammation (Ringheim et al., 1998) induce APP overexpression, and acute APP overexpression leads to synaptic depression via reduced levels of AMPA and NMDA receptors (Kamenetz et al., 2003; Almeida et al., 2005; Ting et al., 2007). In addition, APP gene duplication results in early onset AD with cerebral amyloid angiopathy (Sleegers et al., 2006; Rovelet-Lecrux et al., 2006), further indicating that increased APP expression is a risk factor for AD. The present results suggest that the control of neuronal excitability may be a major function of APP. Abnormal APP expression could have dramatic effects on neuronal function, and APP-mediated depression of synaptic activity may contribute to neurodegeneration, long before Aβ accumulates in senile plaques.
Footnotes
This work was supported by the Belgian Fonds National pour la Recherche Scientifique (FNRS-FRS), by Interuniversity Attraction Poles Programme–Belgian State–Belgian Science Policy, the Communauté Française de Belgique, Actions de Recherche Concertés, The Belgian Fonds de la Recherche Scientifique Médicale, the Fondation pour la Recherche sur la Maladie d'Alzheimer (FRMA), and the Programme d'excellence “Marshall” DIANE convention. We thank L. Mercken for the generous gift from DAPT, J.-P. Brion for providing the B9 anti-MAP2 antibody, and P. Kienlen-Campard and A. Goffinet for critical reading of this manuscript.
References
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J Neurosci Res. 1995;42:674–683. doi: 10.1002/jnr.490420510. [DOI] [PubMed] [Google Scholar]
- Buryi V, Morel N, Salomone S, Kerger S, Godfraind T. Evidence for a direct interaction of thapsigargin with voltage-dependent Ca2+ channel. Naunyn Schmiedebergs Arch Pharmacol. 1995;351:40–45. doi: 10.1007/BF00169062. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Davare MA, Hell JW. Increased phosphorylation of the neuronal L-type Ca(2+) channel Cav1.2 during aging. Proc Natl Acad Sci U S A. 2003;100:16018–16023. doi: 10.1073/pnas.2236970100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- Dewji NN, Do C, Bayney RM. Transcriptional activation of Alzheimer's beta-amyloid precursor protein gene by stress. Brain Res Mol Brain Res. 1995;33:245–253. doi: 10.1016/0169-328x(95)00131-b. [DOI] [PubMed] [Google Scholar]
- Deyo RA, Straube KT, Disterhoft JF. Nimodipine facilitates associative learning in aging rabbits. Science. 1989;243:809–811. doi: 10.1126/science.2916127. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Alterations in intrinsic neuronal excitability during normal aging. Aging Cell. 2007;6:327–336. doi: 10.1111/j.1474-9726.2007.00297.x. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- Dravid SM, Murray TF. Spontaneous synchronized calcium oscillations in neocortical neurons in the presence of physiological [Mg(2+)]: involvement of AMPA/kainate and metabotropic glutamate receptors. Brain Res. 2004;1006:8–17. doi: 10.1016/j.brainres.2004.01.059. [DOI] [PubMed] [Google Scholar]
- Driver JE, Racca C, Cunningham MO, Towers SK, Davies CH, Whittington MA, LeBeau FE. Impairment of hippocampal gamma-frequency oscillations in vitro in mice overexpressing human amyloid precursor protein (APP) Eur J Neurosci. 2007;26:1280–1288. doi: 10.1111/j.1460-9568.2007.05705.x. [DOI] [PubMed] [Google Scholar]
- Ekinci FJ, Malik KU, Shea TB. Activation of the L voltage-sensitive calcium channel by mitogen-activated protein (MAP) kinase following exposure of neuronal cells to beta-amyloid. MAP kinase mediates beta-amyloid-induced neurodegeneration. J Biol Chem. 1999;274:30322–30327. doi: 10.1074/jbc.274.42.30322. [DOI] [PubMed] [Google Scholar]
- Fisher R, Johnston D. Differential modulation of single voltage-gated calcium channels by cholinergic and adrenergic agonists in adult hippocampal neurons. J Neurophysiol. 1990;64:1291–1302. doi: 10.1152/jn.1990.64.4.1291. [DOI] [PubMed] [Google Scholar]
- Flint AC, Dammerman RS, Kriegstein AR. Endogenous activation of metabotropic glutamate receptors in neocortical development causes neuronal calcium oscillations. Proc Natl Acad Sci U S A. 1999;96:12144–12149. doi: 10.1073/pnas.96.21.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Mattson MP. Secreted amyloid precursor protein alpha selectively suppresses N-methyl-D-aspartate currents in hippocampal neurons: involvement of cyclic GMP. Neuroscience. 1998;83:429–438. doi: 10.1016/s0306-4522(97)00398-9. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Barger SW, Blalock EM, Mattson MP. Activation of K+ channels and suppression of neuronal activity by secreted beta-amyloid-precursor protein. Nature. 1996a;379:74–78. doi: 10.1038/379074a0. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Sopher BL, Rydel RE, Begley JG, Pham DG, Martin GM, Fox M, Mattson MP. Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J Neurochem. 1996b;67:1882–1896. doi: 10.1046/j.1471-4159.1996.67051882.x. [DOI] [PubMed] [Google Scholar]
- Gross RA, Uhler MD, Macdonald RL. The cyclic AMP-dependent protein kinase catalytic subunit selectively enhances calcium currents in rat nodose neurones. J Physiol. 1990;429:483–496. doi: 10.1113/jphysiol.1990.sp018268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gu X, Spitzer NC. Distinct aspects of neuronal differentiation encoded by frequency of spontaneous Ca2+ transients. Nature. 1995;375:784–787. doi: 10.1038/375784a0. [DOI] [PubMed] [Google Scholar]
- Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rülicke T, von Kretzschmar H, von Koch C, Sisodia S, Tremml P, Lipp HP, Wolfer DP, Müller U. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000;20:7951–7963. doi: 10.1523/JNEUROSCI.20-21-07951.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellweg R, von Arnim CA, Büchner M, Huber R, Riepe MW. Neuroprotection and neuronal dysfunction upon repetitive inhibition of oxidative phosphorylation. Exp Neurol. 2003;183:346–354. doi: 10.1016/s0014-4886(03)00127-4. [DOI] [PubMed] [Google Scholar]
- Johnston J, O'Neill C, Lannfelt L, Winblad B, Cowburn RF. The significance of the Swedish APP670/671 mutation for the development of Alzheimer's disease amyloidosis. Neurochem Int. 1994;25:73–80. doi: 10.1016/0197-0186(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kienlen-Campard P, Miolet S, Tasiaux B, Octave JN. Intracellular amyloid-beta 1–42, but not extracellular soluble amyloid-beta peptides, induces neuronal apoptosis. J Biol Chem. 2002;277:15666–15670. doi: 10.1074/jbc.M200887200. [DOI] [PubMed] [Google Scholar]
- Kloskowska E, Malkiewicz K, Winblad B, Benedikz E, Bruton JD. APPswe mutation increases the frequency of spontaneous Ca2+-oscillations in rat hippocampal neurons. Neurosci Lett. 2008;436:250–254. doi: 10.1016/j.neulet.2008.03.034. [DOI] [PubMed] [Google Scholar]
- Komuro H, Rakic P. Intracellular Ca2+ fluctuations modulate the rate of neuronal migration. Neuron. 1996;17:275–285. doi: 10.1016/s0896-6273(00)80159-2. [DOI] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Adams PR. Calcium-dependent current generating the afterhyperpolarization of hippocampal neurons. J Neurophysiol. 1986;55:1268–1282. doi: 10.1152/jn.1986.55.6.1268. [DOI] [PubMed] [Google Scholar]
- Lemarchand P, Jaffe HA, Danel C, Cid MC, Kleinman HK, Stratford-Perricaudet LD, Perricaudet M, Pavirani A, Lecocq JP, Crystal RG. Adenovirus-mediated transfer of a recombinant human alpha 1-antitrypsin cDNA to human endothelial cells. Proc Natl Acad Sci U S A. 1992;89:6482–6486. doi: 10.1073/pnas.89.14.6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Arrieta JM, Birks J. Nimodipine for primary degenerative, mixed and vascular dementia. Cochrane Database Syst Rev. 2002 doi: 10.1002/14651858.CD000147. CD000147. [DOI] [PubMed] [Google Scholar]
- Macq AF, Czech C, Essalmani R, Brion JP, Maron A, Mercken L, Pradier L, Octave JN. The long term adenoviral expression of the human amyloid precursor protein shows different secretase activities in rat cortical neurons and astrocytes. J Biol Chem. 1998;273:28931–28936. doi: 10.1074/jbc.273.44.28931. [DOI] [PubMed] [Google Scholar]
- Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Tomaselli KJ, Rydel RE. Calcium-destabilizing and neurodegenerative effects of aggregated beta-amyloid peptide are attenuated by basic FGF. Brain Res. 1993;621:35–49. doi: 10.1016/0006-8993(93)90295-x. [DOI] [PubMed] [Google Scholar]
- Morel N, Buryi V, Feron O, Gomez JP, Christen MO, Godfraind T. The action of calcium channel blockers on recombinant L-type calcium channel alpha1-subunits. Br J Pharmacol. 1998;125:1005–1012. doi: 10.1038/sj.bjp.0702162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Blatter LA, Wier WG, Baraban JM. Spontaneous synchronous synaptic calcium transients in cultured cortical neurons. J Neurosci. 1992;12:4834–4845. doi: 10.1523/JNEUROSCI.12-12-04834.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron. 2004;41:27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Opitz T, De Lima AD, Voigt T. Spontaneous development of synchronous oscillatory activity during maturation of cortical networks in vitro. J Neurophysiol. 2002;88:2196–2206. doi: 10.1152/jn.00316.2002. [DOI] [PubMed] [Google Scholar]
- Pierrot N, Ghisdal P, Caumont AS, Octave JN. Intraneuronal amyloid-beta1–42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J Neurochem. 2004;88:1140–1150. doi: 10.1046/j.1471-4159.2003.02227.x. [DOI] [PubMed] [Google Scholar]
- Pineda JC, Waters RS, Foehring RC. Specificity in the interaction of HVA Ca2+ channel types with Ca2+-dependent AHPs and firing behavior in neocortical pyramidal neurons. J Neurophysiol. 1998;79:2522–2534. doi: 10.1152/jn.1998.79.5.2522. [DOI] [PubMed] [Google Scholar]
- Popa-Wagner A, Schröder E, Walker LC, Kessler C. beta-Amyloid precursor protein and ss-amyloid peptide immunoreactivity in the rat brain after middle cerebral artery occlusion: effect of age. Stroke. 1998;29:2196–2202. doi: 10.1161/01.str.29.10.2196. [DOI] [PubMed] [Google Scholar]
- Ringheim GE, Szczepanik AM, Petko W, Burgher KL, Zhu SZ, Chao CC. Enhancement of beta-amyloid precursor protein transcription and expression by the soluble interleukin-6 receptor/interleukin-6 complex. Brain Res Mol Brain Res. 1998;55:35–44. doi: 10.1016/s0169-328x(97)00356-2. [DOI] [PubMed] [Google Scholar]
- Rose GM, Ong VS, Woodruff-Pak DS. Efficacy of MEM 1003, a novel calcium channel blocker, in delay and trace eyeblink conditioning in older rabbits. Neurobiol Aging. 2007;28:766–773. doi: 10.1016/j.neurobiolaging.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerrière A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- Rui Y, Li R, Liu Y, Zhu S, Yu X, Sheng Z, Xie Z. Acute effect of beta amyloid on synchronized spontaneous Ca2+ oscillations in cultured hippocampal networks. Cell Biol Int. 2006;30:733–740. doi: 10.1016/j.cellbi.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Salmon P, Trono D. Production and titration of lentiviral vectors. Curr Protoc Hum Genet. 2007 doi: 10.1002/0471142905.hg1210s54. Chapter 12: Unit 12.10. [DOI] [PubMed] [Google Scholar]
- Schumacher MA, Rivard AF, Bächinger HP, Adelman JP. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature. 2001;410:1120–1124. doi: 10.1038/35074145. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, Foehring RC, Stafstrom CE, Chubb MC, Crill WE. Multiple potassium conductances and their functions in neurons from cat sensorimotor cortex in vitro. J Neurophysiol. 1988;59:424–449. doi: 10.1152/jn.1988.59.2.424. [DOI] [PubMed] [Google Scholar]
- Scragg JL, Fearon IM, Boyle JP, Ball SG, Varadi G, Peers C. Alzheimer's amyloid peptides mediate hypoxic up-regulation of L-type Ca2+ channels. FASEB J. 2005;19:150–152. doi: 10.1096/fj.04-2659fje. [DOI] [PubMed] [Google Scholar]
- Shi J, Yang SH, Stubley L, Day AL, Simpkins JW. Hypoperfusion induces overexpression of beta-amyloid precursor protein mRNA in a focal ischemic rodent model. Brain Res. 2000;853:1–4. doi: 10.1016/s0006-8993(99)02113-7. [DOI] [PubMed] [Google Scholar]
- Silva LR, Amitai Y, Connors BW. Intrinsic oscillations of neocortex generated by layer 5 pyramidal neurons. Science. 1991;251:432–435. doi: 10.1126/science.1824881. [DOI] [PubMed] [Google Scholar]
- Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, Del-Favero J, Cruts M, van Duijn CM, Van Broeckhoven C. APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain. 2006;129:2977–2983. doi: 10.1093/brain/awl203. [DOI] [PubMed] [Google Scholar]
- Slunt HH, Thinakaran G, Von Koch C, Lo AC, Tanzi RE, Sisodia SS. Expression of a ubiquitous, cross-reactive homologue of the mouse beta-amyloid precursor protein (APP) J Biol Chem. 1994;269:2637–2644. [PubMed] [Google Scholar]
- Solomon PR, Wood MS, Groccia-Ellison ME, Yang BY, Fanelli RJ, Mervis RF. Nimodipine facilitates retention of the classically conditioned nictitating membrane response in aged rabbits over long retention intervals. Neurobiol Aging. 1995;16:791–796. doi: 10.1016/0197-4580(95)00093-t. [DOI] [PubMed] [Google Scholar]
- Sourdet V, Russier M, Daoudal G, Ankri N, Debanne D. Long-term enhancement of neuronal excitability and temporal fidelity mediated by metabotropic glutamate receptor subtype 5. J Neurosci. 2003;23:10238–10248. doi: 10.1523/JNEUROSCI.23-32-10238.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer NC, Olson E, Gu X. Spontaneous calcium transients regulate neuronal plasticity in developing neurons. J Neurobiol. 1995;26:316–324. doi: 10.1002/neu.480260304. [DOI] [PubMed] [Google Scholar]
- Steriade M, Nuñez A, Amzica F. A novel slow (< 1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M. Ca(2+)-activated K+ channels: molecular determinants and function of the SK family. Nat Rev Neurosci. 2004;5:758–770. doi: 10.1038/nrn1516. [DOI] [PubMed] [Google Scholar]
- Stocker M, Krause M, Pedarzani P. An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 1999;96:4662–4667. doi: 10.1073/pnas.96.8.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. An after-hyperpolarization of medium duration in rat hippocampal pyramidal cells. J Physiol. 1989;409:171–190. doi: 10.1113/jphysiol.1989.sp017491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straube KT, Deyo RA, Moyer JR, Jr, Disterhoft JF. Dietary nimodipine improves associative learning in aging rabbits. Neurobiol Aging. 1990;11:659–661. doi: 10.1016/0197-4580(90)90033-v. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Bargas J, Hemmings HC, Jr, Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–397. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272:1017–1020. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- Tilleux S, Hermans E. Down-regulation of astrocytic GLAST by microglia-related inflammation is abrogated in dibutyryl cAMP-differentiated cultures. J Neurochem. 2008;105:2224–2236. doi: 10.1111/j.1471-4159.2008.05305.x. [DOI] [PubMed] [Google Scholar]
- Ting JT, Kelley BG, Lambert TJ, Cook DG, Sullivan JM. Amyloid precursor protein overexpression depresses excitatory transmission through both presynaptic and postsynaptic mechanisms. Proc Natl Acad Sci U S A. 2007;104:353–358. doi: 10.1073/pnas.0608807104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomida T, Hirose K, Takizawa A, Shibasaki F, Iino M. NFAT functions as a working memory of Ca2+ signals in decoding Ca2+ oscillation. EMBO J. 2003;22:3825–3832. doi: 10.1093/emboj/cdg381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomimoto H, Akiguchi I, Wakita H, Nakamura S, Kimura J. Ultrastructural localization of amyloid protein precursor in the normal and postischemic gerbil brain. Brain Res. 1995;672:187–195. doi: 10.1016/0006-8993(94)01160-j. [DOI] [PubMed] [Google Scholar]
- Ueda K, Shinohara S, Yagami T, Asakura K, Kawasaki K. Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J Neurochem. 1997;68:265–271. doi: 10.1046/j.1471-4159.1997.68010265.x. [DOI] [PubMed] [Google Scholar]
- Van Den Heuvel C, Blumbergs P, Finnie J, Manavis J, Lewis S, Jones N, Reilly P, Pereira R. Upregulation of amyloid precursor protein and its mRNA in an experimental model of paediatric head injury. J Clin Neurosci. 2000;7:140–145. doi: 10.1054/jocn.1999.0168. [DOI] [PubMed] [Google Scholar]
- Veng LM, Mesches MH, Browning MD. Age-related working memory impairment is correlated with increases in the L-type calcium channel protein alpha(1D) (Ca. (v) 1.3) in area CA1 of the hippocampus and both are ameliorated by chronic nimodipine treatment. Brain Res Mol Brain Res. 2003;110:193–202. doi: 10.1016/s0169-328x(02)00643-5. [DOI] [PubMed] [Google Scholar]
- Villalobos C, Shakkottai VG, Chandy KG, Michelhaugh SK, Andrade R. SKCa channels mediate the medium but not the slow calcium-activated afterhyperpolarization in cortical neurons. J Neurosci. 2004;24:3537–3542. doi: 10.1523/JNEUROSCI.0380-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gruenstein EI. Mechanism of synchronized Ca2+ oscillations in cortical neurons. Brain Res. 1997;767:239–249. doi: 10.1016/s0006-8993(97)00585-4. [DOI] [PubMed] [Google Scholar]
- Wasco W, Gurubhagavatula S, Paradis MD, Romano DM, Sisodia SS, Hyman BT, Neve RL, Tanzi RE. Isolation and characterization of APLP2 encoding a homologue of the Alzheimer's associated amyloid beta protein precursor. Nat Genet. 1993;5:95–100. doi: 10.1038/ng0993-95. [DOI] [PubMed] [Google Scholar]
- Webster NJ, Ramsden M, Boyle JP, Pearson HA, Peers C. Amyloid peptides mediate hypoxic increase of L-type Ca2+ channels in central neurones. Neurobiol Aging. 2006;27:439–445. doi: 10.1016/j.neurobiolaging.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Ye H, Jalini S, Mylvaganam S, Carlen P. Activation of large-conductance Ca(2+)-activated K(+) channels depresses basal synaptic transmission in the hippocampal CA1 area in APP (swe/ind) TgCRND8 mice. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.05.012. [DOI] [PubMed] [Google Scholar]