

Abstract
We have associated behavioral, pharmacological, and quantitative immunohistochemical study in a rat analog of l-DOPA-induced dyskinesia to understand whether alterations in dopamine receptor fate in striatal neurons may be involved in mechanisms leading to movement abnormalities. Detailed analysis at the ultrastructural level demonstrates specific alterations of dopamine D1 receptor (D1R) subcellular localization in striatal medium spiny neurons in l-DOPA-treated 6-hydroxydopamine-lesioned rats with abnormal involuntary movements (AIMs). This includes exaggerated D1R expression at the plasma membrane. However, D1R retains ability of internalization, as a challenge with the potent D1R agonist SKF-82958 induces a strong decrease of labeling at membrane in animals with AIMs. Since a functional cross talk between D1R and D3R has been suggested, we hypothesized that their coactivation by dopamine derived from l-DOPA might anchor D1R at the membrane. Accordingly, cotreatment with l-DOPA and the D3R antagonist ST 198 restores normal level of membrane-bound D1R. Together, these results demonstrate that AIMs are related to abnormal D1R localization at the membrane and intraneuronal trafficking dysregulation, and suggest that strategies aiming at disrupting the D1R–D3R cross talk might reduce l-DOPA-induced dyskinesia by reducing D1R availability at the membrane.
Introduction
Parkinson's disease (PD) is a neurodegenerative disorder caused by the degeneration of nigral neurons that provide dopamine to the striatum (Ehringer and Hornykiewicz, 1960). The still most effective symptomatic therapy is the dopamine precursor l-3,4-dihydroxyphenylalanine (l-DOPA). Long-term treatment leads to l-DOPA-induced dyskinesia (LID) or involuntary aimless movements (Cotzias et al., 1969). Loss of dopamine in PD induces complex modifications in signaling, with numerous pathways showing exaggerated responses to dopaminergic stimulation in the dopamine-depleted striatum (Bezard et al., 2001a; Jenner, 2008). Chronic l-DOPA treatment further distorts the signaling. Existing evidence suggests that supersensitivity of D1 [dopamine D1 receptor (D1R)] and D2 dopamine receptors is one of the molecular mechanisms underlying LID (Bezard et al., 2001a; Jenner, 2008).
Homologous desensitization effectively terminates signaling by G-protein-coupled receptors (GPCR), thereby controlling their activity. In theory, the acute activation of dopamine receptors by their direct agonist should provoke a dramatic modification of their subcellular distribution in neurons, including plasma membrane depletion and internalization in the cytoplasm (Dumartin et al., 1998). In LID, however, at odds with this predicted behavior, we reported that elevated membrane expression and reduced internalization of D1R occurs in the striatum of dyskinetic monkeys (Guigoni et al., 2007), suggesting that LID are associated with deficiencies in the D1R desensitization and trafficking (Bezard et al., 2005; Guigoni et al., 2007). Interestingly, expression level of the D3R in the dorsal motor-related striatum correlates with experimental mouse (Gross et al., 2003), rat (Bordet et al., 1997), and primate (Bézard et al., 2003) analogs of dyskinesia. D1R and D3R are coexpressed by the striatonigral medium spiny neurons (Le Moine and Bloch, 1996; Ridray et al., 1998) and have recently been shown to directly interact (Fiorentini et al., 2008) through an intramembrane D1R–D3R cross talk (Marcellino et al., 2008).
Therefore, we hypothesized that the lack of l-DOPA-induced D1R internalization may be caused by the concomitant stimulation of both D1R and D3R by their natural ligand dopamine. We here test this hypothesis in the rat analog of LID, i.e., the l-DOPA-induced abnormal involuntary movements (AIMs) in unilaterally 6-hydroxydopamine (6-OHDA)-lesioned animals (Cenci et al., 1998), investigating the cellular and subcellular distribution of D1R at electron microscopic level in a comprehensive behavioral pharmacological study.
Materials and Methods
Experimental design.
Adult male Sprague Dawley rats (Charles River Laboratories), weighing 175–200 g at the beginning of the experiment, were used. They were maintained under standard laboratory conditions. Experiments were performed in accordance with French (87-848, Ministère de l'Agriculture et de la Forêt) and European Economic Community (86-6091 EEC) guidelines for the care of laboratory animals and were approved by the Ethical Committee of Centre National de la Recherche Scientifique, Région Aquitaine.
On day 0 of the protocol (Table 1), unilateral dopamine deprivation of the striatum was obtained by 6-OHDA (3 μg/μl; Sigma) injection in the right medial forebrain bundle (2.5 μl at anterior–posterior = −3.7 mm, medial–lateral = +1.7 mm, and dorso–ventral = −8 mm, relative to bregma) (Meissner et al., 2006; Schuster et al., 2008) in rats pretreated with citalopram (1 mg/kg, i.p.; Sigma), an inhibitor of serotonin reuptake and with desipramine hydrochloride (20 mg/kg, i.p.; Sigma), an inhibitor of noradrenergic reuptake (Torres et al., 2003). Animals displaying both an impaired stepping test (Olsson et al., 1995; Winkler et al., 2002; Pioli et al., 2008), measured on days 18–20 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), and a loss of tyrosine hydroxylase-immunopositive fibers in the striatum >95% (Bezard et al., 2001b; Guigoni et al., 2005), assessed after completion of all experiments (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), were retained for final analysis.
Table 1.
Experimental protocol with timelines, drug treatments, and behavioral consequences
| 6-OHDA | Benserazide | l-DOPA | AIMs | SKF-82958 | ST 198 | AIMs | |
|---|---|---|---|---|---|---|---|
| Timing (day) | 0 | 21–30 | 21–30 | 29 | 30 | 30 | 30 |
| Groups | |||||||
| 1 (n = 4) | + | + | − | No | − | − | − |
| 2 (n = 4) | + | + | + | No | − | − | − |
| 3 (n = 4) | + | + | + | Yes | − | − | + |
| 4 (n = 4) | + | + | − | No | + | − | −/+ |
| 5 (n = 4) | + | + | + | No | + | − | + |
| 6 (n = 4) | + | + | + | Yes | + | − | + |
| 7 (n = 4) | + | + | + | Yes | − | + | + |
On day 30, all animals received the last vehicle injection +/− l-DOPA. D3R antagonist ST 198 was injected 60 min before killing (group 7); D1R agonist SKF-82958 was injected 45 min before killing (groups 4, 5, and 6). All animals were killed 60 min after l-DOPA, the last administration.
From day 21 till day 30, rats were treated once daily with benserazide (15 mg/kg, i.p.; Sigma) and either vehicle (12 rats) or l-DOPA (3 mg/kg, i.p.; Sigma) (65 rats) (Table 1). This dose of l-DOPA was similar to the ED50 value used for AIMs induction in rats [3.2 mg/kg plus benserazide (12.5 mg/kg, i.p.)] (Putterman et al., 2007). In such conditions, l-DOPA induces a gradual development of dyskinetic-like AIMs. On day 29, 21 rats were scored as nondyskinetic (score = 0.71), and 44 rats were scored as dyskinetic (score = 8.3) after observation by a trained investigator as described previously (Meissner et al., 2006; Schuster et al., 2008, 2009) using a validated rating scale (Cenci et al., 1998; Lundblad et al., 2002). On day 30, a subset of the above animals received an additional acute challenge of either the D1R agonist SKF-82958 (2 mg/kg, i.p.; Sigma; 15 min after l-DOPA) or the D3R antagonist ST 198 (30 mg/kg, p.o.; University of Frankfurt) (Mach et al., 2004) together with l-DOPA. Doses were chosen for their respective ability to induce D1R internalization (Dumartin et al., 1998) and to reduce dyskinesia severity (Bézard et al., 2003). AIMs score was rated again during the last 15 min before terminal procedure. All animals were killed 60 min after l-DOPA administration. A total of seven experimental groups was thus considered (Table 1). Four animals were randomly selected in each group for ultrastructural analysis.
Immunohistochemical detection of D1R.
D1R was detected by immunohistochemistry at the light and electron microscopic level according to previously described and validated procedures (Caillé et al., 1996; Dumartin et al., 1998) using a monoclonal antibody raised in rat against a 97 aa sequence corresponding to the C terminus of the human D1R (Sigma) (Levey et al., 1993; Hersch et al., 1995). Chloral hydrate-anesthetized (400 mg/kg, i.p., VWR) rats were perfused with 2% paraformaldehyde (PFA)/0.2% glutaraldehyde in 0.1 m phosphate buffer at pH 7.4. Brains were quickly removed, left overnight in 2% PFA at 4°C, and cut into 60-μm-thick frontal sections with vibratome. To enhance the penetration of the immunoreagents, the sections were equilibrated in a cryoprotectant solution, freeze-thawed, and stored in PBS with 0.03% sodium azide. For light microscopy observation, tissue sections of striatum were treated by immunoperoxidase technique using the tyramide signal amplification method (New England Nuclear) as described previously (Guigoni et al., 2007) or the avidin–biotin complex revealed by the glucose oxidase-DAB-nickel method (Shu et al., 1988). For electron microscopy observation, D1R was detected by the pre-embedding immunogold technique as described previously (Caillé et al., 1996; Dumartin et al., 1998, 2000). After immunodetection, the vibratome sections were postfixed, dehydrated, and included in resin (Durcupan ACM; Fluka). Serial ultrathin sections were cut with a Reichert Ultracut S, contrasted with lead citrate and examined with Hitachi H-7650 microscope.
Quantitative analysis of immunogold experiments.
Abundance of D1R in the different subcellular compartments was analyzed from immunogold-treated sections at the ultrastructural level (Dumartin et al., 1998, 2000; Guigoni et al., 2007). The analysis was performed on digital images obtained with a computer linked directly to charge-coupled device camera on the electron microscope at a final magnification of 8000–12,000 using the MetaMorph software (version 4.6r5; Universal Imaging). The immunogold particles were identified and counted in association with five compartments: plasma membrane, rough endoplasmic reticulum, Golgi apparatus, endosome-like small vesicles (Dumartin et al., 1998), and multivesicular bodies, which are organelles known as marker of the degradative lysosomal pathway (Bernard et al., 2006; Piper and Katzmann, 2007; Hanyaloglu and von Zastrow, 2008). The measures were performed on 25 perikarya per animal with four animals per group and expressed as the number of immunoparticles per 100 μm of membrane, per 100 μm2 of total cytoplasmic surface and the number of immunoparticles associated with each of the five different subcellular compartments per 100 μm2 of cytoplasmic surface.
Statistical analysis.
For multiple comparisons, depending on the number of factors, a one-way ANOVA (Kruskal–Wallis test) or a three-way ANOVA were followed, if allowed, by post hoc t tests corrected for multiple comparisons. All data were normally distributed, and significance levels of t test comparisons were adjusted for inequality of variances when appropriate. These analyses were completed using STATA program (Intercooled Stata 9.0; Stata Corporation). A probability level of 5% (p < 0.05) was considered statistically significant.
Results
Overall D1R distribution in parkinsonian animals
D1R immunostaining at both light and electron microscopic levels showed typical localization in striatal medium spiny neurons in the vehicle-injected 6-OHDA-lesioned animals (Fig. 1). At the light microscopic level, D1R was located mostly at the plasma membrane, whatever group considered, with the noticeable exception of SKF-82958-treated groups, that demonstrated prominent cytoplasmic localization (Fig. 1A–E) as expected from our previous experiments (Caillé et al., 1996; Dumartin et al., 1998, 2000). Ultrastructural analysis confirmed that, in vehicle-injected 6-OHDA-lesioned animals (group 1), the majority of immunogold particles were located at the inner side of the plasma membrane (61.2%) of the cell bodies. D1R was present in low abundance in cytoplasm (38.8%), restricted mostly to the endoplasmic reticulum (30.1%) and the Golgi apparatus (6.2%) (Fig. 1F).
Figure 1.
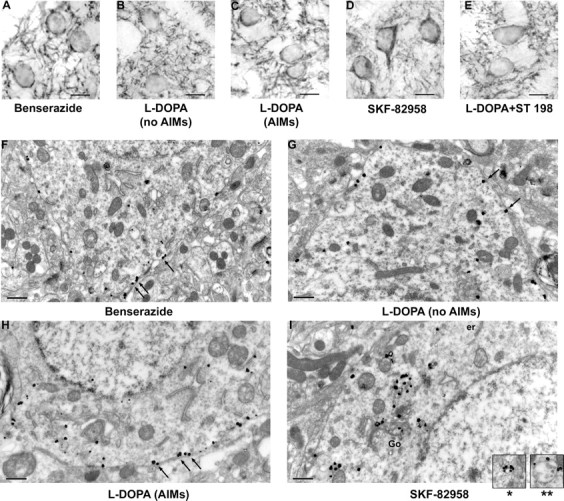
Detection of D1R immunoreactivity at light and electron microscopic levels in striatal medium spiny neurons of 6-OHDA unilaterally lesioned rats. All animals bear a 6-OHDA lesion. A–E, Light microscopy, Avidin–biotin DAB Nickel technique (scale bar, 10 μm). A, Control (benserazide only); B, l-DOPA treated without AIMs; C, l-DOPA-treated with AIMs; D, SKF-82958 treated; E, combination of l-DOPA and ST 198. D1R immunoreactivity is mostly located at plasma membrane of cell bodies and in the neuropil in all situations, except after SKF-82958 where immunoreactivity has prominent cytoplasmic localization. F–I, Electron microscopy, Immunogold technique (scale bar, 0.5 μm). F, Control (benserazide only); G, l-DOPA treated without AIMs; H, l-DOPA treated with AIMs; I, combination of l-DOPA and SKF-82958. D1R immunoreactivity shows prominent localization at plasma membrane in all situations (F–H, arrows) but after combination of l-DOPA and SKF-82958 (I) where D1R immunoreactivity is prominently localized in cytoplasmic organelles (er, endoplasmic reticulum; Go, Golgi apparatus). Insets in I show labeling at the periphery of vesicles (*) and multivesicular bodies (**).
Quantitative analysis of D1R localization at the ultrastructural level in subcellular compartments demonstrated several discrete changes in D1R subcellular distribution depending on pharmacological treatments (Figs. 2, 3).
Figure 2.

Effect of l-DOPA during D1R subcellular distribution in striatal neurons. Ratio L/NL (mean ± SEM) represents the number of immunoparticles in lesioned side (L) versus nonlesioned (NL) side at the plasma membrane [Kruskal–Wallis (KW) = 7.423; p < 0.05], in the cytoplasm [KW = 4.269; not significant (ns; A)], and in organelles (B) (er, endoplasmic reticulum; KW = 0.199, ns; Golgi, KW = 0.105, ns; vesicles, KW = 0.675, ns). * indicates a significant difference between related groups; p < 0.05; Mann–Whitney post hoc test.
Figure 3.

Relationship between D1R subcellular distribution and AIMs: effect of D1R agonist. Data (mean ± SEM) were collected on the 6-OHDA-lesioned side. A, Experimental details of the six groups showing the pharmacological treatments and their behavioral consequences. Number of D1R immunoparticles (B) per neuronal cell body (F(1,23) = 11.6, p < 0.01), per 100 μm of plasma membrane (F(1,23) = 5.5, p < 0.05) (C), and per 100 μm2 of cytoplasm (F(1,23) = 34.4, p < 0.001) (D). The subcellular distribution in cytoplasmic organelles is further broken down into (Di) endoplasmic reticulum (F(1,22) = 39.4, p < 0.001), (Dii) Golgi apparatus (F(1,23) = 8.3, p < 0.01), (Diii) vesicles (F(1,23) = 8.4, p < 0.01), and (Div) multivesicular bodies (F(1,23) = 4.9, p < 0.05). *indicates a significant difference between related groups; p < 0.05, Mann–Whitney post hoc test.
Dramatic increase of D1R at the plasma membrane in lesioned side of rats with AIMs
Quantitative analysis at the ultrastructural level demonstrated a dramatic and specific increase in D1R immunoreactivity at the plasma membrane in lesioned side of l-DOPA-treated rats with AIMs (group 3) compared with their unlesioned side (p < 0.05) (Fig. 2A). Such an increase in D1R abundance at plasma membrane was significantly different from both the vehicle-injected 6-OHDA-lesioned (group 1; p < 0.05) and the nondyskinetic l-DOPA-treated 6-OHDA-lesioned rats (group 2; p < 0.05) (Fig. 2A). Indeed, in these two groups, D1R abundance at membrane was not modified in lesioned side (Fig. 2A). Whatever group considered, D1R abundance did not change in cytoplasm and organelles (Fig. 2B).
AIMs are associated with high abundance of membrane-bound D1R that retains a relative capability of internalization
These data suggested that AIMs are related to a recruitment of D1R at membrane, thereby confirming our previous data collected in monkeys (Guigoni et al., 2007). Such aberrant behavior for a GPCR required further pharmacological analysis. We, therefore, tested the ability of the specific D1R agonist SKF-82958 to induce homologous desensitization in these l-DOPA-treated animals.
Quantitative analysis of D1R localization at the ultrastructural level in lesioned striatum demonstrated several discrete changes in D1R subcellular distribution specifically related to presence of l-DOPA-induced AIMs, after chronic l-DOPA treatment associated or not with acute SKF-82958 injection. Total D1R immunoreactivity in striatal neurons of dopamine-deprived striatum did not change after l-DOPA (groups 2 and 3) or SKF-82958 alone (group 4) but was increased when both drugs were injected (groups 5 and 6) (Fig. 3B). As expected, dopamine receptor stimulation by l-DOPA or by SKF-82958 decreased D1R immunoreactivity at the plasma membrane in all conditions of stimulation, compared with control benserazide group (group 1), with the noticeable exception of the group with l-DOPA-induced AIMs (group 3) (Fig. 3C). Indeed, D1R abundance at the plasma membrane in animals with l-DOPA-induced AIMs was not decreased, whereas it was in animals without l-DOPA-induced AIMs. Interestingly, SKF-82958 challenge unraveled more subtle differences. Animals of groups 5 and 6 differentiated by their AIMs status prior to SKF-82958 challenge, with group 5 animals presenting no AIMs, whereas group 6 did (Table 1). The more dyskinetic they were before SKF-82958, the higher the D1R membrane abundance remains compared with group 4 animals that were challenged with SKF-82958 only (p < 0.05) (Fig. 3C). Together, these data suggest that D1R in l-DOPA-treated brain retains the ability to internalize at a degree that correlates with occurrence of AIMs (the more AIMs the less forced internalization).
AIMs are also associated with alterations of intraneuronal D1R localization and impaired internalization
Analysis of D1R within the cytoplasm expectedly not only mirrored the membrane localization but unraveled further disturbances. Indeed, stand-alone or combined treatment with l-DOPA and SKF-82958 increased cytoplasmic D1R immunoreactivity in all conditions compared with control benserazide group (group 1), with the noticeable exception of group with l-DOPA-induced AIMs (group 3) (Fig. 3D). Greater increase was achieved when SKF-82958 was coadministered (Fig. 3D). Indeed, in the cytoplasm of animals with l-DOPA-induced AIMs, D1R did not increase (compared with benserazide group), whereas it was more abundant in animals without l-DOPA-induced AIMs (p < 0.05). Interestingly, SKF-82958 challenge unraveled more subtle differences. Animals of groups 5 and 6 differentiated by their AIMs status prior to SKF-82958 challenge, with group 5 animals presenting no AIMs, whereas group 6 did (Table 1). The more dyskinetic they were before SKF-82958, the higher the D1R cytoplasm abundance increases compared with group 4 animals (that were challenged with SKF-82958 only) and between them (p < 0.05) (Fig. 3C). Detailed counting in several organelles (Fig. 3D1–D4) confirmed these specific differences related to AIMs. The more dyskinetic they were before SKF-82958, the higher the D1R abundance is in the endoplasmic reticulum (+29.3%; p < 0.05) (Fig. 3D1), the Golgi complex (+88.4%; p < 0.05) (Fig. 3D2), the vesicular compartment (+149.5%; p < 0.05) (Fig. 3D3), and the multivesicular bodies (+424%; p < 0.05) (Fig. 3D4). Together, these data suggest that D1R in l-DOPA-treated brain (1) does not internalize if further challenged with l-DOPA but (2) display greater internalization when challenged with D1R agonist, as D1R overall availability has dramatically increased (Fig. 3B).
Disruption of D1R–D3R cross talk by D3R antagonist normalizes D1R membrane-bound abundance in rats with AIMs
D1R-positive neurons coexpress D3R, which expression levels dramatically increase after l-DOPA treatment in dopamine-depleted dorsal striatum (Bordet et al., 1997; Bézard et al., 2003). In addition, D1R and D3R directly interact through an intramembrane cross talk (Marcellino et al., 2008), suggesting that the natural ligand of those receptor, i.e., dopamine, would coactivate them, whereas receptor-specific ligands would not. We thus hypothesized that the lack of l-DOPA-induced D1R internalization in dyskinesia is caused by the concomitant stimulation of both D1R and D3R. To address that question, we tested the effect of the specific D3R antagonist ST 198 (Bézard et al., 2003) coadministered with l-DOPA during D1R subcellular distribution in dyskinetic 6-OHDA-lesioned rats. ST198, as well as other specific D3R antagonist such as S33084, have been shown to reduce dyskinesia severity in rodent and nonhuman primate analog of Parkinson's disease (Bézard et al., 2003; Visanji et al., 2008), thereby confirming a class effect. Accordingly, ST198 coadministered with l-DOPA reduced AIMs by 22.1% in the 15 min window of behavioral observation immediately before terminal procedure (p = 0.07; paired t test of area under curve). The short duration of the observation period required by the experimental design prevented data from reaching statistical significance, but the AIM reduction is in full agreement with the decrease in dyskinesia observed at the same time point with D3 antagonists (Bézard et al., 2003; Visanji et al., 2008) that then develops in statistical significance at later time points. Quantitative analysis at the ultrastructural level demonstrated that coadministration of ST 198 with l-DOPA decreased significantly D1R abundance at plasma membrane to a level identical to both the nondyskinetic l-DOPA-treated animals and the vehicle-injected animals (p < 0.05) (Fig. 4A). No significant modification was observed in the cytoplasm or in organelles after ST 198 injection (Fig. 4B). These data suggest that D3R anchors D1R at membrane in dyskinetic animals.
Figure 4.

Effect of D3R antagonist ST-198 during D1R subcellular distribution in striatal neurons. Ratio L/NL (mean ± SEM) represents the number of immunoparticles in lesioned side (L) versus nonlesioned (NL) side at the plasma membrane [Kruskal–Wallis (KW) = 7.423, p < 0.05], in the cytoplasm [KW = 3.5, not significant (ns)] (A), and in organelles (B) (er, endoplasmic reticulum; KW = 0.167, ns; Golgi, KW = 0.437, ns; vesicles, KW = 0.716, ns). *indicates a significant decrease compared with l-DOPA-treated dyskinetic rats; p < 0.05, Mann–Whitney post hoc test.
Discussion
The present data show that AIMs are related to D1R overexpression at the plasma membrane associated with intraneuronal trafficking dysregulation including organelles involved in biosynthesis, packaging, and degradation, namely endoplasmic reticulum, Golgi complex, and vesicles. We show that D1R retains its ability to internalize as the potent D1R agonist SKF-82958 promotes membrane depletion in chronically l-DOPA-treated 6-OHDA-lesioned rats, with a magnitude of induced-internalization related to AIMs occurrence prior to SKF-82958 challenge. We further show that this aberrant presence at the plasma membrane of medium spiny neurons is controlled by D1R–D3R interactions as coadministration of the specific D3R antagonist ST 198 with l-DOPA normalizes membrane-bound D1R abundance.
AIMs are linked to D1R overexpression at plasma membrane
Dyskinetic chronically l-DOPA-treated 6-OHDA-lesioned rats showed an overrepresentation of D1R at the plasma membrane of striatal medium spiny neurons. On the contrary, denervation alone or combined with a l-DOPA treatment that does not provoke AIMs has no effect on membrane abundance. These data substantiate previous limited information obtained in dyskinetic monkeys showing both an increased D1R expression at membrane (Guigoni et al., 2007) and a relative decrease in the protein machinery of the homologous desensitization (Bezard et al., 2005). The relationship between D1R membrane abundance and motor abnormalities fits with the main pathophysiological hypothesis of LID placing D1R supersensitivity at the cornerstone of the phenomenon (Bezard et al., 2001a; Jenner, 2008). Such overexpression at membrane is, however, not exclusive of other mechanisms. Further stimulation using the D1R agonist SKF-82958 shows that, if D1R does not internalize spontaneously under l-DOPA in dyskinetic animals, it retains the ability to internalize. Such experimental evidence suggests that AIMs are not caused by the inability of D1R to internalize, and consequently to desensitize, but rather by its active maintenance at membrane.
…and to D1R trafficking dysregulation
Distribution analysis in subcellular compartments shows that specific abnormalities in D1R intraneuronal localization are associated with AIMs. These abnormalities are apparent with the sole l-DOPA treatment but are clearly unraveled by SKF-82958 stimulation suggesting that AIMS are associated with a latent dysregulation of D1R trafficking. Combined treatment with l-DOPA and SKF-92958 has additive effects resulting into an increase in D1R content in all cytoplasmic compartments. In such conditions, the important rise in endoplasmic reticulum, Golgi complex, and vesicular system associated with overexpression of membrane-bound receptor strongly suggest that D1R biosynthetic steps, involving translation and packaging, contribute directly to D1R supersensitivity. The dyskinetic rats have initially more D1R at the membrane. Although D1R internalizes under dopamine agonist stimulation, D1R is nevertheless more retained at the plasma membrane than in the nondyskinetic rats comparably challenged. The dyskinetic rats also present a greater increase in D1R in the endoplasmic reticulum, the Golgi complex, vesicles, and multivesicular bodies under l-DOPA–SKF-82958 association than the nondyskinetic ones. These results are consistent with our previous data showing specific increase in both D1R expression and sensitivity in dyskinetic monkeys (Aubert et al., 2005). A complementary and nonexclusive hypothesis is that AIMs are linked to increased biosynthesis and degradation of D1R (compared with the situation in nondyskinetic rats) associated to a lesser sensitivity to homologous desensitization by its natural agonist, dopamine. That this decreased desensitization capability is caused by decreased bioavailability of the protein machinery, e.g., arrestins and GPCR-regulated kinases (Bezard et al., 2005), or by a modification of the D1R, such as receptor oligomerization (Kong et al., 2006), remains an open question.
Nevertheless, since the electron microscopic detection of cytoplasmic vesicles does not differentiate between endocytic and exocytic vesicles, the important rise of immunogold positive particles in vesicles after l-DOPA–SKF-82958 association may reflect (1) an increased membrane delivery through exocytic vesicles and/or (2) an increased internalization through endocytic vesicles. The dramatic augmentation of multivesicular bodies under l-DOPA–SKF-82958 treatment supports the occurrence of the latter pathway and extends it to increased D1R degradation in the cytoplasm after internalization, since multivesicular bodies are markers of the lysosomal pathway (Bernard et al., 2006; Piper and Katzmann, 2007; Hanyaloglu and von Zastrow, 2008). An all-inclusive concept might well be that dyskinesia are related to an increased D1R turn-over resulting into decreased D1R half-life in medium spiny neurons.
A key role for D3R in maintaining D1R at membrane
Since D1R and D3R directly interact through an intramembrane cross talk (Marcellino et al., 2008) and that the D1R-positive neurons coexpress D3R (Le Moine and Bloch, 1996; Ridray et al., 1998), we hypothesized that the lack of l-DOPA-induced D1R internalization in dyskinesia is caused by the concomitant stimulation of both D1R and D3R. The release of D1R internalization when using D3R antagonist ST 198 in combination with l-DOPA (Fig. 4) firmly substantiates and extends previous pharmacological, behavioral, and anatomical data suggesting functionally relevant important D3R–D1R interactions in neurons of the dorsal striatum. Although a role for the D3R itself in LID pathophysiology has been suggested (Bordet et al., 1997; Bézard et al., 2003) culminating with demonstration that both D3R partial agonist and antagonists decrease dyskinesia in primate (Bézard et al., 2003), the exact mechanism by which modulation of D3R activity was affecting medium spiny activity remained shadowy. The existence of a direct intramembrane D1R–D3R cross talk (Marcellino et al., 2008) offers an explanation both for the previous behavioral data and for the present data. The l-DOPA-derived dopamine would bind to both D1R and D3R, anchoring D1R to membrane in both dyskinetic rats (present data set) and monkeys (Guigoni et al., 2007), two animal models into which D3R is highly increased (Bordet et al., 1997; Bézard et al., 2003). Antagonizing the D3R releases D1R from the plasma membrane. Not only this result demonstrates for the first time the physiological (ensuring dopamine receptor availability) and pathophysiological role (too much D1R at membrane because of increased D3R expression) of the cross talk, but it also provides a mechanistic explanation for the reduction in dyskinesia severity after D3R antagonist treatment.
Together, these results further support the hypothesis that D1R fate in medium spiny neurons during dyskinesia induction and occurrence is under multiple influences and can be modulated by pharmacological approaches. Further studies are now needed for investigating the offered antidyskinetic therapeutic possibilities by disrupting the dopamine receptor cross talks and/or by modulating D1R fate at different levels.
Footnotes
This work was supported by an Agence Nationale de la Recherche grant (B.B., E.B.) and by a Ministère de l'Enseignement et de Recherche grant (A.B.). We thank Service Commun de Microscopie for technical assistance at electron microscope; A. Fayoux, S. Lelgouach, and J.-G. Hervouet for animal care; and P. Gonzalez and A. Estager for general technical assistance. The Université Victor Segalen–Bordeaux 2 and the Centre National de la Recherche Scientifique provided the infrastructural support.
References
- Aubert I, Guigoni C, Håkansson K, Li Q, Dovero S, Barthe N, Bioulac BH, Gross CE, Fisone G, Bloch B, Bezard E. Increased D1 dopamine receptor signalling in levodopa-induced dyskinesia. Ann Neurol. 2005;57:17–26. doi: 10.1002/ana.20296. [DOI] [PubMed] [Google Scholar]
- Bernard V, Décossas M, Liste I, Bloch B. Intraneuronal trafficking of G-protein-coupled receptors in vivo. Trends Neurosci. 2006;29:140–147. doi: 10.1016/j.tins.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Bezard E, Brotchie JM, Gross CE. Pathophysiology of levodopa-induced dyskinesia: potential for new therapies. Nat Rev Neurosci. 2001a;2:577–588. doi: 10.1038/35086062. [DOI] [PubMed] [Google Scholar]
- Bezard E, Dovero S, Prunier C, Ravenscroft P, Chalon S, Guilloteau D, Crossman AR, Bioulac B, Brotchie JM, Gross CE. Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson's disease. J Neurosci. 2001b;21:6853–6861. doi: 10.1523/JNEUROSCI.21-17-06853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bézard E, Ferry S, Mach U, Stark H, Leriche L, Boraud T, Gross C, Sokoloff P. Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat Med. 2003;9:762–767. doi: 10.1038/nm875. [DOI] [PubMed] [Google Scholar]
- Bezard E, Gross CE, Qin L, Gurevich VV, Benovic JL, Gurevich EV. L-DOPA reverses the MPTP-induced elevation of the arrestin2 and GRK6 expression and enhanced ERK activation in monkey brain. Neurobiol Dis. 2005;18:323–335. doi: 10.1016/j.nbd.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Bordet R, Ridray S, Carboni S, Diaz J, Sokoloff P, Schwartz JC. Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc Natl Acad Sci U S A. 1997;94:3363–3367. doi: 10.1073/pnas.94.7.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillé I, Dumartin B, Bloch B. Ultrastructural localization of D1 dopamine receptor immunoreactivity in rat striatonigral neurons and its relation with dopaminergic innervation. Brain Res. 1996;730:17–31. doi: 10.1016/0006-8993(96)00424-6. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Lee CS, Björklund A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur J Neurosci. 1998;10:2694–2706. [PubMed] [Google Scholar]
- Cotzias GC, Papavasiliou PS, Gellene R. Modification of Parkinsonism–chronic treatment with L-dopa. N Engl J Med. 1969;280:337–345. doi: 10.1056/NEJM196902132800701. [DOI] [PubMed] [Google Scholar]
- Dumartin B, Caillé I, Gonon F, Bloch B. Internalization of D1 dopamine receptor in striatal neurons in vivo as evidence of activation by dopamine agonists. J Neurosci. 1998;18:1650–1661. doi: 10.1523/JNEUROSCI.18-05-01650.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumartin B, Jaber M, Gonon F, Caron MG, Giros B, Bloch B. Dopamine tone regulates D1 receptor trafficking and delivery in striatal neurons in dopamine transporter-deficient mice. Proc Natl Acad Sci U S A. 2000;97:1879–1884. doi: 10.1073/pnas.97.4.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehringer H, Hornykiewicz O. [Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system.] Klin Wochenschr. 1960;38:1236–1239. doi: 10.1007/BF01485901. [DOI] [PubMed] [Google Scholar]
- Fiorentini C, Busi C, Gorruso E, Gotti C, Spano P, Missale C. Reciprocal regulation of dopamine D1 and D3 receptor function and trafficking by heterodimerization. Mol Pharmacol. 2008;74:59–69. doi: 10.1124/mol.107.043885. [DOI] [PubMed] [Google Scholar]
- Gross CE, Ravenscroft P, Dovero S, Jaber M, Bioulac B, Bezard E. Pattern of levodopa-induced striatal changes is different in normal and MPTP-lesioned mice. J Neurochem. 2003;84:1246–1255. doi: 10.1046/j.1471-4159.2003.01600.x. [DOI] [PubMed] [Google Scholar]
- Guigoni C, Li Q, Aubert I, Dovero S, Bioulac BH, Bloch B, Crossman AR, Gross CE, Bezard E. Involvement of sensorimotor, limbic, and associative basal ganglia domains in L-3,4-dihydroxyphenylalanine-induced dyskinesia. J Neurosci. 2005;25:2102–2107. doi: 10.1523/JNEUROSCI.5059-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigoni C, Doudnikoff E, Li Q, Bloch B, Bezard E. Altered D(1) dopamine receptor trafficking in parkinsonian and dyskinetic non-human primates. Neurobiol Dis. 2007;26:452–463. doi: 10.1016/j.nbd.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- Hersch SM, Ciliax BJ, Gutekunst CA, Rees HD, Heilman CJ, Yung KK, Bolam JP, Ince E, Yi H, Levey AI. Electron microscopic analysis of D1 and D2 dopamine receptor proteins in the dorsal striatum and their synaptic relationships with motor corticostriatal afferents. J Neurosci. 1995;15:5222–5237. doi: 10.1523/JNEUROSCI.15-07-05222.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci. 2008;9:665–677. doi: 10.1038/nrn2471. [DOI] [PubMed] [Google Scholar]
- Kong MM, Fan T, Varghese G, O'dowd BF, George SR. Agonist-induced cell surface trafficking of an intracellularly sequestered D1 dopamine receptor homo-oligomer. Mol Pharmacol. 2006;70:78–89. doi: 10.1124/mol.105.021246. [DOI] [PubMed] [Google Scholar]
- Le Moine C, Bloch B. Expression of the D3 dopamine receptor in peptidergic neurons of the nucleus accumbens: comparison with the D1 and D2 dopamine receptors. Neuroscience. 1996;73:131–143. doi: 10.1016/0306-4522(96)00029-2. [DOI] [PubMed] [Google Scholar]
- Levey AI, Hersch SM, Rye DB, Sunahara RK, Niznik HB, Kitt CA, Price DL, Maggio R, Brann MR, Ciliax BJ. Localization of D1 and D2 dopamine receptors in brain with subtype-specific antibodies. Proc Natl Acad Sci U S A. 1993;90:8861–8865. doi: 10.1073/pnas.90.19.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA. Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson's disease. Eur J Neurosci. 2002;15:120–132. doi: 10.1046/j.0953-816x.2001.01843.x. [DOI] [PubMed] [Google Scholar]
- Mach UR, Hackling AE, Perachon S, Ferry S, Wermuth CG, Schwartz JC, Sokoloff P, Stark H. Development of novel 1,2,3,4-tetra-hydroisoquinoline derivatives and closely related compounds as potent and selective dopamine D3 receptor ligands. Chembiochem. 2004;5:508–518. doi: 10.1002/cbic.200300784. [DOI] [PubMed] [Google Scholar]
- Marcellino D, Ferré S, Casadó V, Cortés A, Le Foll B, Mazzola C, Drago F, Saur O, Stark H, Soriano A, Barnes C, Goldberg SR, Lluis C, Fuxe K, Franco R. Identification of Dopamine D1–D3 Receptor Heteromers: Indications for a role of synergistic D1–D3 receptor interactions in the striatum. J Biol Chem. 2008;283:26016–26025. doi: 10.1074/jbc.M710349200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner W, Ravenscroft P, Reese R, Harnack D, Morgenstern R, Kupsch A, Klitgaard H, Bioulac B, Gross CE, Bezard E, Boraud T. Increased slow oscillatory activity in substantia nigra pars reticulata triggers abnormal involuntary movements in the 6-OHDA-lesioned rat in the presence of excessive extracellular striatal dopamine. Neurobiol Dis. 2006;22:586–598. doi: 10.1016/j.nbd.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Olsson M, Nikkhah G, Bentlage C, Björklund A. Forelimb akinesia in the rat Parkinson model: differential effects of dopamine agonists and nigral transplants as assessed by a new stepping test. J Neurosci. 1995;15:3863–3875. doi: 10.1523/JNEUROSCI.15-05-03863.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioli EY, Meissner W, Sohr R, Gross CE, Bezard E, Bioulac BH. Differential behavioral effects of partial bilateral lesions of ventral tegmental area or substantia nigra pars compacta in rats. Neuroscience. 2008;153:1213–1224. doi: 10.1016/j.neuroscience.2008.01.084. [DOI] [PubMed] [Google Scholar]
- Piper RC, Katzmann DJ. Biogenesis and function of multivesicular bodies. Annu Rev Cell Dev Biol. 2007;23:519–547. doi: 10.1146/annurev.cellbio.23.090506.123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putterman DB, Munhall AC, Kozell LB, Belknap JK, Johnson SW. Evaluation of levodopa dose and magnitude of dopamine depletion as risk factors for levodopa-induced dyskinesia in a rat model of Parkinson's disease. J Pharmacol Exp Ther. 2007;323:277–284. doi: 10.1124/jpet.107.126219. [DOI] [PubMed] [Google Scholar]
- Ridray S, Griffon N, Mignon V, Souil E, Carboni S, Diaz J, Schwartz JC, Sokoloff P. Coexpression of dopamine D1 and D3 receptors in islands of Calleja and shell of nucleus accumbens of the rat: opposite and synergistic functional interactions. Eur J Neurosci. 1998;10:1676–1686. doi: 10.1046/j.1460-9568.1998.00173.x. [DOI] [PubMed] [Google Scholar]
- Schuster S, Nadjar A, Guo JT, Li Q, Ittrich C, Hengerer B, Bezard E. The 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor lovastatin reduces severity of l-DOPA-induced abnormal involuntary movements in experimental Parkinson's disease. J Neurosci. 2008;28:4311–4316. doi: 10.1523/JNEUROSCI.4720-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster S, Doudnikoff E, Rylander D, Berthet A, Aubert I, Ittrich C, Bloch B, Cenci MA, Surmeier DJ, Hengerer B, Bezard E. Antagonizing L-type Ca(2+) channel reduces development of abnormal involuntary movement in the rat model of L-3,4-dihydroxyphenylalanine-induced dyskinesia. Biol Psychiatry. 2009;65:518–526. doi: 10.1016/j.biopsych.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Shu SY, Ju G, Fan LZ. The glucose oxidase-DAB-nickel method in peroxidase histochemistry of the nervous system. Neurosci Lett. 1988;85:169–171. doi: 10.1016/0304-3940(88)90346-1. [DOI] [PubMed] [Google Scholar]
- Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci. 2003;4:13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- Visanji NP, Fox SH, Johnston T, Reyes G, Millan MJ, Brotchie JM. Dopamine D(3) receptor stimulation underlies the development of L-DOPA-induced dyskinesia in animal models of Parkinson's disease. Neurobiol Dis. 2008 doi: 10.1016/j.nbd.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Winkler C, Kirik D, Björklund A, Cenci MA. L-DOPA-induced dyskinesia in the intrastriatal 6-hydroxydopamine model of parkinson's disease: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis. 2002;10:165–186. doi: 10.1006/nbdi.2002.0499. [DOI] [PubMed] [Google Scholar]