

Abstract
Myelin basic protein (MBP) is an essential structural component of CNS myelin. The electrostatic association of this positively charged protein with myelin-forming membranes is a crucial step in myelination, but the mechanism that regulates myelin membrane targeting is not known. Here, we demonstrate that phosphatidylinositol 4,5-bisphosphate (PIP2) is important for the stable association of MBP with cellular membranes. In oligodendrocytes, overexpression of synaptojanin 1-derived phosphoinositide 5-phosphatase, which selectively hydrolyzes membrane PIP2, causes the detachment of MBP from the plasma membrane. In addition, constitutively active Arf6/Q67L induces the formation of PIP2-enriched endosomal vacuoles, leading to the redistribution of MBP to intracellular vesicles. Fluorescence resonance energy transfer imaging revealed an interaction of the PIP2 sensing probe PH-PLCδ1 with wild-type MBP, but not with a mutant MBP isoform that fails to associate with the plasma membrane. Moreover, increasing intracellular Ca2+, followed by phospholipase C-mediated PIP2 hydrolysis, as well as reduction of the membrane charge by ATP depletion, resulted in the dissociation of MBP from the glial plasma membrane. When the corpus callosum of mice was analyzed in acute brain slices by electron microscopy, the reduction of membrane surface charge led to the loss of myelin compaction and rapid vesiculation. Together, these results establish that PIP2 is an essential determinant for stable membrane binding of MBP and provide a novel link between glial phosphoinositol metabolism and MBP function in development and disease.
Introduction
In the CNS, oligodendrocytes assemble myelin as a multilamellar spiral extension of their cell membrane (for review, see Trapp and Kidd, 2004). The biochemical composition of myelin differs from most plasma membranes (PMs) by its high lipid content and unique protein composition (Taylor et al., 2004; Simons and Trotter, 2007; Werner et al., 2007). Myelin is enriched in cholesterol, glycosphingolipids, galactosylceramide, and its sulfated derivative sulfatide, and contains an unusually high proportion of ethanolamine phosphoglycerides in the plasmalogen form. Surprisingly, among the structural myelin proteins that have been studied in mutant mice, only myelin basic protein (MBP) has emerged as a rate-limiting component of CNS myelination (Harauz et al., 2004; Boggs, 2006). MBP is one of the most abundant myelin proteins. It is thought that the interaction of MBP with the cytoplasmic leaflets of the membrane bilayer causes two opposing layers to physically associate, leading to myelin membrane compaction at the major dense line (MDL) (Omlin et al., 1982; Smith, 1992; Riccio et al., 2000). This function has been concluded both from the localization of MBP in normal myelin and by the myelin ultrastructure of a null mutant mouse (shiverer) that is defined by a deletion in the Mbp gene (Roach et al., 1985; Popko et al., 1987; Readhead et al., 1987; Mikoshiba et al., 1991). However, shiverer mice form only minute amounts of CNS myelin that is noncompacted in the absence of a MDL (Rosenbluth, 1980; Inoue et al., 1981). Thus, the function of MBP in the myelin membrane has remained difficult to study in vivo.
Another hypothesized function of MBP is the clustering of myelin lipids with the myelin membrane, thereby modulating the lipid packing of this membrane (Fitzner et al., 2006; Rosetti and Maggio, 2007; Hu and Israelachvili, 2008). This function may have far-reaching consequences for membrane assembly and signaling events. MBP may indeed act as a lipid coupler, not only by promoting myelin compaction between two membranes, but also by condensing specific lipids in a lateral dimension (Fitzner et al., 2006).
In mice, MBP has four isoforms (14, 17, 18.5, and 21.5 kDa) that are generated by alternative mRNA splicing (Barbarese et al., 1978; de Ferra et al., 1985; Kamholz et al., 1986). In the following, we are using the nomenclature of the “classical” MBP mRNAs that are transcribed from the much larger MBP-golli gene (Campagnoni and Campagnoni, 2004). On translation, the association with membranes is thought to be determined by the high density of basic amino acids that interact with negatively charged lipid head groups (for review, see Boggs, 2006).
Although the interaction of purified MBP with artificial membrane systems has been studied for many years (Smith, 1992; Rivas and Castro, 2002; Haas et al., 2007; Rispoli et al., 2007), the structural features of protein and lipids required for efficient membrane targeting of this protein are not known. Here, we used a molecular cell biological approach to investigate the interactions of MBP with the glial cell membrane. We demonstrate that the lipid phosphatidylinositol 4,5-bisphosphate (PIP2) is a critical molecular target and provide evidence that acutely disturbed MBP–PIP2 interactions are not tolerated and thus relevant for myelin disease.
Materials and Methods
Cell culture and transfections.
Primary oligodendrocytes were prepared as described previously (Trajkovic et al., 2006). After shaking oligodendrocytes from a mixed monolayer, cells were plated onto poly-l-lysine-coated coverslips and cultured in DMEM with B27 supplement and 1% horse serum, l-thyroxine, tri-iodo-thyronine, glucose, glutamine, gentamycine, pyruvate, and bicarbonate. The oligodendroglial precursor cell line, Oli-neu (provided by J. Trotter, University of Mainz, Mainz, Germany), and OLN-93 cells (provided by C. Richter-Landsberg, University of Oldenburg, Oldenburg, Germany) were cultured as described previously (Jung et al., 1995; Richter-Landsberg and Heinrich, 1996). Transient transfections were performed using FuGENE transfection reagent (Roche) or Lipofectamine transfection reagent (Invitrogen) according to the manufacturer's protocol. Transient transfection of COS1 cells was performed using Lipofectamine reagent according to the manufacturer's protocol.
Expression constructs and virus generation.
A MBP-EYFP fusion protein was generated by cloning the PCR product of 14 kDa mouse MBP cDNA, which included an additional BamHI site, into the EYFP-N1 vector (Clontech) using EcoRI–BamHI sites. The PCR product of MBP exon 1 was also cloned into pEYFP-N1 using EcoRI–BamHI sites. The PCR product of exon 7 including an additionally NotI site was cloned into pEYFP-N1 using EcoRI–NotI sites. The GFP-PH-PLCδ1 fusion construct was subcloned into pECFP-N1 (BD Biosciences) using AgeI–NotI sites. Doubly palmitoylated YFPmem vector was purchased from Clontech. The 14 kDa MBP cDNA with additional 3′-untranslated region (UTR) was cloned into pCMV vector (Stratagene) using EcoRI sites. All site-directed mutants were generated by circular amplification with Pfu Turbo DNA polymerase (Stratagene) followed by digestion of methylated and hemimethylated DNA with DpnI (New England Biolabs). All constructs were verified by DNA sequencing. Recombinant Semliki forest virus was generated as described previously (Fitzner et al., 2006). Viral RNA was generated by in vitro transcription of linearized vector plasmid and pSFV-Helper1 plasmid (linearized with SpeI). SFV-Helper RNA and the respective RNA constructs were electroporated into Baby Hamster Kidney (BHK21) packing cells that were cultured at 37°C and 5% CO2. Approximately 24 h after electroporation, supernatant containing virus particles was collected. For transduction, primary oligodendrocytes were incubated with supernatant containing viral particles (for 1 h), before replacing with a conditioned medium. The infection was allowed to continue for additional 7 h.
Antibodies and plasmids.
Plasmid mRFP-LactC2 was provided by S. Grinstein (University of Toronto, Toronto, ON, Canada). Clones GFP-PH-PLCδ1 and GFP-PH-PLCδ1–3xmut, and IPPCAAX-GFP-SFV (SFV-Synj1-GFP) were obtained from I. Milosevic and J. Sørensen (Max Planck Institute for Biophysical Chemistry, Göttingen, Germany), and ARF6/Q67L-HA was provided by J. G. Donaldson (National Institutes of Health, Bethesda, MD). PH-AKT-YFP was provided by T. Meyer (Stanford University, Stanford, CA). MBP cDNA was provided by T. Campagnoni (University of California at Los Angeles, Los Angeles, CA). The following antibodies were used: NG2 (provided by J. Trotter; 1:50), polyclonal MBP (Dako; 1:200), Lamp1 (BD Pharmingen; 1:100), anti-KDEL (BIP; Nventa Biopharmaceuticals; 1:100), and GM130 (BD Biosciences Transduction Laboratories; 1:100).
Immunofluorescence.
Immunofluorescence was performed as described previously (Trajkovic et al., 2006). Briefly, cells were fixed with 4% paraformaldehyde (PFA). For MBP and hemagglutinin (HA) staining, cells were permeabilized with 0.1% Triton X-100 in PBS, followed by incubation with blocking solution, containing Eagle's basal medium supplemented with 10% horse serum (for 30 min at room temperature). Cells were incubated with primary antibodies diluted in blocking solution, washed with PBS, and incubated with the respective secondary antibody.
Life cell imaging and image analysis.
Phospholipase C (PLC) activation experiments were performed as described previously (Várnai and Balla, 1998). Cells were washed twice with modified Krebs–Ringer's solution (containing the following: 120 mm NaCl, 4.7 mm KCl, 1.2 mm CaCl2, 0.7 mm MgSO4, 10 mm glucose, 10 mm Na-HEPES, pH 7.4) before imaging. Coverslips were then placed into a chamber that was mounted on a heat stage and kept at 33°C (temperature control 37-2 digital; Zeiss) during image acquisition. Cells were imaged in modified Krebs–Ringer's solution, and fluorescent images were acquired under oil with an inverse microscope (Leica Axiovert 200M). Images were obtained using AxioVision Software at multiple x–y positions with a high-resolution digital camera with a progressive scan interline CCD chip camera (ORCA ER; C4742-80-12AG; Hamamatsu). Images were acquired every 10 s using the appropriate filters [excitation filter for yellow fluorescent protein (YFP) and green fluorescent protein (GFP), bandpass (BP) 450–490; emission filter, BP 515–565; Carl Zeiss].
For calcium entry, 10 μm ionomycin (Calbiochem) was added to the imaging solution, by removing 0.5 of 2 ml of medium and adding back 0.5 ml of medium containing reagents. For PIP2 blockage, cells were incubated for 10 min at 37°C with 10 mm neomycin (G418; Invitrogen). After cells were treated for 2 min with ionomycin, 5 mm EGTA was added for 30 min. In all conditions, cells were fixed and mounted in Aqua-Poly/Mount (Polysciences), and fluorescent images were analyzed using a modified ImageJ macro. Line scans were taken directly at the cell membrane (n > 10; pixel size, 100 × 100 nm) (see Fig. 8). Statistical analysis was performed using GraphPrism software. For quantification, line scans were taken from confocal images (n > 30; average pixel size, 40 × 40 nm) (see Figs. 7, 9). For the quantification of plasma membrane-associated MBP, we used Oli-neu cells. In contrast to primary oligodendrocytes (containing large membrane sheets and only small volume of cytoplasm), Oli-neu cells display a clear outline of the plasma membrane in confocal images.
Figure 8.

Rapid dissociation of MBP14k-YFP from the plasma membrane of Oli-neu cells after elevating intracellular calcium. Oli-neu cells were transfected with MBP14k-YFP or GFP-PH-PLCδ1 and subjected to live-cell imaging. Cells were bathed in medium with or without Ca2+. Twenty seconds after addition of 10 μm ionomycin, images were obtained every 10 s. Neomycin (10 mm) was added to the culture medium 10 min before acquisition of images and ionomycin treatment. Shown are the line scans of cells presented before and after ionomycin treatment. Error bars indicate SEM.
Figure 7.
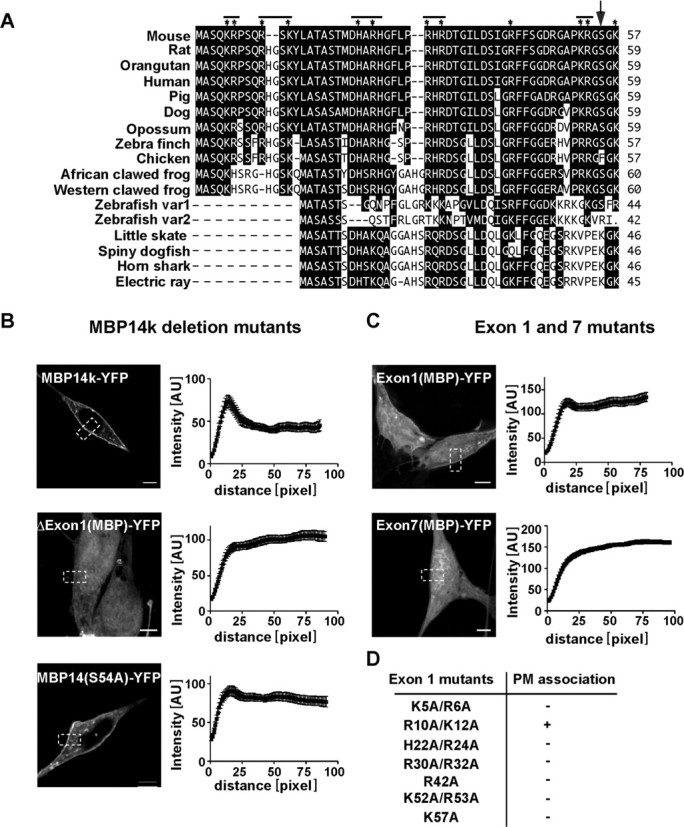
Domain mapping of MBP domains required for plasma membrane association. A, Sequence alignment of the N-terminal domain (encoded by exon1) with selected orthologs. Sequences were retained from NCBI database and clustal-alignment with PAM250. Amino acids identical with mouse MBP are in black boxes. Basic amino acids analyzed by site-directed mutagenesis (in D) are marked by asterisk; a serine residue altered by mutagenesis is marked with an arrow (A). B, C, Quantification of membrane localization of truncated MBP isoform. Shown are cells expressing deletion construct lacking the exon 1-encoded region (ΔExon1-YFP), or expressing the exon 1-encoded region fused to YFP (Exon1-YFP) or expressing the exon 7-encoded region fused to YFP (Exon7-YFP). S54A mutant shows less plasma membrane association than wild-type MBP14k-YFP. Scale bars, 5 μm. D, Various positive amino acids (Arg, His, or Lys) were replaced by Ala. Intensity profiles were generated for each cell. Presented are the mean intensity profiles from 30 different cells with SDs. The table indicates which mutant protein associates with the plasma membrane.
Figure 9.
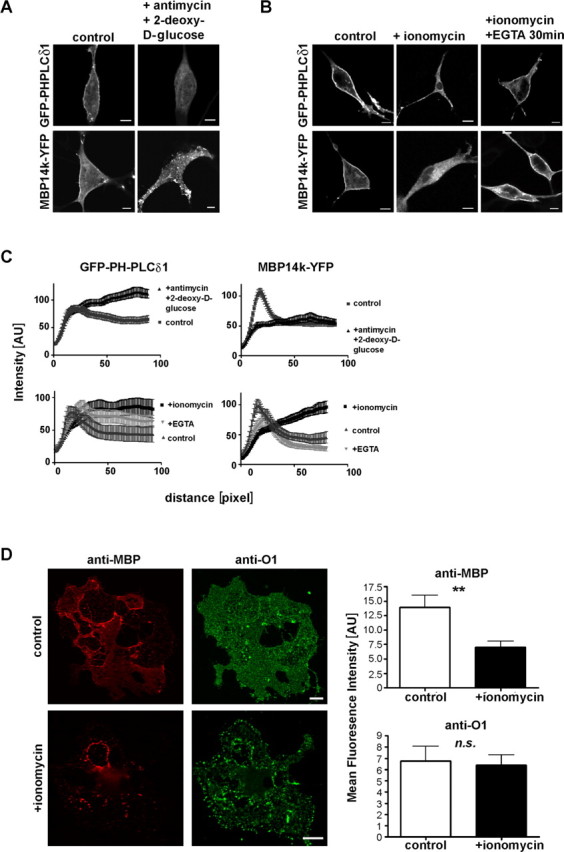
Reduction of surface charge displaces MBP from the plasma membrane of oligodendrocytes. MBP14k-YFP or GFP-PH-PLCδ1 transfected Oli-neu cells were treated with antimycin and 2-deoxy-d-glucose, which reduces surface charge by preventing new synthesis of PIP2 (A) or ionomycin (B). Quantification of ionomycin treatment and ATP depletion is shown in C. MBP localization was quantified from confocal images taken from fixed and mounted Oli-neu cells that were treated with ionomycin, ionomycin and EGTA, antimycin/2-deoxy-d-glucose, or control, respectively. Shown are fluorescence intensity profiles, obtained through line scans over the plasma membrane of at least 10 individual cells with respective SDs (n > 10). Primary oligodendrocytes that were treated with ionomycin or not, were permeabilized with saponin to wash out released proteins, fixed and immunolabeled against MBP and GalC (O1) (D). Fluorescence intensity of MBP-cy3 and O1-cy2 was quantified relative to background (n = 30 cells two independent experiments; p = 0.0039). Scale bars: (A, B, D), 10 μm. Error bars (D, right) indicate SEM.
Membrane sheets.
Sheets were generated as described previously (Milosevic et al., 2005). Briefly, OLN-93 cells were plated onto 25 mm poly-l-lysine-coated coverslips and kept at 37°C, 5% CO2 for 8 h, before transfection of plasmid DNA using Lipofectamine reagent. Membrane sheets were generated from OLN-93 cells 12 h after transfection. For wortmannin (Wm) treatment, cells were treated with 30 nm wortmannin (Sigma-Aldrich) for 4 h before generation of sheets. Coverslips were placed 1–2 cm below the sonication tip in sonication buffer (pH 7.2) (120 mm K-Glu, 20 mm K-acetate, 20 mm HEPES, and 10 mm EGTA) with a total volume of 300 ml, and one single sonication pulse was applied (Sonifier 450; power setting at 2.5; duty cycle, 30%; Branson Ultrasonics).
Sheets were fixed for 1 h in 4% PFA and washed three times with PBS before imaging. Coverslips containing sheets were then placed into the microscope chamber. To identify intact sheets, we visualized the phospholipid bilayer by adding 1-(4-trimethyl-amoniumphenyl)-6-phenyl-1,3,5-hexatriene (TMA-DPH) (Invitrogen) to the imaging solution. For imaging, we used an Axiovert 100 TV fluorescence microscope (Zeiss) equipped with a 100×, 1.4 numerical aperture plan achromate objective using appropriate fluorescence filter sets [excitation filter, G 365 and band suppression (BS) 395, and emission filter, long pass 420, were used for TMA-DPH dye; excitation filter, BP 480/40 and BS 505, and emission filter, BP 527/30, were used for YFP]. The focal position of the objective was controlled using a low-voltage piezo translater driver and a linear variable transformer displacement controller (Physik Instrumente). Images were acquired using a back-illuminated CCD camera (512 × 512 chip with 24 × 24 μm pixel size with a magnifying lens; 2.5× Optovar), to avoid spatial undersampling by the larger pixels. The focal plane was adjusted by using small fluorescent beads as a reference (0.2 μm Tetraspek-beads; Invitrogen), applied to the imaging solution. Digital images were obtained and analyzed using MetaMorph software (Molecular Devices).
For quantification, a randomly selected region of interest (ROI) was defined on the sheet and the average fluorescence intensity in that ROI was background corrected. For each condition, >100 sheets were measured that were taken from at least three independent experiments. Statistical significance was determined using nonparametric Student's t test in GraphPrism. Fluorescence resonance energy transfer measurement. Oli-neu cells were transiently transfected with plasmids CFP-PH-PLCδ1 and MBP14k-YFP (mixed in a 1:1 ratio) using FuGENE transfection reagent. Cells were fixed with 4% PFA 12 h after transfection, and mounted on glass microscope slides in Aqua-Poly/Mount (Polysciences). Fluorescence images were acquired with a Leica DMRXA microscope (Leica).
Fluorescence resonance energy transfer (FRET) was detected by an increase in donor fluorescence after photobleaching of the acceptor using a confocal Leica microscope (TCS SP2 equipped with AOBS) as described previously (Fitzner et al., 2006). Acceptor photobleaching was performed using Leica Microsystems software. YFP was excited at 514 nm and cyan fluorescent protein (CFP) at 458 nm HeNe laser line. Image analysis of FRET data was performed using custom-written MATLAB routines. Fluorescence emission was collected in spectral windows (collected at 525–610 nm for YFP and collected at 468–495 nm for CFP). YFP was bleached in a ROI minimal of its initial fluorescence intensity, representing background signal. All settings were kept constant for all images acquired. FRET was calculated on a pixel-to-pixel basis.
Quantification of protein localization at the plasma membrane.
Oli-neu cells were transiently transfected with plasmids MBP14k-YFP or GFP-PH-PLCδ1 using FuGENE transfection reagent and incubated at 37°C, 5% CO2 for 12 h. For phosphatidylinositol 3-kinase (PI3K) inhibition, cells were treated for 4 h with 30 nm wortmannin. Subsequently, cells were fixed with 4% PFA and stained against plasma membrane localized NG2 and secondary cy5 (1:250; Millipore Bioscience Research Reagents). Fluorescent images were acquired using a Zeiss LSM 510 confocal microscope with a 63× oil Plan-Apochromat objective (numerical aperture, 1.4).
NG2 staining was used as mask to calculate the fluorescence intensity of MBP14k-YFP at plasma membrane compared with the cytosol. For this, an optimal threshold for the mask was calculated and the images of the mask were then converted to black and white. PM localization was determined by multiplying the mask with the YFP channel (corresponding to MBP14k-YFP or YFPmem signal). The PM association constant was then calculated according to the following formula (Heo et al., 2006): (IbeforePM)/(Icyt) * (Iaftercyt)/(IafterPM), with Ibefore and Iafter as the fluorescent intensities before and after PIP2 and phosphatidylinositol 3,4,5-trisphosphate (PIP3) depletion. For each condition, 70 cells were analyzed using custom-written MATLAB routines. Statistical significance was determined using nonparametric Student's t test.
Acute slices of corpus callosum.
Thirty-day-old mice were killed, and the frontal lobes were isolated. Coronal slices were cut using a Leica VT1200S Microtome (Leica) at 300 μm thickness in ice-cold cutting solution containing the following: 130 mm NaCl, 3.5 mm KCl, 10 mm MgSO4, 0.5 mm CaCl2, 1.25 mm NaH2PO4, 24 mm NaHCO3, and 10 mm glucose, and was maintained at pH 7.4 in 5% CO2 atmosphere. Before treatment, slices were equilibrated in artificial CSF (ACSF) containing the following: 130 mm NaCl, 3.5 mm KCl, 1.5 mm MgSO4, 2 mm CaCl2, 1.25 mm NaH2PO4, 24 mm NaHCO3, 10 mm glucose for 2 h at room temperature. ACSF was continuously bubbled with carbogen (95% O2 and 5% CO2) gas. Slices were then incubated at 35°C with ionomycin (10 μm) or antimycin (200 nm) and 2-deoxy-d-glucose (10 mm) for 1 h. Control slices were incubated in 1% DMSO for ionomycin treatment. Acute slices were then fixed for electron microscopy in Karlsson–Schultz fixative for 8 h, containing the following: 2.5% glutaraldehyde, 2% formaldehyde, 0.1 m phosphate buffer, pH 7.4 (Karlsson and Schultz, 1965).
Electron microscopy.
Ultrathin sections were cut using Leica Ultracut S ultramicrotome (Leica) and stained with aqueous 4% uranylacetate followed by lead citrate. The sections were viewed in an electron microscope (Leo EM912AB; Zeiss), and images were taken using on-axis 2048 × 2048 charge-coupled device camera (Proscan; Schering).
Results
MBP is a protein with a high net positive charge that interacts with acidic phospholipids in model membranes. In cellular membranes, phosphatidylserine (PS) is by far the most abundant negatively charged lipid. Two minor lipids, previously shown to interact with signaling proteins, are PIP2 and PIP3, which constitute only a few percent of total membrane lipids (McLaughlin et al., 2002). We expressed a recombinant pleckstrin homology (PH) domain, derived from one of the two signaling proteins PLCδ1 or AKT, as a GFP and YFP fusion protein, to localize PIP2 and PIP3 in oligodendroglial cells. As shown in Figure 1A, fusion proteins GFP-PH-PLCδ1 and PH-AKT-YFP, which recognize PIP2 and PIP3, respectively (Várnai and Balla, 1998, Várnai and Balla, 2007), were exclusively found at the plasma membrane of oligodendroglial Oli-neu cells. Similar results were obtained when we expressed GFP-PH-PLCδ1 in primary oligodendrocytes using the Semliki forest virus vector (Fig. 1B). In primary oligodendrocytes, GFP-PH-PLCδ1 did not only localize to the plasma membrane at the cell soma but also was found in the flat membrane sheets that contain large amounts of MBP. To study, for comparison, the localization of PS, we used Lactadherin, with its major PS-binding motif localized to its C2 domain (Lact-C2), fused to a monomeric red fluorescent protein (mRFP), as a specific PS-sensor (termed mRFP-LactC2) (Yeung et al., 2008). In contrast, mRFP-Lact-C2 was targeted to both the plasma membrane and intracellular membranes (Fig. 1A).
Figure 1.
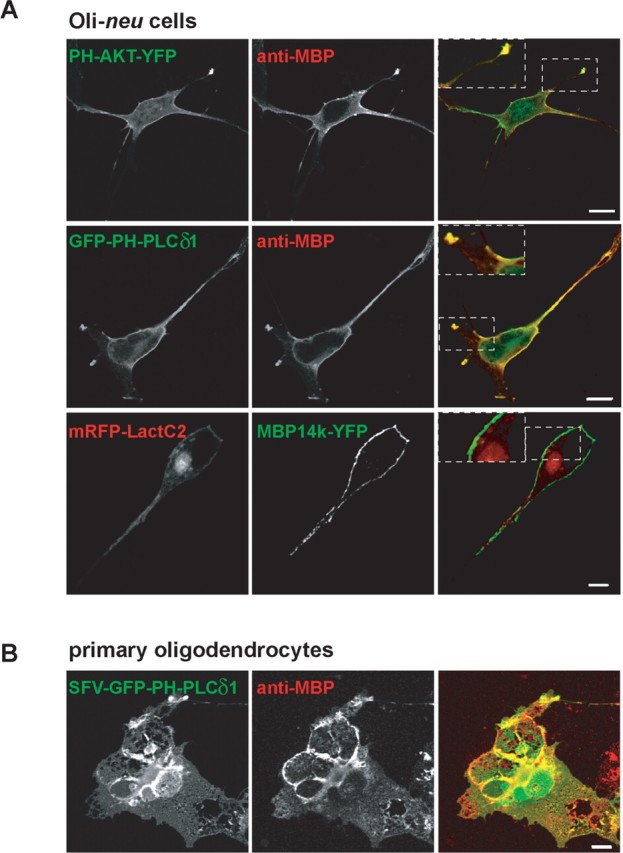
MBP colocalizes with PIP2 and PIP3 at the plasma membrane of oligodendrocytes. A, Oli-neu cells were transiently cotransfected to express GFP-PH-PLδ1, PH-AKT-YFP, or mRFP-LactC2 to visualize PIP2, PIP3, and PS, respectively, with MBP14k or MBP14k-YFP. Colocalization with MBP14k is shown on the right (overlay). B, GFP-PH-PLCδ1 was expressed with the SFV vector for 8 h in primary oligodendrocytes. Cells were fixed and immunolabeled against MBP. Scale bars: A, 5 μm; B, 10 μm.
Next, we coexpressed the 14 kDa isoform of MBP with the fluorescent lipid sensors. Interestingly, MBP staining was most robust at the plasma membrane in regions that also showed an enrichment of PIP2 and PIP3 (Fig. 1A). MBP was not associated with intracellular membranes that contained high levels of PS. This was not attributable to the physical masking of PS by mRFP-Lact-C2, because MBP showed the same distribution in the absence of any lipid sensor. Similar results were observed for the 18.5 kDa MBP isoform (data not shown), whereas expression of the 21.5 kDa MBP resulted in nuclear staining of some cells as described previously (Pedraza et al., 1997). The preferential association of MBP with membranes of the myelin compartment is achieved, in part, by transport of its mRNA into oligodendroglial cell processes and local translation, stimulated by neuronal signals (White et al., 2008). This transport is mediated by a 21 nt RNA transport signal (RTS) in the 3′-UTR of MBP mRNA (Ainger et al., 1997). Importantly, this targeting of MBP to the plasma membrane of Oli-neu cells was independent of the RTS in the 3′-UTR, because expression constructs with or without this mRNA targeting signal led to an indistinguishable MBP localization (data not shown). Together, these data show that MBP does not associate equally well with all membrane surfaces, but has a preference for the plasma membrane, which is enriched in both PIP2 and PIP3.
To confirm the interaction of MBP with PIP2, we performed FRET experiments. CFP-PH-PLCδ1 and YFP-PH-PLCδ1 have previously been used as a FRET pair, to measure PIP2 dynamics (van der Wal et al., 2001). We note that PH-PLCδ1 interacts with PIP2 even if basic proteins are bound to the latter (McLaughlin et al., 2002; Gambhir et al., 2004). If MBP and PIP2 were to interact with each other by forming clusters, a FRET pair of the PH-domain of PLC and MBP should show energy transfer. We therefore expressed CFP-PH-PLCδ1 and MBP14k-YFP in Oli-neu cells, and we were indeed able to detect energy transfer between both proteins (Fig. 2). In contrast, cells expressing a mutant form of 14 kDa MBP that does not bind to the plasma membrane (see also below) showed significantly reduced FRET efficiency (Fig. 2). This confirms that membrane-bound MBP associates with PIP2 and that the loss of plasma membrane binding correlates with the loss of MBP–PIP2 association.
Figure 2.
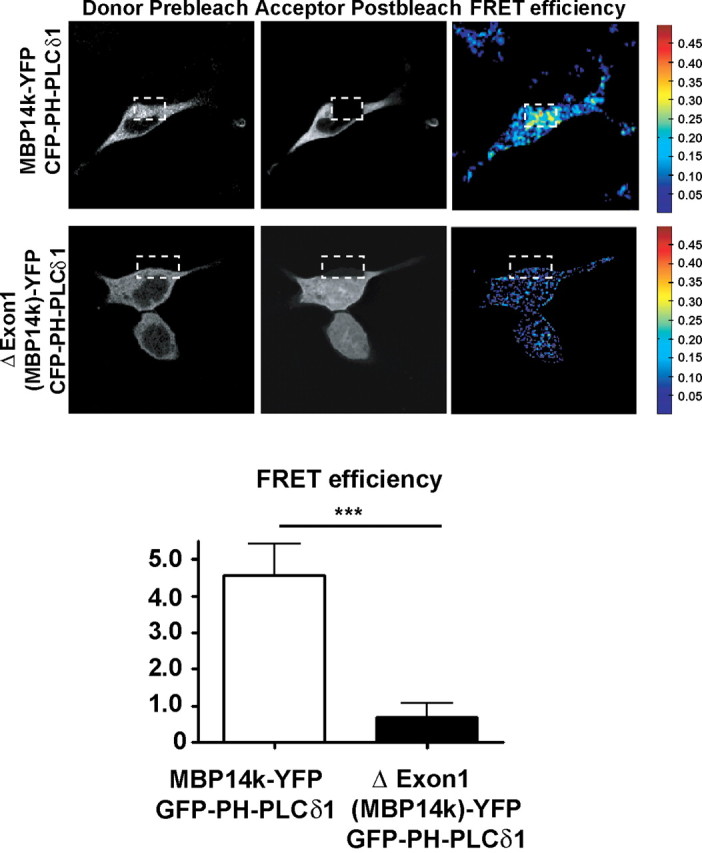
FRET imaging revealed an interaction of PIP2 (sensed by CFP-PH-PLCδ1) with MBP, but not with a mutant form of MBP that is unable to bind to the plasma membrane (see also Fig. 7). Oli-neu cells were transiently transfected to express the indicated plasmids in a 1:1 ratio and fixed after 12 h. FRET was detected by an increase in donor fluorescence after photobleaching of the acceptor. Confocal images of both FRET pairs are shown before (pre) and after (post) photobleaching. FRET efficiency is indicated in pseudocolor (shown from blue to red with increasing FRET efficiency; n > 20 cells; error bars indicate ±SEM; ***p = 0.0002; t test).
To investigate whether PIP2 and PIP3 are indeed required for the plasma membrane association of MBP, we experimentally altered the PIP2 and PIP3 levels in glial cells, as previously described for NIH3T3, HeLa, bovine chromaffin cells, and PC12 cells (Milosevic et al., 2005; Heo et al., 2006). Cells were cotransfected to express a YFP-fluorescent MBP fusion protein together with synaptojanin 1-derived PIP2 degrading phosphatase (Synj1) (Krauss et al., 2003). Cells were also treated with the PI3K inhibitor Wm to inhibit PIP3 synthesis. We quantified the level of MBP14k-YFP at the PM by measuring the fluorescence intensity at the membrane compared with the cytosol (Heo et al., 2006). To normalize these results for a known membrane protein, we immunostained cells for the membrane glycoprotein NG2 and also calculated the ratio of fluorescence intensity between plasma membrane and cytosol. Although plasma membrane localization of MBP was not completely abolished by synaptojanin 1, the results clearly showed reduced MBP signal at the plasma membrane (Fig. 3A). For comparison, a double-palmitoylated (i.e., membrane-anchored) YFP (YFPmem) did not change its membrane localization after coexpression of Synj1 and treatment with Wm (Fig. 3A). To compare the localization of MBP in primary oligodendrocytes after decrease in PIP2, we infected cells with recombinant Semliki forest virus and expressed a Synj1-GFP fusion construct. In oligodendrocytes expressing this PIP2 phosphatase, MBP accumulated in the cytosol, whereas in control oligodendrocytes, MBP staining formed a rim, demarcating the cell membrane (Fig. 3B).
Figure 3.
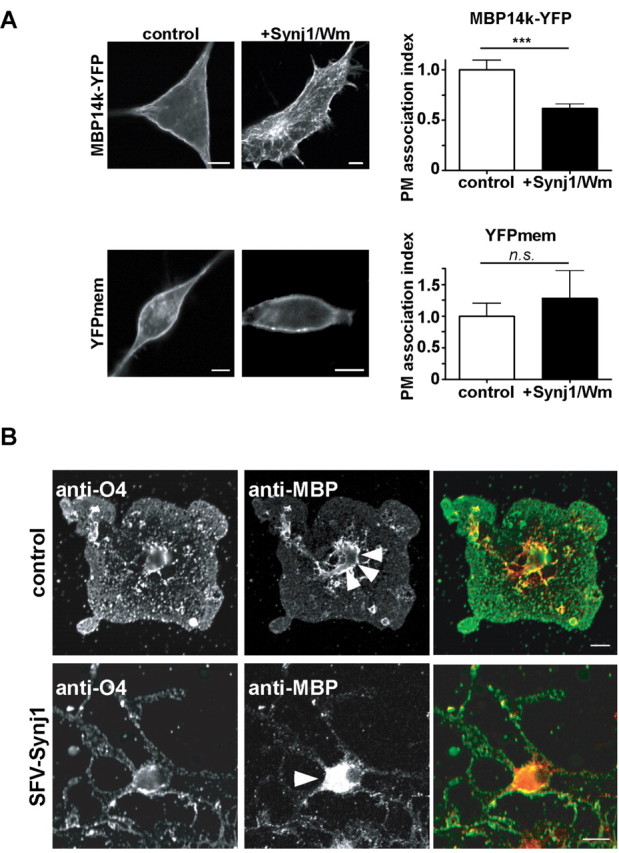
PIP2 phosphatase synaptojanin1 decreases MBP localization at the plasma membrane. A, Oli-neu cells were transiently transfected with Synj1-mRFP and MBP14k-YFP or MBP14k-YFP and vector control in a 1:1 ratio 12 h before treating (for 4 h) with Wm. Cells were fixed and stained against plasma membrane localized NG2. Plasma membrane localization of MBP was quantified by comparing its fluorescence intensity at the plasma membrane relative to the cytosol. We calculated a PM association index as the relative ratio of PM over cytosolic fluorescence. PIP2 and PIP3 depletion reduced MBP at the plasma membrane, whereas the localization of a double-palmitoylated YFP protein (YFPmem) remained unchanged (n > 70 cells; mean values ± SEM; ***p = 0.0006; t test). Scale bars, 10 μm. B, Primary oligodendrocytes were infected with SFV-Synj1-GFP, fixed after 8 h, and immunolabeled with antibodies against MBP and O4. Note that expression of Synj1 in oligodendrocytes causes the dissociation of MBP from the cell membrane (indicated by arrowhead) and redistribution into the oligodendroglial soma. O4 immunoreactivity remained unaltered. Scale bars, 10 μm.
To test directly, whether PIP2 is essential for MBP targeting to membranes, we performed three additional experiments. First, we prepared isolated plasma membrane sheets from transfected cells of the oligodendroglial OLN-93 line by applying an ultrasonic pulse as described previously (Lang et al., 2001). In this preparation, previous overexpression of Synj1 resulted in a decrease in PIP2, which could be monitored as reduced GFP-PH-PLCδ1 binding to these sheets (Milosevic et al., 2005). Again, when OLN-93 cells were cotransfected to express Synj1 and MBP14k-YFP, we observed a significant decrease in fluorescence intensity of MBP14k-YFP (Fig. 4B).
Figure 4.
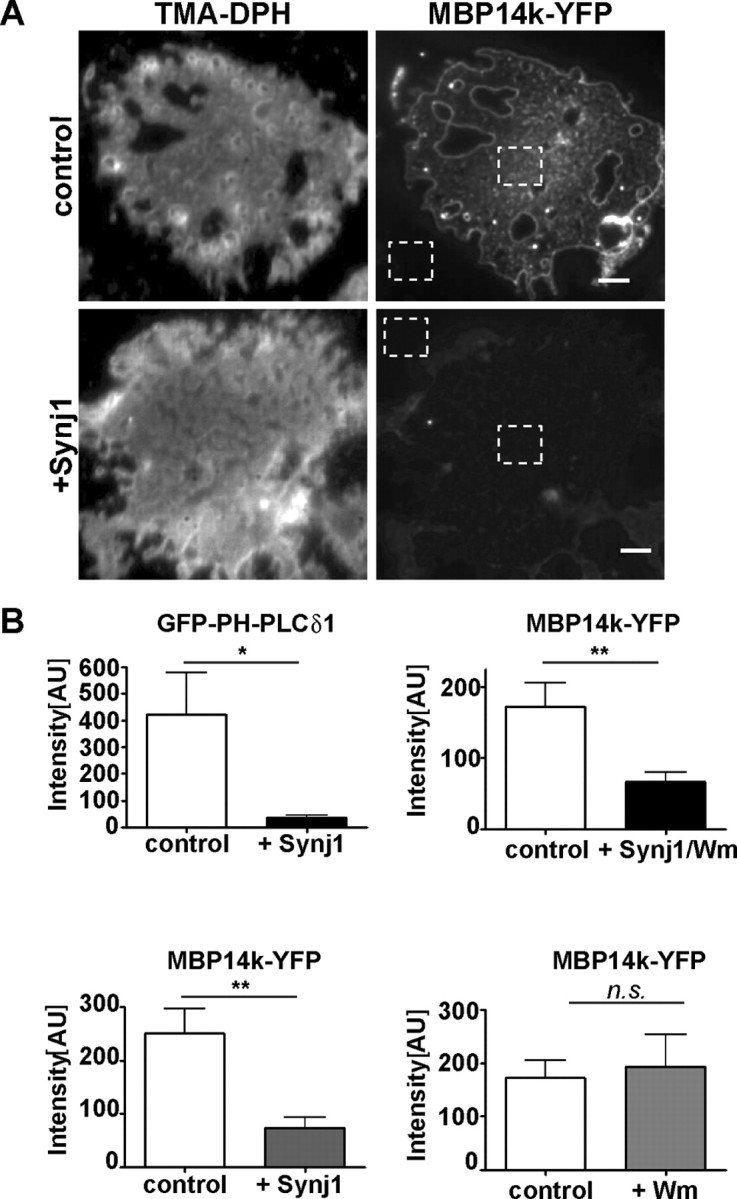
PIP2 depletion reduces the association of MBP with plasma membrane sheets. A, OLN-93 cells were transiently transfected in a 1:1 ratio with either MBP14k-YFP and Synj1 or MBP14k-YFP and a control vector. Membrane sheets were prepared by application of a short ultrasonic pulse 16 h after transfection as described in Materials and Methods. The fluorescent dye, TMA-DPH, was used to visualize intact sheets. Images are shown after contrast enhancement. Images showing MBP14k-YFP fluorescence were not contrast enhanced. Scale bars, 3 μm. B, Quantification of MBP binding to the membrane surface relative to background (A, dotted squares). For PI3K inhibition, cells were treated for 4 h with Wm 12 h after transfection. GFP-PH-PLCδ1 was used as control to indicate the reduced level of PIP2 upon Synj1 coexpression (n > 100 cells from at least three independent experiments; mean values ± SEM; t test, GFP-PH-PLCδ1, *p = 0.022; Synj1 expression with PI3K inhibition, **p = 0.003; PI3K inhibition, ns, p = 0.747; Synj1 and MBP14k-YFP expression, **p = 0.0019).
To test whether reduced PIP3 levels would be sufficient to prevent MBP binding to the plasma membrane, cells were treated with wortmannin as an inhibitor of PI3K activity. This decrease in membrane PIP3 did not cause a loss of MBP binding, suggesting that PIP2 rather than PIP3 is the responsible lipid target (Fig. 4B). Additionally, we noticed that MBP formed small clusters at the isolated plasma membrane sheets (Fig. 4A), similar to clusters previously observed for PIP2 (Milosevic et al., 2005).
Experimental support for PIP2 as a MBP binding lipid came from the overexpression of a constitutively active Arf6 protein (Arf6/Q67L) in COS1 cells, a technique known to cause a redistribution of PIP2 from the plasma membrane to a large pool of intracellular vacuoles (Donaldson, 2003). This assay can be used as a “gain-of-function” approach to visualize the interaction of PIP2 with cellular proteins (Ono et al., 2004). Indeed, cotransfection of COS1 cells and expression of both Arf6/Q67L and MBP resulted in the accumulation of MBP at these (PIP2-enriched) vacuoles (Fig. 5). To show the specificity of this test, we expressed a triple mutant version of GFP-PH-PLCδ1 that no longer binds to PIP2 (Milosevic et al., 2005) and therefore failed to accumulate at intracellular vesicles. Together, these experiments demonstrated that PIP2 is a critical determinant for membrane targeting of MBP.
Figure 5.

COS1 cells overexpressing constitutive active Arf6 (Arf6/Q67L-HA), which induces accumulation of PIP2-enriched vacuoles and recruits MBP from the plasma membrane to intracellular vacuoles. COS1 cells were transiently cotransfected with plasmids encoding Arf6/Q67L-HA, GFP-PH-PLCδ1, MBP14k-YFP, or GFP-PH-PLCδ1–3xmut in a 1:1 ratio and fixed after 44 h. Arf6/Q67L-HA was visualized by staining with antibodies against the HA tag (in red). GFP-PH-PLCδ1 was used to visualize PIP2 in the vacuoles (in green), whereas GFP-PH-PLCδ1–3xmut, which does not bind to PIP2, served as a control. Scale bars, 5 μm.
The plasma membrane is thought to be more negatively charged than endomembranes. Its content of PIP2 and PIP3 discriminates the cell surface from endomembranes that contain less charged phospholipids such as PS (Yeung et al., 2008). In reconstituted systems, MBP also binds to other lipids (e.g., PS) (Hu et al., 2004). To investigate whether MBP would associate with intracellular membranes in the absence of PIP2 (but presence of PS), we performed colocalization studies. Indeed, after selective depletion of PIP2 by Synj1, MBP dissociated from the plasma membrane and was found both in the cytosol and on intracellular membranes. Here, MBP colocalized with the PS-sensor (mRFP-LactC2) on intracellular membranes (Fig. 6). Thus, MBP interacts with membranes in a charge-dependent manner and PIP2 is a major determinant of its preferential cell surface localization.
Figure 6.

PIP2 depletion results in a visible association of MBP to PS containing intracellular membranes. Oli-neu cells were transiently transfected with MBP14k-YFP, LactC2-mRFP, and Synj1 and fixed after 16 h. Those cells that were treated for 2 min with ionomycin before fixation (see also Fig. 8) also show MBP localization at PS-containing membranes. Scale bars, 5 μm.
In contrast to other “natively unfolded” proteins, the positively charged amino acids of MBP are not clustered, but equally distributed within the primary sequence. To investigate whether MBP contains a “critical region” for plasma membrane binding, we created deletion mutants beginning with the smallest MBP isoform (14 kDa), which is sufficient to rescue the shiverer phenotype in vivo (Kimura et al., 1989). The 14 kDa MBP is encoded by five exons (exons 1, 3, 4, 5, 7) that we decided to study individually by deleting them from the corresponding cDNA expression clone. A deletion construct from which exons 1, 3, and 4 were removed led to a truncated protein without plasma membrane association (data not shown). Also, MBP mutants lacking the residues encoded by exon 1 and 3 failed to bind (data not shown). Indeed, already the deletion of exon 1 [in clone ΔExon1(MBP)-YFP] was sufficient to abolish the membrane interaction of MBP (Fig. 7B).
Because the deletion of exon 1 resulted in loss of 57 (from 128) aa, including 15 basic residues, we analyzed whether this polypeptide was sufficient by itself to mediate the interaction with the plasma membrane. We generated an expression construct containing MBP exon 1 fused to YFP [termed Exon1(MBP)-YFP]. This fusion protein was also targeted to the plasma membrane, although not as efficiently as the full-length protein (indicated by the line scans in Fig. 7). For comparison, when only exon 7 was fused to YFP [termed Exon7(MBP)YFP], expression of MBP Exon7(MBP)-YFP in Oli-neu cells did not result in plasma membrane delivery (Fig. 7C).
When the N-terminal sequences of MBP from different species were aligned, we noticed that many residues were strictly conserved (Fig. 7A). To test for the functional significance of these positions in transfected cells, we generated MBP mutants in which one or two (closely spaced) basic amino acids were replaced by alanine (Fig. 7A,D). Unexpectedly, in the majority of cases (K5A/R6A; H22A/R24A; R30A/R32A; R42A; K52A/R53A; K57A), the replacement of only one or two positively charged amino acids was indeed sufficient to prevent protein binding to the membrane, whereas the exchange of two other basic residues within exon 1 (R10A/K12A) did not effect plasma membrane localization (Fig. 7D). This indicates that both the number and localization of basic residues in MBP are important for binding to PIP2.
Previous biochemical studies had suggested that PIP2 can covalently link to Ser54 of MBP, based on the isolation of a lipid-bound fragment of MBP from bovine brain and its subsequent amino acid analysis (Chang et al., 1986; Yang et al., 1986). We therefore generated a 14 kDa MBP(S54A) mutant and a S54A variant of MBP Exon1(MBP)-YFP [termed MBP(S54A)-YFP and Exon1(S54A)-YFP]. Both mutants showed reduced plasma membrane association when compared with the respective wild-type MBP construct (Fig. 7B) (data not shown). Together, our mutagenesis data demonstrate that the N-terminal region of MBP is sufficient to bind to the plasma membrane but requires positively charged amino acids at specific positions as well as Ser54.
Phosphoinositides are good candidates for having a dynamic role in myelination, because the activity of specific kinases and phosphatases can rapidly change their structure and add or remove membrane charges (De Matteis and Godi, 2004). Some of these enzymatic reactions are calcium controlled, but as a divalent cation Ca2+ may by itself influence the association of proteins with membranes. To test the effect of altered surface charges on MBP, we triggered Ca2+ influx by ionomycin as previously described (Várnai and Balla, 1998; Yeung et al., 2006). Primary cultures of oligodendrocytes were first treated with 10 μm ionomycin for 2 min and then permeabilized with 0.005% saponin to wash out the released proteins. We found that ionomycin treatment resulted in a significant loss of MBP, but not of galactosylceramide (recognized by anti-O1 staining) from the cells (see Fig. 9D). When Oli-neu cells were transfected to express MBP14k-YFP and were treated (8 h later) with ionomycin, we observed a rapid dissociation of MBP from the plasma membrane by live fluorescence microscopy (Fig. 8; supplemental Videos S1, S2, available at www.jneurosci.org as supplemental material). The kinetics of plasma membrane dissociation was similar for MBP and GFP-PH-PLCδ1. After dissociation from the plasma membrane, some MBP accumulated in PS-containing endosomal membranes (Fig. 6).
The destabilizing effect of ionomycin and Ca2+ entry on MBP binding to the plasma membrane was more pronounced than that of selective PIP2 depletion. One possible explanation is that Ca2+ influx induces in addition a transfer of PS from the inner to the outer leaflet of the plasma membrane.
To investigate whether a Ca2+ release from intracellular pools would be sufficient to dissociate MBP from the plasma membrane, we performed the same experiment in cells that were maintained in a Ca2+-free modified Krebs–Ringer's solution. Under these conditions, MBP was not released from the plasma membrane when cells were treated with ionomycin (Fig. 8). Also, when ionomycin was added at the same time as EGTA, it did not change the plasma membrane localization of MBP (data not shown). In addition, when ionomycin treatment (for 2 min) was followed by the chelation of Ca2+ with EGTA (for 30 min), the effect was reversed, with MBP binding again to the plasma membrane (Fig. 9).
Ca2+ is known to activate PLC, which in turn hydrolyzes PIP2, thereby releasing IP3 and DAG. To test for a possible role of PLC in the observed dissociation of MBP from the plasma membrane by ionomycin treatment, we added neomycin. Neomycin forms a 1:1 electroneutral complex with PIP2, thereby blocking PLC-dependent hydrolysis of PIP2 (Várnai and Balla, 1998). Indeed, when transfected cells were treated with both ionomycin and neomycin, MBP remained stably at the plasma membrane, suggesting that PLC activation is the underlying cause of Ca2+-dependent MBP release (Fig. 8).
Depletion of the cellular ATP pool is commonly used as another method to reduce membrane surface charge, because ATP depletion alters the phosphoinositide content in cells (McLaughlin, 1989; Yeung et al., 2006). When transfected Oli-neu cells were maintained in an ATP depleting medium (without Ca2+ and glutamate), supplemented with antimycin and 2-deoxy-d-glucose to prevent glycolysis, MBP dissociated from the plasma membrane within 45 min (Fig. 9). Quantification of these results showed that both GFP-PH-PLCδ1 and MBP14k-YFP lost their peak signal of membrane association (Fig. 9). These results confirm that MBP interacts with the plasma membrane in a surface charge-dependent manner.
NMDA-mediated calcium entry has been suggested as a possible mechanism of myelin injury in ischemia (Káradóttir et al., 2005; Micu et al., 2006), but the mechanism leading to myelin disruption is not clear. To verify our in vitro results at the tissue level and in CNS myelin, we prepared acute brain slices from 30-d-old mice that included the corpus callosum. Treating these slices for 1 h with ionomycin led to a rapid delamination and vesiculation of the myelin membrane that could be observed at the EM level within 1 h (Fig. 10) (Smith et al., 1985). Similar results were obtained when acute brain slices were ATP depleted for 1 h (Fig. 10). Importantly, vesiculation was much less dramatic when slices were incubated with both ionomycin and neomycin, supporting the view that PLC activation is the underlying cause of Ca2+-dependent MBP release (Fig. 10). Although these ex vivo experiments cannot be analyzed in the same level of detail as cells in culture, they support the model that specific electrostatic interactions between PIP2 and MBP are required to maintain myelin membrane integrity.
Figure 10.
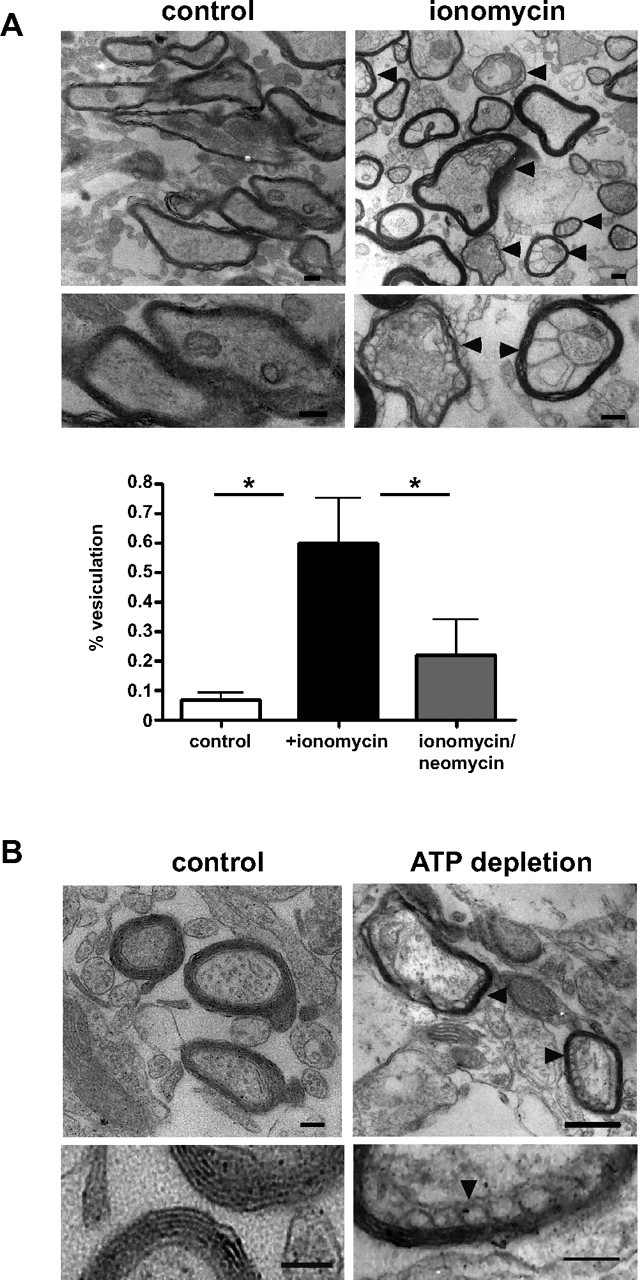
Vesiculation of myelin in acute brain slices after ionomycin treatment and ATP depletion. Acute slices from 30-d-old mice were equilibrated in ACSF for 1 h at room temperature before incubated with ionomycin or ATP depleting reagents for 1 h at 35°C. For ionomycin treatment, control slices were incubated in DMSO (A; scale bar, 0.6 μm). Vesiculation was less pronounced when slices were incubated for 15 min with 10 mm neomycin before and during ionomycin treatment (30 min, control vs ionomycin: p = 0.0129; control vs ionomycin/neomycin: p = 0.0338; n > 100 myelinated axons; 4 independent slices) (A). For ATP depletion acute slices from adult mice were incubated in glucose and Ca2+ free ACSF together with antimycin and 2-deoxy-d-glucose for 1 h. Control slices were incubated in ACSF (B; scale bars, 0.4 μm). After 1 h incubation, acute slices were fixated in and processed for electron microscopy. The arrowheads indicate myelin delamination. Error bars indicate SEM.
Discussion
MBP, one of the major structural proteins of CNS myelin, has been investigated for many years, but mechanistic insight into MBP function has not been reached, except for its rather unspecific interaction with negatively charged lipid head groups. Although mutant mice lacking MBP expression (shiverer) have revealed a protein function in myelin compaction, these in vivo experiments failed to provide mechanistic insight at the molecular level. We therefore turned to an intact cellular system, in which the presumed interaction of MBP with phospholipids can be studied. Specifically, we used different experimental approaches to investigate the role of PIP2 for the stable association of MBP with the plasma membrane. In a loss-of-function approach, we found that coexpression of MBP with the PIP2 hydrolyzing enzyme, Synj1, reduced the binding of MBP to the plasma membrane. Conversely, in a gain-of-function approach, coexpression of MBP with a constitutive active Arf6 variant resulted in the redistribution of MBP from the plasma membrane to intracellular PIP2-enriched endosomal vesicles. Furthermore, we observed FRET between MBP and PIP2-sensing probe, indicating a tight colocalization. A putative PIP2 binding domain of MBP was localized at the N-terminal domain, encoded by exon 1 (contained in all MBP splice isoforms) and harboring critical lysine and serine residues. Finally, we found that increasing the intracellular Ca2+ level causes a rapid dissociation of MBP from the plasma membrane, which involves the PLC-dependent hydrolysis of PIP2. Together, these results provide experimental evidence that PIP2 is the critical membrane lipid required for the membrane association of MBP. Abnormal Ca2+ entry and Ca2+-dependent myelin delamination in white matter tracts, as observed here by EM ex vivo, is thus likely to be caused by the detachment of MBP from myelin membranes, which becomes clinically relevant after hypoxic injury or an autoimmune attack.
We previously found that the interaction of MBP with the plasma membrane is essential for the reorganization of membrane components that occurs when oligodendrocytes come in contact with neuronal processes. We found that MBP increases the lipid packaging of the myelin-forming membrane bilayer in cultured oligodendrocytes (Fitzner et al., 2006). The results presented in this study provide evidence that the critical interaction of MBP is with the signaling lipid PIP2. It is interesting to note that other PIP2 binding proteins, such as MARCKS (a member of the GAP43-like family of proteins), induce the formation of large lipid domains in a PIP2-dependent manner (Laux et al., 2000; McLaughlin et al., 2002; Gambhir et al., 2004). While our study was under review, Musse et al. (2008) demonstrated by fluorescence quenching and electron paramagnetic resonance spectroscopy that MBP sequesters PIP2 in model membranes, providing independent support for our model. The binding of basic proteins (i.e., MBP) to PIP2 reduces the lateral diffusion of this lipid and may also sequester other lipids into membrane microdomains. These domains may act as recruitment sites for myelin membrane lipids, finally resulting in a condensation of the myelin membrane as determined by the fluorescent probe Laudran and two-photon microscopy (Fitzner et al., 2006).
The “classical” function of MBP is the formation of the MDL in compact myelin, as revealed by its absence in the CNS of shiverer mice. This concept was confirmed by the discovery that the MDL in PNS myelin is dependent on myelin protein zero (P0), a single span adhesion protein with a highly basic intracellular domain (Privat et al., 1979; Lemke and Axel, 1985; Martini et al., 1995). It has also been suggested that the adhesion of cytosolic surfaces to MBP are sensitive to changes in surface charge (Inouye and Kirschner, 1988). These fluctuations might involve axonally regulated Ca2+ rises or pH changes in oligodendrocytes (Ro and Carson, 2004), as suggested in other cell types (McLaughlin, 1989). The inner membrane surface potential (i.e., the potential created by negatively charged lipids and their counter ions attracted by them) has been estimated to be ∼105 V/cm (Olivotto et al., 1996). PIP2 contributes significantly to the surface charge having a valance of −4 at physiological pH (McLaughlin et al., 2002). When the two highly negatively charged surfaces of opposing cytosolic surfaces come together in compact myelin (to ∼1.7 nm), their negative charge has to be neutralized, and we suggest that MBP primarily serves this neutralizing function in compact myelin. Acute loss of this interaction should in turn result in a rapid delamination and degeneration of compacted myelin, as seen in the brain slice preparation. We further hypothesize that the binding of MBP to the cytosolic membrane not only reduces the negativity of the inner membrane surface but is likely to also promote the interaction of the membrane leaflets with each other (i.e., in the formation of the MDL) (Smith, 1977; Inouye and Kirschner, 1988).
MBP serves as a molecular “zipper,” but whether or not MBP needs to dimerize to serve such a “zipper” function remains to be determined. Specific posttranslational modifications may also modulate surface binding of MBP. It is interesting to note that an increase in citrulline, which reduces the charge of MBP, has been detected in myelin from multiple sclerosis patients (Boggs et al., 1997; Kim et al., 2003).
We found that both ATP depletion of MBP-expressing cells or their treatment with ionomycin caused a dissociation of MBP from the plasma membrane. The effect of ionomycin treatment is most likely secondary to enzymatic hydrolysis of PIP2, because the presence of neomycin (binding stochiometrically to PIP2) blocked it. Alternatively, Ca2+ regulates the association of MBP with the plasma membrane in a Ca2+/calmodulin-dependent manner. A Ca2+/calmodulin-dependent dissociation of membranes has also been observed for other basic unstructured proteins such as MARCKS (McLaughlin et al., 2002). Although we found that alteration of membrane surface charge resulted in less MBP binding to the plasma membrane in vitro (and to myelin vesiculation in ex vivo experiments), additional in vivo studies are required to better understand the role of MBP binding to the myelin membrane under pathological conditions.
Interestingly, after dissociation from the plasma membrane, MBP was found on intracellular membrane containing PS (but not PIP2). This result shows that MBP binds to many different phospholipids, but the binding to PIP2 might be of higher affinity possibly because of the valence of −4 compared with −1 of PS. We also note that the effect of ionomycin on plasma membrane association was more pronounced than the specific decrease in PIP2. This could be attributable to the additional loss of PS from the inner leaflet of the plasma membrane by flipping that occurs after Ca2+ influx (Yeung et al., 2006).
Importantly, in acute brain slices that included white matter, ionomycin treatment and ATP depletion also led to a rapid myelin “vesiculation.” Such a vesiculation would be expected if MBP, once bound to myelin membranes, serves to neutralize many of the negative surface charges of the closely interacting myelin membrane layers. Abnormal Ca2+ entry is frequent sign of cellular pathology. Thus, the effects of elevated intracellular Ca2+ on MBP–membrane interactions may be relevant to the changes of myelin that occur under various pathological conditions. Recent studies have shown that CNS myelin contains NMDA receptors that could be responsible for a rise in intracellular Ca2+ when the white matter is injured by hypoxia and excitotoxicity (Káradóttir et al., 2005; Micu et al., 2006). Moreover, intracellular accumulation of Ca2+ induced by glycine/glutamate signaling was shown to disrupt the myelin ultrastructure. In addition, it has been reported oxygen–glucose deprivation causes a similar vesiculation after the activation of AMPA/kainate receptors (Tekkök et al., 2005; Micu et al., 2006).
In summary, we provided experimental evidence that PIP2, a highly charged phospholipid, is an essential component for the binding of MBP to the cell membrane, and in extension for the function of MBP in myelination. It is intriguing that PIP2 has been independently implicated in myelination as a signaling lipid and substrate of PI3K, which generates PIP3 and activates AKT/PKB to drive CNS myelination (Flores et al., 2008) (Goebbels et al., unpublished observations), presumably as a response to specific axonal signals. We propose that oligodendroglial PIP2 is equally important as a signaling lipid and as a lipid docking site for MBP to fulfill its function in lipid sorting and myelin compaction. Interfering with this highly specific lipid–protein interaction, for example by abnormal increases of intracellular Ca2+, leads to the destabilization of CNS myelin and may be related to myelin destruction in ischemic conditions and in demyelinating diseases.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 523) (M.S., K.-A.N.), a European Research Council Starting Grant (M.S.), Bundesministerium für Bildung und Forschung (Leukonet), and The Myelin Project. We are thankful to Sehera Nawaz for custom-written MATLAB files; A. T. Campagnoni, J. G. Donaldson, S. Grinstein, R. Jahn, T. Meyer, I. Milosevic, and J. Sørensen for sharing plasmids; and J. Trotter for NG2 antibody. We also thank G. Schulz for excellent technical assistance, T. Ruhwedel for help with electron microscopy, and I. Bormuth for help with image processing.
References
- Ainger K, Avossa D, Diana AS, Barry C, Barbarese E, Carson JH. Transport and localization elements in myelin basic protein mRNA. J Cell Biol. 1997;138:1077–1087. doi: 10.1083/jcb.138.5.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbarese E, Carson JH, Braun PE. Accumulation of the four myelin basic proteins in mouse brain during development. J Neurochem. 1978;31:779–782. doi: 10.1111/j.1471-4159.1978.tb00110.x. [DOI] [PubMed] [Google Scholar]
- Boggs J. Myelin basic protein: a multifunctional protein. Cell Mol Life Sci. 2006;63:1945–1961. doi: 10.1007/s00018-006-6094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggs JM, Yip PM, Rangaraj G, Jo E. Effect of posttranslational modifications to myelin basic protein on its ability to aggregate acidic lipid vesicles. Biochemistry. 1997;36:5065–5071. doi: 10.1021/bi962649f. [DOI] [PubMed] [Google Scholar]
- Campagnoni AT, Campagnoni CW. Myelin biology and disorders (Lazzarini RA) San Diego: Elsevier Academic; 2004. Myelin basic protein gene; pp. 387–395. [Google Scholar]
- Chang PC, Yang JC, Fujitaki JM, Chiu KC, Smith RA. Covalent linkage of phospholipid to myelin basic protein: identification of serine-54 as the site of attachment. Biochemistry. 1986;25:2682–2686. doi: 10.1021/bi00357a059. [DOI] [PubMed] [Google Scholar]
- de Ferra F, Engh H, Hudson L, Kamholz J, Puckett C, Molineaux S, Lazzarini RA. Alternative splicing accounts for the four forms of myelin basic protein. Cell. 1985;43:721–727. doi: 10.1016/0092-8674(85)90245-4. [DOI] [PubMed] [Google Scholar]
- De Matteis MA, Godi A. PI-loting membrane traffic. Nat Cell Biol. 2004;6:487–492. doi: 10.1038/ncb0604-487. [DOI] [PubMed] [Google Scholar]
- Donaldson JG. Multiple roles for Arf6: sorting, structuring, and signaling at the plasma membrane. J Biol Chem. 2003;278:41573–41576. doi: 10.1074/jbc.R300026200. [DOI] [PubMed] [Google Scholar]
- Fitzner D, Schneider A, Kippert A, Möbius W, Willig KI, Hell SW, Bunt G, Gaus K, Simons M. Myelin basic protein-dependent plasma membrane reorganization in the formation of myelin. EMBO J. 2006;25:5037–5048. doi: 10.1038/sj.emboj.7601376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores AI, Narayanan SP, Morse EN, Shick HE, Yin X, Kidd G, Avila RL, Kirschner DA, Macklin WB. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci. 2008;28:7174–7183. doi: 10.1523/JNEUROSCI.0150-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambhir A, Hangyás-Mihályné G, Zaitseva I, Cafiso DS, Wang J, Murray D, Pentyala SN, Smith SO, McLaughlin S. Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys J. 2004;86:2188–2207. doi: 10.1016/S0006-3495(04)74278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas H, Steitz R, Fasano A, Liuzzi GM, Polverini E, Cavatorta P, Riccio P. Laminar order within Langmuir-Blodgett multilayers from phospholipid and myelin basic protein: a neutron reflectivity study. Langmuir. 2007;23:8491–8496. doi: 10.1021/la700733y. [DOI] [PubMed] [Google Scholar]
- Harauz G, Ishiyama N, Hill CM, Bates IR, Libich DS, Farès C. Myelin basic protein-diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron. 2004;35:503–542. doi: 10.1016/j.micron.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Heo WD, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ, Meyer T. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314:1458–1461. doi: 10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Israelachvili J. Lateral reorganization of myelin lipid domains by myelin basic protein studied at the air-water interface. Colloids Surf B Biointerfaces. 2008;62:22–30. doi: 10.1016/j.colsurfb.2007.09.028. [DOI] [PubMed] [Google Scholar]
- Hu Y, Doudevski I, Wood D, Moscarello M, Husted C, Genain C, Zasadzinski JA, Israelachvili J. Synergistic interactions of lipids and myelin basic protein. Proc Natl Acad Sci U S A. 2004;101:13466–13471. doi: 10.1073/pnas.0405665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Nakamura R, Mikoshiba K, Tsukada Y. Fine structure of the central myelin sheath in the myelin deficient mutant Shiverer mouse, with special reference to the pattern of myelin formation by oligodendroglia. Brain Res. 1981;219:85–94. doi: 10.1016/0006-8993(81)90269-9. [DOI] [PubMed] [Google Scholar]
- Inouye H, Kirschner DA. Membrane interactions in nerve myelin: II. Determination of surface charge from biochemical data. Biophys J. 1988;53:247–260. doi: 10.1016/S0006-3495(88)83086-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Krämer E, Grzenkowski M, Tang K, Blakemore W, Aguzzi A, Khazaie K, Chlichlia K, von Blankenfeld G, Kettenmann H. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. Eur J Neurosci. 1995;7:1245–1265. doi: 10.1111/j.1460-9568.1995.tb01115.x. [DOI] [PubMed] [Google Scholar]
- Kamholz J, de Ferra F, Puckett C, Lazzarini R. Identification of three forms of human myelin basic protein by cDNA cloning. Proc Natl Acad Sci U S A. 1986;83:4962–4966. doi: 10.1073/pnas.83.13.4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Káradóttir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. 2005;438:1162–1166. doi: 10.1038/nature04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson U, Schultz RL. Fixation of the central nervous system from electron microscopy by aldehyde perfusion. I. Preservation with aldehyde perfusates versus durect perfusion with osmium tetroxide with special preference to membranes and the extracellular space. J Ultrastruct Res. 1965;12:160–186. doi: 10.1016/s0022-5320(65)80014-4. [DOI] [PubMed] [Google Scholar]
- Kim JK, Mastronardi FG, Wood DD, Lubman DM, Zand R, Moscarello MA. Multiple sclerosis: an important role for post-translational modifications of myelin basic protein in pathogenesis. Mol Cell Proteomics. 2003;2:453–462. doi: 10.1074/mcp.M200050-MCP200. [DOI] [PubMed] [Google Scholar]
- Kimura M, Sato M, Akatsuka A, Nozawa-Kimura S, Takahashi R, Yokoyama M, Nomura T, Katsuki M. Restoration of myelin formation by a single type of myelin basic protein in transgenic shiverer mice. Proc Natl Acad Sci U S A. 1989;86:5661–5665. doi: 10.1073/pnas.86.14.5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss M, Kinuta M, Wenk MR, De Camilli P, Takei K, Haucke V. ARF6 stimulates clathrin/AP-2 recruitment to synaptic membranes by activating phosphatidylinositol phosphate kinase type Iγ. J Cell Biol. 2003;162:113–124. doi: 10.1083/jcb.200301006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Bruns D, Wenzel D, Riedel D, Holroyd P, Thiele C, Jahn R. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 2001;20:2202–2213. doi: 10.1093/emboj/20.9.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laux T, Fukami K, Thelen M, Golub T, Frey D, Caroni P. GAP43, MARCKS, and CAP23 modulate PI(4,5)P(2) at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J Cell Biol. 2000;149:1455–1472. doi: 10.1083/jcb.149.7.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke G, Axel R. Isolation and sequence of a cDNA encoding the major structural protein of peripheral myelin. Cell. 1985;40:501–508. doi: 10.1016/0092-8674(85)90198-9. [DOI] [PubMed] [Google Scholar]
- Martini R, Mohajeri MH, Kasper S, Giese KP, Schachner M. Mice doubly deficient in the genes for P0 and myelin basic protein show that both proteins contribute to the formation of the major dense line in peripheral nerve myelin. J Neurosci. 1995;15:4488–4495. doi: 10.1523/JNEUROSCI.15-06-04488.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin S. The electrostatic properties of membranes. Annu Rev Biophys Biophys Chem. 1989;18:113–136. doi: 10.1146/annurev.bb.18.060189.000553. [DOI] [PubMed] [Google Scholar]
- McLaughlin S, Wang J, Gambhir A, Murray D. PIP(2) and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, McRory JE, Rehak R, Zamponi GW, Wang W, Stys PK. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. 2006;439:988–992. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- Mikoshiba K, Okano H, Tamura T, Ikenaka K. Structure and function of myelin protein genes. Annu Rev Neurosci. 1991;14:201–217. doi: 10.1146/annurev.ne.14.030191.001221. [DOI] [PubMed] [Google Scholar]
- Milosevic I, Sørensen JB, Lang T, Krauss M, Nagy G, Haucke V, Jahn R, Neher E. Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffin cells. J Neurosci. 2005;25:2557–2565. doi: 10.1523/JNEUROSCI.3761-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musse A, Gao W, Homchaudhuri L, Boggs J, Harauz G. Myelin basic protein as a “PI(4,5)P2-Modulin”: a new biological function for a major central nervous system protein. Biochemistry. 2008;47:10372–10382. doi: 10.1021/bi801302b. [DOI] [PubMed] [Google Scholar]
- Olivotto M, Arcangeli A, Carlà M, Wanke E. Electric fields at the plasma membrane level: a neglected element in the mechanisms of cell signalling. Bioessays. 1996;18:495–504. doi: 10.1002/bies.950180612. [DOI] [PubMed] [Google Scholar]
- Omlin FX, Webster HD, Palkovits CG, Cohen SR. Immunocytochemical localization of basic protein in major dense line regions of central and peripheral myelin. J Cell Biol. 1982;95:242–248. doi: 10.1083/jcb.95.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A. 2004;101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedraza L, Fidler L, Staugaitis SM, Colman DR. The active transport of myelin basic protein into the nucleus suggests a regulatory role in myelination. Neuron. 1997;18:579–589. doi: 10.1016/s0896-6273(00)80299-8. [DOI] [PubMed] [Google Scholar]
- Popko B, Puckett C, Lai E, Shine HD, Readhead C, Takahashi N, Hunt SW, 3rd, Sidman RL, Hood L. Myelin deficient mice: expression of myelin basic protein and generation of mice with varying levels of myelin. Cell. 1987;48:713–721. doi: 10.1016/0092-8674(87)90249-2. [DOI] [PubMed] [Google Scholar]
- Privat A, Jacque C, Bourre JM, Dupouey P, Baumann N. Absence of the major dense line in myelin of the mutant mouse “shiverer.”. Neurosci Lett. 1979;12:107–112. doi: 10.1016/0304-3940(79)91489-7. [DOI] [PubMed] [Google Scholar]
- Readhead C, Popko B, Takahashi N, Shine HD, Saavedra RA, Sidman RL, Hood L. Expression of a myelin basic protein gene in transgenic shiverer mice: correction of the dysmyelinating phenotype. Cell. 1987;48:703–712. doi: 10.1016/0092-8674(87)90248-0. [DOI] [PubMed] [Google Scholar]
- Riccio P, Fasano A, Borenshtein N, Bleve-Zacheo T, Kirschner DA. Multilamellar packing of myelin modeled by lipid-bound MBP. J Neurosci Res. 2000;59:513–521. doi: 10.1002/(SICI)1097-4547(20000215)59:4<513::AID-JNR6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Richter-Landsberg C, Heinrich M. OLN-93: a new permanent oligodendroglia cell line derived from primary rat brain glial cultures. J Neurosci Res. 1996;45:161–173. doi: 10.1002/(SICI)1097-4547(19960715)45:2<161::AID-JNR8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Rispoli P, Carzino R, Svaldo-Lanero T, Relini A, Cavalleri O, Fasano A, Liuzzi GM, Carlone G, Riccio P, Gliozzi A, Rolandi R. A thermodynamic and structural study of myelin basic protein in lipid membrane models. Biophys J. 2007;93:1999–2010. doi: 10.1529/biophysj.106.103820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas AA, Castro RM. Interaction of bovine myelin basic protein with triphosphoinositide. J Colloid Interface Sci. 2002;256:290–296. doi: 10.1006/jcis.2002.8689. [DOI] [PubMed] [Google Scholar]
- Ro HA, Carson JH. pH microdomains in oligodendrocytes. J Biol Chem. 2004;279:37115–37123. doi: 10.1074/jbc.M403099200. [DOI] [PubMed] [Google Scholar]
- Roach A, Takahashi N, Pravtcheva D, Ruddle F, Hood L. Chromosomal mapping of mouse myelin basic protein gene and structure and transcription of the partially deleted gene in shiverer mutant mice. Cell. 1985;42:149–155. doi: 10.1016/s0092-8674(85)80110-0. [DOI] [PubMed] [Google Scholar]
- Rosenbluth J. Central myelin in the mouse mutant shiverer. J Comp Neurol. 1980;194:639–648. doi: 10.1002/cne.901940310. [DOI] [PubMed] [Google Scholar]
- Rosetti CM, Maggio B. Protein-induced surface structuring in myelin membrane monolayers. Biophys J. 2007;93:4254–4267. doi: 10.1529/biophysj.107.112441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Trotter J. Wrapping it up: the cell biology of myelination. Curr Opin Neurobiol. 2007;17:533–540. doi: 10.1016/j.conb.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Hall SM, Schauf CL. Vesicular demyelination induced by raised intracellular calcium. J Neurol Sci. 1985;71:19–37. doi: 10.1016/0022-510x(85)90034-6. [DOI] [PubMed] [Google Scholar]
- Smith R. Non-covalent cross-linking of lipid bilayers by myelin basic protein: a possible role in myelin formation. Biochim Biophys Acta. 1977;470:170–184. doi: 10.1016/0005-2736(77)90098-0. [DOI] [PubMed] [Google Scholar]
- Smith R. The basic protein of CNS myelin: its structure and ligand binding. J Neurochem. 1992;59:1589–1608. doi: 10.1111/j.1471-4159.1992.tb10989.x. [DOI] [PubMed] [Google Scholar]
- Taylor CM, Marta CB, Claycomb RJ, Han DK, Rasband MN, Coetzee T, Pfeiffer SE. Proteomic mapping provides powerful insights into functional myelin biology. Proc Natl Acad Sci U S A. 2004;101:4643–4648. doi: 10.1073/pnas.0400922101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekkök SB, Faddis BT, Goldberg MP. AMPA/kainate receptors mediate axonal morphological disruption in hypoxic white matter. Neurosci Lett. 2005;382:275–279. doi: 10.1016/j.neulet.2005.03.054. [DOI] [PubMed] [Google Scholar]
- Trajkovic K, Dhaunchak AS, Goncalves JT, Wenzel D, Schneider A, Bunt G, Nave KA, Simons M. Neuron to glia signaling triggers myelin membrane exocytosis from endosomal storage sites. J Cell Biol. 2006;172:937–948. doi: 10.1083/jcb.200509022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Kidd GJ. Myelin biology and disorders (Larzzraini RA) San-Diego: Elsevier Academic; 2004. Structure of myelinated axons; pp. 3–22. [Google Scholar]
- van der Wal J, Habets R, Várnai P, Balla T, Jalink K. Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer. J Biol Chem. 2001;276:15337–15344. doi: 10.1074/jbc.M007194200. [DOI] [PubMed] [Google Scholar]
- Várnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143:501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Várnai P, Balla T. Visualization and manipulation of phosphoinositide dynamics in live cells using engineered protein domains. Pflugers Arch. 2007;455:69–82. doi: 10.1007/s00424-007-0270-y. [DOI] [PubMed] [Google Scholar]
- Werner HB, Kuhlmann K, Shen S, Uecker M, Schardt A, Dimova K, Orfaniotou F, Dhaunchak A, Brinkmann BG, Möbius W, Guarente L, Casaccia-Bonnefil P, Jahn O, Nave KA. Proteolipid protein is required for transport of sirtuin 2 into CNS myelin. J Neurosci. 2007;27:7717–7730. doi: 10.1523/JNEUROSCI.1254-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R, Gonsior C, Krämer-Albers EM, Stöhr N, Hüttelmaier S, Trotter J. Activation of oligodendroglial Fyn kinase enhances translation of mRNAs transported in hnRNP A2-dependent RNA granules. J Cell Biol. 2008;181:579–586. doi: 10.1083/jcb.200706164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Chang PC, Fujitaki JM, Chiu KC, Smith RA. Colvalent linkage of phospholipid to myelin basic protein: identification of phosphatidylinositol bisphosphate as the attached phospholipid. Biochemistry. 1986;25:2677–2681. doi: 10.1021/bi00357a058. [DOI] [PubMed] [Google Scholar]
- Yeung T, Terebiznik M, Yu L, Silvius J, Abidi WM, Philips M, Levine T, Kapus A, Grinstein S. Receptor activation alters inner surface potential during phagocytosis. Science. 2006;313:347–351. doi: 10.1126/science.1129551. [DOI] [PubMed] [Google Scholar]
- Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. Membrane phosphatidylserine regulates surface charge and protein localization. Science. 2008;319:210–213. doi: 10.1126/science.1152066. [DOI] [PubMed] [Google Scholar]