

Abstract
Studies of naturally occurring mutant mice, wlds, showing delayed Wallerian degeneration phenotype, suggest that axonal degeneration is an active process. We previously showed that increased nicotinamide adenine dinucleotide (NAD)-synthesizing activity by overexpression of nicotinamide mononucleotide adenylyltransferase (NMNAT) is the essential component of the Wlds protein, the expression of which is responsible for the delayed Wallerian degeneration phenotype in wlds mice. Indeed, NMNAT overexpression in cultured neurons provides robust protection to neurites, as well. To examine the effect of NMNAT overexpression in vivo and to analyze the mechanism that causes axonal protection, we generated transgenic mice (Tg) overexpressing NMNAT1 (nuclear isoform), NMNAT3 (mitochondrial isoform), or the Wlds protein bearing a W258A mutation, which disrupts NAD-synthesizing activity of the Wlds protein. Wallerian degeneration delay in NMNAT3-Tg was similar to that in wlds mice, whereas axonal protection in NMNAT1-Tg or Wlds(W258A)-Tg was not detectable. Detailed analysis of subcellular localization of the overexpressed proteins revealed that the axonal protection phenotype was correlated with localization of NMNAT enzymatic activity to mitochondrial matrix. Furthermore, we found that isolated mitochondria from mice showing axonal protection expressed unchanged levels of respiratory chain components, but were capable of increased ATP production. These results suggest that axonal protection by NMNAT expression in neurons is provided by modifying mitochondrial function. Alteration of mitochondrial function may constitute a novel tool for axonal protection, as well as a possible treatment of diseases involving axonopathy.
Introduction
Axonal degeneration is now regarded as a major component of many neurodegenerative diseases, including Parkinson's disease and Alzheimer's disease (Raff et al., 2002). Therefore, the axonal degeneration process could well be a therapeutic target against these diseases. The discovery of a spontaneous dominant mutation in mice that results in delayed axonal degeneration, the Wallerian degeneration slow (wlds) mice, strongly suggests that axonal degeneration is an active process of self-destruction (Lunn et al., 1989). The wlds mutation comprises an 85 kb tandem triplication, which causes overexpression of a chimeric molecule (Wlds protein). This protein is composed of the N-terminal 70 aa of Ufd2a (ubiquitin fusion degradation protein 2a)/Ube4b (ubiquitination factor E4b), a ubiquitin-chain assembly factor, fused to the complete sequence of nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1), an enzyme in the NAD biosynthetic pathway that generates NAD in the nucleus (Conforti et al., 2000; Mack et al., 2001). We previously showed that overexpression of NMNAT enzymatic activity is the essential mechanism that causes the axonal protection phenotype in wlds mice (Araki et al., 2004). NMNAT is a family of proteins that mediates NAD synthesis by using nicotinamide mononucleotide (NMN) and ATP as substrates. In addition to NMNAT1, NMNAT2 (located mostly in Golgi apparatus and expressed most highly in mammalian brain) and NMNAT3 (located mostly in mitochondria) have been identified in mammals (Berger et al., 2005). We previously showed that not only NMNAT1 but also NMNAT3 overexpression in neurons is protective in an in vitro Wallerian degeneration model, which supports our hypothesis that NMNAT enzymatic activity is the key for axonal protection (Sasaki et al., 2006).
There are conflicting results against our hypothesis. Coleman and colleagues showed that the NAD content in wlds mouse tissue is similar to that in wild-type despite the increased NMNAT activity in wlds mice (Mack et al., 2001). Furthermore, they recently showed that NMNAT1-overexpressing transgenic mice do not show delayed Wallerian degeneration observed in wlds mice (Conforti et al., 2007), which supports their hypothesis that N-terminal Ufd2a region of the Wlds protein plays a role (Laser et al., 2006). These reports suggest that NMNAT activity may not be crucial for axonal protection.
In this study, to analyze the requirement of NMNAT activity for axonal protection in vivo, we generated and analyzed the phenotype of mice that overexpressed either NMNAT1 (NMNAT1-Tg), NMNAT3 (NMNAT3-Tg), or the Wlds protein bearing a W258A mutation that disrupts NAD producing activity of the Wlds protein [Wlds(W258A)-Tg]. We found that NMNAT3-Tg mice demonstrated delayed Wallerian degeneration, similar to wlds mice; whereas NMNAT1-Tg or Wlds(W258A)-Tg mice did not show any axonal protection. In both NMNAT3-Tg and wlds mice, significant amounts of NMNAT activity were found to be located in the mitochondrial matrix. We further found that mitochondrial localization of NMNAT activity resulted in increased ATP synthetic capacity without affecting the expression profile of mitochondrial enzymes of the respiratory chain. These data suggest that mitochondrial localization of NMNAT activity plays an important role in NMNAT expression-mediated axonal protection.
Materials and Methods
Generation of transgenic mice.
The transgene was constructed by inserting the cDNA 3′ downstream of the chicken β-actin promoter in the pCAGGS plasmid (a kind gift from Dr. J. Miyazaki, Osaka University, Osaka, Japan) (Niwa et al., 1991). The cDNAs for mouse NMNAT1 and NMNAT3 used as transgenes were obtained from expressed sequence-tagged clones (BC038133 for murine NMNAT1; BC005737 for murine NMNAT3). cDNA for Wlds protein bearing W258A mutation was generated as previously described (Araki et al., 2004). The integrity of each clone was verified by nucleotide sequence analysis. Transgenic constructs were microinjected into fertilized BDF1 mouse oocytes by following standard procedure (Conner, 2004). Genomic integrations of transgenes were examined by Southern blot analysis and/or PCR with the following primers sets: NMNAT1-Tg and Wlds(W258A)-Tg, 5′-gaccggcggctctagagcctctgctaa-3′ and 5′-cctgaggtgcatgttggtgatggggtta-3′; NMNAT3-Tg, 5′-gaccggcggctctagagcctctgctaa-3′ and 5′-atggggttgaaggaaccacaggccagaa-3′. The PCR conditions were 94°C for 10 s, 62°C for 30 s, 72°C for 1 min, performed for 40 cycles. All the transgenic lines were backcrossed to C57BL/6 for three generations when we performed the experiments described here. Animal care and use in our laboratory were in strict accordance with the guidelines for animal and recombinant DNA experiments by National Center of Neurology and Psychiatry.
Antibodies.
Polyclonal antiserum specific for Wlds protein was raised in rabbit by using a peptide containing 18 aa of linker region in Wlds protein (Wld-18, CDNIAVRGLHVGQHHQLLP) as antigen and affinity-purified. Specificity of the antiserum was confirmed by immunoblot analysis (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Animal surgery and analysis of injured nerve.
Surgical procedures to obtain mouse sciatic nerve samples were approved by the Committee for Animal Resources in National Center of Neurology and Psychiatry. Sciatic nerves of 8- to 11-week-old mice were transected and recovered as described previously (Mack et al., 2001). Integrity of the injured nerve was analyzed by immunoblot and immunohistochemistry to detect neurofilament and by electron microscopy as detailed below.
Histological analysis.
For immunohistochemistry, mice were perfused by 4% paraformaldehyde/PBS. Immunohistochemical analysis of mouse tissue was performed on 10 μm cryostat sections of tissues by using standard methods. Sections were incubated with primary antibodies against neurofilament (2H3; Developmental Studies Hybridoma Bank) or His tag (Novagen) at 4°C overnight and were subsequently incubated with biotinylated anti-mouse secondary antibody (Jackson ImmunoResearch) followed by Cy3-conjugated streptavidin (Jackson ImmunoResearch) for visualization under a fluorescent microscope.
For electron-microscopic examination, mice were perfused by 2% paraformaldehyde and 2.5% glutaraldehyde in PBS. The fixed sciatic nerves were embedded in 3% agarose in PBS and 70-μm-thick transverse sections of the location 2 mm distal to the transected site were prepared on the Microslicer (DTK-3000; Dosaka). The transverse sections were rinsed, osmicated, dehydrated, and embedded in epoxy resin. Then, 1-μm-thick semithin sections were cut and stained with toluidine blue for initial light-microscopic analysis. Subsequently, ultrathin sections were prepared, stained with lead citrate and uranyl acetate, and analyzed by a transmission electron microscope (Hitachi H-7000).
For counting protected axons in injured nerve, the numbers of morphologically intact axons (designated by preserved myelin sheath structures) in transected sciatic nerves of indicated genotypes (n = 7 for each genotype) were determined on semithin transverse sections at 7 d after injury. The entire axons in a 100 × 100 μm square field were counted under a light microscopy for each sample.
Assays of NMNAT enzymatic activities.
Whole cerebrum was lysed in a buffer containing 50 mm Tris-HCl at pH 7.4, 300 mm NaCl, 5 mm EDTA, and 1% Triton X-100 and protease inhibitor mixture EDTA-free (Roche). NMNAT activity was measured by SpectraMax M2 (Molecular Devices) at 37°C on 96-well plates, based on the procedure as previously described (Balducci et al., 1995). Briefly, a 100 μl mixture containing 28 mm HEPES at pH 7.4, 46 mm ethanol, 16 mm semicarbazide-HCl, 10 mm MgCl2, 2 mm ATP, 6 U of alcohol dehydrogenase (Sigma-Aldrich), and lysate supernatant (30 μg protein) was made. The reaction was started by adding 10 μl of 20 mm NMN. Optical density at 340 nm (ε340 of NADH: 3.0 mm/100 μl in 96-well plate, one-half area) was measured for 1 h. One unit of activity was defined as the amount of protein that catalyzes the synthesis of 1 mmol of NAD per hour.
Subcellular fractionation.
Fractionation of adult mouse cerebrum to obtain nucleus, mitochondria, lysosome/peroxysome, microsome, and cytosol fractions was performed by sequential centrifugation as described previously (Tateno et al., 2004) (also shown in Fig. 4A). Ten percent of each fraction was subjected to SDS-PAGE and immunoblotting with antibodies against His tag, Wld-18, Nucleoporin p62 (BD Biosciences), Prohibitin (NeoMarkers), Adaptin γ (BD Biosciences), PMP70 (Affinity Bioreagents), and superoxide dismutase 1 (SOD1) (Assay Designs).
Figure 4.

Mitochondrial localization is shared between NMNAT3 and Wlds protein. Subcellular localization of transgene-derived proteins in NMNAT1-Tg, NMNAT3-Tg, and Wlds protein in wlds mice was determined by sequential centrifugation as illustrated in A. B, NMNAT1 and NMNAT3 were mainly detected in P1 (nucleus) and P2 (mitochondria) in each of the transgenic mice. Wlds proteins in wlds mice showed wide distribution including mitochondria.
Isolation of mitochondria.
Mitochondria were isolated from cerebrum as described previously with minor modifications (Sims, 1990; Rajapakse et al., 2001) (also shown in supplemental Fig. S2, available at www.jneurosci.org as supplemental material). To compare mitochondrial quantity in wild-type, NMNAT3-Tg, and wlds mice by immunoblot analysis, the S1 fraction was analyzed to detect complex II 70 kDa Fp (Mitoscience) and complex III subunit core 2 (Mitoscience). To compare expression level of each respiratory enzyme complex by immunoblot analysis, crude mitochondrial fraction (P2) (shown in supplemental Fig. S2A, available at www.jneurosci.org as supplemental material) was analyzed to detect complex I subunit GRIM-19 (Mitoscience), complex II subunit 70 kDa Fp, complex III subunit core 2, complex IV subunit IV (COX IV) (Abcam), and complex V subunit β (Mitoscience). Layer 2 (purified synaptic mitochondrial fraction) (shown in supplemental Fig. S2B, available at www.jneurosci.org as supplemental material) obtained by the last centrifugation was subject to mitochondrial subfractionation and analysis for ATP synthetic activity.
Mitochondria subfractionation.
Purified synaptic mitochondria were subfractionated by using a phosphate swelling–shrinking method described previously (Bijur and Jope, 2003), with minor modifications to obtain fractions containing outer membrane, intermembrane space, inner membrane, and matrix. Separation of each fraction was confirmed by immunoblot analysis with antibodies against His tag, Wld-18, manganese superoxide dismutase (MnSOD) (Nventa Biopharmaceuticals), COX IV, cytochrome c (BD Biosciences), and voltage-dependent anion channel (VDAC) (Cell Signaling).
ATP-synthetic activity.
The citrate-sustained ATP-synthetic rate was assayed by a modified procedure as described previously (Baracca et al., 2000; Shaik et al., 2008). Purified mitochondria were suspended in a buffer containing 0.25 m sucrose, 50 mm HEPES, 0.5 mm EDTA, 2 mm MgSO4, 2 mm KH2PO4, and 10 mm citrate, pH 7.4. The reaction was initiated by adding ADP at 2 mm, maintained for 15 min at 30°C, and terminated by adding trichloroacetic acid. The quantity of ATP was analyzed by using HPLC (Shimadzu) on 125/4 mm Necleosil 4000-7 PEI anion-exchange column as described previously (Suter et al., 2006).
Results
Transgenic overexpression of NMNAT enzymatic activity in neurons is necessary, but not sufficient, for delayed axonal degeneration in vivo
We previously showed that the expression of Wlds protein or NMNAT1 is sufficient to protect axons in an in vitro Wallerian degeneration assay, suggesting that increased NAD production is crucial for axonal protection (Araki et al., 2004). However, Conforti et al. (2007) reported that NMNAT1 overexpression in vivo does not result in axonal protection. To clarify the requirement of NAD producing activity in Wlds- or NMNAT-mediated axonal protection, we generated mice that overexpress NMNAT at different levels.
Since NMNAT2 is constitutively expressed highly in neurons (Raffaelli et al., 2002), we decided to generate mice that overexpress mammalian NMNAT orthologs other than NMNAT2 (i.e., NMNAT1 and NMNAT3, either of which are unexpressed in neurons) (Emanuelli et al., 2001; Zhang et al., 2003). To determine the requirement of NMNAT activity in Wlds-mediated axonal protection, we also generated mice that express Wlds protein with a W258A mutation shown previously to disrupt NMNAT activity of the Wlds protein (Araki et al., 2004). To express molecules of interest at high levels, we used a standard CAG promoter construct to drive expression in vivo in this work (Fig. 1A). We generated the transgenic mice using a standard protocol and obtained 6, 12, and 3 lines for NMNAT1-Tg, NMNAT3-Tg, and Wlds(W258A)-Tg, respectively. Expression of the transgene-derived protein in brain tissue was determined by immunoblot analysis (Fig. 1B) and immunohistochemistry (Fig. 1C), as well as an NAD synthetic activity assay (Fig. 1D). We found that, among multiple mouse lines exhibiting differential expression levels for each molecule, some lines (line 744 of NMNAT1-Tg, lines 821 and 822 of NMNAT3-Tg) revealed NAD synthetic activity higher than that observed in wlds mice, whereas others were at similar levels. We observed that transgene-derived proteins were expressed in most of the cells of the nervous system in different mouse lines having different expression levels (Fig. 1C; supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Figure 1.
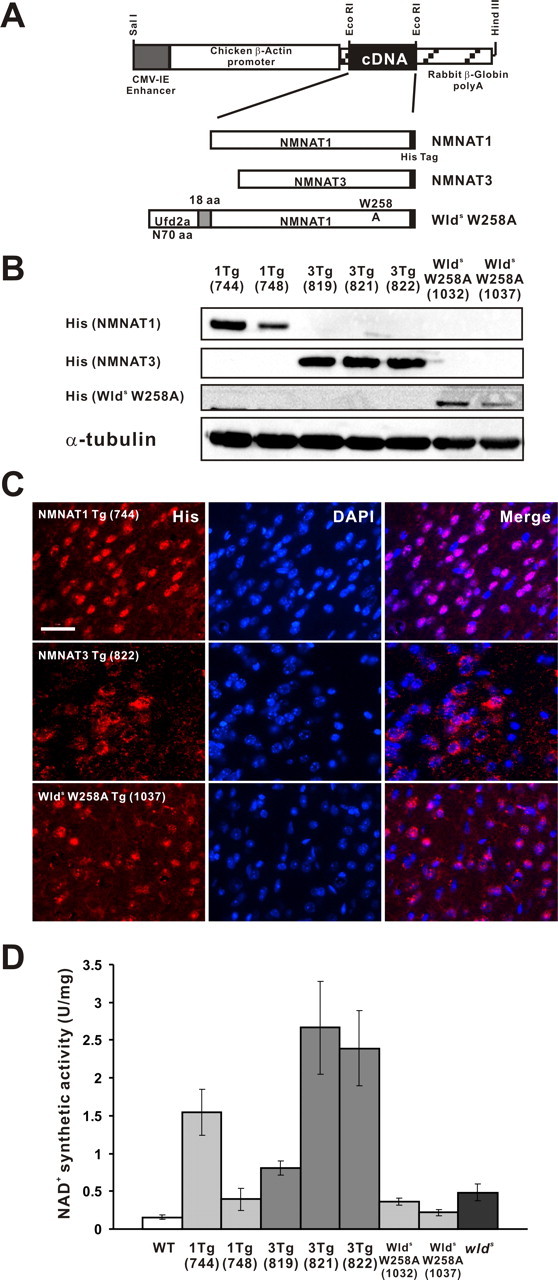
Generation of transgenic mice overexpressing NMNAT1, NMNAT3, and Wlds(W258A). The three transgenic mouse lines were generated using a vector containing CAG promoter. A schematic construction map is presented in A. Transgene-derived proteins were designed to be tagged with hexahistidine. Their expression was confirmed by immunoblot analysis (B) or by immunohistochemistry on cerebral cortex sections (C) using an antibody against His tag. α-Tubulin served as a loading control in B. Note that the majority of cells, including neurons, expressed transgene-derived proteins for all constructs. Scale bar, 50 μm. D, Differential expression of NAD-synthesizing activity observed in NMNAT1-Tg, NMNAT3-Tg, Wlds(W258A)-Tg, and wlds mice. NMNAT enzymatic activity was evaluated by the rate of NAD synthesis in transgenic mice cerebrum lysate. One unit of activity is defined as the amount of protein that catalyzes the synthesis of 1 mmol of NAD per hour. Error bars indicate SD.
Mice overexpressing NMNAT1, NMNAT3, or Wlds(W258A) were born at the expected Mendelian ratio, developed normally, and displayed no overt phenotype when compared with their wild-type littermates up to 1 year of age. Despite the overexpression of enzymatic activity that synthesizes a respiratory coenzyme, NAD, body weight of the mice overexpressing NMNAT1 or NMNAT3 was similar to that of wild-type littermates (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). To examine whether delayed Wallerian degeneration phenotype is observed in any of these different transgenic lines, we established Wallerian degeneration in sciatic nerve by transection and examined, by immunoblot and immunohistochemistry, the integrity of neurofilament in the segment distal to the transection site at different time points. We found that only NMNAT3-Tg mice had axonal protection phenotype similar to that observed in wlds mice among different mice that we generated. Immunoblot analysis showed that injured nerves in NMNAT3-Tg and wlds mice expressed similar levels of neurofilament immunoreactivity at 14 d after transection (Fig. 2A). We also analyzed integrity of the injured nerves extensively by morphological methods. Neurofilament immunoreactivity profile in injured sciatic nerves in NMNAT3-Tg mice (14 d after transection) was similar to that in injured wlds mice (Fig. 2B). Morphological examination of toluidine blue-stained semithin sections of injured sciatic nerves (7 d after injury) showed that axons were mostly preserved in injured nerves of NMNAT3-Tg and wlds mice, whereas they were mostly degraded in injured nerves in wild-type mice (Fig. 2C). In ultrastructural analysis by electron microscopy, mostly intact myelinated and unmyelinated nerves, characterized by organized microtubule structures and intact mitochondria in axoplasm as well as intact myelin sheath, were observed in injured nerves (7 d after injury) of NMNAT3-Tg and wlds mice, whereas degeneration was severe in injured nerves of wild-type mice, as axoplasm was lost from both myelinated and unmyelinated axons and destruction of myelin structures was prominent (Fig. 2D). The numbers of morphologically preserved axons at 7 d after injury indicated the similar degree of axonal protection in NMNAT3-Tg and wlds mice (Fig. 2E). Thus, NMNAT3-Tg mice show significant delay of axonal degeneration, which is very similar to the phenotype of wlds mice.
Figure 2.
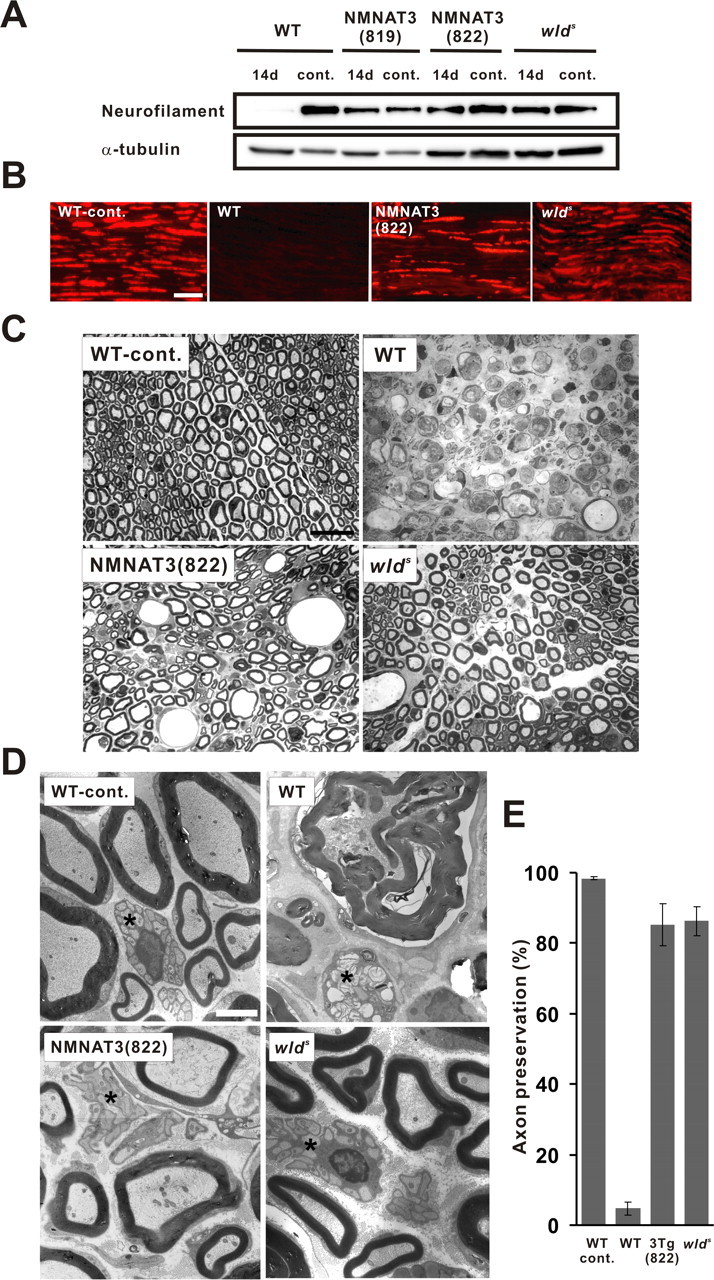
NMNAT3-Tg mice show delayed Wallerian degeneration comparable with wlds mice. Axonal degeneration was examined in a sciatic nerve injury model. Neurofilament immunoreactivity in the distal segment of the transected sciatic nerve was examined in NMNAT3-Tg (lines 819 and 822), wlds mice, as well as in wild-type control at 14 d after nerve lesion and compared with uninjured control by immunoblot analysis (A) and on longitudinal sections by immunohistochemistry 14 d after injury (B). α-Tubulin served as a loading control in A. Note that neurofilament expression remains close to wild-type level even at 14 d after injury. Scale bar, 50 μm. C, Injured nerve morphology in indicated mice was analyzed on toluidine blue-stained semithin sections (1 μm thick) 7 d after transection at 2 mm distal to the injury site. Injured nerves of NMNAT3-Tg and wlds mice exhibited apparently intact myelin structures almost comparable with uninjured control, whereas injured nerves in wild-type mice lacked intact myelin sheath and showed severe degeneration. Scale bar, 20 μm. D, Electron-microscopic images of the transverse sections of injured sciatic nerves at 2 mm distal to the transection site at 7 d after injury. Organized axonal structures and intact myelin sheath were observed in myelinated and unmyelinated (asterisk) axons of injured nerves in NMNAT3-Tg and wlds mice, whereas severe degeneration of both myelinated and unmyelinated (asterisk) axons and destruction of myelin structures was observed in injured nerves of wild-type mice. Scale bar, 2 μm. E, Morphologically preserved axon numbers in transverse sections of the injured sciatic nerve (7 d after transection) were counted for NMNAT3-Tg (line 822), wlds, and wild type mice (n = 7 for each genotype) and shown as percentage to the total number of axons. Error bars indicate SD.
In contrast to the axonal protection phenotype of NMNAT3-Tg, we did not see any delay of Wallerian degeneration in either NMNAT1-Tg or Wlds(W258A)-Tg mice. We first examined transected nerve 7 d after injury in the same way as that for NMNAT3-Tg, but we did not see any remaining neurofilament immunoreactivity (data not shown). In this sciatic nerve transection model, neurofilament immunoreactivity became undetectable by immunoblot analysis at 3 d after transection in wild-type mice (Fig. 3A). By immunohistochemistry, scattering neurofilament-immunoreactive structures can still be observed after 3 d, but become completely undetectable at 4 d after transection (Fig. 3B). Therefore, to examine whether subtle axonal protection occurs in these transgenic mice, we examined transected nerves at 3 and 4 d after injury. The neurofilament expression profile (by immunoblot analysis and immunohistochemistry) in injured nerves of NMNAT1-Tg and Wlds(W258A)-Tg mice is mostly the same as that of wild-type mice (Fig. 3A,B). It is noteworthy that NAD-producing activity in brain tissue of line 744 of NMNAT1-Tg, which showed no axonal protection, was higher than that naturally occurring in wlds mice or line 819 of NMNAT3-Tg mice, both of which displayed significant delays in Wallerian degeneration. The complete loss of axonal protection phenotype in Wlds(W258A)-Tg indicates that NAD-producing activity was indispensable for Wlds-mediated axonal protection, but the lack of axonal protection in NMNAT1-Tg mice suggests that the increased NMNAT enzymatic activity in neuronal cells was not sufficient for axonal protection phenotype in vivo.
Figure 3.

Wallerian degeneration occurs normally in NMNAT1-Tg or Wlds(W258A)-Tg. Axonal degeneration was examined in a sciatic nerve injury model. Neurofilament immunoreactivity in the distal segment of the transected sciatic nerve was examined in NMNAT1-Tg (lines 744 and 748), wlds mice, Wlds(W258A)-Tg (lines 1032 and 1037), as well as in wild-type control, at 3 and 4 d after nerve lesion and compared with uninjured control by immunoblot analysis (A), and on longitudinal sections by immunohistochemistry (B). α-Tubulin served as a loading control in A. Scale bar, 50 μm. Note that even subtle protection of axons was not observed in NMNAT1-Tg or Wlds(W258A)-Tg.
Mitochondrial localization of NMNAT is correlated with axonal protection phenotype
Differential axonal protection phenotype observed in NMNAT1-Tg and NMNAT3-Tg mice suggested that some factor(s) other than neuronal NMNAT activity determines whether axonal protection in vivo is enabled. The major difference between NMNAT1 and NMNAT3 is their subcellular localization; NMNAT1 is located in nucleus, whereas NMNAT3 is mainly in mitochondria (Berger et al., 2005). Also, subcellular localization of Wlds protein is not the same as that of NMNAT1 presumably because of its N-terminal sequence (Laser et al., 2006). To examine whether specific subcellular localization of NMNAT correlates with the axonal protection phenotype, we determined detailed localization of transgene-derived proteins in NMNAT1-Tg and NMNAT3-Tg mice and compared that with the localization of Wlds proteins. We performed a standard subcellular fractionation analysis of brain tissue homogenate by sequential centrifugation (Fig. 4A). Separation of fractions enriched for nucleus, mitochondria, lysosome/peroxysome, microsome (containing endoplasmic reticulum, Golgi), and cytosol was confirmed by identification of nucleoporin p62, prohibitin, PMP70, adaptin γ, and SOD1, respectively. We located NMNAT1 in the nucleus of NMNAT1-Tg mice, and NMNAT3 in mitochondria of NMNAT3-Tg mice (Fig. 4B). Previous reports suggest that NMNAT3 is located mainly, but not exclusively, in mitochondria when it is transiently overexpressed in cultured cells (Zhang et al., 2003; Berger et al., 2005); however, we found that NMNAT3 was located almost exclusively in mitochondria of NMNAT3-Tg mice. Contrary to the previous immunohistochemical analysis, we found that various levels of Wlds protein were located not only in nucleus but also in mitochondria and most other fractions as well (Fig. 4B). The subcellular distribution profile of Wlds(W258A) was mostly similar to that of Wlds protein (data not shown). Comparison of NMNAT1, NMNAT3, and Wlds protein localization showed that the mitochondrial fraction was the only one that contained NMNAT3 and Wlds but lacked NMNAT1. These results suggest that, contrary to our previous expectations, NMNAT activity may need to be located in mitochondria to exert delayed axonal degeneration phenotype in vivo.
NMNAT3 and Wlds protein are transported to mitochondrial matrix
Previous reports have shown that NMNAT3 is located mainly in mitochondria, but it was not identified where in the mitochondria NMNAT3 is located. With regard to Wlds protein, it has been regarded as a nuclear molecule, but if it functions in mitochondria as we showed here, it should be transported across the outer membrane into the mitochondria. To gain more insights on the functions of NMNAT3 and Wlds proteins in mitochondria, we performed mitochondrial subfractionation to locate NMNAT3 and Wlds protein in mitochondrial subcompartments. Synaptic mitochondria were fractionated and purified from brain tissue of NMNAT3-Tg and wlds mice [the purification procedure is summarized in supplemental Fig. S2 (available at www.jneurosci.org as supplemental material)] and subfractionated. Both NMNAT3 and Wlds protein were detected mainly in mitochondrial matrix and inner membrane fractions, but not in outer membrane and intermembrane space fractions (Fig. 5), indicating that NMNAT3 and Wlds protein were both transported to the inside of the mitochondria. These data support our finding that Wlds protein is located in mitochondria, and suggest that NMNAT3 and Wlds protein may affect mitochondrial function in neurons via their enzymatic activity.
Figure 5.

NMNAT3 and Wlds protein are located in mitochondrial matrix. Mitochondria fractionated and purified from cerebrum of NMNAT3-Tg or wlds mice were subject to subfractionation to obtain fractions for outer membrane (OM), intermembrane space (IMS), inner membrane (IM), and matrix (Mx). Separation of each fraction was confirmed by immunoblot analysis to detect VDAC (OM), cytochrome c (IMS), COX IV (IM), and MnSOD (Mx). NMNAT3 or Wlds protein was detected in IM and Mx fractions by anti-His tag or anti-Wld18 antibody, respectively. Wlds protein-specific band is indicated by an arrow.
Mitochondria of NMNAT3-Tg and wlds mice show increased ATP-synthesizing activity
By analyzing delayed Wallerian degeneration phenotype in NMNAT1-Tg, NMNAT3-Tg, and wlds mice, we observed a correlation between the axonal protection phenotype and localization of NMNAT activity in the mitochondrial matrix. To determine how NMNAT activity may affect mitochondrial functions, we examined whether an overall change occurred in the quantity of mitochondria in brain tissues of NMNAT3-Tg and wlds mice compared with wild-type control. For this purpose, we explored the expression of complex II 70 kDa Fp and complex III core 2 proteins in brain tissue homogenate. The expression levels of both proteins in NMNAT3-Tg and wlds mice were similar to those in control mice (Fig. 6A). Furthermore, mitochondrial DNA content relative to nuclear DNA in NMNAT3-Tg and wlds mice was unchanged from that in wild-type mice, indicating that mitochondrial DNA number in NMNAT3-Tg and wlds mice is similar to that in wild-type mice (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). These results suggest that the number of mitochondria in NMNAT3-Tg and wlds mice was comparable with wild type. We also observed that mitochondrial morphology and subcellular distribution in primary dorsal root ganglia neurons of NMNAT3-Tg and wlds mice were comparable with those of wild-type control (data not shown). These results suggest that NMNAT expression in mitochondria did not affect the division/fusion or transport of mitochondria.
Figure 6.

Mitochondrial expression of NMNAT increased ATP synthesis capability without affecting expression of electron transfer chain enzyme expression. Expression of complex II 70 kDa Fp and complex III core 2 proteins was examined in crude lysate of cerebrum from NMNAT3-Tg, wlds, as well as wild-type mice (A). Neurofilament served as a loading control. Expression of representative components of electron transfer complex (complex I subunit GRIM-19, complex II subunit 70 kDa Fp, complex III subunit core 2, COX IV, and complex V subunit β) was examined in mitochondrial pellets (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material) (P2 fraction) obtained from NMNAT3-Tg (line 822), wlds mice, as well as from wild-type mice, by immunoblot analysis (B). Citrate-sustained ATP-synthetic activity was examined by using purified mitochondria obtained from NMNAT3-Tg (line 822), wlds mice, as well as from wild-type mice. ATP-synthetic activity (nanomoles of ATP per minute per microgram of protein) was calculated; mean ± SD is shown for indicated number of animals for each condition (*p < 0.05; **p < 0.01, one-way ANOVA followed by Tukey's test) (C).
NAD is one of the coenzymes for mitochondrial electron transfer system. Therefore, the expression of NMNAT in the mitochondrial matrix would likely affect cellular ATP metabolism. To know whether mitochondrial respiration is modified by the expression of NMNAT within the matrix, we examined the ATP synthesis rate of synaptic mitochondria fractionated and purified from brain tissue of NMNAT3-Tg, wlds, and wild-type mice. Citrate was used as a substrate for respiration, and synthesized ATP was measured by HPLC. The ATP synthesis rate increased significantly in mitochondria derived from NMNAT3-Tg and wlds mice compared with mitochondria from wild-type control (Fig. 6C). Thus, mitochondria expressing NMNAT activity were capable of increased ATP synthesis. As stated above, enzymatic function of NMNAT that modifies metabolism of NAD and ATP could well affect mitochondrial ATP synthesis. However, NMNAT activity in mitochondria may indirectly affect expression of mitochondrial proteins involved in the electron transfer chain, and thereby change the ATP synthesis. To exclude this possibility, we compared by immunoblot analysis the expression of representative proteins of each respiratory chain complex. The expression level of representative proteins in complex I through V was unchanged in mitochondria (Fig. 6B). Furthermore, the enzymatic activity of each mitochondrial respiratory complex in NMNAT3-Tg and wlds mice was comparable with wild-type control (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). Together, our results suggest that NMNAT expression in the mitochondrial matrix increased ATP synthesis rate by a mechanism not directly related to electron transfer chain and thereby has an important role in formation of delayed Wallerian degeneration phenotype.
Discussion
We previously identified that overexpression of NMNAT gave the most robust axonal protection among all other enzymes in the NAD-synthesizing pathway (Sasaki et al., 2006). However, Coleman et al. showed that NMNAT1-overexpressing mice did not show an axonal protection phenotype and speculated that axonal protection by NMNAT overexpression may only be observed in in vitro settings (Conforti et al., 2007). Here, we showed for the first time that overexpression of a native NMNAT enzyme can save axons in vivo and that Wlds protein lacking NMNAT activity cannot save axons. Together, these results suggest that increased NMNAT activity does lead to axonal protection phenotype in vivo.
In this report, we identified that NMNAT3-Tg mice showed robust axonal protection similar to wlds mice, whereas NMNAT1-Tg did not, even when the NAD-synthesizing activity by NMNAT1 overexpression reached a level higher than that in NMNAT3-Tg mice with the axonal protection phenotype. These results suggest that the increased NMNAT activity is necessary, but not sufficient, to see axonal protection phenotype in vivo. Based on the previously reported molecular characteristics of NMNAT1, NMNAT3, and Wlds proteins (Berger et al., 2005; Laser et al., 2006), we hypothesized that their subcellular localization is an important determinant of the axonal protection phenotype. We showed that NMNAT activity was found in the mitochondrial matrix of mice showing Wallerian degeneration delay and that NMNAT in mitochondria increased ATP synthesis capability. These results suggest that NMNAT activity is transported into the mitochondrial matrix of mice showing the axonal protection phenotype, and the subsequent increased ATP-synthesizing capability may play a role in axonal protection.
Roles of NMNAT, NAD, and Sirtuins for axonal protection
We previously screened NAD synthesis pathway enzymes and their substrates/products using the in vitro Wallerian degeneration model (Sasaki et al., 2006). This work not only demonstrated that the most effective method for axonal protection is overexpression of NMNAT, but it also made us realize the uniqueness of NMNAT (i.e., the axonal protection phenotype caused by NMNAT overexpression does not seem to reflect the function of NMNAT as an NAD synthesis enzyme). Overexpression of each NAD-synthesizing enzyme in the presence of its substrate caused axonal protection similar to exogenous application of NAD. NMNAT overexpression, however, gave much more robust protection to axons than NAD application, regardless of the presence of the substrate. This observation made us recognize another previously observed discrepancy. We showed that exogenously applied NAD results in activation of Sirt1, an NAD-dependent protein deacetylase, and discussed that this is the key mechanism that connects increased NMNAT activity and axonal protection (Araki et al., 2004). However, it was not clear how NMNAT overexpression affects Sirt1 activity, because, whereas Sirt1 activity is strictly dependent on intracellular NAD levels (Yamamoto et al., 2007), overexpression of NMNAT activity does not cause major changes in NAD levels (Mack et al., 2001). Researchers, including ourselves, discussed that this apparent contradiction could be explained by rapid degradation of NAD by nuclear enzymes such as Sirtuins or poly(ADP-ribose) polymerases (Araki et al., 2004; Kim et al., 2005). However, our subsequent studies suggested that this speculation may not be correct. NMNAT activity that is located outside of the nucleus protected axons the same way as nuclear NMNAT (Sasaki et al., 2006). In the current report, we confirmed that nuclear NMNAT1 cannot protect axons in vivo, suggesting that nuclear localization of NMNAT is not related to axonal protection. Together with the unique feature of NMNAT overexpression-derived axonal protection, this result made us realize a possibility that Sirt1 activation and resultant transcriptional changes are necessary for axonal protection by NAD, but that NMNAT overexpression may be able to protect axons independent from cellular NAD levels or Sirtuin activation.
Because overexpression of NMNAT3 but not NMNAT1 protects axons in vivo, we hypothesized that subcellular localization of overexpressed NMNAT activity has a significant impact on the axonal protection phenotype. Localization of NMNAT3 and Wlds proteins in the mitochondrial matrix indicates that they are actively transported into the mitochondria and suggests that mitochondrial localization of NMNAT activity may be important for the axonal protection phenotype. Although the Wlds protein is primarily a nuclear molecule determined by the nuclear localization signal in NMNAT1, the N-terminal sequence derived from Ufd2a presumably affects the localization so that Wlds can be located outside of the nucleus. The Wlds protein is likely subjected to the intramitochondrial transport mechanism the same way as NMNAT3 so long as the proximity to mitochondria is close, because the three-dimensional structure of NMNAT1 is remarkably similar to that of NMNAT3 (Zhang et al., 2003). We showed in a previous report that lentiviral NMNAT1 overexpression in primary cultured neurons results in the axonal protection phenotype in the Wallerian degeneration model in vitro (Araki et al., 2004; Sasaki et al., 2006). Because NMNAT1 overexpression was driven so strongly in this in vitro experiment, we suspect that NMNAT1 was located not only in the nucleus but also in the cytoplasm (Fig. 7). As a result, the cytoplasmic NMNAT1 was transported into the mitochondria the same way as the Wlds protein. Together, we currently speculate that the robust axonal protection by NMNAT overexpression is derived from the mitochondrial localization of NMNAT activity and resultant modification of mitochondrial function.
Figure 7.

Schematic illustration of subcellular localization of overexpressed NMNAT1, NMNAT3, and Wlds protein in neurons. Although NMNAT1 is a nuclear protein (A), it can also be located outside of the nucleus and close to mitochondria when highly overexpressed (B). Wlds protein is also located outside of the nucleus presumably because of the N-terminal 70 aa sequence (C). NMNAT1 or Wlds protein, which comes close to mitochondria, can be transported into mitochondrial matrix and causes phenotype inseparable from that observed in D.
NMNAT is reported to show “chaperone-like” activity and thereby protect neurons against degeneration. This chaperone-like activity is independent from NAD-synthesizing activity of NMNAT (Zhai et al., 2008). In our current analysis, we found that the axonal protection activity was not observed in Wlds(W258A)-Tg mice. Our results, together with the previous observations (Araki et al., 2004), suggest that axonal protection activity of NMNAT and Wlds proteins is inseparable from NAD-synthesizing activity and therefore is not related to the chaperone-like activity of NMNAT.
Role of NMNAT in mitochondria
Previous reports on axonal protection observed in wlds mice suggested possible significance of mitochondrial function (Ikegami and Koike, 2003; Wishart et al., 2007; Press and Milbrandt, 2008). However, how mitochondrial functions are modified in neurons with protected axons has never been analyzed. We determined that isolated mitochondria from NMNAT3-Tg and wlds mice show increased ATP-synthesizing potential. This result confirms that NMNAT activity in these mice is incorporated inside of the mitochondria, and is functional, and suggests that this increased ATP-synthesizing potential may be important for the axonal protection phenotype in these mice. We showed that the respiratory chain expression level and activity in NMNAT3-Tg or wlds mice are similar to those in wild type. These results suggest that NMNAT in the mitochondrial matrix is either involved in ATP synthesis or modifies enzymatic activity to increase ATP synthesis or decrease ATP degradation. Among these possibilities, one likely mechanism is direct involvement of NMNAT in ATP generation. Normally, NMNAT enzymes synthesize NAD from NMN and ATP, but all NMNAT family members can also catalyze the inverse reaction in which NAD serves as a substrate to generate ATP (Berger et al., 2005). This inverse NMNAT reaction in the mitochondrial matrix generates extra ATP and thereby increases ATP synthesis efficiency. Therefore, lower glucose is required and less reactive oxygen species is generated to produce the same amount of ATP in neurons. Recent proteomic analysis indicates that a significant amount of mitochondrial proteins are involved in functions other than ATP metabolism (Fukada et al., 2004). Such proteins include components of urea cycles, enzymes that regulate intracellular calcium concentration, and metabolism of ketone bodies. These functions are correlated with cellular energy metabolism and redox status, and thus it is possible that overall ATP production increases via modification of some of these activities in NMNAT-expressing mitochondria.
The relationship between mitochondrial functions and cellular protection is complex. Increased respiration by calorie restriction increases longevity in yeast and Caenorhabditis elegans, but increased respiration in mammals may simply result in acceleration of the aging process. Successful neuronal protection by altering mitochondrial functions would be a novel treatment for neuronal disorders. Additional investigation will be necessary to pursue this possibility.
Footnotes
This work was supported by grants from Ministry of Health, Labour, and Welfare (Research on Psychiatric and Neurological Diseases and Mental Health), National Institute of Biomedical Innovation (Program for Promotion of Fundamental Studies in Health Sciences), Kakenhi (19650089), Terumo Life Science Foundation, Suzuken Memorial Foundation, Kowa Life Science Foundation, Sankyo Foundation for Life Science, Astellas Foundation for Research on Metabolic Disorders, Mishima Kaiun Memorial Foundation, and Takeda Science Foundation (T.A.). pCAGGS is a gift from Dr. J. Miyazaki (Osaka University, Osaka, Japan).
References
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Balducci E, Emanuelli M, Raffaelli N, Ruggieri S, Amici A, Magni G, Orsomando G, Polzonetti V, Natalini P. Assay methods for nicotinamide mononucleotide adenylyltransferase of wide applicability. Anal Biochem. 1995;228:64–68. doi: 10.1006/abio.1995.1315. [DOI] [PubMed] [Google Scholar]
- Baracca A, Barogi S, Carelli V, Lenaz G, Solaini G. Catalytic activities of mitochondrial ATP synthase in patients with mitochondrial DNA T8993G mutation in the ATPase 6 gene encoding subunit a. J Biol Chem. 2000;275:4177–4182. doi: 10.1074/jbc.275.6.4177. [DOI] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J Neurochem. 2003;87:1427–1435. doi: 10.1046/j.1471-4159.2003.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc Natl Acad Sci U S A. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, Magni G, Glass JD, Coleman MP. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ. 2007;14:116–127. doi: 10.1038/sj.cdd.4401944. [DOI] [PubMed] [Google Scholar]
- Conner DA. Current protocols in molecular biology. New York: Wiley; 2004. Transgenic mouse production by zygote injection; pp. 23.9.1–23.9.29. [DOI] [PubMed] [Google Scholar]
- Emanuelli M, Carnevali F, Saccucci F, Pierella F, Amici A, Raffaelli N, Magni G. Molecular cloning, chromosomal localization, tissue mRNA levels, bacterial expression, and enzymatic properties of human NMN adenylyltransferase. J Biol Chem. 2001;276:406–412. doi: 10.1074/jbc.M008700200. [DOI] [PubMed] [Google Scholar]
- Fukada K, Zhang F, Vien A, Cashman NR, Zhu H. Mitochondrial proteomic analysis of a cell line model of familial amyotrophic lateral sclerosis. Mol Cell Proteomics. 2004;3:1211–1223. doi: 10.1074/mcp.M400094-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami K, Koike T. Non-apoptotic neurite degeneration in apoptotic neuronal death: pivotal role of mitochondrial function in neurites. Neuroscience. 2003;122:617–626. doi: 10.1016/j.neuroscience.2003.08.057. [DOI] [PubMed] [Google Scholar]
- Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: “PAR-laying” NAD+ into a nuclear signal. Genes Dev. 2005;19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- Laser H, Conforti L, Morreale G, Mack TG, Heyer M, Haley JE, Wishart TM, Beirowski B, Walker SA, Haase G, Celik A, Adalbert R, Wagner D, Grumme D, Ribchester RR, Plomann M, Coleman MP. The slow Wallerian degeneration protein, WldS, binds directly to VCP/p97 and partially redistributes it within the nucleus. Mol Biol Cell. 2006;17:1075–1084. doi: 10.1091/mbc.E05-04-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Press C, Milbrandt J. Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J Neurosci. 2008;28:4861–4871. doi: 10.1523/JNEUROSCI.0525-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Raffaelli N, Sorci L, Amici A, Emanuelli M, Mazzola F, Magni G. Identification of a novel human nicotinamide mononucleotide adenylyltransferase. Biochem Biophys Res Commun. 2002;297:835–840. doi: 10.1016/s0006-291x(02)02285-4. [DOI] [PubMed] [Google Scholar]
- Rajapakse N, Shimizu K, Payne M, Busija D. Isolation and characterization of intact mitochondria from neonatal rat brain. Brain Res Brain Res Protoc. 2001;8:176–183. doi: 10.1016/s1385-299x(01)00108-8. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaik ZP, Fifer EK, Nowak G. Akt activation improves oxidative phosphorylation in renal proximal tubular cells following nephrotoxicant injury. Am J Physiol Renal Physiol. 2008;294:F423–F432. doi: 10.1152/ajprenal.00463.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NR. Rapid isolation of metabolically active mitochondria from rat brain and subregions using Percoll density gradient centrifugation. J Neurochem. 1990;55:698–707. doi: 10.1111/j.1471-4159.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem. 2006;281:32207–32216. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- Tateno M, Sadakata H, Tanaka M, Itohara S, Shin RM, Miura M, Masuda M, Aosaki T, Urushitani M, Misawa H, Takahashi R. Calcium-permeable AMPA receptors promote misfolding of mutant SOD1 protein and development of amyotrophic lateral sclerosis in a transgenic mouse model. Hum Mol Genet. 2004;13:2183–2196. doi: 10.1093/hmg/ddh246. [DOI] [PubMed] [Google Scholar]
- Wishart TM, Paterson JM, Short DM, Meredith S, Robertson KA, Sutherland C, Cousin MA, Dutia MB, Gillingwater TH. Differential proteomics analysis of synaptic proteins identifies potential cellular targets and protein mediators of synaptic neuroprotection conferred by the slow Wallerian degeneration (Wlds) gene. Mol Cell Proteomics. 2007;6:1318–1330. doi: 10.1074/mcp.M600457-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Schoonjans K, Auwerx J. Sirtuin functions in health and disease. Mol Endocrinol. 2007;21:1745–1755. doi: 10.1210/me.2007-0079. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, Bellen HJ. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–891. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Kurnasov OV, Karthikeyan S, Grishin NV, Osterman AL, Zhang H. Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J Biol Chem. 2003;278:13503–13511. doi: 10.1074/jbc.M300073200. [DOI] [PubMed] [Google Scholar]