

Abstract
Lipid rafts (LRs) are membrane realms characterized by high concentrations of cholesterol and sphingolipids. Often, they are portrayed as scaffolds on which many different signaling molecules can assemble their cascades. The idea of rafts as scaffolds is garnering significant attention as the consequences of LR disruption have been shown to be manifest in multiple signaling pathways. In this study, LRs in the brain of the twitcher (TWI) mouse, a bona-fide model for infant variants of human globoid cell leukodystrophy or Krabbe disease, were investigated. This mouse has deficient activity of GALC (β-galactosylceramidase) that leads to a progressive accumulation of some galactosyl-sphingolipids in the brain. We hypothesized that the accumulation of psychosine (galactosyl-sphingosine) in the TWI CNS may result in the disruption of rafts in different cell populations such as neurons and oligodendrocytes, both cellular targets during disease. In this communication, we demonstrate that psychosine specifically accumulates in LRs in the TWI brain and sciatic nerve and in samples from brains of human Krabbe patients. It is also shown that this accumulation is accompanied by an increase in cholesterol in these domains and changes in the distribution of the LR markers flotillin-2 and caveolin-1. Finally, we show evidence that this phenomenon may provide a mechanism by which psychosine can exert its known inhibitory effect on protein kinase C. This study provides a previously undescribed biophysical aspect for the mechanism of pathogenesis in Krabbe disease.
Introduction
Globoid cell leukodystrophy or Krabbe disease (KD) is a sphingolipidosis that is caused by the genetic deficiency of the enzyme β-galactosylceramidase (GALC). The loss of GALC results in the progressive accumulation of the sphingolipid metabolite galactosylsphingosine (psychosine), demyelination, and early death (Wenger et al., 2001; Suzuki, 2003). Since it is known that psychosine is a highly toxic compound, it has long been assumed that its accumulation is the major driving force behind the progression of KD (Suzuki, 1998). It is of note that, with the exception of few reports, particularly those from Giri et al. (2006) showing psychosine-mediated activation of phospholipase A2 and from Haq et al. (2003) describing psychosine-based upregulation of AP-1 in oligodendrocytes, very little is understood about the molecular mechanism by which psychosine imparts toxicity. As a result of this lack of knowledge, the mechanism by which KD might progress has largely remained undescribed.
Lipid rafts (LRs) are defined as unique regions of the cell membrane that have a characteristically high concentration of cholesterol and sphingolipids (Pike, 2006). These membrane microdomains, as they are often called, are considered to be extremely important facilitators, if not participants, in processes involving cellular signaling. One important example of this phenomenon is the segregation of γ-secretase in different membrane domains in developing mice. It is thought that the association of γ-secretase with lipid rafts is important for the regulation of its proteolytic targeting (Vetrivel et al., 2005). Another example is the apparent targeting of some protein kinase C isozymes to specific membrane regions after activation (Rybin et al., 1999). This phenomenon is thought to facilitate interactions between protein kinase C (PKC) and its downstream targets during important signaling processes. Lipid raft disruption is also suggested as a mechanism important in the progression of several diseases (Simons and Ehehalt, 2002). Included in this list of diseases are several neurodegenerative diseases such as Alzheimer, Niemann-Pick, and metachromatic leukodystrophy (MLD). Interestingly, MLD is a disease caused by the loss of the enzyme arylsulfatase A and the resulting accumulation of sulfatides in the brain (Gieselmann, 2003). Since its etiology is similar to that of KD, it is reasonable to assume that raft disruption may be a mechanism that is also at play in the progression of KD. In this study, the possibility that psychosine accumulation may lead to disruption of lipid rafts and associated signals is considered as one possible mechanism for the progression of Krabbe disease.
Materials and Methods
Samples.
For the twitcher colony, breeder twitcher heterozygous mice (C57BL/6J, twi/+; Jackson Laboratories) were maintained under standard housing conditions with approval of the Animal Care and Use Committee. Mice were allowed to survive as long as humanely possible. When a mouse reached a moribund condition, it was killed. DNA was extracted from clipped tails of newborn mice at postnatal day 1 (P1) to P2 and used for PCR genotyping as described previously (Sakai et al., 1996; Dolcetta et al., 2006). Postmortem samples from brain cortex from a 3-year-old Krabbe patient with the classic infantile variant and from an age-matching control were generously provided by Dr. Bizzozero (University of New Mexico, Albuquerque, NM). Collection time was within 4–6 h after death.
Cell culture.
HeLa cells were obtained from ATCC. Cells were grown in standard conditions with DMEM-F12 medium containing 10% fetal bovine serum (FBS). Before exposure to sphingolipids, cells were briefly trypsinized, centrifuged, and counted. Cells were plated on 10 cm culture dishes (2 × 104 cells per cm2) in serum-free medium and were incubated overnight before addition of 10 μm (final concentration) of either psychosine, d-sphingosine, or vehicle. Cells were incubated for 24 h before being exposed to 20 ng/ml of platelet-derived growth factor (PDGF)-BB for 15 min. After growth factor incubation, cells were scraped (not trypsinized), collected, washed in PBS three times, and processed for raft preparation.
Lipid raft preparation.
Lipid raft preparation was performed as described by Vetrivel et al. (2004). Tissue was dissected and homogenized 15× with a glass-Teflon homogenizer in 2 ml of buffer A (20 mm Tris-HCl pH 7.4, 50 mm NaCl, 250 mm sucrose, 1 mm DTT, mammalian protease inhibitor cocktail, 1 mm PMSF, 1 mm okadaic acid, 2 mm sodium orthovanadate). One ml of homogenate was saved for total brain assays; the other was further processed for LR preparation. The experimental sample was then passed through a 25 gauge needle 5× and centrifuged at 960 g for 10 min at 4°C. The supernatant was then collected and placed in a 15 ml conical tube. The pellet was resuspended in 0.5 ml of homogenization buffer A and again passed through a 25 gauge needle 5× and centrifuged at 960 g for 10 min at 4°C. Both supernatants were pooled and supplemented with 10 μl of 1 m Tris HCl pH 7.4, 200 μl of 1 m NaCl, 20 μl of 0.5 m EDTA, 200 μl of 5% Lubrol, and 200 μl of buffer A. The resulting mixture of ∼2 ml was nutated for 30 min at 4°C. Once completed, the sample was brought to a concentration of 45% sucrose and a volume of 4 ml. Using a Labconco auto densi-flow density gradient fractionator, 4 ml of 35% sucrose and 5% sucrose were loaded on top of the homogenate. Tubes were then transferred to a Beckman-Coulter SW-41 rotor and subjected to ultracentrifugation at 39,000 rpm for 19–22 h at 4°C. After ultracentrifugation, the gradient fractionator was used to collect gradients in 1 ml aliquots. The resultant preparations contained raft markers in fractions 4–5. Raft preparation using the detergent-free optiprep method was performed essentially as described previously (Macdonald and Pike, 2005).
Rafts from cells were prepared by collecting cells in 2 ml of lysis buffer (25 mm Tris-HCl pH 7.4, 150 mm NaCl, 5 mm EDTA, 0.5% Lubrol, mammalian protease inhibitor cocktail, 1 mm PMSF, 1 mm okadaic acid, and 2 mm sodium orthovanadate), and passaging them through a 25 gauge needle 5×. An equal volume of 90% sucrose was added to the lysates to bring them to a concentration of 45% sucrose. Gradients were then poured on to the lysates and ultracentrifuged as above.
Mass spectrometry.
Tissue and lipid raft samples were extracted in chloroform/methanol/water and analyzed using liquid chromatography-tandem mass spectrometry, as described previously (Galbiati et al., 2007). Positive ion electrospray precursor ion scanning was performed using an Applied Biosystems API 4000 triple quadrupole mass spectrometer equipped with a Shimadzu HPLC system and Leap autosampler (Whitfield et al., 2001).
Cholesterol assay.
Cholesterol concentrations were measured in 96-well plates using the fluorometric Amplex Red cholesterol assay kit (Invitrogen). Assay plates were quantified for relative fluorescence using the Beckman Coulter DTX 880 multimode detector.
Western blot.
Proteins were extracted from aliquots of total homogenates used for LR preparation. For LR analysis, LR fractions 4–12 were solubilized (fractions 1–3 are devoid of proteins) in a final concentration of 0.25% SDS. Loading volumes were normalized to the protein content of pooled aliquots of fractions 8–12 from the samples being compared. Gel electrophoresis was performed using the XCell Sure-lock vertical electrophoresis system (Invitrogen) and 4–12% 10-well 1.5 mm precast Nupage gels (Invitrogen). After transferring to PVDF membrane, blots were blocked with milk/BSA solution and then treated with primary antibodies. Antibodies used in this study were the following: anti-flotillin-2, P115 (Beckton Dickson), caveolin-1 (Cell Signaling), PKC (Santa Cruz), phospho-PKC (Cell Signaling), and actin (Millipore Bioscience Research Reagents). Antibody-reactive products were detected using peroxidase-labeled secondary antibodies and ECL chemiluminescent substrate (Pierce). Wild-type and twitcher lipid raft blots were exposed for the same amount of time on the same film to allow for comparison of protein abundance in raft fractions.
Immunocytochemistry.
Immunocytochemical methods are described in detail previously (Givogri et al., 2006, 2008). Briefly, cells were treated experimentally and then fixed using 4% paraformaldehyde. Fixed cells were then incubated with primary antibodies for phosphorylated PKC and/or cholera toxin B for the labeling of GM1 gangliosides. After labeling with secondary antibodies, cells were imaged using a Zeiss LSM 510 meta confocal microscope.
Statistical analysis.
Results are the average from two to four different experiments and are expressed as mean ± SE. Data were analyzed by the Student's t test. p values <0.05 were considered statistically significant.
Results
Psychosine preferentially accumulates in lipid rafts in the twitcher nervous system
The twitcher (TWI) mouse is a natural model for KD. The progression of disease in this animal is severe, with disease-related changes becoming externally noticeable around postnatal day 20 (P20). Despite a well characterized demyelinating phenotype, little is understood regarding the molecular progression of KD. The most obvious candidate as a molecular catalyst for disease progression is psychosine, a sphingolipid that accumulates as patients develop the disease. Figure 1 A depicts the total concentration of psychosine, as determined using mass spectrometry, that was found in wild-type (WT) and TWI brains at P3, P20, and P40. This figure shows significantly higher levels of psychosine in the TWI brain at all time points. Figure 1 B shows representative data acquired during the mass spectrometric analyses of the P3 mouse samples. The labeled peak in this picture represents psychosine. From this data it can clearly be seen that this sphingolipid is detected at much higher levels in samples prepared from TWI mice as early as P3.
Figure 1.

Psychosine accumulates in detergent-free and detergent-based lipid raft fractions. Psychosine accumulations were studied by subjecting lipid raft fractions prepared from both WT and TWI mice at P3, P20, and P40 to mass spectrometric analysis. A, Analysis of total concentrations of psychosine in picomoles per gram of wet tissue shows significant increases in TWI brains when compared with WT brains. Data for P3 is shown in the inset. B, Representative data from mass spectrometric analysis of psychosine in raft fractions shows a significantly larger MS–MS response peak in P3 TWI compared with P3 WT. C, Analysis of LR fractions prepared using a detergent-based method shows preferential distribution of psychosine and cholesterol in raft fractions (4–5). Data for each fraction are presented as a percentage of the total. D, Investigation of fractions prepared using a detergent-free method also shows preferential distribution of psychosine and cholesterol in raft fractions (4–5). Data for each fraction are presented as a percentage of the total. Data for A are expressed as a mean ± SE from two to four mice per time point.
To examine the effects of psychosine on cell membranes in the TWI nervous system, LRs were isolated from whole brains at P3, P20, and P40 and from sciatic nerves at P40. Again, using mass spectrometry, the concentrations of psychosine were assayed in the LRs and non-LR membranes in both genotypes at each time point. First, to determine whether psychosine accumulates in LRs, samples from P40 brains were prepared using both a detergent-based (Lubrol) and a detergent-free (optiprep) method. Figure 1, C and D, shows a comparison of psychosine and cholesterol levels in these preparations and clearly illustrates that psychosine does indeed accumulate in rafts and that this is not a method-dependent phenomenon. Next, a full analysis of fractions from all mouse tissues and time points was carried out. Figure 2 A shows that, in fractions prepared from WT and TWI brains, psychosine in LRs builds up to very high levels. Concentrations measured in the TWI brain LRs at P3 were ∼8 pmol/g of wet tissue, representing an increase of around sevenfold from the WT (Fig. 2 A,B). At P20, psychosine was found to be very highly targeted to rafts and concentrations were found to have risen to ∼700 pmol/g of wet tissue (Fig. 2 A). This trend was continued in P40 samples, where psychosine concentration in the TWI brain LRs was found to have risen to near 1000 pmol/g of wet tissue, or >300-fold with respect to the age-matched WT (Fig. 2 A). Figure 2 C accounts for all parts resulting from the preparation of LRs and shows that in all membrane (nonpellet) fractions at each time point psychosine is preferentially accumulated in rafts. It should be noted that apparent increases in pellet psychosine in P40 TWI samples can be correlated to substantial increases in the total levels of psychosine that are detected in the late stage brains (see also Fig. 2 A), suggesting that at this age, psychosine is saturating all cellular compartments.
Figure 2.

Psychosine preferentially accumulates in TWI lipid rafts. A, Comparison of total psychosine in raft fractions shows a significant increase in levels of the sphingolipid in TWI rafts at P3, P20, and P40. Data are expressed as a mean ± SE from two to four mice per time point and are shown in picomoles per gram. B, Mass spectrometric analysis of fractions prepared from P3 brains are amplified to more clearly convey differences at this age. These data are expressed in picomoles per gram and confirms that psychosine preferentially accumulates in rafts (fractions 4–5) at P3. C, Quantification of psychosine in raft, nonraft membrane, and pellet portions of the preparation indicate that membrane psychosine is preferentially accumulated in lipid rafts. Data are expressed as a percentage with respect to total psychosine. D, Analysis of psychosine accumulation in TWI sciatic nerves at P40 shows that, in peripheral nervous tissue, psychosine is accumulated in lipid rafts at levels comparable to those found in the brain. This figure is also expressed in picomoles per gram.
The peripheral nervous system is highly affected by the genetic defect in the TWI mouse and was also investigated. In Figure 2 D, the distribution of psychosine in LR fractions prepared from P40 sciatic nerves is shown. This graph depicts findings analogous to those in the brain, showing that, in these samples, psychosine is also found to accumulate in LR fractions. These experiments illustrate a never-before-observed finding that psychosine accumulates preferentially in LRs in the TWI mouse. Importantly, this has been demonstrated at several time points throughout the life of the animal and in both the central and the peripheral nervous systems.
Lipid raft cholesterol concentrations and protein marker distributions are altered in the twitcher nervous system
Since the above data clearly establish that psychosine accumulates preferentially in lipid rafts of TWI brains, assays were performed to show whether this results in disruption of raft-associated cholesterol. Figure 3 A shows concentrations of cholesterol in WT and TWI brains at P3, P20, and P40. In agreement with previous studies (Igisu et al., 1983), no differences in total brain cholesterol were observed between WT and TWI. Figure 3 B illustrates the amount of esterified cholesterol found in WT and TWI brains at both time points. Interestingly, there is a slight (∼5% at P3, 6% at P20, and 1% at P40) but observable decrease in the amount of esterified cholesterol found in the TWI brain with respect to the WT. Figure 3, C and D, depicts the distribution of cholesterol in LR fractions at P3 (Fig. 3 C), along with the percentage of total cholesterol that is found in the raft fractions from WT and TWI brains at all time points (Fig. 3 D). The distribution of cholesterol in LR fractions shown for P3 mice confirms that the fractionation method used isolates rafts that are rich in cholesterol while also depicting an interesting trend of slight increases in the cholesterol concentrations in the TWI LRs. During quantification of this increase it was calculated that, for P3 brains, WT LRs contain ∼56% of the total brain cholesterol, and TWI LRs contained ∼63.5% of the total brain cholesterol. At P20, LR cholesterol is shown to be 34% of the total in the WT and 42.7% of the total in the TWI. In the P40 WT brain, LR cholesterol is shown to be ∼13% of the total and the P40 TWI rafts are shown to contain 18% of the total (Fig. 3 D). Interestingly, the increase in free cholesterol in the TWI LR fractions can be correlated to the decrease found in the levels of esterified cholesterol in the mutant brains.
Figure 3.
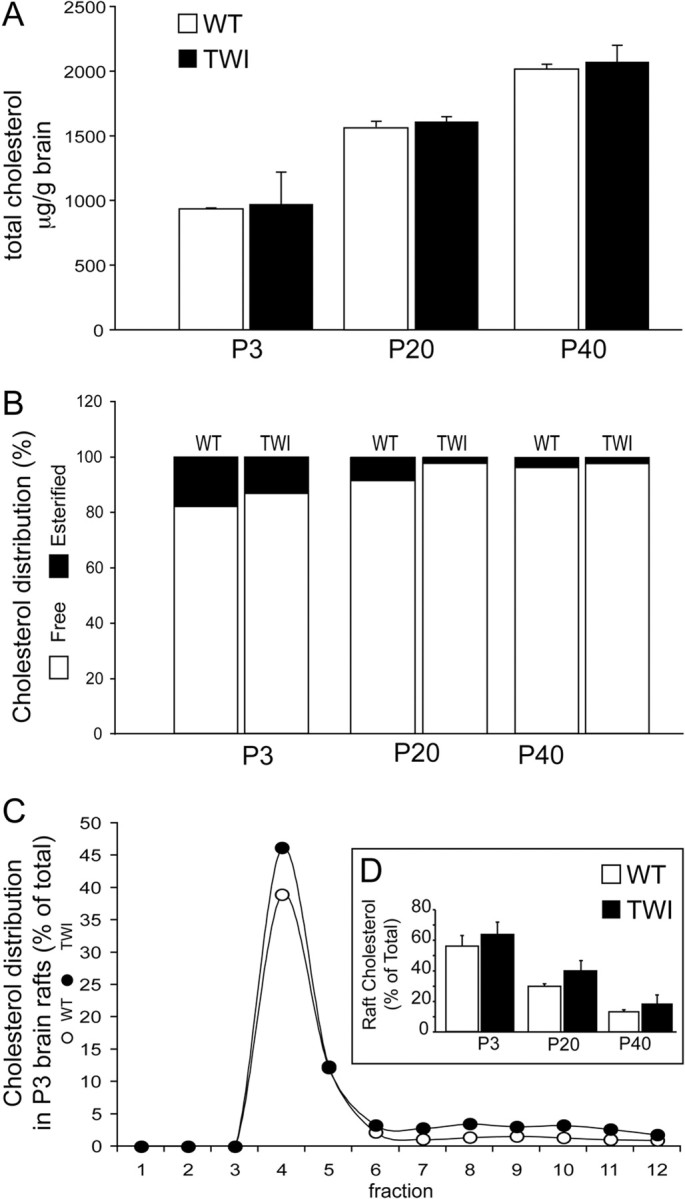
Increase in cholesterol accompanies psychosine accumulation in TWI lipid rafts. A, Fluorometric analysis of total cholesterol concentrations are expressed in microgram per gram of tissue and show no change between WT and TWI at P3, P20, or P40. B, Assay of esterified cholesterol as a percentage of the total shows that less esterified cholesterol is found in TWI brains when compared with WT at P3, P20, and P40. C, Levels of cholesterol in raft fractions are expressed as a percentage of the total. This shows a preferential distribution of cholesterol concentrations in LR samples (fractions 4–5). D, Total levels of cholesterol in LRs (fractions 4–5) are increased in a manner that may be correlated to decreases in esterified cholesterol. These measurements are also conveyed as a percentage of the total. Data are expressed as a mean ± SE from two to four mice per time point.
Next, an experiment was performed to determine whether the accumulation of psychosine in LRs could be correlated to any changes in raft protein marker distribution. To answer this question, Western blots were performed using fractions prepared from WT and TWI brains at P3, P20, and P40. The protocol used for raft preparation in this study isolates LRs in fractions 4–5, whereas fractions 6–12 are considered nonraft and 1–3 are devoid of protein. The pellet, which is also assayed for each sample, is made up of undefined material that is the result of the low-speed centrifugation in the early stages of the preparation. Each sample was assayed for the raft markers caveolin-1, flotillin-2, and the nonraft control P115. In the first panel of Figure 4, the protein distributions in raft fractions prepared from P3 brains of WT and TWI mice are shown. This result indicates that the P3 twitcher brain has an obvious disruption in the form of a significant loss of both flotillin-2 and caveolin-1 from fractions 4 and 5. Figure 4 also shows the results from the LR preparations of P20 brains. This was chosen as a mid-point in disease progression and shows that, in raft fractions 4 and 5, flotillin-2 levels only increase a small amount, whereas levels of caveolin-1 have increased substantially. The analysis of P40 brain samples is found in the third panel of Figure 4. In these mice, results exactly the opposite of those for P3 are observed with flotillin-2 and caveolin-1 accumulating substantially in the LR fractions. These results suggest a scenario in which levels of raft marker proteins fluctuate as psychosine begins to accumulate in these membrane domains. Essentially, markers are displaced from their proper localization early on and then begin to accumulate at high levels as the disease progresses and psychosine levels in rafts go up. Experiments to examine the level of each marker in total homogenates at all time points with respect to the housekeeping gene actin show no significant change in the total amounts of the raft marker proteins. Measurement of protein in the uncharacterized pellets resulting from the raft preparation show that, in most cases, changes in protein levels in raft fractions correlate to changes in those levels in the pellet.
Figure 4.

Redistribution of raft marker proteins accompanies psychosine accumulation in TWI lipid rafts. Lipid raft fractions from wild-type (W) and twitcher (T) nervous tissue were subjected to Western blot analysis. Representative blots are presented and show distributions of both raft Flotillin-2 (Flot 2)- and Caveolin-1 (Cav 1)- and nonraft (P115)-markers. These blots show that raft-marker distribution in the twitcher at all time points and in all tissues is disrupted in conjunction with the accumulation of psychosine. Proteins were also analyzed in total homogenates (To) and pellets (Pe). Actin was used as a house-keeping gene in Pe and To fractions.
The final portion of Figure 4 describes the characterization of protein markers in representative peripheral nervous tissue from P40 mice. For this experiment, sciatic nerves were removed and subjected to LR preparation. Interestingly, the results from the sciatic nerves mimic those seen in the brain at P40 with an increase in both flotillin-2 and caveolin-1. This suggests that the LR defect characterized in the CNS of the TWI mouse is also present in the peripheral nervous tissue.
Distribution of psychosine, cholesterol, and lipid raft marker proteins in samples from human Krabbe disease patients
The above findings suggest an intriguing pattern of LR disruption in the TWI mouse. To determine whether this phenomenon is relevant to human disease, LRs were prepared from brain samples of cortical gray matter from a 3-year-old human Krabbe patient along with an age-matched control. Figure 5 A shows the distribution of psychosine in control versus Krabbe LR fractions. These results indicate an accumulation of psychosine of ∼170 pmol/g tissue in the LRs of Krabbe brain. This is an increase of ∼18-fold when compared with rafts prepared from control tissue. Figure 5 B conveys the results of an assay of cholesterol concentrations in these fractions. As is shown in the TWI experiments, in the human samples, accumulation of psychosine is accompanied by an increased accumulation of cholesterol in LR fractions. In control samples, ∼31% of the total cholesterol is found in LR fractions. In contrast, in the Krabbe samples closer to 39% of total cholesterol is found to be localized in LRs. Finally, Figure 5 C shows the results of assays of raft marker proteins in the human samples. These pictures show a loss of raft markers flotillin-2 and caveolin-1 in the LR fractions, indicating a biochemical alteration in the composition of the LRs in the Krabbe brain.
Figure 5.
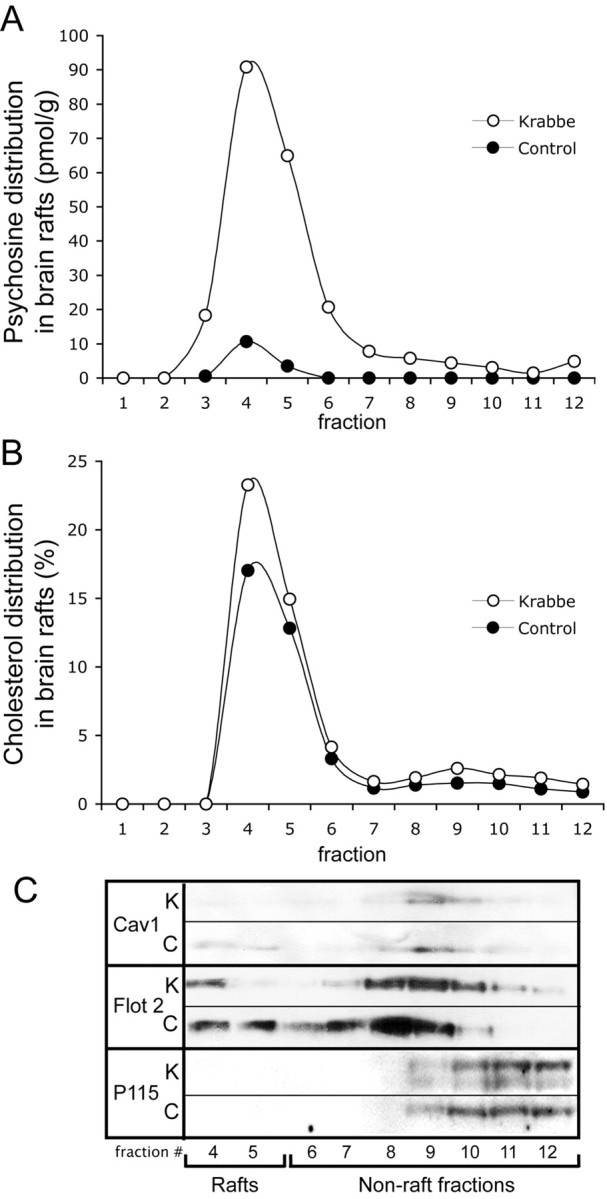
Psychosine accumulation disrupts raft architecture in brains of human Krabbe patients. A, Mass spectrometric analysis of human samples is expressed in picomoles per gram and reveals that psychosine preferentially accumulates in lipid rafts in humans with Krabbe disease. B, Fluorometric cholesterol assays of human samples are expressed as a percentage of the total and show that cholesterol accumulation accompanies the accumulation of psychosine. C, Western blotting was used to analyze fractions prepared from human tissue for distributions of the lipid raft-marker proteins Flotillin-2 (Flot 2) and Caveolin-1 (Cav-1). The results show a disruption of these proteins in a pattern similar to that observed in the TWI mouse. P115 was again used as a marker for nonrafts.
Activation of protein kinase C in lipid rafts is inhibited in twitcher nervous tissue
PKC is an important signaling protein involved in many cellular functions such as proliferation, apoptosis, and differentiation (Nakashima, 2002). It has previously been shown that psychosine can inhibit the activity of protein PKC (Hannun and Bell, 1987), and that this inhibition has implications in cell systems that are relevant to KD. In oligodendrocytes, the major myelinating cell type of the CNS, treatment with psychosine has been shown to alter PKC-dependent phosphorylation of myelin basic protein (Vartanian et al., 1989). When Schwann cells, which produce myelin in the PNS, are cultured directly from the TWI mouse, they are shown to have impaired PKC activity, which results in suppressed proliferation (Yoshimura et al., 1993, Yamada et al., 1996). Importantly, many isozymes of PKC have also been described as LR-dependent signaling proteins (Rybin et al., 1999). These studies in combination suggest the possibility that PKC inhibition in the TWI mouse may be the result of psychosine accumulation in LRs and their subsequent disruption.
The data presented so far in this communication describe clear alterations in the membrane architecture in the TWI and KD nervous systems. Importantly, this disruption is highly correlated to the preferential accumulation of psychosine in the LRs. Furthermore, not only is psychosine a known inhibitor of PKC, but raft disruption has also been shown to inhibit the normal activity of PKC (Wang et al., 2007). To describe the implications of these findings in the realm of the progression of KD, a series of experiments relating to the function of PKC in LRs were carried out. Figure 6 describes the first of these experiments, which was a characterization of phosphorylated (activated) and total PKC in raft fractions from P20 and P40 brains along with P40 sciatic nerves. This was done to provide two important measures of PKC activity in vivo: first, whether PKC is associated with rafts, and second, the level to which it is phosphorylated. Western blot data show that, at both time points and in both tissues, levels of total PKC in the LR fractions are decreased in the TWI when compared with the WT (Fig. 6 A). This figure also suggests that levels of phosphorylation in these tissues are lower in TWI LRs (Fig. 6 A). Levels of phosphorylation as a function of total PKC in LR fractions (fractions 4 and 5) were quantified and are presented in Figure 6 B. This graph confirms that phosphorylated PKC is measurably lower in the TWI rafts in all samples when compared with the WT (Fig. 6 B). To show whether this phenomenon is simply the result of a lack of PKC in the tissues as a whole, levels of total PKC in tissue homogenates were also assayed. The results of this analysis are presented in Figure 6 C and show very little overall change in total levels of PKC (Fig. 6 C). Taken in the context of this communication as a whole, these experiments suggest that psychosine accumulation in LRs is indeed correlated to alterations in the distribution of the functionally important PKC-signaling molecule. Importantly, this provides significant evidence that the previously observed inhibition of PKC in TWI tissues and by psychosine directly may be the result of the effects of this sphingolipid on the architecture of the membrane.
Figure 6.
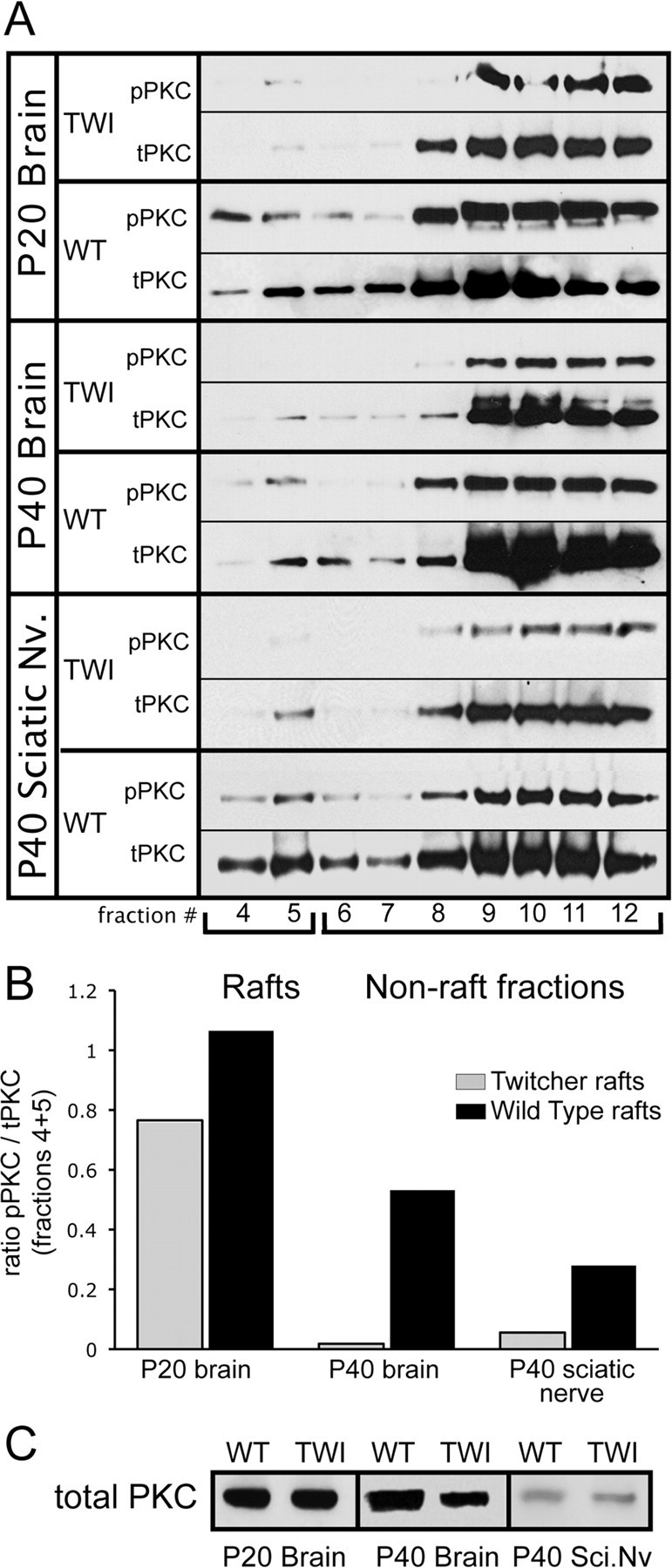
Psychosine accumulation parallels a decrease in protein kinase C in TWI lipid rafts. Lipid raft fractions from WT and TWI nervous tissue were analyzed using antibodies for total protein kinase C (tPKC) and phosphorylated PKC (pPKC). A, Western blots show the distribution of PKC and its active phosphorylated form in the brain at P20 and P40 and in the sciatic nerve at P40. These results show a decrease of total PKC in TWI rafts while also illustrating a much more significant loss of pPKC in these fractions. This indicates a significant reduction in the raft-based localization of PKC that occurs as psychosine concentrations go up in these membrane realms. B, Levels of activated (phosphorylated) PKC in raft fraction (4–5) are quantified with respect to the amount of total PKC in the same fractions. These results are expressed as a ratio of pPKC/tPKC. This analysis confirms a measurable reduction in pPKC in TWI tissues. C, Levels of PKC in total homogenates are presented from P20 and P40 brains and from P40 sciatic nerves.
Psychosine treatment inhibits raft localization of activated PKC
The above data suggests that LR localization and associated activation of PKC are impaired in TWI nervous tissue. The following experiments were designed to more clearly establish whether psychosine may be a direct cause of the aforementioned alterations. To answer this question, an in vitro paradigm was established in which the activation of PKC could be induced in a controlled and measurable manner and the effects of psychosine could be directly assayed. In this system, HeLa cells were cultured and treated with 10 μm psychosine or vehicle alone for 24 h before being stimulated to activate PKC. Since it is well established as an activator of PKC, stimulation of cells was done using PDGF (Nakashima, 2002). It should also be noted that PDGF was used in the above referenced studies that showed an impairment of PKC activity in TWI Schwann cells and that PKC is involved in Schwann cell proliferation (Yamada et al., 1996, Yoshimura et al., 1993), further establishing its relevance to this study. After designing this system, several control experiments were performed to see whether exogenous exposure of cultured cells to psychosine can induce LR alterations similar to those seen in vivo. Figure 7 A shows results from the measurement of endogenous psychosine and cholesterol in LR fractions prepared from untreated HeLa cells. This graph depicts the presence of the expected peaks of accumulation of these lipid species in fractions 4 and 5 (Fig. 7 A). Cells were then subjected to fractionation after 24 h of exposure to psychosine or vehicle. These fractions were assayed using mass spectrometry, which showed that even when cells are exogenously exposed to psychosine, it still preferentially accumulates in the LRs (Fig. 7 B). Measurement of cholesterol and the LR marker caveolin-1 in these same fractions showed that, as expected, both are preferentially localized in fractions 4 and 5 (Fig. 7 C,D). To establish the functionality of the PDGF receptor (PDGFr) in this model in the presence of psychosine, cell cultures were treated with PDGF for 15 min before being lysed and analyzed using Western blots. Figure 7, E and F, shows that levels of phosphorylated (active) PDGFr are increased after exposure to PDGF regardless of the presence of psychosine. Western blot data are also presented for the cytoskeletal proteins actin and tubulin (Fig. 7 E). These results show no changes in levels of either cytoskeletal protein. Together, this data establishes two important pieces of information. First, it is shown that LR preparation from cells provides results that are comparable to those seen in fractions from tissue homogenates. Second, and more importantly, the data also show that psychosine treatment induces changes in LRs that are analogous to those seen in the TWI in vivo. Specifically, these results show a preferential accumulation of psychosine (Fig. 7 B) and a slight increase in cholesterol and caveolin-1 in LR fractions (Fig. 7 C,D). Not only does this validate the utility of the in vitro paradigm, but it also provides compelling evidence that psychosine accumulation is at least partially responsible for the raft disruption observed in TWI nervous tissue.
Figure 7.

Treatment of cells in vitro with psychosine results in lipid raft alterations that mimic those observed in vivo. HeLa cells were cultured and treated with either 10 μm psychosine or vehicle for 24 h before being subjected to lipid raft preparation. A, Rafts from untreated HeLa cells were assayed for cholesterol levels, which are expressed in micrograms per milligram of protein, and endogenous psychosine levels which are expressed in nanomoles per milligram of protein. These analyses show an enrichment of both lipid species in raft fractions. B, Mass spectrometric detection in fractions from treated HeLa cells shows that exogenous addition of psychosine results in its targeted accumulation in rafts (fractions 4–5), which is expressed in nanomoles per milligram of protein. C, Analysis of cholesterol concentration in fractions from treated HeLa cells is expressed in micrograms per milligram of protein and shows an increase in cholesterol in the LR fractions (4–5) after psychosine exposure. D, The level and distribution of the raft marker protein caveolin-1 was assayed and shows an increase in abundance in raft fractions during treatment with psychosine. The nonraft marker P115 was also measured in addition to the measurement of the levels of both proteins in total cell lysates. E, Levels of phosphorylated and total PDGFr (pPDGFr and tPDGFr, respectively) along with levels of tubulin and actin were analyzed by Western blot. Analyses were done using HeLa cells in each of the following conditions: untreated control conditions (control), in the presence of PDGF (+PDGF), in the presence of 10 μm psychosine (+10 μm Psy), and in the presence of both PDGF and 10 μm psychosine (+10 μm Psy +PDGF). F, Quantification of the level of phosphorylation of PDGFr in E is presented. These results indicate that PDGFr can be activated even in the presence of psychosine. Measurements are presented as a ratio of pPDGFr/tPDGFr.
Immunocytochemistry (ICC) was used to visualize the localization of activated PKC in HeLa cells cultured in various conditions after a 15 min stimulation with 20 ng/ml of PDGF. Figure 8 A–D shows images from the first set of ICC analyses using an antibody for the detection of active PKC. As a baseline, unstimulated cells show low positive immunoreactivity for active PKC near or within the membrane (highlighted by arrows) (Fig. 8 D). In contrast, stimulation of cells with PDGF is shown to result in the detection of brightly fluorescent clusters of active PKC (in red and highlighted by arrowheads) that appear to be localized within the membrane (Fig. 8 C). Interestingly, when cells are incubated for 24 h with psychosine before stimulation with PDGF, this phenomenon is abolished and very few clusters of active PKC seem to be found within the membrane (highlighted by arrows) (Fig. 8 A). To show that this observation is specific to psychosine and not a general nonspecific consequence of exposure to any sphingolipid, cells treated for 24 h with d-sphingosine were also stimulated with PDGF. Figure 8 B shows that, similar to the stimulation of untreated cells, these conditions produce highly immunoreactive clusters of phosphorylated PKC that appear to be near or within the membrane (highlighted by arrowheads) (Fig. 8 B). This combination of results establishes that psychosine induces visible alterations in the localization of active PKC. However, they do not clearly show whether this change is associated with the loss of PKC in the membrane.
Figure 8.
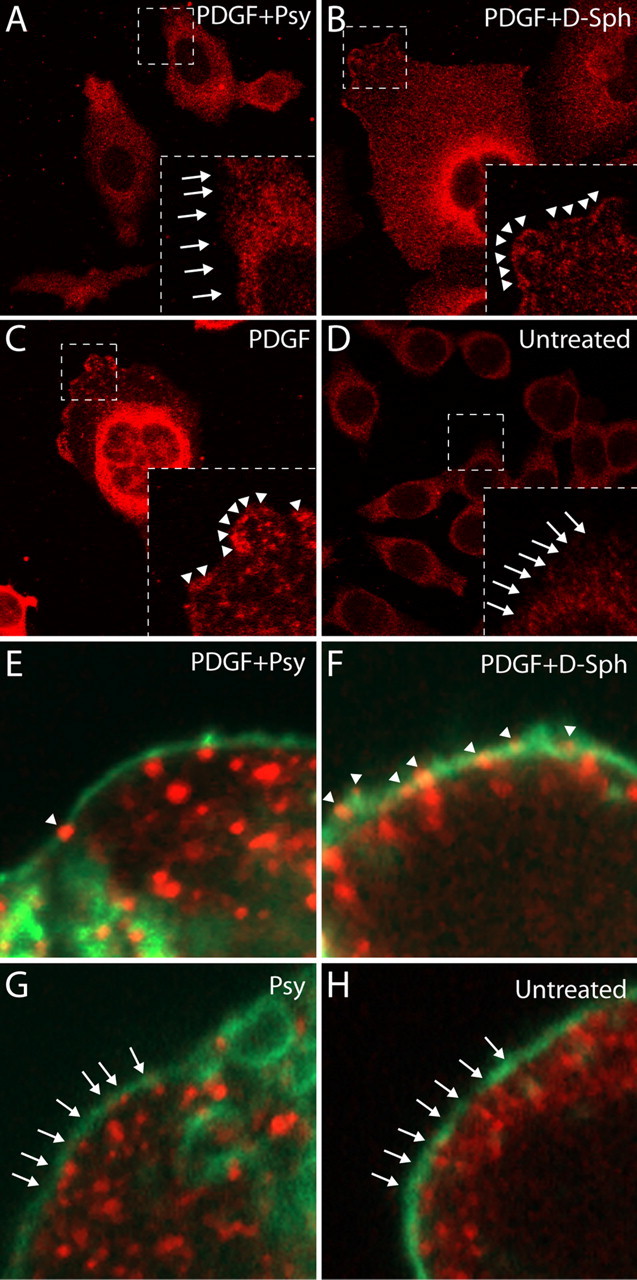
Psychosine reduces PDGF-induced colocalization of active (phosphorylated) PKC with the cell membrane and lipid rafts. A–D, An experiment in which HeLa cells treated with either psychosine, d-sphingosine, or nothing were stimulated with PDGF to activate PKC and subsequently stained with an antibody for active PKC (pPKC) (red). A, HeLa cells treated with psychosine (Psy) for 24 h and subsequently stimulated with PDGF. Under these conditions, very little active PKC is labeled at or around the membrane (see arrows). B, Cells treated with d-sphingosine (d-Sph) for 24 h before PDGF stimulation. These cells showed abundant localization of pPKC near or within the membrane (see arrowheads). C, Cells that were not exposed to any sphingolipid treatment before stimulation with PDGF. These results demonstrate that pPKC can be observed at or near the membrane (see arrowheads) in normal cells after stimulation with PDGF. D, Control cells were not treated and were not stimulated with PDGF, showing their steady state. As expected, no pPKC is observed at or near the membrane (see arrows) before stimulation with PDGF. E–H, The same experiment as above, except that cells are labeled with both pPKC (red) and the lipid raft-marker CTXB (green) to show true colocalization of pPKC with the membrane. E, Cells that were treated for 24 h with Psy followed by stimulation with PDGF. This image shows, under these conditions, minimal colocalization of pPKC and CTXB (see arrowhead). F, Cells treated with d-Sph for 24 h and subsequently stimulated with PDGF. This image shows that these conditions allow for a robust colocalization between pPKC with CTXB (see arrowheads) after stimulation with PDGF. G, Cells treated with Psy for 24 h without PDGF stimulation. As expected, this image shows no colocalization of pPKC and CTXB (see arrows). H, Cells that were not subjected to treatment with sphingolipids or stimulation with PDGF. This image shows the unperturbed basal state of these cells in which no colocalization of pPKC and CTXB can be observed (see arrows).
To more directly approach this question, a second set of ICC experiments was performed. For this set of analyses, the same antibody for active PKC (red) was used in conjunction with fluorescently labeled cholera toxin B (CTXB) (green), which labels the membrane by binding to GM1. This method is designed to allow for the direct imaging of any possible colocalization of clusters of active PKC with the membrane after stimulation with PDGF. The results of these experiments are shown in Figure 8 E–H. As expected, cells grown under normal conditions or in the presence of psychosine do not show large punctuate clusters of active PKC and do not show colocalization of active PKC with the membrane (highlighted by arrows) before stimulation with PDGF (Fig. 8 H,G). Since in the previous experiment it was already established that d-sphingosine does not have significant effects on the activation of PKC by PDGF, these cells were used to show the normal localization of activated PKC. Figure 8 F shows that, during PDGF stimulation, d-sphingosine treated cells exhibit strong punctate-phosphorylated PKC immunoreactivity, and, most importantly, that these clusters are highly colocalized with the CTXB-labeled membrane (highlighted by arrowheads) (Fig. 8 F). In contrast, when cells treated with psychosine are stimulated with PDGF, colocalization of active PKC with the membrane is almost completely abolished (highlighted by arrowheads) (Fig. 8 E). This result supports what is suggested by Figure 8 A, which is that psychosine interferes with the proper localization and activation of PKC within the membrane.
Discussion
In this study, it has been shown that the TWI mouse model of Krabbe disease shows a significant accumulation of psychosine in LR fractions and that this accumulation is correlated to significant disruptions in the architecture and composition of LR domains. It has also been demonstrated that a similar phenomenon can be observed in human patients suffering from Krabbe disease. Furthermore, the accumulation of psychosine in LR fractions has been shown to significantly disrupt the normal activities of PKC. Therefore, for the first time, it is reported that psychosine, the lipid species accumulated in KD, preferentially accumulates in lipid rafts which can subsequently disrupt their organization and drive a functional defect of signaling activity.
These results give rise to several important questions. First, since it is clear that psychosine is preferentially accumulated in rafts, by what mechanism might this occur? Targeting of lipid species to rafts is thought to be a result of their biochemical and biophysical properties (Simons and Ikonen, 1997). It is suggested that these characteristics result in the preferential ordering of cholesterol and sphingolipids based on their highly favorable interaction. Psychosine has similar properties to those of other sphingolipids found in rafts, and this may result in a favorable interaction with cholesterol. This would explain not only the observation of targeted psychosine accumulation but may also account for the increased cholesterol concentrations found in TWI, human Krabbe, and psychosine-treated HeLa cell LRs.
With regard to cholesterol, a few intriguing observations have been presented here. Importantly, cholesterol is highly enriched in the brain and is thought to play a very significant role in the function of its cellular population (for a review, see Vance et al., 2005). Since brain cholesterol is synthesized in situ, it is interesting to note that the observations presented here show an increase in raft cholesterol that is concurrent with a decrease in esterified cholesterol. This would suggest that local stores of cholesterol esters in the brain provide the source for increased levels of the lipid in rafts. This idea is also supported by the fact that total cholesterol concentrations in the TWI and Krabbe brain are not changed during the course of the disease.
Another question that arises from the above data is why LR marker proteins in the mouse show different responses at different time points. A possible explanation is that psychosine may initially disrupt the normal interactions of raft proteins with these domains. This disruption may result in a type of distress signal that may increase the amount of these raft proteins targeted to the appropriate place to reestablish any interrupted protein–protein interactions. The large subsequent increase in psychosine may also play a role in this phenomenon by increasing the pool of sphingolipids in the membrane, resulting in an increase in raft size and stability. This conclusion may be supported by the observation that the human, with psychosine accumulation more comparable to the P3 TWI, shows a protein distribution that is also closer to that of the P3 TWI.
The most important question that is raised by the results presented here is what the potential functional consequences of psychosine-induced LR disruption are. For several years, it has been established that chemical lipid raft disruption can result in loss of function of associated signaling molecules (Keller and Simons, 1998; Mañes et al., 1999). It has also been shown that treatment of cells with short-chain ceramides can induce changes in membrane architecture that disrupt raft-associated signals (Gidwani et al., 2003). It is possible that psychosine accumulation in rafts may have a similar effect, thereby representing a potential molecular mechanism for its toxicity. This idea is supported by the results presented above for experiments regarding PKC. Psychosine has long been known to be an inhibitor of PKC activity (Hannun and Bell, 1987), however, in what way this occurs has remained undescribed. Interestingly, the function of PKCα, the isozyme activated by PDGF (Choudhury et al., 1993), has been shown to be intimately related to the characteristics of the membrane. For example, in a study using liposomes of known composition, PKCα activity was shown to be decreased by alterations in membrane phase/fluidity that resulted in an increase in the overall order of the system (Micol et al., 1999). More specifically related to rafts, the ability of PKCα to exert its normal function on the epidermal growth factor receptor has been shown to be dependent on the raft protein caveolin-1 (Wang et al., 2007). Importantly, in the latter study, the disruption of LRs using methyl-β-cyclodextrin also showed that general raft manipulations can result in the loss of normal function of PKC. A somewhat analogous phenomenon is suggested by the data presented in this study; however, in this case psychosine is the mediator of LR disruption. As stated above, it is well known that psychosine can inhibit PKC and also that PKC activity in the TWI mouse is decreased. Since PKC activity is dependent on the makeup of the membrane, it is logical to conclude that the mechanistic link between these two findings is the psychosine-based disruption of its architecture.
There are several other raft-associated processes that may be disrupted by psychosine, some of which include neurite outgrowth (Niethammer et al., 2002), axonal guidance (Ibáñez et al., 2004), long-term potentiation (Ma et al., 2003) and myelination (Krämer et al., 1999). The disturbance of myelination is of particular interest in KD as it is classically characterized as a demyelinating disease. Disruption of proper raft-mediated signaling may be one of the forces driving the demyelinating process by the lack of proper communication between oligodendrocytes and the environment or disrupted interactions with the axons themselves (Galbiati et al., 2009). Each of these mechanisms provides interesting and important targets for future investigation.
A few other observations from this study should also be mentioned. First, a normal cellular function for psychosine has never been observed or suggested. In fact, it is commonly thought to be an intermediate without a known biological function (Suzuki, 1998). Here, it has been shown that psychosine appears to be present in low concentrations in normal LRs, including the LRs of the human brain and those isolated from HeLa cells. It may be that the normal function of psychosine is somehow related to LRs, an idea that certainly warrants further investigation.
Another important observation from this study is the fact that raft disruption can be detected at P3 in the TWI mouse, a time point well before significant demyelination (Taniike and Suzuki, 1994) and certainly before noticeable phenotypic abnormalities. This may suggest that the molecular pathogenesis of Krabbe disease begins earlier than previously thought. It may also highlight the possibility that other cell populations such as neurons may be adversely effected by psychosine (L. Cantuti and E. R. Bongarzone, unpublished observations). Notably, this idea may provide insight into the general inability of current cell and enzyme replacement therapies for KD to reverse the course of disease (Galbiati et al., 2009).
A final important implication of this investigation is that this type of mechanism may not be unique to the TWI and its accumulation of psychosine. Several other diseases including MLD, Niemann-Pick disease, and the gangliosidoses are the result of aberrant sphingolipid metabolism. Since, by definition, LRs are dependent on specific concentrations of sphingolipids, these metabolic errors, although unique, may all result in an analogous phenomenon of LR disruption.
To conclude, based on the data provided in this examination of both TWI and human Krabbe LRs, the following pathophysiological mechanism for Krabbe disease is proposed. The mutation of GALC and the resultant LR-targeted accumulation of psychosine disrupts the normal architecture of the rafts (Fig. 9). This drives a cascade of events in which there is an interference of protein–protein interactions that normally occur in rafts, resulting in aberrant cell signaling, which then could lead to a decrease in normal overall cellular function and survival (Fig. 9). This series of events should be considered, at the least, as a potential contributing factor in the overall decline in health of those suffering from KD.
Figure 9.

Proposed model of deregulation of various raft-modulated cell functions in Krabbe disease. The illustration proposes a working model where the preferential accumulation of psychosine in the raft domain, consequent to the deficiency of GALC activity, leads to deregulation of raft-associated signals in Krabbe disease. In this hypothetical model, under normal sphingolipid metabolism, two hypothetical partners (A, B) are spatially accommodated to provide functional interaction and hence, normal signal activity. In Krabbe disease, the raft domain increases in size because of the accumulation of psychosine and other components of the raft, leading to the spatial separation of the two partner molecules A and B, disrupting the function of the associated signal. The consequent deregulation in signal activity can influence different dependent cellular processes according to the cell type where this occurs. The proposed model does not illustrate other possible raft alterations such as fragmentation of abnormal rafts and impairment of signaling by spatial separation of raft components.
Footnotes
This study was performed with funds from an intramural seed grant from the University of Illinois at Chicago (E.R.B.). We are thankful to the anonymous reviewers for their insightful comments and to Gustavo Pigino, Gerardo Morfini, and Scott Brady (University of Illinois at Chicago) for fruitful discussions. We are in debt to Oscar Bizzozero (University of New Mexico, Albuquerque, NM) for his generous donation of lipid and protein extracts from human Krabbe samples and to Kulandaivelu S. Vetrivel (University of Chicago, IL) for technical assistance in raft preparation We dedicate this study to the memory of Tito Bongarzone.
References
- Choudhury GG, Biswas P, Grandaliano G, Abboud HE. Involvement of PKC-alpha in PDGF-mediated mitogenic signaling in human mesangial cells. Am J Physiol. 1993;265:F634–F642. doi: 10.1152/ajprenal.1993.265.5.F634. [DOI] [PubMed] [Google Scholar]
- Dolcetta D, Perani L, Givogri MI, Galbiati F, Amadio S, Del Carro U, Finocchiaro G, Fanzani A, Marchesini S, Naldini L, Roncarolo MG, Bongarzone E. Design and optimization of lentiviral vectors for transfer of GALC expression in Twitcher brain. J Gene Med. 2006;8:962–971. doi: 10.1002/jgm.924. [DOI] [PubMed] [Google Scholar]
- Galbiati F, Basso V, Cantuti L, Givogri MI, Lopez-Rosas A, Perez N, Vasu C, Cao H, van Breemen R, Mondino A, Bongarzone ER. Autonomic denervation of lymphoid organs leads to epigenetic immune atrophy in a mouse model of Krabbe disease. J Neurosci. 2007;27:13730–13738. doi: 10.1523/JNEUROSCI.3379-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbiati F, Givogri MI, Cantuti L, Lopez Rosas A, Cao H, van Breemen R, Bongarzone ER. Combined hematopoietic and lentiviral gene-transfer therapies in newborn Twitcher mice reveal contemporaneous neurodegeneration and demyelination in Krabbe disease. J Neurosci Res. 2009;87:1748–1759. doi: 10.1002/jnr.22006. [DOI] [PubMed] [Google Scholar]
- Gidwani A, Brown HA, Holowka D, Baird B. Disruption of lipid order by short-chain ceramides correlates with inhibition of phospholipase D and downstream signaling by FcεRI. J Cell Sci. 2003;116:3177–3187. doi: 10.1242/jcs.00621. [DOI] [PubMed] [Google Scholar]
- Gieselmann V. Metachromatic leukodystrophy: recent research develoments. J Child Neurol. 2003;18:591–595. doi: 10.1177/08830738030180090301. [DOI] [PubMed] [Google Scholar]
- Giri S, Khan M, Rattan R, Singh I, Singh AK. Krabbe Disease: psychosine-mediated activation of phospholipase A2 in oligodendrocyte cell death. J Lipid Res. 2006;47:1478–1492. doi: 10.1194/jlr.M600084-JLR200. [DOI] [PubMed] [Google Scholar]
- Givogri MI, Galbiati F, Fasano S, Amadio S, Perani L, Superchi D, Morana P, Del Carro U, Marchesini S, Brambilla R, Wrabetz L, Bongarzone E. Oligodendroglial progenitor cell therapy limits central neurological deficits in mice with metachromatic leukodystrophy. J Neurosci. 2006;26:3109–3119. doi: 10.1523/JNEUROSCI.4366-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givogri MI, Bottai D, Zhu HL, Fasano S, Lamorte G, Brambilla R, Vescovi A, Wrabetz L, Bongarzone E. Multipotential neural precursors transplanted into the metachromatic leukodystrophy brain fail to generate oligodendrocytes but contribute to limit brain dysfunction. Dev Neurosci. 2008;30:340–357. doi: 10.1159/000150127. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Bell RM. Lysosphingolipids inhibit protein kinase C: implications for the sphingolipidoses. Science. 1987;235:670–674. doi: 10.1126/science.3101176. [DOI] [PubMed] [Google Scholar]
- Haq E, Giri S, Singh I, Singh AK. Molecular mechanism of psychosine-induced cell death in human oligodendrocyte cell line. J Neurochem. 2003;86:1428–1440. doi: 10.1046/j.1471-4159.2003.01941.x. [DOI] [PubMed] [Google Scholar]
- Ibáñez CF. Lipid rafts as organizing platforms for cell chemotaxis and axon guidance. Neuron. 2004;42:3–5. doi: 10.1016/s0896-6273(04)00194-1. [DOI] [PubMed] [Google Scholar]
- Igisu H, Shimomura K, Kishimoto Y, Suzuki K. Lipids of developing brain of Twitcher mouse: an authentic murine model of human Krabbe disease. Brain. 1983;106:405–417. doi: 10.1093/brain/106.2.405. [DOI] [PubMed] [Google Scholar]
- Keller P, Simons K. Cholesterol Is Required for Surface Transport of Influenza Virus Hemagglutinin. J Cell Biol. 1998;140:1357–1367. doi: 10.1083/jcb.140.6.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer EM, Klein C, Koch T, Boytinck M, Trotter J. Compartmentation of Fyn kinase with glycosylphosphatidylinositol-anchored molecules in oligodendrocytes facilitates kinase activation during myelination. J Biol Chem. 1999;274:29042–29049. doi: 10.1074/jbc.274.41.29042. [DOI] [PubMed] [Google Scholar]
- Ma L, Huang YZ, Pitcher GM, Valtschanoff JG, Ma YH, Feng LY, Lu B, Xiong WC, Salter MW, Weinberg RJ, Mei L. Ligand-dependent recruitment of the ErbB4 signaling complex into neuronal lipid rafts. J Neurosci. 2003;23:3164–3175. doi: 10.1523/JNEUROSCI.23-08-03164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald JL, Pike LJ. A simplified method for the preparation of detergent-free lipid rafts. J Lipid Res. 2005;46:1061–1067. doi: 10.1194/jlr.D400041-JLR200. [DOI] [PubMed] [Google Scholar]
- Mañes S, Mira E, Gómez-Moutón C, Lacalle RA, Keller P, Labrador JP, Martínez-A C. Membrane raft microdomains mediate front–rear polarity in migrating cells. EMBO J. 1999;18:6211–6220. doi: 10.1093/emboj/18.22.6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micol V, Sánchez-Piñera P, Villalaín J, de Godos A, Gómez-Fernández JC. Correlation between protein kinase C alpha activity and membrane phase behavior. Biophys J. 1999;76:916–927. doi: 10.1016/S0006-3495(99)77255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima S. Protein kinase Cα (PKCα): regulation and biological function. J Biochem. 2002;132:669–675. doi: 10.1093/oxfordjournals.jbchem.a003272. [DOI] [PubMed] [Google Scholar]
- Niethammer P, Delling M, Sytnyk V, Dityatev A, Fukami K, Schachner M. Cosignaling of NCAM via lipid rafts and the FGF receptor is required for neuritogenesis. J Cell Biol. 2002;157:521–532. doi: 10.1083/jcb.200109059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LJ. Rafts defined: a report on the Keystone Symposium on Lipid Rafts and Cell Function. J Lipid Res. 2006;47:1597–1598. doi: 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- Rybin VO, Xu X, Steinberg SF. Activated protein kinase c isoforms target to cardiomyocyte caveolae. Circ Res. 1999;84:980–988. doi: 10.1161/01.res.84.9.980. [DOI] [PubMed] [Google Scholar]
- Sakai N, Inui K, Tatsumi N, Fukushima H, Nishigaki T, Taniike M, Nishimoto J, Tsukamoto H, Yanagihara I, Ozono K, Okada S. Molecular cloning and expression of cDNA for murine galactocerebrosidase and mutation analysis of the twitcher mouse, a model of Krabbe's disease. J Neurochem. 1996;66:1118–1124. doi: 10.1046/j.1471-4159.1996.66031118.x. [DOI] [PubMed] [Google Scholar]
- Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest. 2002;110:597–603. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Suzuki K. Twenty five years of the “psychosine hypothesis”: a personal perspective of its history and present status. Neurochem Res. 1998;23:251–259. doi: 10.1023/a:1022436928925. [DOI] [PubMed] [Google Scholar]
- Suzuki K. Evolving perspective of the pathogenesis of globoid cell leukodystrophy (Krabbe disease) Proc Jpn Acad Ser B. 2003;79:1–8. [Google Scholar]
- Taniike M, Suzuki K. Spacio-temporal progression of demyelination in twitcher mouse: with clinico-pathological correlation. Acta Neuropathol. 1994;88:228–236. doi: 10.1007/BF00293398. [DOI] [PubMed] [Google Scholar]
- Vance JE, Hayashi H, Karten B. Cholesterol homeostasis in neurons and glial cells. Semin Cell Dev Biol. 2005;16:193–212. doi: 10.1016/j.semcdb.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Vartanian T, Dawson G, Soliven B, Nelson DJ, Szuchet S. Phosphorylation of myelin basic protein in intact oligodendrocytes: inhibition by galactosylsphingosine and cyclic AMP. Glia. 1989;2:370–379. doi: 10.1002/glia.440020509. [DOI] [PubMed] [Google Scholar]
- Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, Wong PC, Xu H, Thinakaran G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J Biol Chem. 2004;279:44945–44954. doi: 10.1074/jbc.M407986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivel KS, Cheng H, Kim SH, Chen Y, Barnes NY, Parent AT, Sisodia SS, Thinakaran G. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J Biol Chem. 2005;280:25892–25900. doi: 10.1074/jbc.M503570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XQ, Yan Q, Sun P, Liu JW, Go L, McDaniel SM, Paller AS. Suppression of epidermal growth factor receptor signaling by protein kinase C-α Activation requires CD82, caveolin-1, and ganglioside. Cancer Res. 2007;67:9986–9995. doi: 10.1158/0008-5472.CAN-07-1300. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Suzuki K, Suzuki Y, Suzuki K. In: The metabolic and molecular bases of inherited disease. Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. New York: McGraw-Hill; 2001. p. 3669.p. 3670.p. 3687. [Google Scholar]
- Whitfield PD, Sharp PC, Taylor R, Meikle P. Quantification of galactosylsphingosine in the twitcher mouse using electrospray ionization-tandem mass spectrometry. J Lipid Res. 2001;42:2092–2095. [PubMed] [Google Scholar]
- Yamada H, Martin P, Suzuki K. Impairment of protein kinase C activity in twitcher Schwann cells in vitro. Brain Res. 1996;718:138–144. doi: 10.1016/0006-8993(96)00098-4. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Goda S, Kobayashi T, Goto I. Involvement of protein kinase C in the proliferation of cultured Schwann cells. Brain Res. 1993;617:55–60. doi: 10.1016/0006-8993(93)90612-q. [DOI] [PubMed] [Google Scholar]