

Abstract
Sodium channel blockers are known for reducing pain and hyperalgesia. In the present study we investigated changes in cerebral processing of secondary mechanical hyperalgesia induced by pharmacological modulation with systemic lidocaine. An experimental electrical pain model was used in combination with functional magnetic resonance imaging. After induction of pin-prick hyperalgesia lidocaine or placebo was administered systemically using a double-blinded design. A 2 × 2 factorial analysis was performed. The factors were (1) sensitization to pain (levels: pin-prick hyperalgesia and normal pin-prick pain) and (2) pharmacological modulation (levels: lidocaine and placebo). A main effect of (1) sensitization was found in bilateral secondary somatosensory cortex (S2), insula, anterior cingulate gyrus (ACC), medial prefrontal cortex (mPFC), dorsolateral prefrontal cortex (dlPFC), parietal association cortex (PA), thalamus and contralateral midbrain. A main effect of (2) pharmacological modulation was found in bilateral S2, insula, ACC, mPFC, dlPFC, PA, midbrain and contralateral primary motor cortex, and thalamus. Interaction of pharmacological modulation and sensitization to pin-prick pain with activity increase during hyperalgesia and placebo was found in mPFC, posterior cingulate gyrus and thalamus. However, only activity in mPFC was inversely correlated to area of hyperalgesia during placebo and antihyperalgesic treatment effect. Furthermore, the difference of mPFC activity during hyperalgesia and placebo versus hyperalgesia and lidocaine correlated inversely with the change of the weighted hyperalgesic area (as a factor of area and rated pain intensity). We conclude that activity in mPFC correlates inversely with individual extent of central hyperalgesia and predicts individual pharmacological antihyperalgesic treatment response.
Introduction
Neuropathy or inflammation can result in spontaneous or evoked pain (Woolf and Mannion, 1999). Evoked pain can be divided into hyperalgesia or allodynia. Hyperalgesia means increased painfulness toward noxious stimuli and can be classified as primary or secondary hyperalgesia. Primary hyperalgesia results from peripheral sensitization in the area of tissue damage. In contrast, secondary mechanical hyperalgesia occurs in neighboring tissue because of central sensitization. As revealed by neuroimaging studies in experimental and clinical pain states, sensitization to noxious stimuli has specific correlates in cerebral pain processing (Apkarian et al., 2005; Borsook et al., 2007; Moisset and Bouhassira, 2007; Tracey and Mantyh, 2007; Seifert and Maihöfner, 2009). The two main mechanisms are (1) an increase of activity in pain processing brain areas [primary somatosensory cortex (S1), secondary somatosensory cortex (S2), insula, anterior cingulate cortex (ACC), prefrontal cortex (PFC)] and (2) an additional recruitment of other brain regions [additional PFC areas, parietal association cortex (PA), brainstem nuclei) reflecting mainly cognitive, autonomic and motor processing or modulatory networks (Apkarian et al., 2005; Maihöfner and Handwerker, 2005; Borsook et al., 2007; Moisset and Bouhassira, 2007; Seifert et al., 2008). However, only two studies have investigated the neural correlates of pharmacological antihyperalgesia by gabapentin (Iannetti et al., 2005) and nonsteroidal anti-inflammatory drugs (Maihöfner et al., 2007).
Sodium channel blockers administered intravenously are beneficial in the treatment of neuropathic pain states (Attal et al., 2000; Challapalli et al., 2005). Clinical pain studies revealed analgesic effects of intravenous sodium channel blockers especially in pain states dominated by hyperalgesia (Tremont-Lukats et al., 2005). Sodium channel blockade by systemically applied low-dose lidocaine has been shown to result in analgesia and lasting reduction of pin-prick hyperalgesia (Koppert et al., 2000; Koppert et al., 2001). The analgesic effects are believed to be achieved by both a peripheral and a central mode of action (Woolf and Wiesenfeld-Hallin, 1985; Koppert et al., 2000). However, there is evidence for an isolated central mode of action relative to the antihyperalgesic effect (Koppert et al., 2000). To investigate the effects of systemic sodium channel blockade by lidocaine on pain related cerebral activation during normal and central sensitization states we performed the present placebo-controlled, double-blinded and randomized cross-over functional magnetic resonance imaging (fMRI) study in healthy subjects using the model of electrically induced hyperalgesia.
Materials and Methods
Subjects.
A total of 12 healthy subjects (7 male, 5 female, mean age 25.1 years ± 0.5 years) participated in the study. The volunteers were informed about the procedures of the study but were unaware of the specific experimental goals. Informed consent was obtained from all participants before the experiments and the study adhered to the tenets of the Declaration of Helsinki. The study was approved by the local ethics committee. All subjects were already experienced in psychophysical studies. The stimulation site was the middle aspect of the right dorsal foot in all subjects.
Experimental design.
To explore the effects of systemically administered lidocaine we performed a randomized, double-blinded, placebo-controlled cross-over study. Each volunteer participated on two experimental days, one for lidocaine and one for placebo application. The time interval between the experimental sessions was 14 d. The experimental procedures were identical on both days. At the beginning of the experiment the volunteer was transferred into the MRI scanner. Consecutively anatomical MRI data were acquired. Stable areas of pin-prick hyperalgesia and dynamic tactile allodynia were induced over 20 min using the model of electrical induced pin-prick hyperalgesia (see below). Consecutively, placebo (isotonic saline) or lidocaine in a dose of 12 mg per kg body weight was infused intravenously for 10 min (= 2 mg/kg), followed by 2 mg/kg/h for 15 min. Based on a data sheet from literature (Rowland et al., 1971), lidocaine blood levels between 2 and 3 μl/ml were achieved with this regimen (Fig. 1). Fifteen minutes after beginning the infusion the following conditions were tested by functional MRI using a classical block design: (1) pin-prick stimulation outside the hyperalgesic area (normal pin-prick pain) and (2) pin-prick stimulation inside the hyperalgesic area (pin-prick hyperalgesia). The stimulation blocks of the two different conditions were applied in a pseudo-randomized order, thus, each subject received the same sequence in the two experimental sessions. Psychophysical data were obtained inside the scanner during the experimental procedure. After each run the subjects rated the intensity of (1) electrical pain and (2) pin-prick stimuli inside the hyperalgesic area. Finally, the areas of the hyperalgesia were determined. A schematic diagram and a time-line of the experimental procedures are depicted in Figure 1A.
Figure 1.

Experimental procedure. A, Schematic diagram and time-line of the experimental procedures. B, Lidocaine pharmacokinetics based on a data sheet from literature (Rowland et al., 1971). Lidocaine in a dose of 12 mg per kg body weight was infused intravenously for 10 min (= 2 mg/kg), followed by 2 mg/kg/h for 15 min. Lidocaine blood levels between 2 and 3 μl/ml during the fMRI measurement and the psychophysical testing were achieved with this regimen.
Induction of electrically induced pin-prick hyperalgesia.
Transcutaneous electrical stimulation was used to induce secondary mechanical hyperalgesia as described previously (Koppert et al., 2001; Koppert et al., 2005; Filitz et al., 2008). Briefly, two electrodes were mounted on the skin 4 mm apart on the dorsum of the right foot. Monophasic, rectangular electrical pulses of 0.5 ms duration were applied via a constant current stimulator (Digitimer S7) at 1 Hz. The current was gradually adjusted during the first 20 min of stimulus administration, targeting a pain rating of 6 on an 11-point numeric rating scale (NRS; 0 = no pain and 10 = maximum tolerable pain), and was then kept constant for the remaining time of the experiment (during the fMRI measurement). This experimental approach has been proven to provoke stable areas of secondary hyperalgesia to punctate stimuli caused by an activation of primarily mechano-insensitive C-nociceptors (Schmelz et al., 2000). This class of nociceptors was shown to be electrically activated preferentially at high current densities as used in this model (Weidner et al., 1999).
Psychophysical testing.
Psychophysical ratings were obtained after application of the respective stimulus series inside the scanner. The following parameters were assessed: (1) intensity of electrical pain, (2) intensity of pin-prick stimuli inside the hyperalgesic area, and (3) areas of pinprick hyperalgesia. Pain intensity was measured on a numeric rating scale (NRS) ranging from 0 (no pain) to 10 (maximum pain). The hyperalgesic area was assessed using a 256 mN pin-prick stimulator. The area of hyperalgesia was determined along eight orthogonal trajectories by testing pinprick sensitivity radially each 0.5 cm from the center in both directions.
Stimulus application during the fMRI measurements.
For stimulus application during fMRI measurements the stimuli were applied by mechanically silent devices. Pin-prick pain was induced during the stimulation periods using a calibrated 256 mN pin-prick stimulator. Stimulation frequency was 1 Hz. The site of stimulation was the area of hyperalgesia and an area 3 cm distant from the border of the hyperalgesic area, respectively.
Functional magnetic resonance imaging.
Echoplanar images were collected on a 1.5 Tesla MRI scanner (Sonata, Siemens Medical Solutions) using the standard head coil. For each of the two sessions two time series (pin-prick hyperalgesia and normal pin-prick stimulation) of 93 whole-brain images were obtained with a gradient-echo, echo-planar scanning sequence (EPI; TR 3 s, time to echo 40 ms, flip angle 90°; field of view 220 mm2, acquisition matrix 64 × 64, 16 axial slices, slice thickness 4 mm, gap 1 mm). The first 4 images were discarded to account for spin saturation effects. A T1-weighted three-dimensional magnetization prepared rapid acquisition gradient echo sequence (MPRAGE) scan (voxel size = 1.0 × 1.0 × 1.0 mm3) was recorded in the same session as the functional measurements for the recording of the individual brain anatomy. During each of the two sessions MRI sequences were assessed in the following order: anatomical scout, MPRAGE, EPI in randomized order (pin-prick hyperalgesia, normal pin-prick stimulation). Data analysis, registration and visualization were performed with the fMRI software package BrainVoyager QX. Data were motion-corrected using sinc interpolation. Preprocessing furthermore included Gaussian spatial [full-width at half-maximum (FWHM) = 4 mm] and temporal (FWHM = 3 volumes) smoothing of the functional data. Afterward, the functional data were transformed into a standard stereotactic space and linear-interpolated to 3 × 3 × 3 mm (Talairach and Tournoux, 1988). A block design with two conditions (stimulus, baseline) was applied with each block lasting 21 s in which seven images were acquired. Each stimulation protocol served to obtain appropriate reference functions reflecting experimental and baseline conditions (stimulus = 1, baseline condition = 0). The stimulation protocols were convoluted with a canonical hemodynamic response function. The reference functions served as independent predictors for a general linear model (GLM). As implemented in the BrainVoyager software package, a z-transformation of the functional volume time courses for each subject was applied to take account for different baseline signal levels. Group analysis was performed resulting in T-statistical activation maps for the conditions (1) normal pin-prick pain and placebo, (2) pin-prick hyperalgesia and placebo, (3) normal pin-prick pain and lidocaine, and (4) pin-prick hyperalgesia and lidocaine. Analysis of the baseline blood oxygenation level-dependent (BOLD) signal was performed by introduction of an additional predictor into the GLM, which allowed direct comparison of the baseline signal on the whole brain level. The 2 × 2 factorial design allowed us the separation of (1) brain areas with a main effect of sensitization, (2) brain areas with a main effect of pharmacological treatment, and (3) brain areas with an interaction of sensitization and pharmacological treatment. For that purpose, the following contrasts were calculated: (1) effect of sensitization, averaged across both treatment groups (placebo and lidocaine), (2) effect of pharmacological treatment, averaged across both sensitization states, and (3) the interaction contrast of sensitization and pharmacological treatment. Those areas with an interaction of both factors were analyzed further to delineate the nature of the interaction by means of the parameter estimates of all four conditions. For all contrasts, corresponding p-values were corrected for multiple comparisons using Bonferroni correction over all voxels. Activation maps were thresholded at p < 0.025 (Bonferroni corrected, two-tailed) and T >5. Contrast maps were thresholded at p < 0.005 (uncorrected, two-tailed) and T >2.8 as indicated. A minimum cluster size of 300 mm3 (except for the activation map for the condition normal pin-prick pain and lidocaine, in which no cluster threshold was used to show any activation) was applied. The cluster size criterion was used as a conservative measure to minimize false positive activations (Maihöfner and Handwerker, 2005).
Statistical and correlation analysis.
Psychophysical data are presented as mean ± SEM. Statistical evaluation was performed using the STATISTICA software package. To assess statistically significant differences between pain thresholds the Wilcoxon matched pairs test was used. p values <0.05 were considered to be statistically significant. Correlation analysis of individual psychophysical data with individual BOLD responses was conducted using Pearson product-moment correlation coefficient. p values <0.05 were considered to be statistically significant.
Results
Psychophysics
Psychophysical ratings were obtained after application of the respective stimulus series inside the scanner. The pain induced by electrical stimulation was rated 4.92 ± 0.19 (NRS 0–10) during placebo and 3.42 ± 0.19 (NRS 0–10) during lidocaine. The pain intensity of the repetitive electrical stimuli applied to maintain the hyperalgesia was significantly higher during placebo compared with lidocaine (p < 0.05). Pin-prick stimulation inside the hyperalgesic area was rated 5.83 ± 0.55 during placebo and 3.50 ± 0.48 during lidocaine. The pain intensity of the pin-prick stimuli applied inside the hyperalgesic area was significantly higher during placebo compared with lidocaine (p < 0.05). The hyperalgesic area was 46.76 ± 9.06 cm2 during placebo and 20.31 ± 4.94 cm2 during lidocaine. The hyperalgesic area was significantly higher during placebo compared with lidocaine (p < 0.05). Therefore, the psychophysical testings clearly indicated antihyperalgesic effect of systemically applied lidocaine in the model of electrically induced hyperalgesia.
Calculation of stimulus-induced cerebral activations
Functional magnetic resonance imaging analysis revealed brain areas activated by pin-prick stimuli during the four different conditions: (1) normal pin-prick pain and placebo, (2) pin-prick hyperalgesia and placebo (3) normal pin-prick pain and lidocaine, and (4) pin-prick hyperalgesia and lidocaine. The activated brain areas with corresponding Talairach-coordinates, cluster size, T-scores and p-levels are listed in Table 1. The T-statistic contrast maps depicting these activations are shown in Figure 2A–D. During condition (1) “normal pin-prick pain and placebo” activations were found in the bilateral S2 cortex, bilateral IFC and bilateral insula. (Fig. 2A). During condition (2) “pin-prick hyperalgesia and placebo” activation was found in contralateral S1, bilateral S2, bilateral insula, bilateral ACC, bilateral medial prefrontal cortex (mPFC), bilateral dorsolateral prefrontal cortex (dlPFC), bilateral dorsal prefrontal cortex (dPFC), bilateral PA, bilateral thalamus and contralateral midbrain (Fig. 2B). During condition (3) “normal pin-prick pain and lidocaine” activation was found only in contralateral S2 (Fig. 2C). During condition (4) pin-prick hyperalgesia and lidocaine activations were observed in bilateral S2 and contralateral insula (Fig. 2D).
Table 1.
Regions of cerebral activations
| Region | Side | x | y | z | BA | T-score | p value (corr.) | Size (mm3) |
|---|---|---|---|---|---|---|---|---|
| Normal pin-prick pain and placebo | ||||||||
| S2 | Contralateral | −54 | −22 | 20 | – | 3.545 | 0.0003 | 4264 |
| S2 | Ipsilateral | 50 | −27 | 19 | – | 3.182 | 0.0014 | 1652 |
| INS | Contralateral | −41 | −26 | 19 | 13 | 3.216 | 0.0013 | 5471 |
| INS | Ipsilateral | 32 | 9 | 3 | 13 | 3.668 | 0.0002 | 537 |
| IFC/PC | Contralateral | −51 | −3 | 22 | 9/6 | 3.298 | 0.0009 | 290 |
| IFC | Ipsilateral | 52 | 2 | 24 | 9 | 3.646 | 0.0002 | 739 |
| Pin-prick hyperalgesia and placebo | ||||||||
| S2 | Contralateral | −51 | −20 | 19 | – | 5.191 | 0.0001 | 13,738 |
| S2 | Ipsilateral | 48 | −19 | 21 | – | 4.841 | 0.0001 | 15,688 |
| INS | Contralateral | −40 | 2 | 12 | 13 | 4.063 | 0.0001 | 23,878 |
| INS | Ipsilateral | 38 | 5 | 11 | 13 | 4.099 | 0.0001 | 14,684 |
| ACC | Contralateral | −8 | 22 | 29 | 24/32 | 4.785 | 0.0001 | 11,427 |
| ACC | Ipsilateral | 4 | 18 | 31 | 24/32 | 5.345 | 0.0001 | 13,872 |
| MPFC | Contralateral | −11 | 33 | 40 | 8 | 3.875 | 0.0001 | 820 |
| MPFC | Ipsilateral | 6 | 31 | 40 | 8 | 4.258 | 0.0001 | 755 |
| DLPFC | Contralateral | −23 | 29 | 36 | 9 | 4.650 | 0.0001 | 2244 |
| DLPFC | Ipsilateral | 40 | 24 | 27 | 9 | 4.646 | 0.0001 | 685 |
| DPFC | Contralateral | −20 | −15 | 51 | 6 | 5.098 | 0.0001 | 6538 |
| DPFC | Ipsilateral | 33 | −4 | 37 | 6 | 4.938 | 0.0001 | 10,962 |
| PA/S1 | Contralateral | −26 | −58 | 41 | 7 | 4.520 | 0.0001 | 5942 |
| IPL | Ipsilateral | 34 | −45 | 42 | 40 | 3.183 | 0.0015 | 159 |
| TH | Contralateral | −9 | −13 | 6 | – | 4.824 | 0.0001 | 6689 |
| TH | Ipsilateral | 10 | −11 | 6 | – | 4.288 | 0.0001 | 2937 |
| MB | Contralateral | −9 | −18 | −3 | – | 4.449 | 0.0001 | 473 |
| PCC | Contralateral | −6 | −42 | 17 | 29/30 | 4.635 | 0.0001 | 3108 |
| PCC | Ipsilateral | 7 | −44 | 19 | 29/30 | 5.720 | 0.0001 | 5963 |
| Normal pin-prick pain and lidocaine | ||||||||
| S2* | Contralateral | −49 | −27 | 20 | – | 1.281 | 0.2003 | 24 |
| M1 | Ipsilateral | 14 | −33 | 61 | 4 | −4.701 | 0.0001 | 883 |
| Pin-prick hyperalgesia and lidocaine | ||||||||
| S2 | Contralateral | −47 | −29 | 21 | – | 2.516 | 0.0118 | 6139 |
| S2* | Ipsilateral | 46 | −28 | 24 | – | 1.949 | 0.0513 | 449 |
| INS | Contralateral | −40 | −6 | 13 | 13 | 2.231 | 0.02567 | 459 |
| Main effect of sensitization across both pharmacological treatments | ||||||||
| Areas more activated during hyperalgesia | ||||||||
| S2 | Contralateral | −47 | −27 | 28 | – | 2.650 | 0.0081 | 6416 |
| S2 | Ipsilateral | 47 | −24 | 32 | – | 2.364 | 0.0181 | 5992 |
| pINS | Contralateral | −41 | −3 | 6 | 13 | 2.245 | 0.0247 | 14,653 |
| pINS* | Ipsilateral | 41 | −7 | 8 | 13 | 1.807 | 0.0704 | 346 |
| aINS | Contralateral | −29 | 24 | 11 | 13 | 2.403 | 0.0163 | 3427 |
| aINS | Ipsilateral | 31 | 18 | 8 | 13 | 2.037 | 0.0417 | 2129 |
| ACC | Contralateral | −5 | 34 | 28 | 32 | 2.692 | 0.0071 | 938 |
| ACC | Ipsilateral | 6 | 33 | 26 | 32 | 2.867 | 0.0041 | 3679 |
| VMPFC | Contralateral | −6 | 37 | 30 | 9 | 2.497 | 0.0126 | 803 |
| VMPFC | Ipsilateral | 8 | 32 | 45 | 8/9 | 2.753 | 0.0059 | 3902 |
| DMPFC | Contralateral | −5 | 36 | 47 | 8/9 | 2.662 | 0.0078 | 879 |
| DLPFC | Contralateral | −33 | 17 | 31 | 9 | 2.868 | 0.0041 | 3308 |
| DLPFC | Ipsilateral | 34 | 12 | 33 | 9 | 3.289 | 0.0010 | 4440 |
| DPFC | Contralateral | −17 | −11 | 52 | 6 | 3.176 | 0.0015 | 8697 |
| DPFC | Ipsilateral | 15 | −24 | 48 | 6 | 2.640 | 0.0083 | 1522 |
| PA | Contralateral | −26 | −60 | 42 | 7 | 3.518 | 0.0004 | 17,956 |
| PA | Ipsilateral | 29 | −57 | 44 | 7 | 2.215 | 0.0268 | 2730 |
| TH | Contralateral | −11 | −13 | 5 | – | 2.415 | 0.0157 | 2579 |
| TH | Ipsilateral | 8 | −11 | 5 | – | 2.212 | 0.0269 | 1135 |
| MB | Contralateral | −11 | −16 | −2 | – | 2.053 | 0.0401 | 438 |
| PCC | Ipsilateral | 1 | −41 | 26 | 31 | 2.832 | 0.0046 | 703 |
| Areas more activated during normal pain | ||||||||
| None | ||||||||
| Main effect of pharmacological treatment averaged across both sensitization states | ||||||||
| Areas more activated during placebo | ||||||||
| S2 | Contralateral | −52 | −19 | 26 | – | 3.978 | 0.0001 | 3207 |
| S2 | Ipsilateral | 50 | −16 | 19 | – | 3.672 | 0.0002 | 4521 |
| aINS | Contralateral | −42 | 11 | 3 | 13 | 3.603 | 0.0003 | 11,963 |
| aINS | Ipsilateral | 37 | 4 | 5 | 13 | 3.763 | 0.0001 | 8711 |
| pINS | Ipsilateral | 40 | −22 | 14 | 13 | 3.648 | 0.0002 | 7026 |
| ACC | Contralateral | −4 | 18 | 29 | 24/32 | 2.129 | 0.0332 | 2799 |
| ACC | Ipsilateral | 5 | 17 | 33 | 24/32 | 2.853 | 0.0043 | 4194 |
| MPFC | Contralateral | −13 | 45 | 35 | 8 | 2.655 | 0.0079 | 1696 |
| MPFC | Ipsilateral | 10 | 25 | 45 | 8 | 3.144 | 0.0016 | 676 |
| DLPFC | Contralateral | −22 | 38 | 35 | 9 | 3.666 | 0.0002 | 11,814 |
| DPFC | Contralateral | −42 | −1 | 34 | 6 | 3.464 | 0.0005 | 12,747 |
| DPFC | Ipsilateral | 46 | −10 | 35 | 6 | 2.862 | 0.0042 | 6076 |
| PA | Contralateral | −25 | −58 | 46 | 7 | 2.772 | 0.0055 | 3786 |
| PA | Ipsilateral | 22 | −58 | 54 | 7 | 2.626 | 0.0086 | 447 |
| TH | Ipsilateral | 13 | −12 | 0 | – | 3.032 | 0.0024 | 369 |
| MB | Contralateral | −10 | −18 | −7 | – | 2.840 | 0.0045 | 113 |
| MB | Ipsilateral | 16 | −15 | −4 | – | 3.155 | 0.0016 | 393 |
| Areas more activated during lidocaine | ||||||||
| None | ||||||||
| Interaction of sensitization and pharmacological treatment | ||||||||
| VMPFC/ACC | Contralateral | −5 | 46 | 3 | 10/32 | 2.843 | 0.0045 | 2309 |
| VMPFC/ACC | Ipsilateral | 16 | 48 | 16 | 10/32 | 2.968 | 0.0030 | 2239 |
| DMPFC | Ipsilateral | 2 | 57 | 31 | 9 | 2.317 | 0.0205 | 927 |
| PCC | Contralateral | −4 | −49 | 17 | 29/30 | 2.914 | 0.0036 | 3074 |
| PCC | Ipsilateral | 2 | −49 | 18 | 29/30 | 2.978 | 0.0029 | 3171 |
| TH | Contralateral | −4 | −48 | 8 | – | 2.313 | 0.0208 | 206 |
| BG | Ipsilateral | 15 | 20 | 5 | – | 2.535 | 0.0113 | 387 |
*These clusters are not significant (p > 0.05) after Bonferroni correction (corr.). INS, Insula; IPL, inferior parietal lobulus; TH, thalamus; MB, midbrain.
Figure 2.
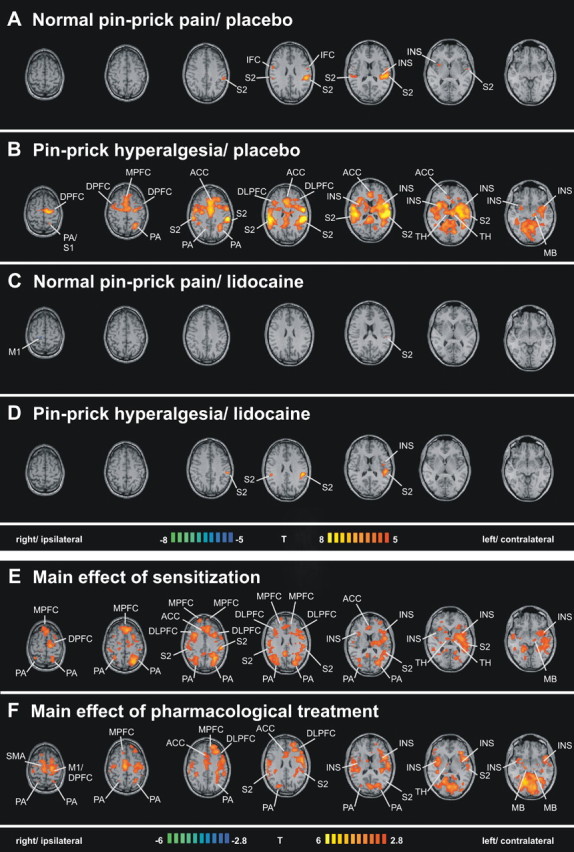
A–D, Regions of cerebral activations related to the tested conditions: normal pin-prick pain/placebo (A), pin-prick hyperalgesia/placebo (B), normal pin-prick pain/lidocaine (C), and pin-prick hyperalgesia/lidocaine (D). Group activations were registered onto a Talairach-transformed brain in axial view, thresholded at T >5, p < 0.025, corrected for multiple comparisons. E, F, Main effects of the factors sensitization and pharmacological treatment. The T-statistic contrast maps show the main effect of sensitization, averaged across both pharmacological treatment groups (E), and the main effect of pharmacological treatment, averaged across both sensitization states (F). Areas that are coded red/yellow in E showed a significantly greater response during the sensitized state compared with the nonsensitized state. Areas that are coded in red/yellow in F showed a significantly greater response to pin-prick stimuli during the placebo treatment compared with the lidocaine treatment. The group statistic contrast maps are registered onto a Talairach-transformed brain, thresholded at T >2.8, p < 0.005 uncorrected for multiple comparisons. The Talairach-coordinates, T-scores, Bonferroni-corrected p values and cluster sizes are depicted in Table 1. INS, Insula; M1, primary motor cortex; TH, thalamus; MB, midbrain.
Calculation of factor main effects
Factorial analysis of the fMRI data separated (1) brain areas with a main effect of sensitization, and (2) brain areas with a main effect of pharmacological treatment. A (1) main effect of sensitization, averaged across both treatment groups (placebo and lidocaine), was found in bilateral S2, bilateral insula, bilateral ACC, bilateral mPFC, bilateral dlPFC, bilateral dPFC, bilateral PA, bilateral thalamus and contralateral midbrain (Fig. 2E). A (2) main effect of pharmacological treatment, averaged across both sensitization states, was found in bilateral S2, bilateral insula, bilateral ACC, bilateral mPFC, bilateral dlPFC, contralateral dPFC, contralateral primary motor cortex, bilateral PA, contralateral thalamus, and bilateral midbrain (Fig. 2F).
Calculation of factor interactions
The interaction contrast of the factorial fMRI analysis revealed those brain areas with an interaction of the factors sensitization to pin-prick pain and pharmacological treatment with lidocaine. Interaction of pharmacological modulation and sensitization with an increased activity for hyperalgesia only during placebo was found in bilateral mPFC, the posterior cingulate gyrus and the thalamus. Those areas with significant interaction of both factors and correlation to psychophysical parameters are shown in Figure 3.
Figure 3.

A–C, Interaction contrast. The T-statistic contrast map shows interaction of the factors “sensitization” and “pharmacological treatment” in the subregions of the mPFC: A, left vmPFC; B, right vmPFC; and C, right dmPFC. The group statistic contrast maps are registered onto a Talairach-transformed brain, thresholded at T > 2.8, p < 0.005 uncorrected for multiple comparisons. The Talairach-coordinates and cluster sizes are depicted in Table 1. The mean parameter estimates of the four conditions reveal the nature of sensitization–treatment interaction: only pin-prick stimuli on sensitized skin lead to increased activation in those mPFC areas. Correlation analysis shows that the activity in the bilateral vmPFC correlated significantly inverse with the difference in the hyperalgesic area between placebo and lidocaine. The activity in the right dmPFC correlated significantly inverse with (1) the difference in hyperalgesic area between placebo and lidocaine, (2) the hyperalgesic area during placebo, (3) the weighted hyperalgesic area during placebo, and (4) the difference in weighted hyperalgesic area between placebo and lidocaine. Moreover, the difference in dmPFC BOLD response between placebo and lidocaine correlated significantly inverse with (1) the weighted hyperalgesic area during placebo and (2) the difference of weighted hyperalgesic area between placebo and lidocaine.
Analysis of baseline MR signal
Analysis of the baseline BOLD signal was performed by introduction of an additional predictor into the GLM, which allowed comparison of the baseline signal during placebo and lidocaine condition. No differences between the baselines in both conditions were found.
Correlation of cerebral activations with psychophysical data
We tested those areas with significant interaction of the factors pharmacological modulation and sensitization for correlation with individual psychophysical parameters. Only the activity of three brain regions was significantly correlated to psychophysical parameters. All three clusters were located in the mPFC [left ventromedial PFC (vmPFC), right vmPFC and right dorsomedial PFC (dmPFC)] during hyperalgesia and placebo. The activity in the left vmPFC correlated significantly negative with the difference in hyperalgesic area between placebo and lidocaine (r = −0.62, p = 0.030). Also, the activity in the right vmPFC correlated significantly negative with the difference in hyperalgesic area between placebo and lidocaine (r = −0.63, p = 0.027). The activity in the right dmPFC correlated significantly negative with (1) the difference in hyperalgesic area between placebo and lidocaine (r = −0.74, p = 0.005), (2) hyperalgesic area during placebo (r = −0.63, p = 0.029), (3) the weighted hyperalgesic area during placebo (r = −0.58, p = 0.048), and (4) the difference in weighted hyperalgesic area between placebo and lidocaine (r = −0.73, p = 0.006). Moreover, the difference in dmPFC activity between hyperalgesia placebo and hyperalgesia lidocaine correlated significantly negative with (1) the weighted hyperalgesic area during placebo (r = −0.60, p = 0.037), and (2) the difference of weighted hyperalgesic area between placebo and lidocaine (r = −0.65, p = 0.021). Thus, individual mPFC activity is negatively correlated with the individual extent of hyperalgesia and predictive for the individual treatment effect. Furthermore, a highly significant correlation was found for the hyperalgesic area during placebo with the decrease of hyperalgesic area resulting from lidocaine treatment (r = 0.87, p = 0.0001) and for the weighted hyperalgesic area during placebo with the decrease of weighted hyperalgesic area resulting from lidocaine treatment (r = 0.9, p = 0.0001). However, there was no significant correlation of the hyperalgesic area during lidocaine with the decrease of hyperalgesic area resulting from lidocaine treatment (r = 0.41, p = 0.18) and of the weighted hyperalgesic area during lidocaine with the decrease of weighted hyperalgesic area resulting from lidocaine treatment (r = 0.14, p = 0.66).
Discussion
In the present placebo-controlled, double-blinded and randomized cross-over fMRI study we investigated the effects of systemic sodium channel blockade by lidocaine on the cerebral processing of pin-prick hyperalgesia and normal pin-prick pain. We showed that lidocaine reduced the activity in the medial prefrontal cortex only during central sensitization. Moreover, we demonstrated that the activity in the medial prefrontal cortex during central hyperalgesia correlates inversely with the individual extent of central hyperalgesia and is predictive for the individual pharmacological treatment response to systemic sodium channel blockade by lidocaine.
Electrically induced hyperalgesia and systemic low-dose lidocaine
The high current density electrical paradigm has been previously shown to provoke stable areas of secondary hyperalgesia to punctate stimuli (Koppert et al., 2001) caused by activation of primarily mechano-insensitive C-nociceptors (Schmelz et al., 2000). This class of nociceptors was shown to be preferentially activated at high current densities as used in this model (Weidner et al., 1999). This facilitatory mechanism is paralleled by an acute activation of an endogenous naloxone-sensitive inhibitory system reducing pain and hyperalgesia and a naloxone-insensitive inhibitory system that mainly acts antihyperalgesic (Koppert et al., 2001; Koppert et al., 2005). Thus, the model is adequate to assess the cerebral processing of pin-prick hyperalgesia and pharmacological antihyperalgesia in the presence of both, activated facilitatory and inhibitory systems. In human, there is evidence that systemic lidocaine induces analgesia as a result of peripheral and central effects, whereas reduction of experimentally induced secondary hyperalgesia (the antihyperalgesic effect) is the result of an isolated central mode of action (Koppert et al., 2000).
Large-scale brain effects of hyperalgesia and lidocaine
The factorial analysis of our fMRI data revealed large-scale brain effects of both factors sensitization and lidocaine treatment. Sensitization resulted in significantly increased activity in the classical pain processing brain regions (bilateral ACC, PFC, insula, S2, Thalamus, but not S1) and a recruitment of additional brain areas (bilateral subareas of the PFC, bilateral PA, contralateral midbrain, PCC). The differences are not an effect of different BOLD MR signal baseline levels. These findings corroborate the findings of previous studies of our group (Maihöfner et al., 2005; Seifert et al., 2008) and others (Zambreanu et al., 2005) showing increased activity in these areas during mechanical hyperalgesia.
Lidocaine treatment led to activity decrease in all pin-prick pain activated areas (bilateral ACC, PFC, insula, S2, PA, Thalamus, midbrain). A recent study by Iannetti et al. (2005) investigated the effects of gabapentin on capsaicin-induced secondary hyperalgesia. The authors found that gabapentin reduced the activations in the bilateral insula, independently of the presence of central sensitization. Furthermore, gabapentin reduced the activation in the brainstem and suppressed stimulus-induced deactivations, both only during central sensitization. The suppression of stimulus-induced brain deactivations was more robust than the effect on brain activation. In our study, using electrically induced secondary hyperalgesia, we did not observe such stimulus-induced deactivations as described in the study of Iannetti et al. (2005). This could be because of the different surrogate models used. In agreement with our results there are other functional imaging studies that did not report stimulus induced deactivations during different kinds of experimentally induced hyperalgesia (Maihöfner and Handwerker, 2005; Zambreanu et al., 2005; Seifert et al., 2008). When further comparing our results to those of Iannetti et al. (2005), there is a more large-scale decrease of activity in pain activated areas during lidocaine. Moreover, we found sensitization–treatment interactions, i.e., decreases of activity during lidocaine only in the presence of hyperalgesia, in the mPFC, whereas Iannetti et al. (2005) found such interactions in the brainstem. It can be concluded that central mechanisms of antihyperalgesia induced by lidocaine differ from those induced by gabapentin.
Medial prefrontal cortex predicts pharmacological antihyperalgesia
To unravel those brain areas selectively involved in hyperalgesia as well as pharmacological antihyperalgesia we first identified areas with an interaction of the factors sensitization and pharmacological modulation. We found this interaction in the bilateral mPFC, the PCC and the thalamus. In these regions significantly increased activity was present only in the condition placebo treatment and hyperalgesia. We then tested individual BOLD responses in these regions for correlation with individual psychophysical parameters. We found that only mPFC activity during placebo was correlated significantly to the degree of secondary hyperalgesia (dmPFC) and also to the antihyperalgesic treatment effect of lidocaine (vmPFC and dmPFC). During lidocaine this correlation was abolished. For the interpretation of these results it is important that there was a highly significant correlation of the hyperalgesic area during placebo with the antihyperalgesic treatment effect. Thus, the highest antihyperalgesic treatment effect was present in those subjects with the highest hyperalgesic area during placebo. In turn, there was no significant correlation of the hyperalgesic area during lidocaine with the antihyperalgesic treatment effect. This reveals that it is not the magnitude of the individual antihyperalgesic treatment effect that determines the hyperalgesic area during lidocaine, but that it is the degree of hyperalgesia during placebo that determines the individual antihyperalgesic treatment effect of lidocaine. With regard to the mPFC activity this means that subjects with a high mPFC activity during placebo had a small hyperalgesic area and a low antihyperalgesic lidocaine effect, whereas those subjects with low mPFC activity during placebo had a great hyperalgesic area but also displayed a great antihyperalgesic effect to lidocaine. What does this mean to the function of the mPFC during hyperalgesia? In our study this area is selectively activated during hyperalgesia, however, the higher the activation, the smaller the hyperalgesic area. This hints at a role of the mPFC in endogenous hyperalgesia-specific pain modulatory networks. That is, individuals with a high capacity of endogenous descending hyperalgesia modulation (i.e., high mPFC activity), exhibit small areas of hyperalgesia, whereas those with a low capacity of endogenous descending hyperalgesia modulation (i.e., low mPFC activity) exhibit larger areas. If lidocaine is applied, nociceptive input is reduced as shown by our psychophysical data, and consecutively the activity in the mPFC reduces. Interestingly, the antihyperalgesic potential of lidocaine seems to be greater in those subjects with a low capacity of mPFC-originating descending pain modulation. These effects seem to be hyperalgesia-specific because of a lack of correlation of normal pin-prick pain activity in mPFC with the degree of hyperalgesia. Accordingly, it was shown previously that the PFC exerts active control on pain perception by modulating corticosubcortical and corticocortical pathways (Lorenz et al., 2003). However, in the study by Lorenz et al. (2003), the dorsolateral part of the PFC was found to have a key role in cortical mechanisms of pain modulation. It may be speculated that the different location of the pain modulatory areas within the PFC in our study is the result of a different stimulus modality (heat pain versus pin-prick pain) and a different hyperalgesia model used (capsaicin versus electrically induced hyperalgesia, thus, primary versus secondary hyperalgesia). Moreover, there is evidence from the literature that the orbitofrontal, medial prefrontal and cingulofrontal cortex exhibits a top-down pain modulation during cognitive interference of nociceptive input (Petrovic et al., 2002; Wager et al., 2004; Bingel et al., 2007; Tracey and Mantyh, 2007). This was demonstrated for placebo cognition (Bingel et al., 2006) and distraction from pain (Bantick et al., 2002; Valet et al., 2004). The pain modulatory content seems to be mediated via brainstem nuclei like the PAG (Valet et al., 2004) that forms together with the RVM the backbone of a descending pain modulatory system projecting on spinal dorsal horn neurons (Mason, 2005). But also in the absence of cognitive interferences the mPFC seems to be relevant for pain modulation. In a study with graded heat stimuli mPFC activity has been shown to decrease during pain intensity increase (Derbyshire et al., 1997). In context with the literature a key role of the mPFC in pain-modulatory processes is suggested, and this function seems to be disrupted or superseded by systemic sodium channel blockade.
Conclusion
We show that systemically applied low-dose lidocaine reduces the activity in the medial prefrontal cortex only during central sensitization. We further show that the activity in the medial prefrontal cortex during central hyperalgesia correlates inversely with the individual extent of central hyperalgesia and is predictive for the individual pharmacological treatment response to systemic sodium channel blockade by lidocaine. We hypothesize that this brain area has a key role in pain-modulatory processes that were affected by systemic sodium channel blockade.
Footnotes
This work was supported by the “German Research Network on Neuropathic Pain” [German Federal Ministry of Education and Research (BMBF)] and the German Research Foundation (KFO130).
The authors declare no competing financial interests.
References
- Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain. 2005;9:463–484. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Attal N, Gaudé V, Brasseur L, Dupuy M, Guirimand F, Parker F, Bouhassira D. Intravenous lidocaine in central pain: a double-blind, placebo-controlled, psychophysical study. Neurology. 2000;54:564–574. doi: 10.1212/wnl.54.3.564. [DOI] [PubMed] [Google Scholar]
- Bantick SJ, Wise RG, Ploghaus A, Clare S, Smith SM, Tracey I. Imaging how attention modulates pain in humans using functional MRI. Brain. 2002;125:310–319. doi: 10.1093/brain/awf022. [DOI] [PubMed] [Google Scholar]
- Bingel U, Lorenz J, Schoell E, Weiller C, Büchel C. Mechanisms of placebo analgesia: rACC recruitment of a subcortical antinociceptive network. Pain. 2006;120:8–15. doi: 10.1016/j.pain.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Bingel U, Schoell E, Büchel C. Imaging pain modulation in health and disease. Curr Opin Neurol. 2007;20:424–431. doi: 10.1097/WCO.0b013e328259c34d. [DOI] [PubMed] [Google Scholar]
- Borsook D, Moulton EA, Schmidt KF, Becerra LR. Neuroimaging revolutionizes therapeutic approaches to chronic pain. Mol Pain. 2007;3:25. doi: 10.1186/1744-8069-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challapalli V, Tremont-Lukats IW, McNicol ED, Lau J, Carr DB. Systemic administration of local anesthetic agents to relieve neuropathic pain. Cochrane Database Syst Rev. 2005:CD003345. doi: 10.1002/14651858.CD003345.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire SW, Jones AK, Gyulai F, Clark S, Townsend D, Firestone LL. Pain processing during three levels of noxious stimulation produces differential patterns of central activity. Pain. 1997;73:431–445. doi: 10.1016/S0304-3959(97)00138-3. [DOI] [PubMed] [Google Scholar]
- Filitz J, Ihmsen H, Gunther W, Troster A, Schwilden H, Schuttler J, Koppert W. Supra-additive effects of tramadol and acetaminophen in a human pain model. Pain. 2008;136:267–270. doi: 10.1016/j.pain.2007.06.036. [DOI] [PubMed] [Google Scholar]
- Iannetti GD, Zambreanu L, Wise RG, Buchanan TJ, Huggins JP, Smart TS, Vennart W, Tracey I. Pharmacological modulation of pain-related brain activity during normal and central sensitization states in humans. Proc Natl Acad Sci U S A. 2005;102:18195–18200. doi: 10.1073/pnas.0506624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppert W, Ostermeier N, Sittl R, Weidner C, Schmelz M. Low-dose lidocaine reduces secondary hyperalgesia by a central mode of action. Pain. 2000;85:217–224. doi: 10.1016/s0304-3959(99)00268-7. [DOI] [PubMed] [Google Scholar]
- Koppert W, Dern SK, Sittl R, Albrecht S, Schüttler J, Schmelz M. A new model of electrically evoked pain and hyperalgesia in human skin: the effects of intravenous alfentanil, S(+)-ketamine, and lidocaine. Anesthesiology. 2001;95:395–402. doi: 10.1097/00000542-200108000-00022. [DOI] [PubMed] [Google Scholar]
- Koppert W, Filitz J, Tröster A, Ihmsen H, Angst M, Flor H, Schüttler J, Schmelz M. Activation of naloxone-sensitive and -insensitive inhibitory systems in a human pain model. J Pain. 2005;6:757–764. doi: 10.1016/j.jpain.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Lorenz J, Minoshima S, Casey KL. Keeping pain out of mind: the role of the dorsolateral prefrontal cortex in pain modulation. Brain. 2003;126:1079–1091. doi: 10.1093/brain/awg102. [DOI] [PubMed] [Google Scholar]
- Maihöfner C, Handwerker HO. Differential coding of hyperalgesia in the human brain: a functional MRI study. Neuroimage. 2005;28:996–1006. doi: 10.1016/j.neuroimage.2005.06.049. [DOI] [PubMed] [Google Scholar]
- Maihöfner C, Forster C, Birklein F, Neundörfer B, Handwerker HO. Brain processing during mechanical hyperalgesia in complex regional pain syndrome: a functional MRI study. Pain. 2005;114:93–103. doi: 10.1016/j.pain.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Maihöfner C, Ringler R, Herrndobler F, Koppert W. Brain imaging of analgesic and antihyperalgesic effects of cyclooxygenase inhibition in an experimental human pain model: a functional MRI study. Eur J Neurosci. 2007;26:1344–1356. doi: 10.1111/j.1460-9568.2007.05733.x. [DOI] [PubMed] [Google Scholar]
- Mason P. Ventromedial medulla: pain modulation and beyond. J Comp Neurol. 2005;493:2–8. doi: 10.1002/cne.20751. [DOI] [PubMed] [Google Scholar]
- Moisset X, Bouhassira D. Brain imaging of neuropathic pain. Neuroimage 37 Suppl. 2007;1:S80–S88. doi: 10.1016/j.neuroimage.2007.03.054. [DOI] [PubMed] [Google Scholar]
- Petrovic P, Kalso E, Petersson KM, Ingvar M. Placebo and opioid analgesia—imaging a shared neuronal network. Science. 2002;295:1737–1740. doi: 10.1126/science.1067176. [DOI] [PubMed] [Google Scholar]
- Rowland M, Thomson PD, Guichard A, Melmon KL. Disposition kinetics of lidocaine in normal subjects. Ann N Y Acad Sci. 1971;179:383–398. doi: 10.1111/j.1749-6632.1971.tb46915.x. [DOI] [PubMed] [Google Scholar]
- Schmelz M, Schmid R, Handwerker HO, Torebjörk HE. Encoding of burning pain from capsaicin-treated human skin in two categories of unmyelinated nerve fibres. Brain. 2000;123:560–571. doi: 10.1093/brain/123.3.560. [DOI] [PubMed] [Google Scholar]
- Seifert F, Maihöfner C. Central mechanisms of experimental and chronic neuropathic pain: findings from functional imaging studies. Cell Mol Life Sci. 2009;66:375–390. doi: 10.1007/s00018-008-8428-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert F, Jungfer I, Schmelz M, Maihöfner C. Representation of UV-B-induced thermal and mechanical hyperalgesia in the human brain: a functional MRI study. Hum Brain Mapp. 2008;29:1327–1342. doi: 10.1002/hbm.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talairach J, Tournoux P. New York: Thieme Medical; 1988. Co-planar stereotaxic atlas of the human brain; pp. 1–122. [Google Scholar]
- Tracey I, Mantyh PW. The cerebral signature for pain perception and its modulation. Neuron. 2007;55:377–391. doi: 10.1016/j.neuron.2007.07.012. [DOI] [PubMed] [Google Scholar]
- Tremont-Lukats IW, Challapalli V, McNicol ED, Lau J, Carr DB. Systemic administration of local anesthetics to relieve neuropathic pain: a systematic review and meta-analysis. Anesth Analg. 2005;101:1738–1749. doi: 10.1213/01.ANE.0000186348.86792.38. [DOI] [PubMed] [Google Scholar]
- Valet M, Sprenger T, Boecker H, Willoch F, Rummeny E, Conrad B, Erhard P, Tolle TR. Distraction modulates connectivity of the cingulo-frontal cortex and the midbrain during pain—an fMRI analysis. Pain. 2004;109:399–408. doi: 10.1016/j.pain.2004.02.033. [DOI] [PubMed] [Google Scholar]
- Wager TD, Rilling JK, Smith EE, Sokolik A, Casey KL, Davidson RJ, Kosslyn SM, Rose RM, Cohen JD. Placebo-induced changes in FMRI in the anticipation and experience of pain. Science. 2004;303:1162–1167. doi: 10.1126/science.1093065. [DOI] [PubMed] [Google Scholar]
- Weidner C, Schmelz M, Schmidt R, Hansson B, Handwerker HO, Torebjörk HE. Functional attributes discriminating mechano-insensitive and mechano-responsive C nociceptors in human skin. J Neurosci. 1999;19:10184–10190. doi: 10.1523/JNEUROSCI.19-22-10184.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353:1959–1964. doi: 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Wiesenfeld-Hallin Z. The systemic administration of local anaesthetics produces a selective depression of C-afferent fibre evoked activity in the spinal cord. Pain. 1985;23:361–374. doi: 10.1016/0304-3959(85)90006-5. [DOI] [PubMed] [Google Scholar]
- Zambreanu L, Wise RG, Brooks JC, Iannetti GD, Tracey I. A role for the brainstem in central sensitisation in humans. Evidence from functional magnetic resonance imaging. Pain. 2005;114:397–407. doi: 10.1016/j.pain.2005.01.005. [DOI] [PubMed] [Google Scholar]